

Abstract
Purpose
Mutations in the visual system homeobox 1 (VSX1) gene have been described at a low frequency in keratoconus and posterior polymorphous corneal dystrophy (PPCD). The putative role is controversial for several reasons, including a lack of mutations detected in other population cohorts. This study aims to determine whether VSX1 contributes to the genetic pathogenesis of keratoconus and PPCD in a New Zealand population, and includes analysis of a Polynesian population.
Methods
Recruitment of patients with keratoconus and PPCD, comprehensive clinical examination including corneal topography and pachymetry, and collection of biologic samples for DNA extraction were undertaken. Mutational analysis of VSX1 (exons 1–7) with PCR and sequencing with bioinformatic assessment of variants was performed. Probable pathogenic variants were screened for in a control population using high-resolution melting analysis.
Results
Forty-seven patients with keratoconus, including 15 familial cases, and ten unrelated patients with PPCD were recruited. Two pathogenic changes were detected; a novel change c.173C>T (p.Pro58Leu) was found in a patient with PPCD, predicted to be pathogenic, and not seen in 200 ethnically matched control alleles. The previously reported c.731A>G (p.His244Arg) was detected in a patient with sporadic keratoconus, and not present in the controls. No family members were available for segregation analysis.
Conclusions
This study reports the presence of pathogenic mutations in VSX1 in PPCD and keratoconus, including a novel disease-causing variant. The affected numbers are small, but given the growing body of evidence of pathogenic segregating changes in VSX1 in disease cohorts, the expression in keratocytes as part of wound healing, and the documented association of PPCD and keratoconus, it seems likely that the role of VSX1 as a genetic factor contributing to disease is real.
Introduction
The visual system homeobox 1 (VSX1) gene is a member of the “paired-like” homeodomain transcription factors. This family plays a role in craniofacial and ocular development. Human VSX1 has been mapped to 20p11.2. It was initially reported as containing five exons and approximately 6.2 kb in size [1] with an additional two exons characterized [2,3] that encode isoforms of the VSX1 transcript. The expression of VSX1 has been detected in embryonic craniofacial tissues, adult retinas, and adult corneas [1,4]. Mutations in VSX1 were reported associated with craniofacial abnormalities, empty sella tunica, and abnormal retinal cells [5], but more frequently and controversially with several corneal dystrophies and ectasias, specifically keratoconus and posterior polymorphous corneal dystrophy (PPCD).
VSX1 was initially implicated in the pathogenesis of PPCD in 2002 [6]. PPCD is a frequently asymmetric autosomal dominant corneal dystrophy with characteristic involvement of Descemet’s membrane and the endothelium. In three families, mutations of VSX1 were reported to segregate with the disease [6,7], but this was not replicated in other studies [8,9].
PPCD is genotypically heterogeneous: The largest percentage of PPCD (approximately one third) is associated with mutations in ZEB1, at the PPCD3 locus [10]. Haploinsufficiency results in ectopic collagen type IV, alpha 3 (COL4A3) expression in the cornea. A mutation in another gene, COL8A2, was reported in one family with PPCD [11] as well as in Fuchs endothelial corneal dystrophy. No further PPCD reports have been described with COL8A2 mutations suggesting this association is tenuous or of a low frequency.
The relationship between keratoconus and VSX1 was first reported in the study by Heon et al. [6]. The phenotypic heterogeneity of VSX1 with involvement in keratoconus and PPCD is feasible as the disorders share a potential common mode of involvement of the posterior surface of the cornea, specifically Descemet’s membrane. The association of PPCD with keratoconus is also well documented with many cases described cooccurring in the same cornea [12-16].
Several mutations linked to keratoconus have since been identified [2,6,17-20]. The role of VSX1 in the pathogenesis of keratoconus has also been controversial. Several other studies have failed to identify an association between VSX1 variants/polymorphisms and keratoconus [21-24]. These contradictory results may be partly attributed to the low frequency of changes, ethnic variation, and the mounting evidence that keratoconus is likely a multifactorial and polygenic disease [25].
The variety of genetic techniques used to identify keratoconus genes has included family-based linkage studies, identity by descent, genome-wide scans, and genome-wide association studies. These approaches have identified a host of genetic loci and candidate genes [26], which appear to account for only a small number of those affected. Recently, association of keratoconus with the hepatocyte growth factor, HGF [27], and the microRNA MIR184 [28] genes was identified.
Although anecdotally it is widely believed that keratoconus is more prevalent and aggressive in New Zealand, especially in the Maori and Pacific Island population, exact figures are not available [29,30]. However, keratoconus is the leading indication for corneal transplantation in adults and children in New Zealand [31,32]. It is plausible that a genetic factor is responsible for the ethnic predisposition of keratoconus in New Zealand. This study examines whether VSX1 plays a role in the pathogenesis of keratoconus and PPCD in a New Zealand population.
Methods
Patient recruitment
Patients were recruited from the Department of Ophthalmology, Greenlane Clinical Centre, Auckland District Health Board with a clinical diagnosis of keratoconus or PPCD, and reviewed at the University Clinic, Department of Ophthalmology, University of Auckland. The protocol of this study adhered to the tenets of the Declaration of Helsinki with Institutional Ethics and Maori Research Review Board approval (Ministry of Health NTX/06/12/161 and ADHB A+3657).
Clinical
Forty-seven healthy subjects (demographics provided in Table 1) underwent extensive clinical examination, including Snellen visual acuity, autorefraction, corneal topography, and pachymetry using a combined Placido/slit-scanning elevation tomography system (Orbscan II; Bausch & Lomb Surgical, Rochester, NY) and/or Pentacam Schiempflug analysis (Oculus, Wetzlar, Germany), slit-lamp examination and photography, and laser scanning in vivo confocal microscopy (IVCM) using the HRTII (Heidelberg Retina Tomograph II, Rostock Corneal Module [RCM]; Heidelberg Engineering GmbH, Heidelberg, Germany).
Table 1. Demographics of cohort studied.
| Disease | Number | Age(years) | Sex | Familial | Caucasian | Polynesian | Indian |
|---|---|---|---|---|---|---|---|
| Keratoconus |
47 |
41.5
(15 - 83) |
20F:27M |
15 |
17 |
26 |
4 |
| PPCD |
10 |
50.77
(16 - 86) |
5F:5M |
1 |
8 |
2 |
|
| Total | 57 | 41.42 | 28F:34M | 16 | 25 | 28 | 4 |
The table shows the distribution of Age in years, female:male ratio, familial cases and ethnicity as well as disease type of the cohorts investigated. F=female, M=male.
DNA collection
After informed consent was received, biologic samples (10 mls of peripheral venous blood) were obtained through venesection with an ethylenediamine tetraacetic acid (EDTA)-coated Vacutainer (Greiner bio-one, Austria) and stored in a 4 °C refrigerator, or saliva specimen) were collected for DNA extraction using the salt extraction method from blood [33], and according to the manufacturer’s instructions for saliva kits (Oragene, DNAGenotek, Ottawa, Canada). For controls, DNA samples were collected from randomly selected and ethnically matched individuals attending the Ophthalmology Department who did not exhibit any clinical evidence of corneal abnormality in terms of appearance or topographic parameters.
Mutational analysis of genes
DNA samples were screened for mutations in all coding exons of VSX1 (NT_011387.8), including intron–exon boundaries (which included exons 6 and 7). Further details of primers and PCR conditions provided in Appendix 1. Following column purification with the HighPure PCR purification kit (Roche Diagnostic, Mannheim, Germany), the product was sequenced directly according to the protocols accompanying the ABI BigDye Terminator kit v3.1 (Applied Biosystems Inc., Foster City, CA). Bidirectional sequencing of amplicons was undertaken on an ABI 3700 prism genetic analyzer (Applied Biosystems). Nucleotide sequences were compared with the published VSX1 sequence and polymorphic variation data in electronic databases to determine pathogenicity. For any uncharacterized or equivocal sequence change, a control population was screened with high-resolution melting analysis (HRMA).
To determine the frequency of the VSX1 variants c.173C>T (p.Pro58Leu), and c.731A>G (p.His244Arg), 100 ethnically matched control individuals (200 alleles) were screened. Screening for the detected VSX1 sequence variants used HRMA on the RotorGene6000 (Corbett Life Sciences, San Francisco, CA), using the High Resolution Melting Master kit (Roche Diagnostic). Further details of primers and conditions are provided in Appendix 1. Each reaction included a positive and negative control based on the sequencing confirmation. Any sample on the melt curve that produced an equivocal reading was subject to further PCR and sequencing to confirm or exclude the presence of the sequence variation.
For the sequence variants, homology and predicted destruction or creation of exonic splicing enhancers, or effects on splicing, were evaluated using various publicly available software. PolyPhen2 and Sorting Intolerant From Tolerant (SIFT) analysis was used to predict the impact of the c.173C>T (p.Pro58Leu) and c.731A>G (p.His244Arg) missense variants on protein structure and function. Potential pathogenicity was based on positive family segregation, an allele frequency of <1/100 control chromosomes, homology, and bioinformatic prediction of biologic significance.
PolyPhen2 analyzes an amino acid variant at the structural level, to assess any functional implication of an amino acid change [34].
SIFT is another sequence homology-based tool, which works on the philosophy that conservation of proteins throughout evolution is tightly correlated with the function [35]. Therefore, SIFT aims to predict the phenotypic effect any amino acid substitution will have, based on whether it is tolerated.
Results
Forty-seven patients with keratoconus, including 15 familial cases, and ten patients with PPCD were included; their demographic features summarized in Table 1. One novel variation in exon 1 in the proline-rich area (44–127) c.173C>T (p.Pro58Leu; Figure 1) was found in a patient with PPCD. The variation was not present in 200 control alleles (Figure 2 HRMA analysis), and homology showed the variation is highly conserved. Protein prediction with PolyPhen2 suggests this mutation is “probably damaging” with a position-specific independent count (PSIC) score of 2.299 and a SIFT score of 0.04 also labeled as “damaging.” Population screening has detected this allele in 3/8,075 alleles (Exome Variant Server [EVS], accessed September 20, 2012) with a minor allele frequency of 0.0371%, but the allele is not listed in the 1000 Genomes Database (accessed September 20, 2012).
Figure 1.
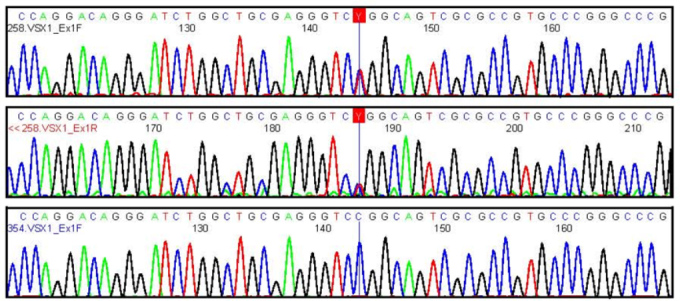
The electropherogram of the proband with posterior polymorphous corneal dystrophy shows the heterozygous sequence variant c.173C>T, which results in the protein change p.Pro58Leu. The top panel shows the forward sequence, the middle panel shows the reverse sequence (reverse complement shown) and the bottom panel in a control shows the wild type sequence c.173 C/C.
Figure 2.
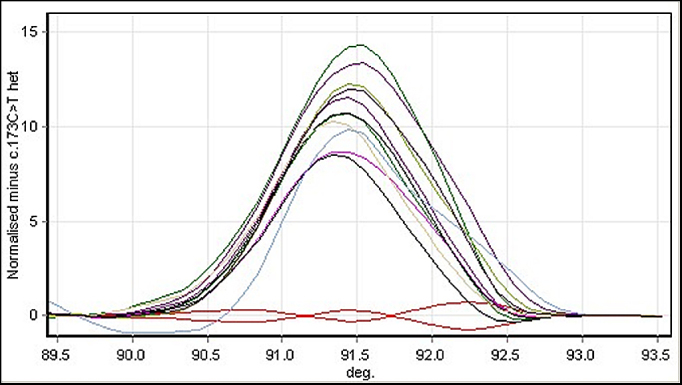
High-resolution melting analysis normalized difference graph of screening in the control population (200 alleles) for the VSX1 c.173C>T variant. The melt profiles shown in red at the bottom are duplicate samples of the positive control (affected c.173C>T heterozygous) samples. All other samples with no variants are represented by the bell-shaped melting curves.
This female subject was a 57-year-old Caucasian, with no family history of PPCD or keratoconus. She had a history of open angle glaucoma and had previously undergone a left trabeculectomy. Visual acuity was 6/5 (right eye) and 6/6 (left eye). Corneal tomography (Orbscan II) demonstrated minor asymmetry with minimal inferior steepening; however, all parameters were within normal limits regarding features of keratoconus: right simulated keratometry 43.8 diopter (D)/42.7 D with 1.1 D of astigmatism at 95° and thinnest pachymetry 519 μm; left simulated keratometry 45.9 D/44.7 D with 1.2 D of astigmatism at 97° and thinnest pachymetry 524 μm. The right eye demonstrated no features of PPCD with a mean endothelial cell density of 2,351 cell/mm2, whereas the left cornea, although entirely clear, demonstrated classical “vesicular” PPCD features of multiple endothelial vesicles (Figure 3A) and curvilinear ridges with a mean endothelial cell density of 1,323 cells/mm2 (HRTII; Figure 3B). The anterior segments of both eyes were otherwise normal.
Figure 3.

Clinical images of Case 1. A: A slit-lamp photograph of Case 1 showing a posterior polymorphous dystrophy band lesion (arrows). B: In vivo confocal microscopy in this patient shows undulation of Descemet’s membrane and the endothelial surface, with needle-shaped hyper-reflectivity at the level of Descemet’s membrane. (Scale bar=100 μm)
The previously described c.731A>G (p.His244Arg) was detected in one patient with sporadic keratoconus. This variant lies within the conserved Chx10/Vsx-1 and ceh-10 (CVC) domain (224–277). The variant was not detected in the 200 control alleles (Figure 4), and protein prediction with PolyPhen2 suggests that this variant is probably damaging, with a PSIC score of 2.532. SIFT analysis also calls this change “deleterious.” Population screening has detected this allele in 27/12,059 alleles (Exome Variant Server, accessed September 20, 2012).
Figure 4.

High-resolution melting analysis normalized difference graph of screening in the control population (200 alleles) for the visual system homeobox 1 gene c.731A>G, p.His244Arg heterozygous variant. The duplicate red lines running parallel to the x-axis at zero are the positive control (affected) samples, while the samples with no variants are represented by the sine-shaped melting curves.
This male subject was Caucasian, 42 years of age with asymmetric keratoconus, right worse than left, and corrected visual acuities with contact lenses were 6/9 (right eye) and 6/6 (left eye). Orbscan II computerized keratometry-confirmed bilateral keratoconus. A right asymmetric bowtie appearance with inferior steepening was associated with simulated keratometry of 53.3 D/46.4 D with 6.9 D of astigmatism at 100° and thinnest pachymetry of 425 μm on Pentacam analysis. Left Orbscan tomography highlighted atypical inferior steepening of early keratoconus with simulated keratometry 44.6 D/43.2 D with 1.4 D of astigmatism at 119° and thinnest pachymetry of 467 μm on Pentacam Scheimpflug analysis. No evidence of PPCD was identified by an experienced corneal subspecialist, and laser scanning in vivo confocal microscopy of the cornea (HRTII) confirmed normal endothelial morphology (Figure 5). Unfortunately, no family members were available for segregation analysis.
Figure 5.

In vivo confocal microscopy in this patient with keratoconus, heterozygous for the visual system homeobox 1 p.His244Arg variant shows a healthy endothelium. (Scale bar=100 μm)
Discussion
This study investigated VSX1 changes in all seven exons in a population with PPCD and keratoconus, with a significant Polynesian ethnic proportion. Two likely pathogenic changes were detected in two Caucasian individuals. The c.173C>T occurs in the proline-rich domain causing the amino acid change p.Pro58Leu, which is highly conserved, not seen in our control population, and predicted to be pathogenic.
The second observed change c.731A>G, p.His244Arg was identified in a Caucasian patient with keratoconus and has previously been described in several papers [6,24]. Heon et al. [6] observed this change in 1/63 patients with keratoconus and segregated with the disease, but also observed it in 2/277 controls. Tang screened a case control panel (70 controls and 77 patients with sporadic keratoconus) but did not observe His244Arg in this panel [24]. That study also screened 444 individuals from 75 families: Two affected and one unaffected had the variant, although it is not clear from this paper if forme fruste keratoconus was included as a parameter for affectation status. The presence in a control population has led to doubts about its pathogenicity.
The role of VSX1 in the pathogenesis of keratoconus and posterior polymorphous dystrophy has been debated in the literature since the gene was first described in 2002 [6]. Several factors contribute to this debate, but one point is the small number of cases of PPCD and keratoconus in which changes in VSX1 are described, despite probable pathogenic changes being described since 2002 in multiple population cohorts. Other factors that have placed doubt upon the validity of the role of this gene in these disorders include changes in the gene not detected in other cohorts, non-segregation of variants, presence of supposed pathogenic alleles in the control population, and questionable corneal expression. Only one study to date has examined exons 6 and 7 [2].
Although VSX1 expression was initially reported in the cornea [1], further studies clearly demonstrated expression only in the retina [36,37]. Indeed, Vsx1 expression was not determined in the mouse cornea or in adult human corneas [6], and a mutant Vsx1 mouse model had no corneal phenotype [38]. Subsequently, however, VSX1 expression in keratocytes has been characterized in vitro and in vivo using real-time PCR, immunohistochemistry, and in situ hybridization [4]. Although not observed in resting or quiescent human keratocytes, in wounded corneas, or when cultured in serum to mimic wounded conditions, the keratocytes express VSX1, and this is associated with fibroblastic transformation [4]. These observations add strength to the hypothesis that VSX1 is involved in the wound healing response and thus may contribute to the underlying pathology in corneal disease. Interestingly, recent work in damaged and normal mouse corneas failed to demonstrate any Vsx1 expression, with the authors concluding there may be a species-specific role for VSX1 [39].
In addition, the original PPCD1 locus (MIM# 122000) was mapped to a 30 cM pericentromeric locus on 20p11-q11 [40]. When the locus was refined in Czech families in 2005, VSX1 sat outside this interval [9]. The PPCD1 locus was further reduced to 2.4 cM [41] and subsequently probed with Sanger and next-generation sequencing [41,42]. The underlying genetic cause within this locus appears to remain elusive. A recent publication further explored this locus demonstrating a founder haplotype in the Czech population, but no causative mutation was identified [43].
Regarding the role of VSX1 in keratoconus, one of the most recent publications on this subject looked at an Italian cohort of 302 patients with keratoconus (the largest series published to date) and found probable pathogenic changes in VSX1 in 3.2% of the affected population. In addition, the authors emphasized the possibility of variable expressivity and incompletely penetrant mutations, as well as the possibility the VSX1 changes are a genetic predisposing factor in this multifactorial disease [44]. These concepts (variable expressivity, incomplete penetrance, and genetic risk alleles) are well documented and accepted in a host of other diseases affecting the eye such as neurofibromatosis type 1, retinoblastoma, and age-related macular degeneration. A study of Iranian patients with keratoconus also showed p.His244Arg segregating with disease in a two-generation pedigree; four affected were heterozygous whereas five unaffected were not, and it was not present in extensively phenotyped controls [45]. Analysis of the pedigrees demonstrated a 58% reduced penetrance in general among the Iranian families with keratoconus, which could explain Tang et al.’s finding [24]. Other recent studies also demonstrate segregation of other VSX1 probable pathogenic changes with keratoconus [44,46].
A recent paper characterized the cornea in patients with PPCD showing the topographic parameters were significantly steeper, but with no clinical or topographical evidence of keratoconus [47]. In this paper, of the 18 individuals characterized, 14 were from six families, and eight patients were below the age of 14 years. Twin studies have demonstrated a genetic component to corneal curvature, and the younger members may be too young to assess whether they truly have keratoconus as the disease may not manifest until later teens. Alternatively, this group may have described a subgroup of individuals in whom PPCD and steep corneal curvature are inextricably genetically linked. This group of patients was also not genetically characterized. In another study of six patients with ZEB1 mutations, three had steep corneas but no evidence of keratoconus [48].
A limitation of our study is the small number of patients, particularly in the PPCD cohort. In addition, family members were not available for those with mutations to prove or disprove segregation. As the greatest percentage of our patients with keratoconus were of a unique Polynesian ethnicity, this may suggest that VSX1 does not play a causative role within this ethnic group.
This study confirms the presence of pathogenic mutations in PPCD and keratoconus. The affected numbers are small, but given the growing body of evidence of pathogenic segregating changes in VSX1 in these cohorts, the expression in keratocytes as part of wound healing, and the documented association of PPCD and keratoconus, it seems likely that the role of VSX1 is real, but not significant in terms of actual numbers. Genetic heterogeneity is almost the norm in a large number of complex eye conditions such as the retinal dystrophies and glaucomas, with these corneal disorders also demonstrating a similar complexity in genetic causation.
Acknowledgments
Jian Li for technical work during a Summer Studentship, Amanda Richards, and Bryan Hay for excellent technical assistance. Funding. Save Sight Society of New Zealand, Auckland Medical Research Foundation.
Appendix 1. VSX1 Primers and conditions.
To access the data, click or select the words “Appendix 1.”
References
- 1.Semina EV, Mintz-Hittner HA, Murray JC. Isolation and characterization of a novel human paired-like homeodomain-containing transcription factor gene, VSX1, expressed in ocular tissues. Genomics. 2000;63:289–93. doi: 10.1006/geno.1999.6093. [DOI] [PubMed] [Google Scholar]
- 2.Dash DP, George S, O'Prey D, Burns D, Nabili S, Donnelly U, Hughes AE, Silvestri G, Jackson J, Frazer D, Heon E, Willoughby CE. Mutational screening of VSX1 in keratoconus patients from the European population. Eye (Lond) 2010;24:1085–92. doi: 10.1038/eye.2009.217. [DOI] [PubMed] [Google Scholar]
- 3.Hosseini SM, Herd S, Vincent AL, Heon E. Genetic analysis of chromosome 20-related posterior polymorphous corneal dystrophy: genetic heterogeneity and exclusion of three candidate genes. Mol Vis. 2008;14:71–80. [PMC free article] [PubMed] [Google Scholar]
- 4.Barbaro V, Di Iorio E, Ferrari S, Bisceglia L, Ruzza A, De Luca M, Pellegrini G. Expression of VSX1 in human corneal keratocytes during differentiation into myofibroblasts in response to wound healing. Invest Ophthalmol Vis Sci. 2006;47:5243–50. doi: 10.1167/iovs.06-0185. [DOI] [PubMed] [Google Scholar]
- 5.Mintz-Hittner HA, Semina EV, Frishman LJ, Prager TC, Murray JC. VSX1 (RINX) mutation with craniofacial anomalies, empty sella, corneal endothelial changes, and abnormal retinal and auditory bipolar cells. Ophthalmology. 2004;111:828–36. doi: 10.1016/j.ophtha.2003.07.006. [DOI] [PubMed] [Google Scholar]
- 6.Héon E, Greenberg A, Kopp KK, Rootman D, Vincent AL, Billingsley G, Priston M, Dorval KM, Chow RL, McInnes RR, Heathcote G, Westall C, Sutphin JE, Semina E, Bremner R, Stone EM. VSX1: a gene for posterior polymorphous dystrophy and keratoconus. Hum Mol Genet. 2002;11:1029–36. doi: 10.1093/hmg/11.9.1029. [DOI] [PubMed] [Google Scholar]
- 7.Valleix S, Nedelec B, Rigaudiere F, Dighiero P, Pouliquen Y, Renard G, Le Gargasson JF, Delpech M. H244R VSX1 is associated with selective cone ON bipolar cell dysfunction and macular degeneration in a PPCD family. Invest Ophthalmol Vis Sci. 2006;47:48–54. doi: 10.1167/iovs.05-0479. [DOI] [PubMed] [Google Scholar]
- 8.Aldave AJ, Yellore VS, Principe AH, Abedi G, Merrill K, Chalukya M, Small KW, Udar N. Candidate gene screening for posterior polymorphous dystrophy. Cornea. 2005;24:151–5. doi: 10.1097/01.ico.0000141235.26096.1d. [DOI] [PubMed] [Google Scholar]
- 9.Gwilliam R, Liskova P, Filipec M, Kmoch S, Jirsova K, Huckle EJ, Stables CL, Bhattacharya SS, Hardcastle AJ, Deloukas P, Ebenezer ND. Posterior polymorphous corneal dystrophy in Czech families maps to chromosome 20 and excludes the VSX1 gene. Invest Ophthalmol Vis Sci. 2005;46:4480–4. doi: 10.1167/iovs.05-0269. [DOI] [PubMed] [Google Scholar]
- 10.Krafchak CM, Pawar H, Moroi SE, Sugar A, Lichter PR, Mackey DA, Mian S, Nairus T, Elner V, Schteingart MT, Downs CA, Kijek TG, Johnson JM, Trager EH, Rozsa FW, Mandal MN, Epstein MP, Vollrath D, Ayyagari R, Boehnke M, Richards JE. Mutations in TCF8 cause posterior polymorphous corneal dystrophy and ectopic expression of COL4A3 by corneal endothelial cells. Am J Hum Genet. 2005;77:694–708. doi: 10.1086/497348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Biswas S, Munier FL, Yardley J, Hart-Holden N, Perveen R, Cousin P, Sutphin JE, Noble B, Batterbury M, Kielty C, Hackett A, Bonshek R, Ridgway A, McLeod D, Sheffield VC, Stone EM, Schorderet DF, Black GC. Missense mutations in COL8A2, the gene encoding the alpha2 chain of type VIII collagen, cause two forms of corneal endothelial dystrophy. Hum Mol Genet. 2001;10:2415–23. doi: 10.1093/hmg/10.21.2415. [DOI] [PubMed] [Google Scholar]
- 12.Bechara SJ, Grossniklaus HE, Waring GO, 3rd, Wells JA., 3rd Keratoconus associated with posterior polymorphous dystrophy. Am J Ophthalmol. 1991;112:729–31. doi: 10.1016/s0002-9394(14)77284-8. [DOI] [PubMed] [Google Scholar]
- 13.Blair SD, Seabrooks D, Shields WJ, Pillai S, Cavanagh HD. Bilateral progressive essential iris atrophy and keratoconus with coincident features of posterior polymorphous dystrophy: a case report and proposed pathogenesis. Cornea. 1992;11:255–61. [PubMed] [Google Scholar]
- 14.Cremona FA, Ghosheh FR, Rapuano CJ, Eagle RC, Jr, Hammersmith KM, Laibson PR, Ayres BD, Cohen EJ. Keratoconus associated with other corneal dystrophies. Cornea. 2009;28:127–35. doi: 10.1097/ICO.0b013e3181859935. [DOI] [PubMed] [Google Scholar]
- 15.Lam HY, Wiggs JL, Jurkunas UV. Unusual presentation of presumed posterior polymorphous dystrophy associated with iris heterochromia, band keratopathy, and keratoconus. Cornea. 2010;29:1180–5. doi: 10.1097/ICO.0b013e3181d007e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weissman BA, Ehrlich M, Levenson JE, Pettit TH. Four cases of keratoconus and posterior polymorphous corneal dystrophy. Optom Vis Sci. 1989;66:243–6. doi: 10.1097/00006324-198904000-00012. [DOI] [PubMed] [Google Scholar]
- 17.Bisceglia L, Ciaschetti M, De Bonis P, Campo PA, Pizzicoli C, Scala C, Grifa M, Ciavarella P, Delle Noci N, Vaira F, Macaluso C, Zelante L. VSX1 mutational analysis in a series of Italian patients affected by keratoconus: detection of a novel mutation. Invest Ophthalmol Vis Sci. 2005;46:39–45. doi: 10.1167/iovs.04-0533. [DOI] [PubMed] [Google Scholar]
- 18.Eran P, Almogit A, David Z, Wolf HR, Hana G, Yaniv B, Elon P, Isaac A. The D144E substitution in the VSX1 gene: a non-pathogenic variant or a disease causing mutation? Ophthalmic Genet. 2008;29:53–9. doi: 10.1080/13816810802008242. [DOI] [PubMed] [Google Scholar]
- 19.Mok JW, Baek SJ, Joo CK. VSX1 gene variants are associated with keratoconus in unrelated Korean patients. J Hum Genet. 2008;53:842–9. doi: 10.1007/s10038-008-0319-6. [DOI] [PubMed] [Google Scholar]
- 20.Paliwal P, Singh A, Tandon R, Titiyal JS, Sharma A. A novel VSX1 mutation identified in an individual with keratoconus in India. Mol Vis. 2009;15:2475–9. [PMC free article] [PubMed] [Google Scholar]
- 21.Aldave AJ, Yellore VS, Salem AK, Yoo GL, Rayner SA, Yang H, Tang GY, Piconell Y, Rabinowitz YS. No VSX1 gene mutations associated with keratoconus. Invest Ophthalmol Vis Sci. 2006;47:2820–2. doi: 10.1167/iovs.05-1530. [DOI] [PubMed] [Google Scholar]
- 22.Liskova P, Ebenezer ND, Hysi PG, Gwilliam R, El-Ashry MF, Moodaley LC, Hau S, Twa M, Tuft SJ, Bhattacharya SS. Novel mutations in the ZEB1 gene identified in Czech and British patients with posterior polymorphous corneal dystrophy. Hum Mutat. 2007;28:638. doi: 10.1002/humu.9495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stabuc-Silih M, Strazisar M, Hawlina M, Glavac D. Absence of pathogenic mutations in VSX1 and SOD1 genes in patients with keratoconus. Cornea. 2010;29:172–6. doi: 10.1097/ICO.0b013e3181aebf7a. [DOI] [PubMed] [Google Scholar]
- 24.Tang YG, Picornell Y, Su X, Li X, Yang H, Rabinowitz YS. Three VSX1 gene mutations, L159M, R166W, and H244R, are not associated with keratoconus. Cornea. 2008;27:189–92. doi: 10.1097/ICO.0b013e31815a50e7. [DOI] [PubMed] [Google Scholar]
- 25.McGhee CN. 2008 Sir Norman McAlister Gregg Lecture: 150 years of practical observations on the conical cornea–what have we learned? Clin Experiment Ophthalmol. 2009;37:160–76. doi: 10.1111/j.1442-9071.2009.02009.x. [DOI] [PubMed] [Google Scholar]
- 26.Nielsen K, Hjortdal J, Pihlmann M, Corydon TJ. Update on the keratoconus genetics. Acta Ophthalmol (Copenh) 2012 doi: 10.1111/j.1755-3768.2012.02400.x. [DOI] [PubMed] [Google Scholar]
- 27.Burdon KP, Macgregor S, Bykhovskaya Y, Javadiyan S, Li X, Laurie KJ, Muszynska D, Lindsay R, Lechner J, Haritunians T, Henders AK, Dash D, Siscovick D, Anand S, Aldave A, Coster DJ, Szczotka-Flynn L, Mills RA, Iyengar SK, Taylor KD, Phillips T, Montgomery GW, Rotter JI, Hewitt AW, Sharma S, Rabinowitz YS, Willoughby C, Craig JE. Association of polymorphisms in the hepatocyte growth factor gene promoter with keratoconus. Invest Ophthalmol Vis Sci. 2011;52:8514–9. doi: 10.1167/iovs.11-8261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hughes AE, Bradley DT, Campbell M, Lechner J, Dash DP, Simpson DA, Willoughby CE. Mutation altering the miR-184 seed region causes familial keratoconus with cataract. Am J Hum Genet. 2011;89:628–33. doi: 10.1016/j.ajhg.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jordan CA, Zamri A, Wheeldon C, Patel DV, Johnson R, McGhee CN. Computerized corneal tomography and associated features in a large New Zealand keratoconic population. J Cataract Refract Surg. 2011;37:1493–501. doi: 10.1016/j.jcrs.2011.03.040. [DOI] [PubMed] [Google Scholar]
- 30.Owens H, Gamble GD, Bjornholdt MC, Boyce NK, Keung L. Topographic indications of emerging keratoconus in teenage New Zealanders. Cornea. 2007;26:312–8. doi: 10.1097/ICO.0b013e31802f8d87. [DOI] [PubMed] [Google Scholar]
- 31.Edwards M, McGhee CN, Dean S. The genetics of keratoconus. Clin Experiment Ophthalmol. 2001;29:345–51. doi: 10.1046/j.1442-9071.2001.d01-16.x. [DOI] [PubMed] [Google Scholar]
- 32.Patel HY, Ormonde S, Brookes NH, Moffatt SL, Sherwin T, Pendergrast DG, McGhee CN. The New Zealand National Eye Bank: survival and visual outcome 1 year after penetrating keratoplasty. Cornea. 2011;30:760–4. doi: 10.1097/ICO.0b013e3182014668. [DOI] [PubMed] [Google Scholar]
- 33.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Altshuler DM, Gibbs RA, Peltonen L, Dermitzakis E, Schaffner SF, Yu F, Bonnen PE, de Bakker PI, Deloukas P, Gabriel SB, Gwilliam R, Hunt S, Inouye M, Jia X, Palotie A, Parkin M, Whittaker P, Chang K, Hawes A, Lewis LR, Ren Y, Wheeler D, Muzny DM, Barnes C, Darvishi K, Hurles M, Korn JM, Kristiansson K, Lee C, McCarrol SA, Nemesh J, Keinan A, Montgomery SB, Pollack S, Price AL, Soranzo N, Gonzaga-Jauregui C, Anttila V, Brodeur W, Daly MJ, Leslie S, McVean G, Moutsianas L, Nguyen H, Zhang Q, Ghori MJ, McGinnis R, McLaren W, Takeuchi F, Grossman SR, Shlyakhter I, Hostetter EB, Sabeti PC, Adebamowo CA, Foster MW, Gordon DR, Licinio J, Manca MC, Marshall PA, Matsuda I, Ngare D, Wang VO, Reddy D, Rotimi CN, Royal CD, Sharp RR, Zeng C, Brooks LD, McEwen JE. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–8. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chow RL, Snow B, Novak J, Looser J, Freund C, Vidgen D, Ploder L, McInnes RR. Vsx1, a rapidly evolving paired-like homeobox gene expressed in cone bipolar cells. Mech Dev. 2001;109:315–22. doi: 10.1016/s0925-4773(01)00585-8. [DOI] [PubMed] [Google Scholar]
- 37.Chow RL, Volgyi B, Szilard RK, Ng D, McKerlie C, Bloomfield SA, Birch DG, McInnes RR. Control of late off-center cone bipolar cell differentiation and visual signaling by the homeobox gene Vsx1. Proc Natl Acad Sci USA. 2004;101:1754–9. doi: 10.1073/pnas.0306520101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ohtoshi A, Wang SW, Maeda H, Saszik SM, Frishman LJ, Klein WH, Behringer RR. Regulation of retinal cone bipolar cell differentiation and photopic vision by the CVC homeobox gene Vsx1. Curr Biol. 2004;14:530–6. doi: 10.1016/j.cub.2004.02.027. [DOI] [PubMed] [Google Scholar]
- 39.Watson T, Chow RL. Absence of Vsx1 expression in the normal and damaged mouse cornea. Mol Vis. 2011;17:737–44. [PMC free article] [PubMed] [Google Scholar]
- 40.Héon E, Mathers WD, Alward WL, Weisenthal RW, Sunden SL, Fishbaugh JA, Taylor CM, Krachmer JH, Sheffield VC, Stone EM. Linkage of posterior polymorphous corneal dystrophy to 20q11. Hum Mol Genet. 1995;4:485–8. doi: 10.1093/hmg/4.3.485. [DOI] [PubMed] [Google Scholar]
- 41.Aldave AJ, Yellore VS, Vo RC, Kamal KM, Rayner SA, Plaisier CL, Chen MC, Damani MR, Pham MN, Gorin MB, Sobel E, Papp J. Exclusion of positional candidate gene coding region mutations in the common posterior polymorphous corneal dystrophy 1 candidate gene interval. Cornea. 2009;28:801–7. doi: 10.1097/ICO.0b013e31819672fb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lai IN, Yellore VS, Rayner SA, D'Silva NC, Nguyen CK, Aldave AJ. The utility of next-generation sequencing in the evaluation of the posterior polymorphous corneal dystrophy 1 locus. Mol Vis. 2010;16:2829–38. [PMC free article] [PubMed] [Google Scholar]
- 43.Liskova P, Gwilliam R, Filipec M, Jirsova K, Reinstein Merjava S, Deloukas P, Webb TR, Bhattacharya SS, Ebenezer ND, Morris AG, Hardcastle AJ. High prevalence of posterior polymorphous corneal dystrophy in the czech republic; linkage disequilibrium mapping and dating an ancestral mutation. PLoS ONE. 2012;7:e45495. doi: 10.1371/journal.pone.0045495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Bonis P, Laborante A, Pizzicoli C, Stallone R, Barbano R, Longo C, Mazzilli E, Zelante L, Bisceglia L. Mutational screening of VSX1, SPARC, SOD1, LOX, and TIMP3 in keratoconus. Mol Vis. 2011;17:2482–94. [PMC free article] [PubMed] [Google Scholar]
- 45.Saee-Rad S, Hashemi H, Miraftab M, Noori-Daloii MR, Chaleshtori MH, Raoofian R, Jafari F, Greene W, Fakhraie G, Rezvan F, Heidari M. Mutation analysis of VSX1 and SOD1 in Iranian patients with keratoconus. Mol Vis. 2011;17:3128–36. [PMC free article] [PubMed] [Google Scholar]
- 46.Paliwal P, Tandon R, Dube D, Kaur P, Sharma A. Familial segregation of a VSX1 mutation adds a new dimension to its role in the causation of keratoconus. Mol Vis. 2011;17:481–5. [PMC free article] [PubMed] [Google Scholar]
- 47.Raber IM, Fintelmann R, Chhabra S, Ribeiro MP, Eagle RC, Jr, Orlin SE. Posterior polymorphous dystrophy associated with nonkeratoconic steep corneal curvatures. Cornea. 2011;30:1120–4. doi: 10.1097/ICO.0b013e3182114452. [DOI] [PubMed] [Google Scholar]
- 48.Liskova P, Filipec M, Merjava S, Jirsova K, Tuft SJ. Variable ocular phenotypes of posterior polymorphous corneal dystrophy caused by mutations in the ZEB1 gene. Ophthalmic Genet. 2010;31:230–4. doi: 10.3109/13816810.2010.518577. [DOI] [PubMed] [Google Scholar]