

Abstract
Although NADPH oxidase 4 (Nox4) is the most abundant Nox isoform in systemic vascular endothelial and smooth muscle cells, its function in the vascular tissue is not entirely known. The literature describes a pathophysiological role for Nox4 in cardiovascular disease; however, some studies have reported that it has a protective role. To date, specific Nox4 inhibitors are not available; hence, the development of a pharmacologic tool to assess Nox4's pathophysiological role garners intense interest. In this study, we selected peptides corresponding to regions in the Nox4 oxidase complex critical to holoenzyme activity and postulated their utility as specific competitive inhibitors. Previous studies in our laboratory yielded selective inhibition of Nox2 using this strategy. We postulated that peptides mimicking the Nox4 B-loop and C-terminus and regions on p22phox inhibit Nox4 activity. To test our hypothesis, the inhibitory activity of Nox4 B-loop and C-terminal peptides as well as N-terminal p22phox peptides was assessed in a reconstituted Nox4 system. Our findings demonstrate that Nox4 inhibition is not achieved by preincubation with this comprehensive array of peptides derived from previously identified active regions. These findings suggest that Nox4 exists in a tightly assembled and active conformation which, unlike other Noxes, cannot be disrupted by conventional means.
1. Introduction
NADPH-oxidase- (Nox-) derived reactive oxygen species (ROS) play a central role in the destruction of pathogenic organisms by phagocytes. The phagocyte Nox complex is composed of flavocytochrome b558, an integral membrane heterodimer composed of gp91phox (a.k.a. Nox2) and p22phox, four cytoplasmic protein subunits, p47phox, p67phox, p40phox, and the regulatory low molecular weight GTPase Rac. In resting phagocytes, the Nox enzyme is in a dormant state. Upon activation, the cytosolic Nox subunits translocate to Nox2 and p22phox followed by the transfer of one electron from NADPH to molecular oxygen, resulting in the formation of superoxide anion (O2 •−) and microbicidal activity [1].
Nox2 is also expressed in cells other than phagocytes, [2, 3] and excessive ROS generation by nonphagocytic Nox2 contributes to a wide variety of disorders [3–5]. Over the past decade since the discovery of Nox2 homologs Nox1, Nox3, Nox4, Nox5, DUOX1 and DUOX2 [4], interest has greatly increased in Nox enzymes and the development of isoform-specific Nox inhibitors. Although numerous chemical compounds have been shown to inhibit Nox enzymes, none of these to our knowledge is specific for one isoform [6, 7]. Importantly, rationally designed, sequence-specific peptide-based inhibitors have the potential to be among the most selective and effective inhibitors of Nox because of their potential to selectively target unique protein interactions within the enzyme. A previous study by our group demonstrated that a peptide sequence mimicking amino acids 86–94 in the first intracellular loop of Nox2 (B-loop) specifically inhibits Nox2 activation in vitro [8, 9]. The effectiveness of this peptide to inhibit ROS production in vivo has been widely shown, and has led to its wide use in numerous studies [10–15]. To date, peptidic inhibitors have been reserved for Nox2; that is, no prior studies tested whether inhibition of other homologs can be achieved by this strategy.
Nox4 is the most abundant Nox isoform in endothelial cells, vascular smooth muscle cells, and the kidney [16, 17], but it is also expressed in the heart, central nervous system, airways, and skeletal muscle [4]. Animal and human studies have shown that Nox4 plays an important role in the pathophysiology of a wide variety of disorders, including systemic hypertension [18], diabetes mellitus [19], vascular injury [20], atherosclerosis [21], ischemic stroke [22], pulmonary fibrosis [23], and diabetic nephropathy [24]. Collectively, these data suggest that Nox4 oxidase is a major contributor to oxidative stress in these pathologic conditions, and blocking the undesirable actions of Nox4 could become a therapeutic strategy to attenuate oxidative stress in patients with these disorders. Unlike other Nox isoforms, numerous studies have also described that Nox4 is involved in a variety of physiological processes, including cell differentiation, survival, and migration [16, 25–27]. Moreover, a few studies have reported that Nox4 has a protective role in cardiovascular tissue, although this is still somewhat controversial [19, 28, 29]. Presently, no specific inhibitors of Nox4 (small molecule or peptidic) are available to the scientific community [30] to elucidate the pathophysiological and/or physiological roles of Nox4.
Nox4 oxidase is a unique Nox isozyme as it differs from the usual model of multimeric Nox assembly found in Nox1, Nox2, and Nox3. Indeed, Nox4 does not require interaction with any of the conventional cytosolic Nox subunits for ROS generation and the membrane-bound subunit p22phox is, to date, the only known classical subunit associated with Nox4. Recently, Poldip2 has been described as a modulator of Nox4 [31]; however, in this study we aimed to target the core of the enzyme.
A previous study reported that mutagenesis of arginine residues in the Nox4 B-loop impedes activity of Nox4 [32]. Moreover, it was suggested that the B-loop of Nox4 serves as a binding sequence facilitating interaction of C-terminal NADPH-binding domain of Nox4 with its membrane-spanning region [32]. In addition, deletion of the first 11 amino acids at the p22phox N-terminus completely abrogated Nox4 activity [33]. These data suggest that interaction of Nox4 B-loop with the C-terminal domain as well as association of Nox4 with the N-terminus of p22phox is important for Nox4 activity. We postulated that recombinant excess of Nox4 B-loop and key p22phox N-terminal peptide mimics would disrupt intramolecular B-loop-C-terminal and Nox4-p22phox interactions, respectively, leading to inhibition of Nox4-derived ROS production. Accordingly, the aim of the present study was to investigate whether targeting the Nox4 B-loop and C-terminal domain with sequential 15-mer and nonamer peptide sequences disrupts their interaction and inhibits Nox4 activity. In the present study, p22phox N-terminal tail peptides were also tested for their ability to inhibit Nox4 activity.
2. Materials and Methods
Catalase, diphenyleneiodonium chloride (DPI), flavin adenine dinucleotide (FAD), horseradish peroxidase (HRP), and phenylmethanesulfonyl fluoride (PMSF) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Amplex Red was purchased from Invitrogen (Eugene, OR, USA). Protease inhibitor cocktail was purchased from Roche Diagnostics GmbH (Mannheim, Germany). All peptides were synthesized by the Tufts University Core Facility (Boston, MA, USA). The purity of peptides was over 97%. Since the current studies were carried out using COS-Nox4 cell lysates, it is important to note that the peptides used in this study did not require chimeric design containing tat peptide for cell permeation.
2.1. Cell Lines and Cell Culture
COS-22 cells (COS-7 cells stably expressing human p22phox) were kindly provided by Dr. Mary C. Dinauer (Indiana University, School of Medicine) [34]. COS-22 cells were maintained in Dulbecco's Modified Eagle Medium (Mediatech, Inc., Manassas, VA, USA) with 4.5 g/L glucose, L-glutamine, and sodium pyruvate containing 10% heat-inactivated fetal bovine serum (FBS), 100 units/mL penicillin, and 100 μg/mL streptomycin.
2.2. Plasmid Preparation, Amplification, and Purification
Plasmid encoding full-length human cDNAs for Nox4 (pcDNA3-hNox4) was kindly provided by Dr. David Lambeth (Emory University, GA) [35]. For human Nox4 expression, the BglII/NotI restriction fragment from the pcDNA3-hNox4 was subcloned into the plasmid pcDNA3.1/Hygro(−) (Invitrogen, Carlsbad, CA) to generate pcDNA3.1/Hygro-hNox4. The fragment sequence, in-frame insertion, and orientation were validated by DNA sequencing after PCR amplification. pcDNA3.1/Hygro-hNox4 was amplified into Escherichia coli strain TOP10 (Invitrogen, Carlsbad, CA) and purified with a QIA filter plasmid purification kit (QIAGEN Inc., Valencia, CA).
2.3. Transfection
Cell transfection was carried out using Lipofectamine LTX and Plus reagent (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's instructions. COS-22 cells were transiently transfected with pcDNA3.1/Hygro-hNox4 (COS-Nox4 cells). Western blot experiments were performed to validate the expression of Nox4 as we reported previously [8]. Twenty-four hours after transfection, cells were harvested by incubating with 0.05% trypsin/EDTA for 5 min at 37°C. Following addition of DMEM/10% FBS to neutralize the trypsin, the cells were pelleted by centrifugation at 1000 ×g for 5 min at 4°C and used for the experiments.
2.4. Hydrogen Peroxide- (H2O2-) Generating Activity
H2O2 production was quantified in lysed COS-Nox4 cells as described previously [36]. It is important to note that COS-Nox4 cells do not produce O2 •− [8]. COS-Nox4 and COS-22 cells were suspended to a concentration of 5 × 107 cells/mL in ice-cold disruption buffer (PBS containing 0.1 mM EDTA, 10% glycerol, protease inhibitor cocktail, and 0.1 mM PMSF). The cells were lysed by five freeze/thaw cycles and passed through a 30-gauge needle five times to further lyse the cells. Throughout all these procedures, extreme care was taken to maintain the lysate at a temperature close to 0°C. Incubation of COS-Nox4 cell lysate (10 μg/100 μL) with peptides was performed in assay buffer (25 mM Hepes, pH 7.4, containing 120 mM NaCl, 3 mM KCl, 1 mM MgCl2, 25 μM FAD, 0.1 mM Amplex Red, and 0.32 U/mL of HRP) for 15 or 60 min at room temperature on an orbital shaker (120 movements/min), before the addition of 36 μM NADPH, to initiate H2O2 production. This relatively low concentration of NADPH was used because it was found that higher concentrations interfered with Amplex Red fluorescence. Fluorescence measurements were made using a Biotek Synergy 4 Hybrid Multi-Mode Microplate Reader (excitation wavelength: 560 nm; emission wavelength: 590 nm). A standard curve of known H2O2 concentrations was developed using the Amplex Red assay (as per the manufacturer's instructions) and was used to quantify H2O2 production in lysed COS-Nox4 cells. The reaction was monitored at room temperature for 40 min. The emission increase was linear during this interval. The rate of H2O2 production was interpolated from the standard curve and is 609 ± 0.01 nmol H2O2/min/mg protein. The effect of a peptide on Nox4-derived H2O2 production was expressed as percent inhibition of Nox4, which was calculated by considering H2O2 production by control mixtures in the absence of peptide as 100%.
2.5. Statistical Analysis
All results are expressed as means ± SEM. Significance of the differences was assessed by two-way ANOVA followed by Bonferroni post hoc test. P < 0.05 was considered to be statistically significant.
3. Results
3.1. Nox4 Catalytic Activity Is Not Inhibited by Preincubation with Nox4 B-Loop Peptides
The first intracellular loop of Nox4 (B-loop) is essential for catalytic activity, that is, hydrogen peroxide (H2O2) generation, as previously demonstrated by point mutations targeting this region (Figure 1(a)) [32]. Amino acid sequence alignment revealed three subregions of Nox B-loops where two conserved regions flank a highly variable central region (Figure 1(b)). The two flanking conserved regions are shared by all Nox isoforms, whereas the central region is variable across Nox isoforms [32]. Previous data indicated that B-loop peptides bind to the C-terminal of the enzyme and that this interaction is important for activity [32]. We postulated then that this interaction would be competitively blocked by peptides derived from the B-loop, as we showed for Nox2 [8]. In the current study, peptide sequences derived from the variable and conserved regions of Nox4 B-loop were tested for their ability to inhibit Nox4 activity. As shown in Figure 1(c), the rate of H2O2 generation was significantly higher in COS-Nox4 cell lysates as compared with nontransfected controls, and ~90% of the Nox4-dependent activity was inhibited by the flavin-containing enzyme inhibitor diphenyleneiodonium (DPI; 50 μM). Detection of H2O2 was confirmed by inhibition of fluorescence with catalase (3000 U/mL). As demonstrated in Figure 1(d), peptide B90–98 (0.1–100 μM) did not inhibit Nox4-derived H2O2 production. As substitution of Arg-96 to glutamic acid previously resulted in almost complete loss of Nox4 activity [32], we tested whether the R96E mutant version of B90–98 (PSRRTRELL) is capable of inhibiting Nox4 activity. Similar to the wild-type peptide, the mutant peptide did not inhibit Nox4 activity (Figure 1(e)).
Figure 1.
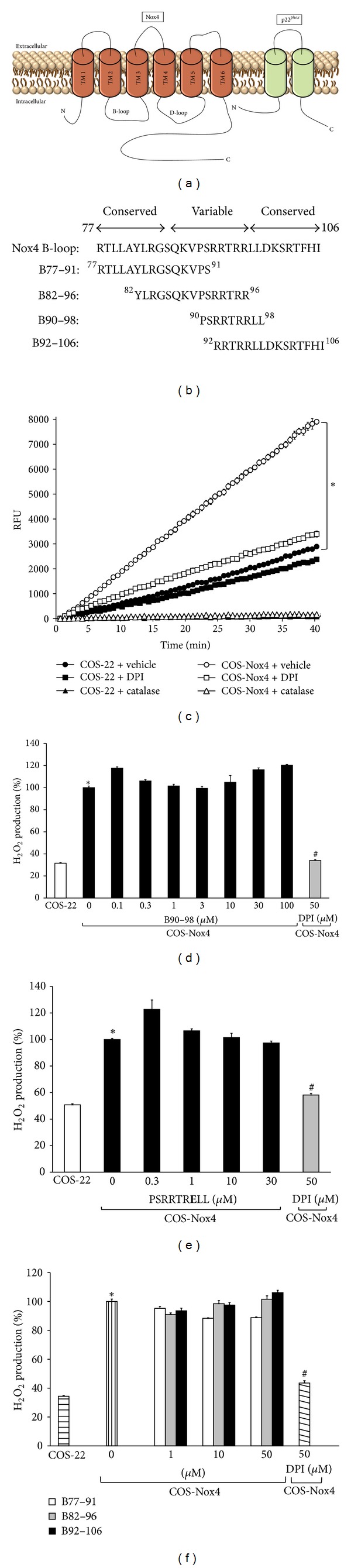
Nox4 B-loop peptides and Nox4 activity. (a) A simplified model of Nox4 oxidase with B- and D-loops located intracellularly. p22phox, the only known protein required for Nox4 activity, is also shown. (b) Amino acid sequence of Nox4 B-loop and list of overlapping Nox4 B-loop region synthetic 15-mer and nonamer peptides used to target Nox4 activity. The numbers at the N- and C-terminus of each peptide indicate the location of the corresponding residues in the amino acid sequence of Nox4. (c) H2O2 generated by Nox4 was measured using Amplex Red fluorescence. H2O2 production was initiated by the addition of 36 μM NADPH. The reaction was monitored at 24°C for 40 min. The flavin-containing enzyme inhibitor diphenyleneiodonium (DPI; 50 μM) was used as a positive control. Identification of H2O2 was confirmed by inhibition of fluorescence with catalase (3000 U/mL). For comparison, H2O2 production in nontransfected COS-22 cell lysate is shown. Data represent the mean ± SEM of 3 experiments. *P < 0.05 indicates significant difference between COS-22 and COS-Nox4 cell lysates. (d) COS-Nox4 cell lysates were preincubated with various concentrations of B90–98 (from 0.1 to 100 μM) for 15 min at 24°C, and H2O2 was measured using Amplex Red fluorescence. Data represent the mean ± SEM of 3 experiments. (e) COS-Nox4 cell lysates were preincubated with the R96E mutant version of B90–98 (PSRRTRELL; 0.3 to 30 μM) for 15 min at 24°C, and H2O2 was measured. Data represent the mean ± SEM of 3 experiments. (f) COS-Nox4 cell lysates were preincubated with peptides from N- and C-terminal ends of the Nox4 B-loop (B77–91, B82–96, and B92–106; 1 to 50 μM) for 15 min at 24°C, and H2O2 was measured. Data represent the means ± SEM of 3 experiments. *P < 0.05 indicates a significant difference between COS-22 and COS-Nox4 cell lysates. # P < 0.05 indicates a significant difference between COS-Nox4 + 0 μM peptide and COS-Nox4 + DPI.
Mutation of Arg-84 to alanine on the N-terminal conserved Nox4 B-loop region [32] and replacement of Ser-101 with acidic residues in the C-terminal Nox4 B-loop conserved region were previously shown to inhibit Nox4 [37]. Thus, we tested whether inhibition of Nox4 activity can be achieved by proximal and distal Nox4 B-loop peptides. Our data demonstrated that preincubation of COS-Nox4 cell lysates with neither peptides B77–91, B82–96, nor B92–106 inhibited H2O2 production (Figure 1(f)).
3.2. Nox4 C-Terminal Tail Peptides Do Not Inhibit Nox4 NADPH Oxidase
A previous study demonstrated that the last 22 amino acids of the whole Nox4 protein are critical for catalytic activity (Figure 2(a)) [38]. The presence of charged residues in this flexible region of Nox4 may suggest that electrostatic effects could promote interaction between the C-terminus and B-loop and/or p22phox. Thus, three overlapping octameric peptides (C555–562, C560–567, and C565–572) and a nonameric peptide (C570–578) were synthesized to cover the last 22 amino acids of Nox4, and these were tested for their ability to inhibit Nox4 activity. Application of these C-terminal tail peptides did not affect Nox4 activity (Figure 2(b)).
Figure 2.
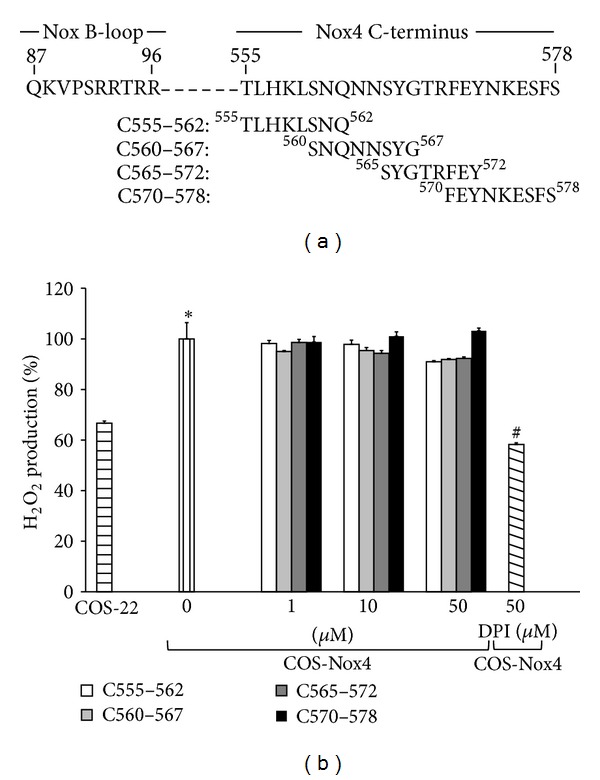
Examination of Nox4 C-terminal tail peptides as Nox4 inhibitors. (a) Map of overlapping Nox4 C-terminal region synthetic octa- and nonapeptides used to inhibit Nox4 activity. Numbers correspond to location and span of the N- and C-terminus for each peptide. (b) COS-Nox4 cell lysates were preincubated with various concentrations of C555–562, C560–567, C565–572, and C570–578 for 15 min at 24°C, and H2O2 was measured using Amplex Red. Data represent the mean ± SEM of 3 experiments. *P < 0.05 indicates a significant difference between COS-22 and COS-Nox4 cell lysates. # P < 0.05 indicates a significant difference between COS-Nox4 + 0 μM peptide and COS-Nox4 + DPI.
3.3. p22phox N-Terminal Tail Peptides Do Not Inhibit Nox4 NADPH Oxidase
Expression of p22phox is required for Nox4 activity, and, to date, p22phox is the only known classical core protein associated with Nox4 [39]. Mutagenesis studies provided evidence that deletion of a large span of the p22phox C-terminus (terminal 130 amino acids) did not affect Nox4 activity [33]. In contrast, deletion of the first 11 amino acids at the p22phox N-terminus attenuated Nox4 activity. Deletion of the first 5 amino acids did not affect Nox4 activity, suggesting that the N-terminal region of p22phox between amino acids 6 and 11 is sensitive to modification. A peptide sequence between amino acids 6 and 11 (p22 WT 6–11 (WAMWAN)) was thus synthesized and tested for its ability to inhibit Nox4 (Figure 3(a)). As demonstrated in Figure 3(b), peptide p22 WT 6–11 did not inhibit Nox4 activity. Mutation of tryptophans within this sequence to arginine (p22 W6/9R (RAMRAN)) also abolished Nox4 activity [33]. Similar to the native peptide, the p22 W6/9R mutant did not inhibit Nox4-derived H2O2 production.
Figure 3.
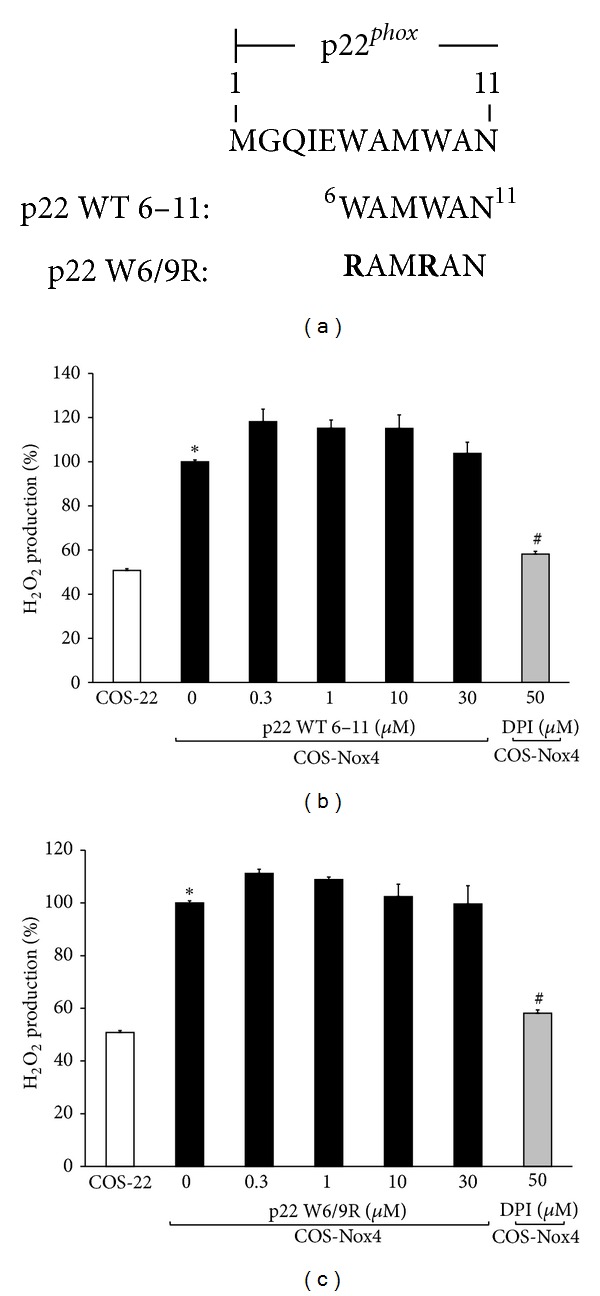
Testing whether p22phox N-terminal tail peptides inhibit Nox4 activity. (a) The first 11 amino acids of p22phox are displayed. The peptide sequence of p22phox between amino acids 6 and 11 is shown (p22 WT 6–11; WAMWAN); point mutations of W6/9R (p22 W6/9R; RAMRAN) are indicated in boldface type. The numbers at the N- and C-terminus of WAMWAN indicate the location of the corresponding residues in the amino acid sequence of p22phox. (b) COS-Nox4 cell lysates were preincubated with various concentrations of p22 WT 6–11 for 15 min at 24°C, and H2O2 was measured using Amplex Red. Data represent the mean ± SEM of 3 experiments. (c) COS-Nox4 cell lysates were preincubated with various concentrations of p22 W6/9R for 15 min at 24°C, and H2O2 was measured using Amplex Red. Data represent the mean ± SEM of 3 experiments. *P < 0.05 indicates a significant difference between COS-22 and COS-Nox4 cell lysates. # P < 0.05 indicates a significant difference between COS-Nox4 + 0 μM peptide and COS-Nox4 + DPI.
3.4. Increased Incubation Time and Temperature Do Not Facilitate Peptide-Induced Inhibition
Our data demonstrate that preincubation of COS-Nox4 cell lysates with Nox4 B-loop, Nox4 C-terminal tail, and N-terminal p22phox peptides for 15 min at 24°C did not inhibit Nox4 activity. To allow more time for the peptides to disrupt the targeted domain interactions, we increased incubation time up to 60 min. Our results demonstrated that none of the Nox4 B-loop, Nox4 C-terminal tail, and N-terminal p22phox peptides inhibit Nox4 activity after 60 min of incubation (data not shown). Moving to a new approach, we tested whether providing more kinetic energy, which we proposed to be more favorable for peptide interference with the tightly assembled conformation of Nox4, would facilitate peptide-induced inhibition. Thus, in an attempt to induce temporary perturbations in the structure of Nox4, these experiments were also performed at 37°C. Again, no inhibition of Nox4 was achieved (data not shown).
4. Discussion
This is the first study to our knowledge that seeks to inhibit Nox4 using a peptidic strategy. Indeed, synthetic peptides mimicking key residues in Nox2, p22phox, p47phox, and Rac1 have been shown to interfere with the activation process of Nox2 and inhibit Nox2-derived O2 •− production [8, 9, 40–43]. Those studies identified domains of functional importance in the assembly of Nox2 oxidase and provided key information about the transformation of the enzyme complex from the dormant, inactive conformation to its active state of the enzyme. Nox1, Nox3, and Duox require cytosolic subunits for activation, while Nox5 does not. Nox4, with p22phox, appears to constitutively generate H2O2 without the requirement of activating cytosolic subunits, with the exception of Poldip2 [31]. With a desire to target the core of the enzyme, the present study was designed to (a) target multiple residues in the Nox4 sequence with the purpose of determining whether prior information gleaned from mutational studies [32, 33] translates to the potential for peptide mimics disrupting key intramolecular interactions for Nox4 activity and (b) develop isoform-specific peptidic Nox4 inhibitors using such findings.
Previous studies showed that multiple residues in the B-loop and the C-terminal end are critical for the catalytic activity of Nox4 [32, 37]. A recent study using fluorescence polarization demonstrated binding between the Nox4 B-loop and Nox4 dehydrogenase domain and showed that this interaction is weakened by mutation of arginine residues in the B-loop variable region [32]. With this in mind, in the current study, we tested whether application of a peptide mimic (B90–98) from the variable region of Nox4 B-loop interferes with the activity of Nox4. An important premise for this work was our previous findings illustrating that mimicking the corresponding B-loop region in Nox2, known as Nox2ds, selectively and potently inhibits Nox2-derived O2 •− production [8]. We went on to provide evidence that Nox2ds, but not its scrambled control, acts as a competitive inhibitor of the enzyme. The theory then for this study was that these B-loop peptides would likewise act as competitive inhibitors of previously proposed key intramolecular interactions occurring between domains of Nox4. Surprisingly, peptide B90–98 up to 100 μM did not inhibit Nox4-derived H2O2 production as measured by Amplex Red fluorescence. Incidentally, Amplex Red was chosen as the most logical method for detecting H2O2, which is widely considered the primary Nox4 product [44].
Next, in the interest of pursuing a comprehensive approach to these studies, we progressed to testing whether peptides derived from the conserved regions of Nox4 B-loop were capable of inhibiting Nox4. We found that incubation of COS-Nox4 cell lysates with mimics of the N- and C-terminal conserved B-loop regions (B77–91, B82–96, and B92–106) did not inhibit activity. With these results, we postulated that a B-loop peptide mimic might instead be replicating a normal intrinsic function of the B-loop. For this reason, we tested whether peptide B90–98 mutated at residue 96 (R96E), which previously was identified to be critical for Nox4 activity [32], inhibits Nox4-derived H2O2 production. We postulated that this mutated peptide could act as a dominant negative in that regard. Similar to the wild-type peptide, the mutant peptide did not inhibit Nox4-derived H2O2 production. One possible explanation for the lack of effect could have been that a multidimensional intramolecular interaction of the enzyme is at play, and thus targeting only one region could be insufficient to interfere with enzymatic activity. A second possibility is that Nox4 exists in a tightly assembled conformation and its unique tertiary structure permits an active electron transferring arrangement that cannot be disrupted by targeting discrete binding sites with either the native or mutant peptide.
A previous study demonstrated that a C-terminal region downstream of the NADPH-binding motif is important for Nox4, but not Nox2, activity [38]. With further analysis, von Löhneysen et al. identified the last 22 amino acids of Nox4 as essential to activity of the isozyme. Based on these data, we postulated that small peptides targeting this more flexible region would inhibit Nox4 activity. To test the hypothesis, sequential octameric and nonameric peptides were synthesized to encompass the last 22 amino acids of Nox4 and tested for their ability to inhibit Nox4. Likewise, our data demonstrate that none of these C-terminal tail peptides inhibited Nox4 activity. Taken together, these data appear to suggest that Nox4 exists in a tightly assembled, active conformation, thus explaining why peptides targeting intramolecular interactions of the enzyme are not able to interfere with its activity. This observation would be consistent with the reported constitutive and high capacity Nox4 activity.
Shifting to a new approach, we tested whether small peptides targeting the Nox4-p22phox intermolecular interaction could inhibit Nox4. A previous study showed that a peptide (175–194) derived from p22phox inhibits ROS production by Nox2 [42]. Subsequent studies by Dahan et al. using peptide walking identified domains throughout the p22phox protein sequence (9–23, 31–45, 47–61, 85–99, and 113–127) that are important for Nox2 activity [45]. It is, however, completely unknown whether introduction of p22phox peptides can disrupt Nox4-p22phox interaction and inhibit Nox4-derived H2O2 production. Importantly, a large part of the p22phox C-terminus does not seem to be important for Nox4 activity as deletion of amino acids up to and including the last 130 amino acids does not affect Nox4 activity [33]. Thus, targeting the C-terminal domain of p22phox is not likely to inhibit Nox4 activity. In contrast, deletion of the first 11, but not the first 5, amino acids at the p22phox N-terminus reportedly abolished Nox4-derived H2O2 production [33]. This suggested to us that the N-terminal region of p22phox between amino acids 6 and 11 might be sensitive to intervention. We targeted this region using a peptide sequence between amino acids 6 and 11 (p22 WT 6–11 (WAMWAN)) and measured Nox4-derived H2O2 production. Once again, the data demonstrated that p22 WT 6–11 did not inhibit Nox4 activity. We next considered previous work showing that mutation of tryptophans within this region to charged residues, such as arginine, abolished Nox4 activity [33]. Thus, in an attempt to mimic this inactive catalytic site, we tested whether the W6/9R mutant version of p22 6-11 (p22 W6/9R) could inhibit Nox4 activity. Similar to the wild-type peptide, the mutant peptide did not inhibit Nox4-derived H2O2 production.
As we observed, preincubation of COS-Nox4 cell lysates with Nox4 B-loop, Nox4 C-terminal tail, and N-terminal p22phox peptides for 15 min at 24°C did not inhibit H2O2 production. To allow more time for the peptides to disrupt the targeted domain interactions, the incubation time was increased up to 60 min. Notably, previous studies illustrate that 60 min is more than sufficient to inhibit Nox activity [40]. In our hands, 60 min incubation did not reveal inhibitory activity. To go one step further, these experiments were performed at 37°C in an attempt to increase access and likelihood of interaction of peptides with the Nox4 complex. Again, no inhibition of Nox4 was achieved using these peptides. Our data suggest that alternative strategies to improve access or penetrability of peptides into the Nox4-p22phox complex may be necessary to achieve inhibition of Nox4.
In conclusion, our findings suggest that the Nox4-p22phox complex is unperturbed by a wide array of rationally selected peptide mimics. This is not to say that other to-date unidentified active regions of the enzyme could not eventually be devised as inhibitors. Moreover, a more comprehensive study using peptide walking of the entire Nox4 protein could be warranted. It is also plausible that various combinations of peptide mimics may be effective. This is currently an area of active investigation in our laboratory. That notwithstanding, it is our tentative conclusion that the tightly assembled Nox4 complex creates a formidable barrier to peptidic interference and thus greater access could be viewed as the sine qua non of these strategies. In that vein, we are actively pursuing other strategies to temporarily improve access (unfolding) or penetrability of peptides into the Nox4 complex. Application of peptidic inhibitors targeting domain interactions of Nox4 while in the endoplasmic reticulum (before its native folding is complete or associates with p22phox) may be another viable strategy. Of course, small molecule inhibitors that specifically target the above-identified interactions are likely to elude limitations of access and are currently a focus of intense interest in our laboratory.
Conflict of Interests
The authors declare that they have no conflict of intrests.
Acknowledgments
This work was supported by the National Institutes of Health Grants R01HL079207 and P01HL103455-01, by the American Heart Association Fellowship 10POST3030009 (GC), and by an Established Investigator Award (PJP). The study was supported by the Institute for Transfusion Medicine and the Hemophilia Center of Western Pennsylvania.
References
- 1.Dale DC, Boxer L, Liles WC. The phagocytes: neutrophils and monocytes. Blood. 2008;112(4):935–945. doi: 10.1182/blood-2007-12-077917. [DOI] [PubMed] [Google Scholar]
- 2.Csányi G, Taylor WR, Pagano PJ. NOX and inflammation in the vascular adventitia. Free Radical Biology and Medicine. 2009;47(9):1254–1266. doi: 10.1016/j.freeradbiomed.2009.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pagano PJ, Clark JK, Eugenia Cifuentes-Pagano M, Clark SM, Callis GM, Quinn MT. Localization of a constitutively active, phagocyte-like NADPH oxidase in rabbit aortic adventitia: enhancement by angiotensin II. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(26):14483–14488. doi: 10.1073/pnas.94.26.14483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiological Reviews. 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 5.Murdoch CE, Alom-Ruiz SP, Wang M, et al. Role of endothelial Nox2 NADPH oxidase in angiotensin II-induced hypertension and vasomotor dysfunction. Basic Research in Cardiology. 2011;106(4):527–538. doi: 10.1007/s00395-011-0179-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jaquet V, Scapozza L, Clark RA, Krause KH, Lambeth JD. Small-molecule nox inhibitors: ROS-generating NADPH oxidases as therapeutic targets. Antioxidants and Redox Signaling. 2009;11(10):2535–2552. doi: 10.1089/ars.2009.2585. [DOI] [PubMed] [Google Scholar]
- 7.Laleu B, Gaggini F, Orchard M, et al. First in class, potent, and orally bioavailable NADPH oxidase isoform 4 (Nox4) inhibitors for the treatment of idiopathic pulmonary fibrosis. Journal of Medicinal Chemistry. 2010;53(21):7715–7730. doi: 10.1021/jm100773e. [DOI] [PubMed] [Google Scholar]
- 8.Csanyi G, Cifuentes-Pagano E, Al Ghouleh I, et al. Nox2 B-loop peptide, Nox2ds, specifically inhibits the NADPH oxidase Nox2. Free Radical Biology & Medicine. 2011;51(6):1116–1125. doi: 10.1016/j.freeradbiomed.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rey FE, Cifuentes ME, Kiarash A, Quinn MT, Pagano PJ. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O2− and systolic blood pressure in mice. Circulation Research. 2001;89(5):408–414. doi: 10.1161/hh1701.096037. [DOI] [PubMed] [Google Scholar]
- 10.Jacobson GM, Dourron HM, Liu J, et al. Novel NAD(P)H oxidase inhibitor suppresses angioplasty-induced superoxide and neointimal hyperplasia of rat carotid artery. Circulation Research. 2003;92(6):637–643. doi: 10.1161/01.RES.0000063423.94645.8A. [DOI] [PubMed] [Google Scholar]
- 11.Liu J, Ormsby A, Oja-Tebbe N, Pagano PJ. Gene transfer of NAD(P)H oxidase inhibitor to the vascular adventitia attenuates medial smooth muscle hypertrophy. Circulation Research. 2004;95(6):587–594. doi: 10.1161/01.RES.0000142317.88591.e6. [DOI] [PubMed] [Google Scholar]
- 12.Norton CE, Broughton BR, Jernigan NL, Walker BR, Resta TC. Enhanced depolarization-induced pulmonary vasoconstriction following chronic hypoxia requires EGFR-dependent activation of NAD(P)H oxidase 2. Antioxidants & Redox Signaling. 2012 doi: 10.1089/ars.2012.4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schiffrin EL, Touyz RM. Inflammation and vascular hypertrophy induced by angiotensin II: role of NADPH oxidase-derived reactive oxygen species independently of blood pressure elevation? Arteriosclerosis, Thrombosis, and Vascular Biology. 2003;23(5):707–709. doi: 10.1161/01.ATV.0000069907.12357.7E. [DOI] [PubMed] [Google Scholar]
- 14.Touyz RM, Chen X, Tabet F, et al. Expression of a functionally active gp91phox-containing neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries: regulation by angiotensin II. Circulation Research. 2002;90(11):1205–1213. doi: 10.1161/01.res.0000020404.01971.2f. [DOI] [PubMed] [Google Scholar]
- 15.Zhang QG, Laird DM, Han D, et al. Critical role of NADPH oxidase in neuronal oxidative damage and microglia activation following traumatic brain injury. PLoS One. 2012;7(4) doi: 10.1371/journal.pone.0034504.e34504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geiszt M, Kopp JB, Várnai P, Leto TL. Identification of Renox, an NAD(P)H oxidase in kidney. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(14):8010–8014. doi: 10.1073/pnas.130135897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Griendling KK. Novel NAD(P)H oxidases in the cardiovascular system. Heart. 2004;90(5):491–493. doi: 10.1136/hrt.2003.029397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peterson JR, Burmeister MA, Tian X, et al. Genetic silencing of Nox2 and Nox4 reveals differential roles of these nadph oxidase homologues in the vasopressor and dipsogenic effects of brain angiotensin II. Hypertension. 2009;54(5):1106–1114. doi: 10.1161/HYPERTENSIONAHA.109.140087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tong X, Hou X, Jourd’Heuil D, Weisbrod RM, Cohen RA. Upregulation of Nox4 by TGFβ1 oxidizes SERCA and inhibits NO in arterial smooth muscle of the prediabetic zucker rat. Circulation Research. 2010;107(8):975–983. doi: 10.1161/CIRCRESAHA.110.221242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Szocs K, Lassègue B, Sorescu D, et al. Upregulation of Nox-based NAD(P)H oxidases in restenosis after carotid injury. Arteriosclerosis, Thrombosis, and Vascular Biology. 2002;22(1):21–27. doi: 10.1161/hq0102.102189. [DOI] [PubMed] [Google Scholar]
- 21.Sorescu D, Weiss D, Lassègue B, et al. Superoxide production and expression of Nox family proteins in human atherosclerosis. Circulation. 2002;105(12):1429–1435. doi: 10.1161/01.cir.0000012917.74432.66. [DOI] [PubMed] [Google Scholar]
- 22.Kleinschnitz C, Grund H, Wingler K, et al. Post-stroke inhibition of induced NADPH Oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biology. 2010;8(9) doi: 10.1371/journal.pbio.1000479.e1000479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carnesecchi S, Deffert C, Donati Y, et al. A key role for NOX4 in epithelial cell death during development of lung fibrosis. Antioxidants and Redox Signaling. 2011;15(3):607–619. doi: 10.1089/ars.2010.3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sedeek M, Callera G, Montezano A, et al. Critical role of Nox4-based NADPH oxidase in glucose-induced oxidative stress in the kidney: implications in type 2 diabetic nephropathy. American Journal of Physiology. 2010;299(6):F1348–F1358. doi: 10.1152/ajprenal.00028.2010. [DOI] [PubMed] [Google Scholar]
- 25.Cucoranu I, Clempus R, Dikalova A, et al. NAD(P)H oxidase 4 mediates transforming growth factor-β1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circulation Research. 2005;97(9):900–907. doi: 10.1161/01.RES.0000187457.24338.3D. [DOI] [PubMed] [Google Scholar]
- 26.Li J, Stouffs M, Serrander L, et al. The NADPH oxidase NOX4 drives cardiac differentiation: role in regulating cardiac transcription factors and MAP kinase activation. Molecular Biology of the Cell. 2006;17(9):3978–3988. doi: 10.1091/mbc.E05-06-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vaquero EC, Edderkaoui M, Pandol SJ, Gukovsky I, Gukovskaya AS. Reactive oxygen species produced by NAD(P)H oxidase inhibit apoptosis in pancreatic cancer cells. Journal of Biological Chemistry. 2004;279(33):34643–34654. doi: 10.1074/jbc.M400078200. [DOI] [PubMed] [Google Scholar]
- 28.Schröder K, Zhang M, Benkhoff S, et al. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circulation Research. 2012;110(9):1217–1225. doi: 10.1161/CIRCRESAHA.112.267054. [DOI] [PubMed] [Google Scholar]
- 29.Touyz RM, Montezano AC. Vascular Nox4: a multifarious NADPH oxidase. Circulation Research. 2012;110(9):1159–1161. doi: 10.1161/CIRCRESAHA.112.269068. [DOI] [PubMed] [Google Scholar]
- 30.Borbély G, Szabadkai I, Horváth Z, et al. Small-molecule inhibitors of NADPH oxidase 4. Journal of Medicinal Chemistry. 2010;53(18):6758–6762. doi: 10.1021/jm1004368. [DOI] [PubMed] [Google Scholar]
- 31.Lyle AN, Deshpande NN, Taniyama Y, et al. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circulation Research. 2009;105(3):249–259. doi: 10.1161/CIRCRESAHA.109.193722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jackson HM, Kawahara T, Nisimoto Y, Smith SME, Lambeth JD. Nox4 B-loop creates an interface between the transmembrane and dehydrogenase domains. Journal of Biological Chemistry. 2010;285(14):10281–10290. doi: 10.1074/jbc.M109.084939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Von Löhneysen K, Noack D, Jesaitis AJ, Dinauer MC, Knaus UG. Mutational analysis reveals distinct features of the Nox4-p22 phox complex. Journal of Biological Chemistry. 2008;283(50):35273–35282. doi: 10.1074/jbc.M804200200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu L, Zhen L, Dinauer MC. Biosynthesis of the phagocyte NADPH oxidase cytochrome b558. Role of heme incorporation and heterodimer formation in maturation and stability of gp91(phox) and p22(phox) subunits. Journal of Biological Chemistry. 1997;272(43):27288–27294. doi: 10.1074/jbc.272.43.27288. [DOI] [PubMed] [Google Scholar]
- 35.Cheng G, Lambeth JD. NOXO1, regulation of lipid binding, localization, and activation of Nox1 by the Phox homology (PX) domain. Journal of Biological Chemistry. 2004;279(6):4737–4742. doi: 10.1074/jbc.M305968200. [DOI] [PubMed] [Google Scholar]
- 36.Nisimoto Y, Jackson HM, Ogawa H, Kawahara T, David Lambeth J. Constitutive NADPH-dependent electron transferase activity of the Nox4 dehydrogenase domain. Biochemistry. 2010;49(11):2433–2442. doi: 10.1021/bi9022285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.von Lohneysen K, Noack D, Hayes P, Friedman JS, Knaus UG. Constitutive NADPH oxidase 4 activity resides in the composition of the B-loop and the penultimate C terminus. The Journal of Biological Chemistry. 2012;287(12):8737–8745. doi: 10.1074/jbc.M111.332494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Von Löhneysen K, Noack D, Wood MR, Friedman JS, Knaus UG. Structural insights into Nox4 and Nox2: motifs involved in function and cellular localization. Molecular and Cellular Biology. 2010;30(4):961–975. doi: 10.1128/MCB.01393-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ambasta RK, Kumar P, Griendling KK, Schmidt HHHW, Busse R, Brandes RP. Direct interaction of the novel Nox proteins with p22phox is required for the formation of a functionally active NADPH oxidase. Journal of Biological Chemistry. 2004;279(44):45935–45941. doi: 10.1074/jbc.M406486200. [DOI] [PubMed] [Google Scholar]
- 40.Dahan I, Molshanski-Mor S, Pick E. Inhibition of NADPH oxidase activation by peptides mapping within the dehydrogenase region of Nox2-A "peptide walking" study. Journal of Leukocyte Biology. 2012;91(3):501–515. doi: 10.1189/jlb.1011507. [DOI] [PubMed] [Google Scholar]
- 41.Joseph G, Pick E. ’Peptide walking’ is a novel method for mapping functional domains in proteins. Its application to the Rac1-dependent activation of NADPH oxidase. Journal of Biological Chemistry. 1995;270(49):29079–29082. doi: 10.1074/jbc.270.49.29079. [DOI] [PubMed] [Google Scholar]
- 42.Nakanishi A, Imajoh-Ohmi S, Fujinawa T, Kikuchi H, Kanegasaki S. Direct evidence for interaction between COOH-terminal regions of cytochrome b558 subunits and cytosolic 47-kDa protein during activation of an O2−-generating system in neutrophils. Journal of Biological Chemistry. 1992;267(27):19072–19074. [PubMed] [Google Scholar]
- 43.Nauseef WM, McCormick S, Renee J, Leidal KG, Clark RA. Functional domain in an arginine-rich carboxyl-terminal region of p47phox. Journal of Biological Chemistry. 1993;268(31):23646–23651. [PubMed] [Google Scholar]
- 44.Dikalov SI, Dikalova AE, Bikineyeva AT, Schmidt HHHW, Harrison DG, Griendling KK. Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radical Biology and Medicine. 2008;45(9):1340–1351. doi: 10.1016/j.freeradbiomed.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dahan I, Issaeva I, Gorzalczany Y, Sigal N, Hirshberg M, Pick E. Mapping of functional domains in the p22phox subunit of flavocytochrome b559 participating in the assembly of the NADPH oxidase complex by ‘peptide walking’. Journal of Biological Chemistry. 2002;277(10):8421–8432. doi: 10.1074/jbc.M109778200. [DOI] [PubMed] [Google Scholar]