

Abstract
Epidermal growth factor receptor (EGFR) targeted nanoparticle are developed by conjugating a single-chain anti-EGFR antibody (ScFvEGFR) to surface functionalized quantum dots (QDs) or magnetic iron oxide (IO) nanoparticles. The results show that ScFvEGFR can be successfully conjugated to the nanoparticles, resulting in compact ScFvEGFR nanoparticles that specifically bind to and are internalized by EGFR-expressing cancer cells, thereby producing a fluorescent signal or magnetic resonance imaging (MRI) contrast. In vivo tumor targeting and uptake of the nanoparticles in human cancer cells is demonstrated after systemic delivery of ScFvEGFR-QDs or ScFvEGFR-IO nanoparticles into an orthotopic pancreatic cancer model. Therefore, ScFvEGFR nanoparticles have potential to be used as a molecular-targeted in vivo tumor imaging agent. Efficient internalization of ScFvEGFR nanoparticles into tumor cells after systemic delivery suggests that the EGFR-targeted nanoparticles can also be used for the targeted delivery of therapeutic agents.
Keywords: antibodies, drug delivery, nanoparticles, proteins, quantum dots
1. Introduction
The epidermal growth factor receptor (EGFR) signaling pathway plays an important role in the regulation of cell proliferation, survival, and differentiation.[1,2] Upregulation of EGFR is found in many cancer types, which provides an opportunity for designing receptor-targeted approaches for cancer detection and treatment.[3,4] The difference in the level of EGFR expression between normal cells and tumor cells, coupled with the phenomenon of the cellular internalization of EGFR–antibody complexes, suggests that EGFR is a potential marker for in vivo receptor-targeted molecular imaging with excellent tumor-to-background contrast, and that EGFR is a good mediator for the targeted drug delivery.[5,6] Previous studies have examined the feasibility of conjugating imaging contrast agents or nanoparticles with EGF, the natural ligand to EGFR, and the monoclonal antibody to EGFR in vitro.[7-10] However, the use of a growth stimulating ligand to target EGFR has limitations when used in developing an application for cancer patients. Although several anti-EGFR monoclonal antibodies have been used for cancer treatment in patients, they have a relatively large size, which limits the number of ligands that can be linked to the surface of a nanoparticle and impedes intratumoral distribution due to interstitial tumor pressure. For example, an immunoglobulin G (IgG) antibody has an average size of 14.5 × 8.5 × 4 nm3 and a molecular weight of 160 kDa.[11,12] As an alternative for generating EGFR-targeted nanoparticles, a single-chain anti-EGFR antibody (ScFvEGFR) provides a much smaller targeting ligand. A single-chain Fv (scFv) fragment consists of antibody heavy- and light-chain variable domains connected with a flexible peptide linker. The resulting antibody fragment (25 to 28 kDa) is smaller than 20% of an intact antibody but maintains a high binding affinity and specificity.[6,13]
Recently, several types of nanoparticles including quantum dots (QDs), magnetic iron oxide (IO), gold, and polymer-based nanoparticles have been developed for cancer applications.[14-18] The concept of designing and synthesizing tumor-targeted or multi-functional nanoparticles for cancer imaging and therapy has been demonstrated and the results show that nanotechnology may provide new means for in vivo tumor-targeted imaging and drug delivery.[17,19,20] However, the challenges for the development of tumor-targeted nanoparticles for in vivo applications are also recognized. For instance, it is important to take into account that the nanoparticles should not only be stable enough to generate strong imaging signals, but should also have a modified surface with reactive functional groups for efficient conjugation of tumor targeting ligands and therapeutic agents. At present, fluorescence emitting quantum dots (QDs), a class of light-emitting nanoparticles, have been used for biomarker-targeted in vivo tumor imaging.[14,15] Another type of nanoparticle, magnetic iron oxide (IO) nanoparticles, is particularly attractive and feasible for molecular imaging in the clinical setting because of prior applications of magnetic resonance imaging (MRI), their biocompatibility and surface chemistry allowing for the introduction of functional biomolecules.[16,21,22] Non-targeted magnetic IO nanoparticles have been used in imaging liver tumors and lymph node metastasis in human prostate cancer patients.[23,24] Several studies have used dextran or poly(ethylene glycol) (PEG)-coated IO nanoparticles to develop targeted imaging contrast agents by attaching targeting ligands, such as antibodies against Her-2/Neu or transferrin, folate acid, or tumor targeting short peptides.[16,17,22,25-27] Results of these studies demonstrated the feasibility of using targeted nanoparticle probes for MRI of subcutaneously implanted tumors in animal models. However, several issues remain to be addressed to increase the sensitivity and specificity of the receptor-targeted tumor imaging agents for future use in cancer patients. Typical obstacles encountered for in vivo applications include heterogeneous levels of expression of the targeted receptor in human tumor cells, various physiological barriers preventing the nanoparticle from reaching the targeted cells, and lack of information on the intratumoral distribution and imaging capability of targeted nanoparticles within tumor sites that are relevant to the locations of most human primary and metastatic tumors.
In this study, we developed EGFR-targeted nanoparticles that specifically bind to and are internalized by EGFR-expressing tumor cells, which are present in a high percentage of the epithelial tumor types. To determine the specificity of ScFvEGFR to the tumor after conjugation to nanoparticles, we used two nanoparticle systems: QDs for direct visualization of binding, internalization, and tissue distribution, and magnetic IO nanoparticles for in vivo MRI of the tumor. The combination of a high affinity and small size single-chain anti-EGFR antibody as a tumor targeting ligand with a compact surface modification of the nanoparticle results in an EGFR-targeted nanoparticle that is able to selectively accumulate in the tumor with a high efficiency to delineate the location and size of the orthotopically xenografted human tumors in the pancreas of the nude mice by MRI. Our results demonstrated that ScFvEGFR-IO nanoparticles can function as a receptor-targeted MRI contrast agent for in vivo detection of pancreatic cancer.
2. Results and Discussion
2.1. Development and Characterization of ScFvEGFR-Conjugated QDs
We have developed a new strategy for conjugating recombinant proteins or peptides to nanoparticles, which utilizes the interactions between histidine and nickel molecules. The addition of six histidine residues to a protein is a commonly used method for the production and purification of recombinant proteins. Nickel (II) nitrilotriacetic acid (Ni-NTA) was first conjugated to QDs using a covalent linkage of the amine group of Ni-NTA with the carboxyl group (–COOH) of the amphiphilic polymer on the surface of the QDs (Figure 1A).[14] His-tagged ScFvEGFR proteins (25 kDa) were mixed with Ni-NTA-QDs. Interaction of the His-tag with the nickel molecule of NTA leads to the stable conjugation of the ScFvEGFR single-chain antibody to the surface of QDs. This approach provides a relatively simple procedure for conjugating tumor targeting peptides, protein ligands, or engineered antibodies to nanoparticles. The dissociation constant of six His-tagged protein to Ni-NTA has been measured as 10−13 m, suggesting stronger binding than those of most antibodies.[28] This indirect “His-tag” coupling method has an advantage over direct conjugation of targeting ligands to nanoparticles through covalent links that are formed randomly between functional groups of a protein and a nanoparticle since the His-tag can be engineered in a specific region of the targeting ligand so that the specificity and binding affinity of the targeted nanoparticles can be preserved.
Figure 1.
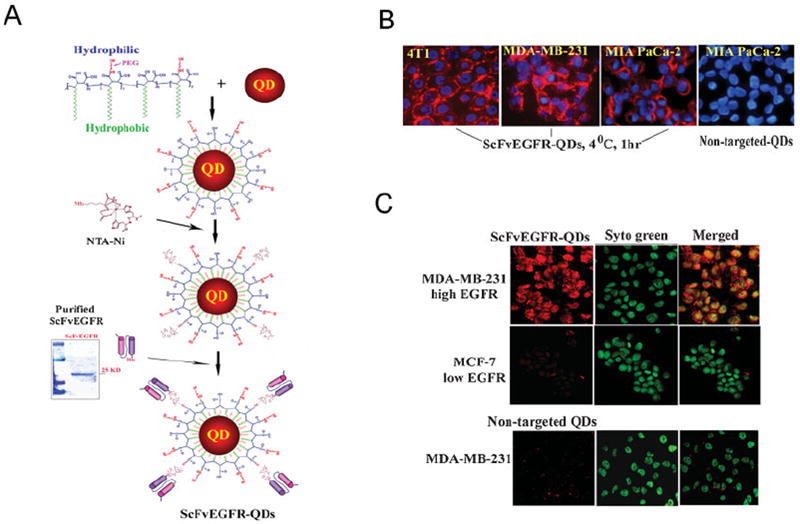
Conjugation of ScFvEGFR to QDs and examination of specificity of EGFR targeting to tumor cell lines. A) QDs were coated with the amphiphilic polymer modified with short PEG chains and conjugated to Ni-NTA. Recombinant ScFvEGFR protein has a high purity showing as a single band protein with a molecular weight around 25 kDa. His-tagged ScFvEGFR was conjugated to QDs through the interaction of nickel with histidine residues located at the C-terminal of the protein. B) Specific binding of ScFvEGFR-QDs to targeted tumor cells was determined by incubating ScFvEGFR-QDs with 4T1, MDA-MB-231, and MIA PaCa-2 tumor cell lines at 4 °C for 1 h. ScFvEGFR-QDs, but not non-targeted QDs, were found on the cell surface of all tumor types (red). Blue: Hoechst 33342 counterstaining. C) Selective internalization of ScFvEGFR-QDs in tumor cells was determined using cancer cell lines expressing a high (MDA-MB-231) or low (MCF-7) level of EGFR. Strong red fluorescent signal was detected inside MDA-MB-231 cells incubated with ScFvEGFR-QDs but not with non-targeted QDs. A very low level of red fluorescence was seen in MCF-7 cells after incubating with ScFvEGFR-QDs. Cell nuclei were counterstained with Sytogreen (Invitrogen).
To determine the specificity of ScFvEGFR nanoparticles, we tested the binding of such nanoprobes to EGFR-expressing cancer cell lines, including breast cancer 4T1 and MDA-MB-231 cells and pancreatic cancer MIA PaCa-2 cells. We found that ScFvEGFR-QDs selectively bound to the cell surface when incubated at 4 °C, whereas the non-targeted QDs did not bind to the cells (Figure 1B). Using confocal microscopy, we further observed the uptake of ScFvEGFR-QDs into MDA-MB-231 cells, which overexpress EGFR, but not in the MCF-7 cells, which express a low level of EGFR after incubation at 37 °C (Figure 1C).[29] Positive reactivity of ScFvEGFR-conjugated nanoparticles to both human and mouse cancer cells is important for adequately evaluating target specificity and biodistribution of the EGFR-targeted nanoparticles in human tumor xenograft models in nude mice.
2.2. ScFvEGFR-QDs Targeting Pancreatic Tumors in an Orthotopic Human Pancreatic Cancer Xenograft Model
To determine if the systemic delivery of ScFvEGFR nanoparticles leads to the target specific accumulation of the nanoparticles into the EGFR-expressing tumors in vivo, we injected the EGFR-targeted QDs through the tail vein into the mice bearing orthotopic pancreatic cancer from an EGFR positive human pancreatic cancer cell line, MIA PaCa-2. We found large numbers of QD-bound tumor cells in the tumor areas after examination of frozen tissue sections (Figure 2A). High magnification of the tissue sections revealed that QDs resided in the cytoplasm of the tumor cells, suggesting uptake of ScFvEGFR-QDs by the cells in the tumor mass possibly via receptor mediated internalization. The results of confocal microscopy examination confirmed the cytoplasmic localization of the targeted QDs as small scattered fluorescent clusters (Figure 2A). In contrast, frozen tissue sections of the pancreatic tumors obtained from the mice that received non-targeted QDs did not have detectable QDs (Figure 2A).
Figure 2.

In vivo tumor targeting and biodistribution of ScFvEGFR-QD. ScFvEGFR-QDs or non-targeted QDs were injected through the tail vein into nude mice bearing intra-pancreatic human xenograft tumor. The mice were sacrificed 5 h after the injection. Frozen tissue sections were counterstained with Hoechst 33342 (blue) and examined under a fluorescence microscope. A) Strong QD fluorescent signal (red) was detected throughout the tumor tissue sections of pancreatic cancer xenograft obtained from the mouse that received ScFvEGFR-QDs but not non-targeted QDs. Under a higher magnification (40 × lens), QDs were located in the cytoplasm of the tumor cells. Confocal microscopy confirmed internalization of the QDs into the tumor cells, showing as small fluorescent QD clusters scattered in the cytoplasmic region of the cell. B) Examination of frozen sections obtained from normal tissues of the mouse that received non-targeted QDs revealed strong QD signal in the liver and spleen and low levels of QDs in the lung and kidney (red). The liver and spleen tissue sections obtained from the mouse that received ScFvEGFR-QDs had markedly lower levels of QDs compared to non-targeted QD injected tissues.
Examination of frozen sections of normal tissues revealed strong QD signals in the livers and spleens obtained from the mice that received non-targeted QDs (Figure 2B), but markedly low signals of ScFvEGFR-QDs were detected in the above normal organs. Non-targeted QDs were found in the lungs and kidneys of the mice, but only a few scattered ScFvEGFR-QDs were found in these normal tissues (Figure 2B). On the other hand, we did not detect either non-targeted QDs or ScFvEGFR-QDs in the hearts (Figure 2B).
2.3. Engineering and Characterization of ScFvEGFR-Conjugated Magnetic IO Nanoparticles
To develop tumor-targeted nanoparticles for in vivo tumor imaging using MRI, we synthesized magnetic IO nanoparticles using an established method.[30] The amphiphilic triblock polymer was used to stabilize and functionalize the surface of the IO nanoparticles (Figure 3A). ScFvEGFR was conjugated to IO nanoparticles by crosslinking carboxyl groups to amino groups of the ScFvEGFR proteins mediated by ethyl-3-dimethyl amino propyl carbodiimide (EDAC) (Figure 3A). Transmission electron microscopy (TEM) analysis showed that the obtained IO nanoparticle has a uniform core size of 10 nm. Examination of the particle sizes using dynamic light scattering measurements (DLS) showed a particle size of (15.7 ± 2.1) nm after coating with an amphiphilic polymer layer and (25.4 ± 4.2) nm after conjugation with ScFvEGFR (Figure 3A). The thickness of the polymer layer is estimated as 2 nm or a total of 4 nm added to the diameter of the nanoparticle size. This surface coating is stable in a physiological pH buffer solution at room temperature for over 16 months.
Figure 3.

Construction of ScFvEGFR-IO nanoparticles and examination of specificity and magnetic signal strength of the IO nanoparticles in tumor cells in vitro. A) 10nm size uniformed IO nanoparticles were coated with amphiphilic copolymers modified with short PEG chains. ScFvEGFR proteins were conjugated to the IO nanoparticles mediated by EDAC. The particle sizes were determined by TEM and hydrodynamic light scattering measurement. The number is the mean of three measurements. B) Specific binding of the ScFvEGFR-IO to tumor cells was determined by incubating the viable cells with the targeted-IO nanoparticles followed by Prussian blue staining. C) MRI scan showed significant T2 signal decrease in the cells incubated with ScFvEGFR-IO but not with GFP-IO or IO nanoparticles. The first well of the upper panel of MDA-MB-231 cells shows a decrease in T2 contrast with darker MRI. The lower panel of MDA-MB-231 and result of MIA PaCa-2 cells display the level of T2 contrast decrease measured by T2 relaxometry mapping method. [41] A low T2 value correlates with a higher iron concentration (red color), indicating higher level of specific binding of ScFvEGFR-IO nanoparticles to tumor cells. D) Multi-echo T2 weighted fast spin echo imaging sequences further confirmed the fastest T2 value drop in MDA-MB-231 cells after incubation with ScFvEGFR-IO but not with control GFP-IO nanoparticles.
The efficiency of conjugating the recombinant proteins to the amphiphilic polymer of the nanoparticles was determined using a recombinant His-tagged green fluorescence protein (GFP). His-tagged GFPs (27 kDa) were conjugated to QDs or IO nanoparticles as described above. The number of proteins conjugated to each nanoparticle was determined by measuring the fluorescence intensity of a diluted sample of GFP-nanoparticles at an emission wavelength of 505 nm. Fluorescence intensity of the conjugated nanoparticles was compared with a standard curve generated from different numbers of free GFP molecules to obtain a total number of GFPs in the nanoparticle preparation. The average number of GFPs on each nanoparticle was calculated by dividing the total number of GFP molecules with the number of nanoparticles. We found that about 8 to 10 GFPs were conjugated to each nanoparticle.
2.4. Tumor Targeting Ability and MRI Contrast Effect of ScFvEGFR-IO Nanoparticles
To determine whether the resulting ScFvEGFR-IO nanoparticles retain the target specificity to EGFR-expressing cells, we incubated non-targeting IO or ScFvEGFR-IO nanoparticles with MIA PaCa-2 cancer cells or the normal human embryonic kidney cell line HEK 293, which has a low level of EGFR expression,[31] at 37 °C for 2 h. We detected a large number of iron-positive cells in the MIA PaCa-2 cells and very few positively stained cells in the HEK 293 cells using Prussian blue staining. In contrast, a few iron-positive cells were detected after incubating EGFR-expressing MIA PaCa-2 cancer cells with non-targeted IO nanoparticles (Figure 3B).
We further demonstrated that the specific internalization of magnetic IO nanoparticles into cells results in a MRI contrast change due to the fact that a significant signal drop in T1 and T2 weighted gradient echo imaging as well as T2 weighted spin echo imaging was detected in the cells incubated with ScFvEGFR-IO nanoparticles but not with non-targeted IO or GFP-IO nanoparticles (Figure 3C). The T2 relaxometry measurement confirmed a significant decrease of T2 value in MDA-MB-231 cells treated with the ScFvEGFR-IO nanoparticles compared to those treated with GFP-IO nanoparticles (Figure 3D). Since the decrease of T2 value is proportional to the increase of iron concentration, T2 relaxometry data suggests that the change of T2 weighted MRI contrast is induced by the specific binding of ScFvEGFR-IO nanoparticles to the EGFR-expressing cells.
2.5. EGFR-Targeted MRI in vivo Using ScFvEGFR-IO Nanoparticles in an Orthotopic Pancreatic Cancer Xenograft Model
Receptor targeting and MRI of human pancreatic cancer in vivo using ScFvEGFR-IO nanoparticles were investigated. The EGFR-targeted or non-targeted IO nanoparticles were administrated via the tail vein and followed by MRI at different post-contrast time points. Results of the T2 weighted fast spin echo imaging showed that the ScFvEGFR-IO nanoparticles selectively accumulated within the pancreatic tumors, as evidenced by a decrease in MRI signal in the area of the tumor. This change in the MRI signal was noted from 5 to 30 h after the tail vein injection of the targeted nanoparticles. Furthermore, tumor lesions, found by observing the MRI signal change from ScFvEGFR-IO targeting (Figure 4A), was confirmed by histological examination of the pancreatic tissue. Two tumor nodules 3 to 4 mm in diameter found within the pancreas correlated very well in sizes measured from the MRI of this tumor-bearing mouse (Figure 4A, lower right). However, a pancreatic tumor in the mouse that received non-targeted IO nanoparticles did not have detectable MRI signal changes (Figure 4B). To estimate the level of IO nanoparticles in the tumor tissues, we measured the MRI signal changes in the representative areas of the tumor in the mice that received either targeted or non-targeted IO nanoparticles. Using the MRI signal level of muscle tissue as a baseline, we found that there is a 4.8 fold change in the MRI signal within the pancreatic tumor of the mouse that received ScFvEGFR-IO nanoparticles compared to the mouse that received non-targeted IO nanoparticles (Figure 5A). To further confirm the accumulation of non-targeted or ScFvEGFR-IO nanoparticles in tumor tissues, Prussian blue staining was performed on the frozen tissue sections harvested 48 h after the injection of nanoparticles. Prussian blue stained cells were found in the tumor sections from the mice that received ScFvEGFR-IO nanoparticles but not in the tissue sections of the animals that received non-targeted IO nanoparticles (Figure 5C). Examination of the microscope images under high magnification showed the intracellular localization of the Prussian blue iron stain within the cells (Figure 5C). The uptake of targeted IO nanoparticles is selective for pancreatic tumor cells, as evidenced by the negative Prussian blue stain in the normal pancreatic ductal epithelial cells and other normal cell types in the neighboring normal pancreatic tissues (Figure 5C). Furthermore, the blue iron-stained cells were not detected in the frozen tumor sections collected from the mice that received 8 nmol kg−1 of body weight of non-targeted IO nanoparticles, which is the same amount as the targeted IO nanoparticles used in the mice. Previous studies have shown that passive tumor targeting due to leaky tumor vasculatures exhibit the enhanced permeation and retention (EPR) effect leading to nanoparticle delivery to the tumor without active targeting.[14] We further increased the amount of non-targeted IO nanoparticles to determine the effect of the IO nanoparticle dosage on the accumulation of the particles in the tumor. We found weak blue iron stain in the extracellular space of the tumor areas but not in the normal pancreatic tissue areas of the mice that received a higher dosage of the non-targeted IO nanoparticles, that is, 16nmol kg−1 of body weight of IO nanoparticles instead of 8nmol kg−1 of body weight (Figure 5C, lower right). This result supports that the ScFvEGFR-IO nanoparticles were delivered to the tumor by way of receptor specific active targeting.
Figure 4.

Examination of target specificity of ScFvEGFR-IO nanoparticles by MRI using an orthotopic human pancreatic xenograft model. A) MRI of a tumor-bearing mouse. ScFvEGFR-IO nanoparticles (8 nmol kg−1 body weight) were injected into the mouse through the tail vein. Pre- and post-contrast MRI at 5 and 30 h were collected. Upper and lower panels showed the MRI from different sectional levels of the same mouse. The areas of the pancreatic tumor were marked as a dash-lined circle (pink). The pancreatic tumor area showed a bright signal before receiving the nanoparticle. After injection of the targeted IO nanoparticles, a marked MRI contrast decrease was detected in the tumor (darker), which delineated the area of the tumor lesion. MRI contrast change is also found in the liver (green arrow) and spleen. These MRIs are representative results of five mice that received ScFvEGFR-IO nanoparticles. Lower right is the picture of tumor and spleen tissues, showing sizes and locations of two intra-pancreatic tumor lesions (arrows) that correspond with the tumor images of MRI. B) MRI of a mouse that received non-targeted IO nanoparticles. The tumor area (pink dash-lined circle) did not show MRI signal decrease at 5 and 30 h after the injection of nanoparticles. The areas representing the liver and spleen had marked signal decrease due to the T2 contrast. Shown are representative MRIs of three mice that received control IO nanoparticles.
Figure 5.

Determination of the biodistribution of ScFvEGFR-IO nanoparticles by MRI and histological analysis. Profiling of MRI signal change. MRI contrast intensity changes in the mouse that received IO or ScFvEGFR-IO nanoparticles for 5 and 28 h were measured in the regions of tumor (A) or various normal tissues (B). Relative intensity was calculated using the intensity in the leg muscle as unity. Fold decreases of the relative intensity in MRI were compared between pre- and post-ScFvEGFR-IO injection and plotted in the figure. The higher level of the relative contrast intensity change suggests that more IO nanoparticles accumulated in the tissue. The figure represents one of two pair of mice that received ScFvEGFR-IO or non-targeted IO nanoparticles and had their MRI data analyzed. The number is the mean value of three measurements of randomly selected regions in each organ. C) Examination of tissue distribution of the IO nanoparticles by Prussian blue staining. Cells with blue staining are found in tumor tissue sections obtained from a mouse that received ScFvEGFR-IO nanoparticles. Upper panel pictures a and b were taken from the same field with either a 20× or a 40× lens. Upper picture c was from a different area of the tissue section showing that not all tumor cells were positive for blue iron staining. Background of the tissue sections was stained with nuclear fast red. The epithelial cells lining a normal pancreatic duct in nearby normal pancreatic tissues were negative for Prussian blue staining (upper panel, d). On the other hand, tumor sections obtained from a mouse that received a tail vein injection of the same amount of non-targeted IO nanoparticles (8 nmol kg−1 of body weight) did not have detectable iron staining positive cells (lower panel, pictures e and f). However, in the tumor sections of a mouse that received a higher concentration of the non-targeted IO nanoparticles (16 nmol kg−1 of body weight), weak Prussian blue positive stained areas were detected in the tumor (green arrows), but not in the normal pancreatic tissue (yellow arrows) (lower panels, g and h).
It is well known that non-targeted nanoparticles easily accumulate in the liver and spleen after systemic delivery.[32,33] In the present study, we also observed the MRI signal decrease in the liver and spleen in mice injected with ScFvEGFR-IO nanoparticles, indicating the uptake of ScFvEGFR-IO nanoparticles by the liver and spleen. However, the reduction of the MRI signal in the animals receiving ScFvEGFR-IO nanoparticle were 25% less (liver) and 52% less (spleen) when compared to the animals received non-targeted IO nanoparticles, suggesting that the uptake of the nanoparticles in the liver and spleen were reduced using ScFvEGFR-IO nanoparticles (Figure 5B).
MRI offers clinical feasibility of molecular imaging because it provides superb anatomic resolution and tissue contrast for visualizing the tissue morphology and anatomical details of organs in vivo. Because magnetic IO nanoparticles generate the strong susceptibility effect that results in strong T2 and T2* contrast for contrast-enhanced MRI, magnetic IO nanoparticles are becoming increasingly attractive for applications of in vivo molecular and cellular MRI.[34] Polymer-coated IO nanoparticles have a long blood retention time, low toxicity, and can be made to be biodegradable.[32,33] Therefore, the development of the EGFR-targeted IO nanoparticles has the potential to be translated into clinical applications for in vivo tumor imaging and targeted therapy. Since our long-term goal for developing the ScFvEGFR-IO nanoparticle is to apply the nanoparticle-based contrast agent in cancer patients, our in vivo and in vitro experiments with magnetic IO nanoparticles were mainly carried out at the current clinical available field strength of 3T.
Important aspects for the development of a targeted nanoimaging probe are the target specificity, the magnitude of the imaging signal or contrast, and the stability in vivo. Using an orthotopic human pancreatic xenograft model in nude mice, we demonstrated a marked decrease in the MRI signal in the pancreatic tumor areas, suggesting specific accumulation of the IO nanoparticles within the tumor. This T2 weighted contrast delineated the tumor region very well and was still detectable at 30 h after the nanoparticle injection. Our MRI data also showed a decrease in the MRI signal in the livers and spleens of the mice that received either targeted or non-targeted IO nanoparticles. However, analysis of MRI contrast change in tumor and normal tissues before and after the IO nanoparticle administration and histological examination of tumor and normal tissues suggest that targeted nanoparticles accumulated in the liver and spleen are less than that of mice receiving non-targeted nanoparticles. We believe that this reduction of non-specific uptake by the organs may allow more ScFvEGFR-conjugated nanoparticles to reach the tumor instead of being trapped by the liver and spleen.
3. Conclusion
By using a high-affinity single-chain anti-EGFR antibody (scFv B10, KD = 3.36 × 10−9 m) and nanoparticles, we have developed a compact-sized, EGFR-targeted nanoparticle for in vivo tumor imaging and delivery of therapeutic agents. We showed that the ScFvEGFR nanoparticles specifically bind to EGFR, which is a cell surface receptor expressed in a high percentage of human tumor cells. The EGFR-mediated internalization of the targeted nanoparticles in tumor cells increases the retention time and amount of the nanoparticles inside the tumor mass. Both of these properties fit very well with the criteria of a molecular target for receptor-targeted tumor imaging. We have also shown that ScFvEGFR nanoparticles are stable, retain target specificity to EGFR-expressing tumor cells, and enable EGFR-targeted MRI of human pancreatic cancer orthotopically implanted in the pancreas of nude mice. Therefore, our results demonstrate the potential applications of ScFvEGFR-conjugated nanoparticles for the detection of EGFR-expressing tumors using in vivo imaging approaches.
4. Experimental Section
Cancer Cell Lines
Human breast cancer MDA-MB-231 and MCF-7, human pancreatic cancer MIA PaCa-2, and human embryonic kidney epithelial HEK 293 cell lines were purchased from American Type Culture Collection (ATCC, Rockville, MD). Mouse mammary carcinoma cell line 4T1 was kindly provided by Dr. Fred R. Miller in the Barbara Ann Karmanos Cancer Institute, Detroit, MI.
Isolation of EGFR-Specific scFv B10 Clone from YUAN-FCCC Phage Display Library
The human EGFR-specific scFv B10 was isolated from the YUAN-FCCC human naive phage display library using established solid phase biopanning methods. HEK 293 cells were stably transfected with a recombinant extracellular domain (ECD) of human EGFR.[6,35] Positive clones were sequenced and expressed from TG1 E. coli competent cells (Biochain Institute, Inc, Hayward, CA) for surface plasmon resonance (SPR) (Biacore 2000, GE Healthcare, Piscataway, NJ) and FACScan characterizations (BD Bioscience, San Jose, CA).[6,36] The scFv B10 clone was one of the strongest clones isolated. Affinity of scFv B10 for human EGFR was determined by SPR to be 3.36 × 10−9 m, with a Ka at 1.04 × 105 (Ms−1) and a Kd at 3.51 × 10−4 (s−1). Recombinant ScFvEGFR proteins with a molecular weight of 25 kDa were obtained from the bacterial lysates of scFv B10 transformed TG1 competent cells after Ni-NTA-agarose column separation under native conditions (Qiagen, Valencia, CA).
Production of ScFvEGFR-QDs or ScFvEGFR-IO Nanoparticles
QDs with an emission of 580 nm were synthesized using the protocol reported previously and then coated with the amphiphilic copolymers as described.[14,37,38] Ni-NTA was conjugated to QDs using a covalent link of the amine group of Ni-NTA with the carboxyl group (−COOH) of the amphiphilic polymer on the surface of QDs. ScFvEGFR proteins tagged with a sequence of six histidine residues were mixed with Ni-NTA-QDs at a molar ratio of 1 to 20 in PBS and incubated at 4 °C for overnight. Interaction of the His-tag with the nickel molecule leads to the stable conjugation of the ScFvEGFR single chain antibody to the surface of QDs.
ScFvEGFR was conjugated to the magnetic IO nanoparticle via a covalent link of the amine group of the protein with the carboxyl group (−COOH) of the amphiphilic polymer. The final ScFvEGFR nanoparticle conjugates were purified using a Nanosep 100k OMEGA filter column (Pall Corp, Ann Arbor, MI). The core size and hydrodynamic size of the magnetic IO nanoparticles were determined using TEM and Zetasizer Nano (Malvern Instruments Inc., Southborough, MA).
His-tagged recombinant GFP was produced from a pGFP-His-tag plasmid that was constructed in our laboratory using the polymerase chain reaction (PCR)-directed insertion of oligonucleotides encoding six histidine residues into a bacterial-expressing pGFP plasmid vector (Clonetech, Mountain View, CA). Recombinant GFP was produced and purified using the same procedure as used for the ScFvEGFR protein.
Determination of Specificity of ScFvEGFR Using QDs
ScFvEGFR-QDs or non-targeted QDs were incubated with cells cultured in chamber slides at 4 °C for 1 h. The slides were examined under an inverted Olympus fluorescence microscope. To further determine the ScFvEGFR-mediated specific binding and internalization of QDs, ScFvEGFR-QDs or non-targeted QDs were incubated with breast cancer cell lines expressing a high level (MDA-MB-231) or low level (MCF-7) of EGFR at 37 °C for 2 h. After washing, cells were fixed with ice-cold acetone and slides were then examined under Zeiss LSM 510 META confocal microscope (Carl Zeiss Micro-Imaging, Inc, Thornwood, NY).
In vitro Assays for Specificity of ScFvEGFR-IO Nanoparticles
Cells in a 24-well tissue culture plate were incubated with IO nanoparticles for 3 h at 37 °C, and then washed and fixed with formaldehyde (2%). Prussian blue staining was performed to determine the presence of the IO nanoparticles inside the cells using a staining solution containing a 1:1 mixture of potassium ferrocyanide (5%) and HCl (5%) at 37 °C for 60 min.[39]
Orthotopic Human Pancreatic Cancer Xenograft Model
A pancreatic tumor model was established using an EGFR-expressing human pancreatic cancer cell line, MIA PaCa-2. Compared to other tumor cell lines, such as the breast cancer cell line MDA-MB-231 that has a level of EGFR expression at 4.8 × 105 receptor cell−1, the MIA PaCa-2 cell line has a moderate level of expression of 1.7 × 105 receptor cell−1.[6,40] This should allow us to determine whether in vivo targeting and tumor imaging can be achieved in a tumor without the need to have a very high level of EGFR on the tumor cells. 1 × 107 of MIA PaCa-2 cells were injected directly into the pancreas of 6 to 8 week old female nude mice (Taconic, Hudson, NY). In 3 to 4 weeks, orthotopically xenografted pancreatic tumors typically reached 5 mm in diameter and were ready for experiments.
Target Specificity and Tissue Distribution Detected by ScFvEGFR-QDs
200 pmol of either ScFvEGFR-QDs or control non-targeted QDs was administrated to the tumor-bearing mice via a tail vein injection. The mice were sacrificed 5 h after the injection. Xenografted pancreatic tumor and normal tissues were removed and frozen in the liquid nitrogen. Frozen tissue sections were counterstained with Hoechst 33342 and examined directly under fluorescence microscopy or confocal microscopy.
MRI Scan in vitro and in vivo
1 × 107 of tumor cells were harvested from the cell culture after incubating with 10 nM of various IO nanoparticles at 37 °C for 3 h. Cells were washed with phosphate buffer saline (PBS) and then embedded in 1% agarose in multi-well plates. These cell plates were scanned using a 3T MRI scanner (Philips Medical Systems, Bothell, WA) and a multi-echo T2 weighted fast spin echo imaging sequence to collect a series of echo time (TE) dependent data points simultaneously (32 different TE values ranging from 6 ms to 200 ms). ScFvEGFR-IO induced MRI signal changes in tumor cells were measured in the selected region of interest (ROI). T2 values of each sample or well were calculated from the captured MRI by fitting the decay curve on a pixel-by-pixel basis using the non-linear mono-exponential algorithm of Mi = M0 × exp(−TEi/T2).
Tumor-bearing mice were scanned using a 3T MRI scanner with a customised rodent coil to collect pre- and post-contrast enhanced MRI data. After the mice were injected with ScFvEGFR-IO or non-targeted IO nanoparticles (8 or 16 nmol kg−1 of body weight) in PBS (100 μl) though the tail vein, they were imaged at different time points to follow the MRI contrast changes. Images from pre- and post-contrast administration were compared to evaluate the contrast enhancement by the target-specific contrast agent. The imaging sequences include: T1 and T2 weighted spin echo or gradient echo imaging. A multi-echo T2 weighted fast spin echo sequence (8 different TE values ranging from 10–120 ms) was used to obtain T2 relaxometry of the whole mouse. The ROI method was used to evaluate and quantify the contrast agent induced signal or T2 value changes in the tumor and other organs. The signal of the leg muscle was used to normalize the signals in ROIs.
Histological Analysis
The mice were sacrificed at 48 h after the injection of contrast agents or after MRI. Tumor and normal tissues were collected and frozen tissue sections were examined by Prussian blue staining to confirm the presence of IO nanoparticles in the tissue sections. After counterstaining with nuclear fast red (Vector Laboratories, Burlingame, CA, USA), the slides were examined under bright field microscopy.
Acknowledgments
We would like to thank Dr. Fred Miller for kindly providing us with 4T1 cell line, and Drs. Adam Marcus and Katherine Schafer-Hales in the Cell Imaging Core of the Winship Cancer Institute for their assistance with confocal microscopy. This research project is supported by the Emory-Georgia Tech Nanotechnology Center for Personalized and Predictive Oncology of NIH, the NCI Center of Cancer Nanotechnology Excellence (CCNE, U54 CA119338-01), and in part by the Idea Award of the Breast Cancer Research Program of the Department of Defense (BC 021952), a seed grant from the Golfers Against Cancer, and the Nancy Panoz Endowed Chair (Dr. Lily Yang).
Contributor Information
Dr. Lily Yang, Department of Surgery, Winship Cancer Institute, Emory University School of Medicine, 1365 C Clifton Road NE Atlanta, GA 30322 (USA), lyang02@emory.edu
Dr. Hui Mao, Department of Radiology, Emory University School of Medicine, EUH AG11, 1364 Clifton Road, Atlanta, GA 30322 (USA)
Dr. Y. Andrew Wang, Ocean Nanotech, LLC, Fayetteville, AR 72701 (USA)
Zehong Cao, Department of Surgery, Winship Cancer Institute, Emory University School of Medicine, 1365 C Clifton Road NE Atlanta, GA 30322 (USA).
Dr. Xianghong Peng, Department of Surgery, Winship Cancer Institute, Emory University School of Medicine, 1365 C Clifton Road NE Atlanta, GA 30322 (USA)
Xiaoxia Wang, Department of Radiology, Emory University School of Medicine, EUH AG11, 1364 Clifton Road, Atlanta, GA 30322 (USA).
Dr. Hongwei Duan, Department of Biomedical Engineering, Emory University School of Medicine, 101 Woodruff Circle, Suite 2007B, Atlanta, GA 30322 (USA)
Chunchun Ni, Department of Radiology, Emory University School of Medicine, EUH AG11, 1364 Clifton Road, Atlanta, GA 30322 (USA).
Dr. Qingan Yuan, Fox Chase Cancer Center, Philadelphia, PA 19111 (USA)
Dr. Gregory Adams, Fox Chase Cancer Center, Philadelphia, PA 19111 (USA)
Dr. Mark Q. Smith, Department of Surgery, Winship Cancer Institute, Emory University School of Medicine, 1365 C Clifton Road NE Atlanta, GA 30322 (USA)
Dr. William C. Wood, Department of Surgery, Winship Cancer Institute, Emory University School of Medicine, 1365 C Clifton Road NE Atlanta, GA 30322 (USA)
Dr. Xiaohu Gao, Department of Bioengineering, University of Washington, Seattle, WA 98195 (USA)
Dr. Shuming Nie, Email: snie@emory.edu, Department of Biomedical Engineering, Emory University School of Medicine, 101 Woodruff Circle, Suite 2007B, Atlanta, GA 30322 (USA).
References
- 1.Tsutsui S, Ohno S, Murakami S, Hachitanda Y, Oda S. Breast Cancer Res Treat. 2002;71:67. doi: 10.1023/a:1013397232011. [DOI] [PubMed] [Google Scholar]
- 2.Yarden Y, Sliwkowski MX. Nat Rev Mol Cell Biol. 2001;2:127. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 3.Gschwind A, Fischer OM, Ullrich A. Nat Rev Cancer. 2004;4:361. doi: 10.1038/nrc1360. [DOI] [PubMed] [Google Scholar]
- 4.Arteaga CL, Truica CI. Semin Oncol. 2004;31:3. doi: 10.1053/j.seminoncol.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 5.Sorkin A. Biochem Soc Trans. 2001;29:480. doi: 10.1042/bst0290480. [DOI] [PubMed] [Google Scholar]
- 6.Zhou Y, Drummond DC, Zou H, Hayes ME, Adams GP, Kirpotin DB, Marks JD. J Mol Biol. 2007;371:934. doi: 10.1016/j.jmb.2007.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tseng CL, Wang TW, Dong GC, Yueh-Hsiu Wu S, Young TH, Shieh MJ, Lou PJ, Lin FH. Biomaterials. 2007;28:3996. doi: 10.1016/j.biomaterials.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 8.Iznaga-Escobar N, Torres LA, Morales A, Ramos M, Alvarez I, Perez N, Fraxedas R, Rodriguez O, Rodriguez N, Perez R, Lage A, Stabin MG. J Nucl Med. 1998;39:15. [PubMed] [Google Scholar]
- 9.Ke S, Wen X, Gurfinkel M, Charnsangavej C, Wallace S, Sevick-Muraca EM, Li C. Cancer Res. 2003;63:7870. [PubMed] [Google Scholar]
- 10.Ramos-Suzarte M, Rodriguez N, Oliva JP, Iznaga-Escobar N, Perera A, Morales A, Gonzalez N, Cordero M, Torres L, Pimentel G, Borron M, Gonzalez J, Torres O, Rodriguez T, Perez R. J Nucl Med. 1999;40:768. [PubMed] [Google Scholar]
- 11.Amit AG, Mariuzza RA, Phillips SE, Poljak RJ. Science. 1986;233:747. doi: 10.1126/science.2426778. [DOI] [PubMed] [Google Scholar]
- 12.Ouerghi O, Touhami A, Othmane A, Ouada HB, Martelet C, Fretigny C, Jaffrezic-Renault N. Biomol Eng. 2002;19:183. doi: 10.1016/s1389-0344(02)00046-1. [DOI] [PubMed] [Google Scholar]
- 13.Adams GP, Schier R. J Immunol Methods. 1999;231:249. doi: 10.1016/s0022-1759(99)00161-1. [DOI] [PubMed] [Google Scholar]
- 14.Gao X, Cui Y, Levenson RM, Chung LW, Nie S. Nat Biotechnol. 2004;22:969. doi: 10.1038/nbt994. [DOI] [PubMed] [Google Scholar]
- 15.Cai W, Shin DW, Chen K, Gheysens O, Cao Q, Wang SX, Gambhir SS, Chen X. Nano Lett. 2006;6:669. doi: 10.1021/nl052405t. [DOI] [PubMed] [Google Scholar]
- 16.Lee JH, Huh YM, Jun YW, Seo JW, Jang JT, Song HT, Kim S, Cho EJ, Yoon HG, Suh JS, Cheon J. Nat Med. 2007;13:95. doi: 10.1038/nm1467. [DOI] [PubMed] [Google Scholar]
- 17.Nasongkla N, Bey E, Ren J, Ai H, Khemtong C, Guthi JS, Chin SF, Sherry AD, Boothman DA, Gao J. Nano Lett. 2006;6:2427. doi: 10.1021/nl061412u. [DOI] [PubMed] [Google Scholar]
- 18.Oh P, Borgstrom P, Witkiewicz H, Li Y, Borgstrom BJ, Chrastina A, Iwata K, Zinn KR, Baldwin R, Testa JE, Schnitzer JE. Nat Biotechnol. 2007;25:327. doi: 10.1038/nbt1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li KC, Guccione S, Bednarski MD. J Cell Biochem Suppl. 2002;39:65. doi: 10.1002/jcb.10401. [DOI] [PubMed] [Google Scholar]
- 20.Reddy GR, Bhojani MS, McConville P, Moody J, Moffat BA, Hall DE, Kim G, Koo YE, Woolliscroft MJ, Sugai JV, Johnson TD, Philbert MA, Kopelman R, Rehemtulla A, Ross BD. Clin Cancer Res. 2006;12:6677. doi: 10.1158/1078-0432.CCR-06-0946. [DOI] [PubMed] [Google Scholar]
- 21.de Vries IJ, Lesterhuis WJ, Barentsz JO, Verdijk P, van Krieken JH, Boerman OC, Oyen WJ, Bonenkamp JJ, Boezeman JB, Adema GJ, Bulte JW, Scheenen TW, Punt CJ, Heerschap A, Figdor CG. Nat Biotechnol. 2005;23:1407. doi: 10.1038/nbt1154. [DOI] [PubMed] [Google Scholar]
- 22.Moore A, Josephson L, Bhorade RM, Basilion JP, Weissleder R. Radiology. 2001;221:244. doi: 10.1148/radiol.2211001784. [DOI] [PubMed] [Google Scholar]
- 23.Harisinghani MG, Barentsz J, Hahn PF, Deserno WM, Tabatabaei S, van de Kaa CH, de la Rosette J, Weissleder R. N Engl J Med. 2003;348:2491. doi: 10.1056/NEJMoa022749. [DOI] [PubMed] [Google Scholar]
- 24.Schultz JF, Bell JD, Goldstein RM, Kuhn JA, McCarty TM. Ann Surg Oncol. 1999;6:691. doi: 10.1007/pl00021736. [DOI] [PubMed] [Google Scholar]
- 25.Medarova Z, Pham W, Kim Y, Dai G, Moore A. Int J Cancer. 2006;118:2796. doi: 10.1002/ijc.21672. [DOI] [PubMed] [Google Scholar]
- 26.Choi H, Choi SR, Zhou R, Kung HF, Chen IW. Acad Radiol. 2004;11:996. doi: 10.1016/j.acra.2004.04.018. [DOI] [PubMed] [Google Scholar]
- 27.Simberg D, Duza T, Park JH, Essler M, Pilch J, Zhang L, Derfus AM, Yang M, Hoffman RM, Bhatia S, Sailor MJ, Ruoslahti E. Proc Natl Acad Sci USA. 2007;104:932. doi: 10.1073/pnas.0610298104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmitt J, Hess H, Stunnenberg HG. Mol Biol Rep. 1993;18:223. doi: 10.1007/BF01674434. [DOI] [PubMed] [Google Scholar]
- 29.Zajchowski D, Band V, Pauzie N, Tager A, Stampfer M, Sager R. Cancer Res. 1988;48:7041. [PubMed] [Google Scholar]
- 30.Yu WW, Falkner JC, Yavuz CT, Colvin VL. Chem Commun. 2004:2306. doi: 10.1039/b409601k. [DOI] [PubMed] [Google Scholar]
- 31.Sakai K, Arao T, Shimoyama T, Murofushi K, Sekijima M, Kaji N, Tamura T, Saijo N, Nishio K. FASEB J. 2006;20:311. doi: 10.1096/fj.05-4034fje. [DOI] [PubMed] [Google Scholar]
- 32.Moore A, Marecos E, Bogdanov A, Jr, Weissleder R. Radiology. 2000;214:568. doi: 10.1148/radiology.214.2.r00fe19568. [DOI] [PubMed] [Google Scholar]
- 33.Jain TK, Reddy MK, Morales MA, Leslie-Pelecky DL, Labhasetwar V. Mol Pharm. 2008;5:316. doi: 10.1021/mp7001285. [DOI] [PubMed] [Google Scholar]
- 34.Bulte JW, Kraitchman DL. NMR Biomed. 2004;17:484. doi: 10.1002/nbm.924. [DOI] [PubMed] [Google Scholar]
- 35.Yuan QA, Robinson MK, Simmons HH, Russeva M, Adams GP. Cancer Immunol Immunother. 2008;57:367. doi: 10.1007/s00262-007-0376-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Horak E, Heitner T, Robinson MK, Simmons HH, Garrison J, Russeva M, Furmanova P, Lou J, Zhou Y, Yuan QA, Weiner LM, Adams GP, Marks JD. Cancer Biother Radiopharm. 2005;20:603. doi: 10.1089/cbr.2005.20.603. [DOI] [PubMed] [Google Scholar]
- 37.Han M, Gao X, Su JZ, Nie S. Nat Biotechnol. 2001;19:631. doi: 10.1038/90228. [DOI] [PubMed] [Google Scholar]
- 38.Smith A, Duan H, Rhyner MN, Ruan G, Nie S. Phys Chem Chem Phys. 2006;8:3895. doi: 10.1039/b606572b. [DOI] [PubMed] [Google Scholar]
- 39.Bulte JW, Douglas T, Witwer B, Zhang SC, Strable E, Lewis BK, Zywicke H, Miller B, van Gelderen P, Moskowitz BM, Duncan ID, Frank JA. Nat Biotechnol. 2001;19:1141. doi: 10.1038/nbt1201-1141. [DOI] [PubMed] [Google Scholar]
- 40.Smith JJ, Derynck R, Korc M. Proc Natl Acad Sci USA. 1987;84:7567. doi: 10.1073/pnas.84.21.7567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kircher MF, Allport JR, Graves EE, Love V, Josephson L, Lichtman AH, Weissleder R. Cancer Res. 2003;63:6838. [PubMed] [Google Scholar]