

Over the last few decades, the carbon nanomaterial-based electrical detection of chemical and biological molecules has gained much attention with its extensive applications to genomic, proteomic and environmental analysis as well as for clinical diagnosis.[1–12] Typical carbon nanomaterial-based electrical detection systems are composed of two components: a biological recognition element and a signal transduction element. These devices can detect minute concentrations of target molecules by measuring the change in electrical conductance that results from the binding of target molecules to the surface of the carbon nanomaterial.[13–17] However, the sensitivity as well as selectivity of these devices depends heavily on the electrical properties of the nanomaterial used in the system and on the interfacial chemistry that exists between the nanomaterial and the receptor molecules. Carbon nanotube field-effect transistors (CNT-FETs) currently lead the field and are one of the most extensively developed systems.[24–29] This is due to the excellent electrical properties and the relatively well-developed surface chemistry of CNTs. However, certain limitations do exist with CNT-based electrical detection devices. These include inconsistencies in the assembly of CNTs resulting in device unreliability, limits in its surface area, and a high-cost of fabrication.
An alternative nanomaterial, which has the potential to overcome the aforementioned limitations of CNTs, is graphene. Graphene consists of a single-atom-thick planar sheet of carbon atoms that are arranged in a perfect honeycomb lattice and has a zero-band gap. It forms ideal two-dimensional carbon crystals that show high electrical conductivity in the absence of any doping even at room temperature. Graphene also demonstrates superb mechanical flexibility and high chemical/thermal stability. Furthermore, due to its low noise level, graphene is extremely sensitive to perturbations in electrical conductance that are induced by biological reactions. These biological reactions can include, but are not limited to, the enzyme-mediated degradation of a target substrate and the binding of proteins to other biomolecules. There have been a number of graphene-based protein sensors with detection limits on the μM scale.[15–17] However, there have been limited demonstrations utilizing graphene for the detection of enzymes, with most demonstrations relying on antibody-mediated recognition of their target proteins.[17,31,32] Enzyme detection is an especially clinically relevant issue as it has been reported that the enzyme levels found in vivo are strongly correlated with the diagnosis, prognosis, and treatment of many diseases such as HIV, cancer, and diabetes. Therefore, to improve our ability to detect enzyme levels and to enhance the clinical potential of graphene-based biosensors, it is possible to take advantage of graphene’s sensitivity to changes in electrical conductance in order to detect the enzymatic cleavage of target peptides that are assembled on the sensor surface.
Herein, we report a novel approach for the detection of enzymes using a graphene-nanoparticle (NP) hybrid biosensor. This system can detect the activity of an enzyme and determine its concentration by measuring the change in electrical hysteresis that results from the interaction of the target enzyme with its corresponding substrate. In addition, we demonstrate its ability to achieve the sensitive and selective detection of the target enzyme. Similar electrical hysteretic behavior in carbon nanomaterial-based devices with gate have been observed previously and is presumably caused by charge transfer from neighboring adsorbates such as water molecules or from charge injection into the trap sites on the dielectric substrate.[18] However, this is the first report of NP-mediated electrical hysteresis in a graphene sensor. Moreover, the utilization of a hysteresis change for sensing is advantageous because the immobilization of specific enzymes on the graphene surface, which is required for field effect transistor-type sensors, is unnecessary in our system allowing us to preserve the superb electrical properties of graphene. The use of hysteresis as a detection method also allows graphene-based devices to overcome the low on-off ratio caused by the zero band gap of graphene. In the majority of previous studies,[13–17] a Dirac shift or a conductance change at the fixed gate voltage that is associated with this Dirac shift when the specific target material was attached selectively on the graphene channel was utilized. When using only a Dirac shift for sensing, non-specific binding such as the physical attachment of positively- or negatively-charged materials can also result in a Dirac voltage shift or conductance change. However, in our process, even though nonspecific binding on the channel occurred, the amount of electrical hysteresis of our devices did not change. Only when these devices were exposed to a specific condition, did the hysteresis phenomenon start to decrease. Specifically, our graphene-NP hybrid biosensor is composed of gold nanoparticles (AuNPs) that are assembled on a graphene surface using a functional polypeptide linker, which concomitantly functions as the substrate for the target enzyme. Proteolysis of the functional polypeptide linker by the target enzyme leads to disassembly of the NP layer and a measurable shift in electrical hysteresis that can be visualized in the gate sweep curve. This can then be used to quantify the enzyme concentration. AuNPs were chosen because they are able to store charge, resulting in hysteresis upon their release from the device surface.[18] In our case, as a proof-of-concept, we detected the activity of Carboxypeptidase B in order to demonstrate the high achievable sensitivity and selectivity of our graphene-NP hybrid biosensor.[30] This enzyme has been reported as a predictor of severe acute pancreatitis, which is an acute inflammatory condition of the pancreas. However, it is important to note that the reported bio-sensing method can be extended to detect any number of key enzymes, pH changes, and UV with the appropriate modification to the functional linker substrate.
A schematic of our graphene-NP hybrid biosensor is depicted in Figure 1a. The device consists of gold source and drain electrodes that are connected by graphene channels (Figure 1b(i)). The graphene channels are coated with a functional polypeptide linker that is specific for our target enzyme, Carboxypeptidase B, followed by a layer of AuNPs that have a negative charge on their surface, and a peripheral ionic liquid, which functions as the gate dielectric (Figure 1b(ii–iv)). Introduction of Carboxypeptidase B would then initiate the hydrolysis of the specific enzyme substrate, which in this case is the functional polypeptide linkers. The degradation of the polypeptide linkers result in the release of AuNPs, which causes a hysteresis shift that can be measured using a silver gate electrode. The range of hysteresis shift has been found to be proportional to the enzyme concentration and can therefore be used to determine the level of enzyme in the solution.
Figure 1.

Schematic of the device. (a) Graphene-nanoparticle hybrid devices for enzyme sensing. (b) Fabrication process of the hybrid biosensor. Fabrication of the graphene channel between the Au electrodes (i). Functionalization of the graphene surface with hydrophilic molecules (ii). Assembly of the functional peptide linker molecules and AuNPs on this polypeptide layer (iii). Sensing target biomolecules by measuring the change in electrical hysteresis (iv). (c) Chemical structure of the functional polypeptide linker molecule.
To fabricate the graphene-NP hybrid biosensor, the source and drain Au electrodes were first fabricated on a SiO2 substrate via the lift-off process. Graphene was deposited and patterned as previously described[2] (See Supporting Information) in order to form graphene channels between the Au electrodes (Figure 1b(i)). Before assembling the AuNPs and the functional polypeptide linker molecules on the graphene surface, the surface was made hydrophilic through functionalization with 4-(pyren-1-yl)butanal, which occurs via π–π interactions. Specifically, functionalization was achieved by incubating the device in a methanol solution (1:500) containing 4-(pyren-1-yl)-butanal (See Supporting Information for synthesis) for 30 minutes (Figure 1b(ii)). Next, the aldehyde groups of 4-(pyren-1-yl) butanal were coupled to the amine groups of the functional polypeptide linker through reductive amination for 6 hours. Once the graphene device, functionalized with polypeptide linker, was placed in the AuNP solution, the negatively charged AuNPs were able to self-assemble on the positively charged functional polypeptide linker molecules via electrostatic interactions (Figure 1b(iii)). Specifically, this is a result of the remaining positively charged amine groups on the functional polypeptide linkers. Finally, with the AuNPs assembled on the graphene surface, we were able to detect the electrical hysteresis and gate effect of our device by utilizing an ionic liquid as the gate dielectric.
The fabrication method developed in our graphene-NP hybrid biosensor has several advantages over conventional graphene-based biosensors. First, we were able to fabricate high performance graphene-NP hybrid devices that have a large surface area available for biodetection and a high conductivity. This was accomplished at a high yield and uniformity by combining the properties of graphene with the nanoscopic features of the AuNPs.[2] Specifically, these properties allow for the development of biosensors that have high achievable current levels sufficient for the measurement of minute conductance changes regardless of the channel length between two electrodes in the devices (Figure 1 and Figure 2a1). As a result, our device provides a larger number of sites that can be conjugated with receptor molecules (e.g. antibody or functional polypeptide linker) in comparison with previous biosensors that used reduced graphene oxide. In addition, these reduced graphene oxide-based biosensors were limited by their low conductivity and limited achievable channel length (e.g. only a few micrometers).[4] Second, the functionalization of the graphene surface with our functional polypeptide linker followed by assembly of AuNPs was accomplished using π–π interactions (graphenepyrene aldehyde) and electrostatic interactions (polypeptides-AuNPs). This method prevents the need to covalently bond the linker to the graphene surface, therefore preserving the superb electrical properties of graphene.[21] Finally, our fabrication method allows for the precise control of the number of graphene layers and can be used to deposit a large quantity of graphene on different substrates such as SiO2, metallic surfaces, or glass due to the flexible and stretchable properties of graphene. The devices can also be fabricated with conventional microfabrication techniques, and the entire process can be accomplished using only conventional microfabrication facilities. Therefore, our method allows us to fabricate graphene-based devices with present Si-based microelectronics, and would be readily accessible to the present semiconductor industry. Collectively, our study presents a novel method that avoids the use of graphene oxide for the development of biosensors and provides a novel approach that combines the advantages provided by graphene and inorganic nanoparticles (e.g. metallic and semiconductive NPs) with the area of biosensing.
Figure 2.

Characterization data of the biosensor using AFM and SEM. (A1) A SEM image of the junction between the Au electrodes, shows a graphene channel (dark region) between two Au electrodes (bright area). AFM topography images of (A2) the graphene surface before the assembly of NPs and (A3) the graphene surface after the functionalization with linker molecules. (A4) An AFM image of AuNPs on the graphene surface. The height profile of the AFM image is shown below the image. (B1) An AFM topography image of the graphene surface before the assembly of NPs. (B2) An AFM image of AuNPs on the graphene surface. The height profile of the AFM image is shown below the image. (B3) An AFM topography image of graphene after exposure to a 10 μM enzyme solution for 6 hours. (C1)-(C6) AFM topography images of graphene after exposure to a 1 μM enzyme solution for 30 sec to 50 hours, respectively. (D1)-(D4) AFM images after exposure to a 10 nM, 100 nM, 1 μM and 10 μM enzyme solution for 3 hours, respectively.
Confirmation of graphene deposition followed by AuNP assembly in the junction between the Au electrodes was obtained using scanning electron microscopy (SEM) (Figure 2A1). The image shows that the two Au electrodes, separated by a 50 μm gap, are connected by a graphene channel that is 420 μm wide. In addition, after coating the graphene surface with the functional polypeptide linker, we confirmed that the negatively charged AuNPs (15 nm in diameter) successfully self-assembled as a monolayer on the graphene surface at a high density (Figure 2 A4 and 2 B2). This is mainly due to the large positive charge and the two-dimensional planar structure of the functional polypeptide linker. The negatively charged surface of the AuNPs also ensures that additional NPs are not assembled onto the underlying functional polypeptide linker. Therefore, the deposition of a single layer of AuNPs with high density was achieved on the graphene surface. It was also found that the complete disassembly of AuNPs was achieved after the device was exposed to a 1 μM Carboxypeptidase B enzyme solution for 50 hours (Figure 2C6) or a 10 μM solution for 3 hours (Figure 2D4).
The gating effect of our device was measured by employing an ionic liquid as the gate dielectric and a silver gate electrode. The use of an ionic liquid, which has a high ionic concentration, as the gate dielectric allowed the thickness of the diffusion layer to be considered negligible. It was found that the effect was similar to that observed in graphene transistors that have ambipolar conduction and no electrical hysteresis (Figure 3a upper graph).[22] In our case, the top-gate bias was swept with a ~0.01 V/sec sweep speed under a source-drain bias of 0.1 V. An ambipolar field effect was also observed with a Dirac point voltage (VDirac) of 0.25 V, where charge carriers change polarity. After functionalization of the graphene surface with our functional polypeptide linker, it was observed that the gate curve shifted to a lower voltage level. This negative Dirac voltage shift is approximately 0.05 V and we hypothesize that this is due to the positively charged amine groups of the functional polypeptide linker. Furthermore, the graphene device with functional polypeptide linker is characterized by a gate curve that is similar to that seen previously with no observable electrical hysteresis.[18] However, when negatively charged AuNPs were assembled, a positive Dirac voltage shift occurred resulting in a field effect due to the negative charge of the AuNPs. Interestingly, a large electrical hysteresis was achieved due to the memory effect during gate sweep. The difference in the Dirac voltage between the forward and backward gate voltage sweeps was defined as the hysteresis gap (ΔVDirac). In terms of the underlying mechanism, graphene has both holes and electrons as carriers, and the density of states in graphene near the Fermi level is low. Thus, it exhibited ambipolar behaviors of some gating effects depending on the gate bias voltages. Note that, in the case without AuNPs (pristine graphene FET or graphene FET with only linker molecules) (Figure 3a(i) and 3a(ii)), the graphene FETs exhibit only a small amount of hysteresis indicating that our devices do not have many charge trap defects. On the other hand, in the case of devices with AuNPs (Figure 3a(iii)), a large hysteresis is evident in the gate sweep curve due to the charge stored in the AuNPs.
Figure 3.

Measurement of the hysteresis change of the device after exposure to enzyme solutions. (a) ΔVDirac change and gate effect of an initial graphene transistor (i), after the functionalization of graphene surface with functional polypeptide linker molecule (ii), after NP assembly on the graphene surface (iii), and after exposure to 1 μM enzyme solution for 30 sec, 5 min, 1 hr and 50 hrs (iv ~vii). (b) Changes in ΔVDirac of graphene device (i), after functionalization with linker molecules (ii), after NP assembly (iii), after the exposure to Carboxypeptidase B in PBS solution with the concentration of 10 nM (iv), 100 nM (v), 1 μM (vi) and 10 μM (vii) for 3 hours.
As stated above, Carboxypeptidase B has been proposed as a predictor for severe acute pancreatitis.[30] This enzyme was used in our proof-of-concept due to the fact that our functional polypeptide linker contains a high number of lysines. Therefore, it acts as a substrate for this enzyme, which specifically cleaves basic amino acids such as lysine. When our graphene-NP hybrid device was exposed to the enzyme solution, the functional polypeptide linker degraded, resulting in the release of AuNPs into the surrounding media. To characterize the effect of the release of AuNPs from the graphene surface on the electrical hysteresis of this device, we measured the ΔVDirac as a function of time using a 1 μM enzyme solution (Figure 4b). We found that increasing the exposure time of the enzyme resulted in an expected decrease in NP density (Figure 2(c1–c6)). This decrease in NP density resulted in a decrease in the measured ΔVDirac, which became saturated after 50 hours. Furthermore, due to the fact that the release of functional polypeptide linker caused the detachment of NPs from the graphene surface, the ΔVDirac was very similar to the release profiles found for polypeptide films.[23]
Figure 4.
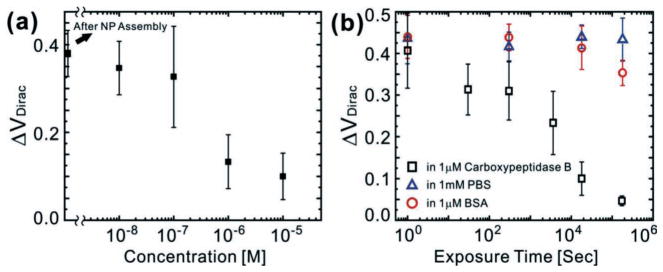
Enzyme detection using the graphene-nanoparticle hybrid sensor (a) Sensitivity (ΔVDirac) of the graphene sensor as a function of enzyme concentration [n =10]. (b) ΔVDirac change under various periods of exposure to a 1 μM solution of Carboxypeptidase B in PBS (black square), a 1 mM PBS solution (blue triangle) and 1 mM solution of BSA in the PBS (red circles) [n =10].
The sensitivity of our graphene-NP hybrid sensor with functional polypeptide and NPs was determined by measuring the Δ VDirac as the concentration of Carboxypeptidase B in phosphate buffered saline (PBS) solution was varied from 10 nM to 10μM (Figure 3b). Representative sensing data shows that the difference between the ΔVDirac for 10 nM and 100 nM solutions of Carboxypeptidase B after 3 hours of exposure was statistically similar. However, a statistically significant difference in ΔVDirac was observed when the concentration of Carboxypeptidase B was increased to 1 μM. Furthermore, as the concentration was subsequently increased, a concentration dependent decrease in the ΔVDirac of our sensors was observed. Figure 4a illustrates the sensitivity (ΔVDirac) of our biosensors as a function of the enzyme concentration. For these experiments, we used ten graphene junctions for each step and repeated sensing experiments ten times demonstrating the repeatability of our results. From these results, we can conclude that the lowest concentration that can be detected using the current functional polypeptide linker is approximately 1 μM and it was found that this concentration correlates with a decrease in the change ratio of ΔVDirac (59.2%). The results also confirm that there is a direct relationship between the ΔVDirac and the concentration of enzyme. Specifically, similar to the non-linear behavior of other graphene-based sensors, the responses of our sensor increase non-linearly with an increase in the enzyme concentration from 10 nM to 10 μM.[13,24–26] Finally, we tested the selectivity of this biosensor towards the target enzyme by using control samples such as PBS and different proteins such as bovine serum albumin (BSA). Results showed that PBS and BSA did not react with the functional polypeptide substrate (Figure 4b).
Our findings show that quantifying the change in electrical hysteresis of a graphene-NP hybrid biosensor is a viable method to measure enzyme activity sensitively and selectively. However, it is apparent that the critical factor that determines this sensitivity and selectivity is the functional polypeptide linker whose degradation results in the release of AuNPs and subsequent shift in hysteresis. Therefore, the performance of our sensor, in terms of its sensitivity and selectivity, can be further improved by modifying the functional polypeptide linker and changing the properties of the metallic nanoparticle system. In the current study, our novel graphene-based biosensor with functional polypeptide linker and NPs can confer a sensitivity of up to 1 μM. However, to improve sensitivity and selectivity, we are synthesizing and screening a variety of functional polypeptide linker molecules that will be self-assembled with different metallic NPs of varying compositions and sizes.
In summary, we have successfully developed a novel graphene-NP hybrid biosensor that uses an electrical hysteresis change to detect the enzymatic activity and concentration of a target enzyme (e.g., Carboxypeptidase B). Our results indicate that our novel graphene-NP hybrid biosensor has the ability to sense concentrations of Carboxypeptidase B down to the μM-scale, which is comparable to previously reported graphene-based biosensors.[15–17] More importantly, to the knowledge of the authors, this is the first demonstration of a graphene-based biosensor that utilizes a hysteresis change resulting from metallic NPs assembled on a graphene surface for enzyme detection. The current configuration developed in our graphene-NP hybrid biosensor holds a number of advantages over other graphene-based devices established to detect enzymes and other biomolecules. First, AuNP-mediated electrical hysteresis is a phenomenon that has not been studied in graphene-based electrical detection methods, even though it can overcome the limitations faced by current graphene-based sensing approaches. Second, the novel method of detection reported here provides an alternative to the low on-off ratio caused by the zero band gap of graphene. Third, in terms of the components used in our device, the functional polypeptide linker acts as the key-sensing component and can be modified to enhance sensitivity and selectivity for any target enzyme or to confer pH and UV sensitivity. In addition, by combining the properties of graphene with inorganic NPs, our device provides a larger number of sites that can be conjugated with receptor molecules and a higher achievable current level. Fourth, the methods used to fabricate the device allow for the homogenous deposition of graphene, resulting in uniform electrical properties. Finally, the use of conventional microfabrication techniques allows for compatibility with facilities found in device industries and allows mass production of our device. Therefore, the results and developed methods presented here warrant the further study of graphene-NP hybrid biosensors for the sensitive and selective detection of enzymes and other biomolecules.
Experimental Section
Metal deposition and measurement of graphene devices
For the electrode fabrication, the photoresist was first patterned on the substrate. Ti/Au (10/30 nm) was then deposited on the substrate and the remaining photoresist was removed with acetone for the lift-off process. A Keithley-4200 semiconductor parameter analyzer was used for measurement and data collection.
Synthesis of 1-butyl-3-methylimidazolium hexafluorophosphate (BMIM-PF6)
The BMIM-PF6 was prepared according to a modified procedure reported by Dupont et al.33 72.5 mL (64.2 g, 0.69 mol) of 1-chlorobutane and 50 mL of 1-methylimidazole (51.8 g, 0.63 mol) were added to a dry round-bottom flask fitted with a reflux condenser and a magnetic stirring bar. The reaction mixture was stirred at 80 °C for 48 h. The unreacted 1-chlorobutane was evaporated at reduced pressure. 1-methylimidazole was washed three times by adding small amount of ethyl acetate to the mixture and decanted. 1-butyl-3-methylimidazolium chloride (BMIM-Cl) was obtained by evaporating the remaining ethyl acetate at reduced pressure and drying in a vacuum oven. 69.6 g (0.38 mol) of KPF6 and 150 mL of H2O were added into a round-bottom flask, followed by the addition of 50 mL (0.32 mol) of as-prepared BMIM-Cl. After vigorous stirring for 24 h at room temperature, undissolved KPF6 disappeared and two phases were formed. The lower ionic liquid layer was separated and dissolved in 50 mL of dichloromethane. The dichloromethane solution was washed with water until no chloride (Cl−) anion in the water layer could be detected by AgNO3 solution. Dichloromethane was evaporated and 1-butyl-3-methylimidazolium hexafluorophosphate (BMIM-PF6) was obtained as a colorless or slightly yellow viscous liquid after drying in a vacuum oven. 1-chlorobutane, 1-methylimidazole, ethyl acetate and dichloromethane were purchased from Sigma Aldrich Inc.
The synthesized BMIM-PF6 was analyzed with 1 H NMR. Spectra were recorded at 600 MHz. The following chemical shifts reported in ppm downfield to TMS (δ =0 ppm) were observed: 1H NMR (600 MHz, DMSO-D6): δ 0.91(t, 3H), δ 1.27(m, 2H), δ 1.77(m, 2H), δ 3.85(s, 3H), δ 4.16 (t, 2H), δ 7.67(s, 1H), δ 7.74(s, 1H), δ 9.08(s, 1H).
Synthesis of 4-(pyren-1-yl)butanal
10 mL of methylene chloride was added to 1.097 g (4 mmol) of 4-(pyren-1-yl) butyl alcohol and stirred vigorously at room temperature for 10 min. The reaction was followed by the addition of 1.292 g (1.5 Equilibrium) of PCC (Pyridinium chlorochromate) in 10 mL methylene chloride and the reaction mixture was stirred for 2 h. The reaction mixture was then diluted with 5 volumes of anhydrous ether (100 mL) and washed with 1:1 brine: water, saturated aq. Na2SO3 solution, and brine, respectively, dried over anhydrous Na2SO4 and concentrated to give the aldehyde. The crude product was purified by silica gel flash chromatography (hexanes: ethyl acetate, 9:1, rf: 0.4) to yield 1.06 g (97%). Most of the crude products were very clean and could be used directly for further applications.
Supplementary Material
Acknowledgments
This work was supported by the NIH Director’s Innovator Award [(1DP20D006462–01), K.-B. L.] and the N. J. Commission on Spinal Cord Injury grant [(09–3085-SCR-E-0), K.-B. L.]. We are grateful to Dr. Barman for his valuable suggestions and scientific comments with regard to the manuscript.
Footnotes
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Dr. Sung Myung, Department of Chemistry and Chemical Biology, Institute for Advanced Materials, Devices, and Nanotechnology (IAMDN), Rutgers, The State University of New Jersey, Piscataway, NJ 08854, USA, http://rutchem.rutgers.edu/~kbleeweb/
Perry T. Yin, Department of Biomedical Engineering, Rutgers, The State University of New Jersey, Piscataway, NJ 08854, USA
Dr. Cheoljin Kim, Department of Chemistry and Chemical Biology, Institute for Advanced Materials, Devices, and Nanotechnology (IAMDN), Rutgers, The State University of New Jersey, Piscataway, NJ 08854, USA, http://rutchem.rutgers.edu/~kbleeweb/
Jaesung Park, Center for Superfunctional Materials, Department of Chemistry, Pohang University of Science and Technology, Pohang, Korea.
Aniruddh Solanki, Department of Chemistry and Chemical Biology, Institute for Advanced Materials, Devices, and Nanotechnology (IAMDN), Rutgers, The State University of New Jersey, Piscataway, NJ 08854, USA, http://rutchem.rutgers.edu/~kbleeweb/.
Pavel Ivanoff Reyes, Department of Electrical and Computer Engineering, Rutgers, The State University of New Jersey, Piscataway, NJ 08854-8058, USA.
Prof. Yicheng Lu, Department of Electrical and Computer Engineering, Rutgers, The State University of New Jersey, Piscataway, NJ 08854-8058, USA
Prof. Kwang S. Kim, Center for Superfunctional Materials, Department of Chemistry, Pohang University of Science and Technology, Pohang, Korea
Prof. Ki-Bum Lee, Email: kblee@rutgers.edu, Department of Chemistry and Chemical Biology, Institute for Advanced Materials, Devices, and Nanotechnology (IAMDN), Rutgers, The State University of New Jersey, Piscataway, NJ 08854, USA, http://rutchem.rutgers.edu/~kbleeweb/. Department of Biomedical Engineering, Rutgers, The State University of New Jersey, Piscataway, NJ 08854, USA
References
- 1.Novoselov KS, Geim AK, Morozov SV, Jiang D, Zhang Y, Dubonos SV, Grigorieva IV, Firsov AA. Science. 2004;306:666. doi: 10.1126/science.1102896. [DOI] [PubMed] [Google Scholar]
- 2.Kim KS, Zhao Y, Jang H, Lee SY, Kim JM, Kim KS, Ahn JH, Kim P, Choi JY, Hong BH. Nature. 2009;457:706. doi: 10.1038/nature07719. [DOI] [PubMed] [Google Scholar]
- 3.Li X, Cai W, An J, Kim S, Nah J, Yang D, Piner R, Velamakanni A, Jung I, Tutuc E, Banerjee SK, Colombo L, Ruoff RS. Science. 2009;324:1312. doi: 10.1126/science.1171245. [DOI] [PubMed] [Google Scholar]
- 4.Novoselov KS, Geim AK, Morozov SV, Jiang D, Katsnelson MI, Grigorieva IV, Dubonos SV, Firsov AA. Nature. 2005;438:197. doi: 10.1038/nature04233. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Y, Tan YW, Stormer HL, Kim P. Nature. 2005;438:201. doi: 10.1038/nature04235. [DOI] [PubMed] [Google Scholar]
- 6.Han M, Özyilmaz B, Zhang Y, Kim P. Phys Rev Lett. 2007;98:206805. doi: 10.1103/PhysRevLett.98.206805. [DOI] [PubMed] [Google Scholar]
- 7.Geim AK, Novoselov KS. Nat Mater. 2007;6:183. doi: 10.1038/nmat1849. [DOI] [PubMed] [Google Scholar]
- 8.Jiao L, Zhang L, Wang X, Diankov G, Dai H. Nature. 2007;458:877. doi: 10.1038/nature07919. [DOI] [PubMed] [Google Scholar]
- 9.Oosinga JB, Heersche HB, Liu X, Morpurgo AF, Vandersypen LMK. Nat Mater. 2007;7:151. doi: 10.1038/nmat2082. [DOI] [PubMed] [Google Scholar]
- 10.Du X, Barker A, Andrei E. Nat Nanotechnol. 2008;3:491. doi: 10.1038/nnano.2008.199. [DOI] [PubMed] [Google Scholar]
- 11.Bolotin KI, Sikes KJ, Jiang Z, Klima M, Fudenberg G, Hone J, Kim P, Stormer HL. Solid State Commun. 2008;146:351. [Google Scholar]
- 12.Lin Y-M, Avouris Ph. Nano Lett. 2008;8:2119. doi: 10.1021/nl080241l. [DOI] [PubMed] [Google Scholar]
- 13.Myung S, Solanki A, Kim C, Park J, Kim KS, Lee KB. Adv Mater. 2011;23:2221. doi: 10.1002/adma.201100014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mao S, Lu G, Yu K, Bo Z, Chen J. Adv Mater. 2010;22:3521. doi: 10.1002/adma.201000520. [DOI] [PubMed] [Google Scholar]
- 15.Lu J, Do I, Drzal LT, Worden RM, Lee I. ACS Nano. 2008;2:1825. doi: 10.1021/nn800244k. [DOI] [PubMed] [Google Scholar]
- 16.Ohno Y, Maehashi K, Yamashiro Y, Matsumoto K. Nano Lett. 2009;9:3318. doi: 10.1021/nl901596m. [DOI] [PubMed] [Google Scholar]
- 17.Shan CS, Yang HF, Song JF, Han DX, Ivaska A, Niu L. Anal Chem. 2009;81:2378. doi: 10.1021/ac802193c. [DOI] [PubMed] [Google Scholar]
- 18.Myung S, Park J, Lee H, Kim KS, Hong S. Adv Mater. 2010;22:2045. doi: 10.1002/adma.200903267. [DOI] [PubMed] [Google Scholar]
- 19.Kobayashi T, Kimura N, Chi J, Hirata S, Hobara D. Small. 2010;6:1210. doi: 10.1002/smll.200902407. [DOI] [PubMed] [Google Scholar]
- 20.Jung I, Dikin D, Piner R, Ruoff R. Nano Lett. 2008;8:4283. doi: 10.1021/nl8019938. [DOI] [PubMed] [Google Scholar]
- 21.Farmer D, Golizadeh-Mojarad R, Perebeinos V, Lin Y, Tulevski G, Tsang J, Avouri Ph. Nano Lett. 2009;9:388. doi: 10.1021/nl803214a. [DOI] [PubMed] [Google Scholar]
- 22.Chen F, Qing Q, Zia J, Li J, Tao N. J Am Chem Soc. 2009;131:9908. doi: 10.1021/ja9041862. [DOI] [PubMed] [Google Scholar]
- 23.Bajzar L, Morser John, Nesheim Michael. J Biol Chem. 1996;271:16603. doi: 10.1074/jbc.271.28.16603. [DOI] [PubMed] [Google Scholar]
- 24.Star A, Gabriel JCP, Bradley K, Gruner G. Nano Lett. 2003;3:459. [Google Scholar]
- 25.Abe M, Murata K, Kojima A, Ifuku Y, Shimizu M, Ataka T, Matsumoto K. J Phys Chem C. 2007;111:8667. [Google Scholar]
- 26.Allen BL, Kichambare PD, Star A. Adv Mater. 2007;19:1439. [Google Scholar]
- 27.Ganzhorn M, Vijayaraghavan A, Dehm S, Hennrich F, Green AA, Fichtner M, Voigt A, Rapp M, Löhneysen Hv, Hersam MC, Kappes MM, Krupke R. ACS Nano. 2011;5:1670. doi: 10.1021/nn101992g. [DOI] [PubMed] [Google Scholar]
- 28.Diehl MR, Steuerman DW, Tseng H, Vignon SA, Star A, Celestre PC, Stoddart JF, Heath JR. Chem Phys Chem. 2003;4:1335. doi: 10.1002/cphc.200300871. [DOI] [PubMed] [Google Scholar]
- 29.Li B, Cao XH, Ong HG, Cheah JW, Zhou XZ, Yin ZY, Li H, Wang JL, Boey F, Huang W, Zhang H. Adv Mater. 2010;22:3058. doi: 10.1002/adma.201000736. [DOI] [PubMed] [Google Scholar]
- 30.Hjalmarsson C, Stenflo J, Borgström A. Pancreatology. 2009;9:700. doi: 10.1159/000215577. [DOI] [PubMed] [Google Scholar]
- 31.Kang X, Wang J, Wu H, Aksay I, Liu J, Lin Y. Biosens Bioelectron. 2009;25:901. doi: 10.1016/j.bios.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 32.Alwarappan S, Liu C, Kumar A, Li C. J Phys Chem C. 2010;114:12920. [Google Scholar]
- 33.Dupont J, Consorti CS, Suarez PAZ, de Souza RF, Fulmer SL, Richardson DP, Smith TE, Wolff S. Org Synth. 2002;79:236. [Google Scholar]
- 34.Liu J, Li Y, Li Y, Li J, Deng Z. J Mater Chem. 2010;20:900. [Google Scholar]
- 35.Wang Y, Li Z, Wang J, Li J, Lin Y. Trends Biotechnol. 2011;29:205. doi: 10.1016/j.tibtech.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.