

Abstract
Objective
Acadesine, an adenosine-regulating agent and activator of AMP-activated protein kinase, has been shown to possess antiinflammatory activity. This study investigated whether and how acadesine inhibits tissue factor (TF) expression and thrombus formation.
Methods and Results
Human umbilical vein endothelial cells and human peripheral blood monocytes were stimulated with lipopolysaccharide to induce TF expression. Pretreatment with acadesine dramatically suppressed the clotting activity and expression of TF (protein and mRNA). These inhibitory effects of acadesine were unchanged for endothelial cells treated with ZM241385 (a specific adenosine A2A receptor antagonist) or AMP-activated protein kinase inhibitor compound C, and in macrophages lacking adenosine A2A receptor or α1–AMP-activated protein kinase. In endothelial cells and macrophages, acadesine activated the phosphoinositide 3-kinase/Akt signaling pathway, reduced the activity of mitogen-activated protein kinases, and consequently suppressed TF expression by inhibiting the activator protein-1 and NF-κB pathways. In mice, acadesine suppressed lipopolysaccharide-mediated increases in blood coagulation, decreased TF expression in atherosclerotic lesions, and reduced deep vein thrombus formation.
Conclusion
Acadesine inhibits TF expression and thrombus formation by activating the phosphoinositide 3-kinase/Akt pathway. This novel finding implicates acadesine as a potentially useful treatment for many disorders associated with thrombotic pathology, such as angina pain, deep vein thrombosis, and sepsis.
Keywords: anticoagulants, atherosclerosis, thrombus, tissue factor
Acadesine, also called aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside, is an adenosine-regulating agent that acts to increase the bioavailability of adenosine. In clinical trials, treatment with acadesine was found to decrease the incidence of perioperative myocardial infarction, cardiac death, and a combined outcome of perioperative myocardial infarction, stroke, and cardiac death.1,2 The mechanisms underlying these beneficial effects remain ill-defined but may involve the inhibitory effects of acadesine on platelet aggregation3,4 neutrophil activation,5 and thrombus formation.6
Acadesine is phosphorylated by adenosine kinase (ADK) to 5-aminoimidazole-4-carboxamide-1-β4-ribonucleotide. The 5-aminoimidazole-4-carboxamide-1-β4-ribonucleotide mimics AMP, activating AMP-activated protein kinase (AMPK) without altering the cellular levels of ATP, ADP, or AMP.7 The activation of AMPK has been associated with many antiinflammatory and antidiabetic effects,8 and many of the effects of acadesine are thought to occur through AMPK activation. These effects include, for example, the ability of acadesine to suppress the expression of adhesion molecules on endothelial cells9 and to inhibit the production of proinflammatory factors (tumor necrosis factor-α, IL-1β, IL-6, and nitric oxide) in astrocytes, microglia, and macrophages via inhibition of NF-κB and C/EBP pathways.10,11 Consequently, acadesine treatment protects animals from experimental autoimmune encephalomyelitis.9,12
Tissue factor (TF) is a critical initiator of the physiological and pathological coagulation cascade.13 In the TF-initiated extrinsic coagulation pathway, prothrombin is converted to thrombin, thereby inducing fibrin formation, platelet activation, and thrombus formation.13 Endothelial cells and monocytes are major cellular origins of TF undergoing pathological circumstances. In response to various inflammatory stimuli, all of the following are activated: mitogen-activated protein kinase (MAPK) p38, extracellular signal-regulated kinase 1/2 (p44/42), and c-jun terminal NH2-kinase. These, in turn, activate transcription factors such as AP-1, NF-κB, and EGR-1, which signal the TF promoter to induce TF expression in endothelial cells and monocytes.14–16 In contrast, the phosphoinositide 3-kinase (PI3K) pathway negatively regulates TF expression.17,18 This effect occurs through cross-talk between PI3K and MAPK.17,18
We hypothesized that acadesine inhibited TF expression and thrombus formation. To test this hypothesis, we determined the effect of acadesine on TF expression in endothelial cells and monocytes. We also explored the intracellular signaling pathways involved in this process and examined the effect of acadesine on TF expression in vivo by using mouse models of sepsis, deep vein thrombosis, and atherosclerosis.
Results
Acadesine Suppresses LPS-Induced or Cytokine-Induced TF Expression in HUVEC and Human Monocytes
To determine whether acadesine regulates TF expression in HUVEC, we first pretreated cells with acadesine at various concentrations for 60 minutes and then stimulated cells with LPS to induce TF expression. Using a 1-stage clotting assay of cell lysates, we found that acadesine downregulated the clotting activity of TF in a dose-dependent manner (Figure 1A). The suppression of clotting activity of TF was further confirmed by an assay that specifically examines cell-surface TF activity (Supplementary Figure I). Consistent with these data, acadesine decreased TF expression at the mRNA and protein levels, as shown by real-time reverse-transcription polymerase chain reaction and Western blots in a dose-dependent manner (Figure 1B,C). In addition, acadesine suppressed IL-1β–induced or tumor necrosis factor-α–induced TF expression in HUVEC, and murine tumor necrosis factor-α–stimulated TF expression in murine bEND.3 endothelial cells (Supplementary Figures II, III). Similar declines in TF clotting activity and expression were found in human blood monocytes that were pretreated with acadesine, followed by stimulation with LPS (Figure 1D-F).
Figure 1.

Acadesine suppresses LPS-induced or cytokine-induced TF expression in HUVEC and human monocytes. HUVEC (A–C) or monocytes (D–F) were pretreated with acadesine for 1 hour at various concentrations, followed by stimulation with LPS at 1 μg/mL for 4 hours. A and D, TF activity of cell lysates was examined with a 1-stage clotting assay. B and E, Total cellular RNA was extracted, and TF mRNA expression was analyzed by real-time reverse-transcription polymerase chain reaction. C and F, TF expression at the protein level was analyzed by Western blotting. Data are shown as means±SEM of 6 independent experiments. *P<0.05. **P<0.01.
AMPK Is Not Involved in the Inhibitory Effect of Acadesine on TF Expression
Acadesine is an activator of AMPK, which is associated with many antiinflammatory and antidiabetic effects.8 Some of the antiinflammatory effects of acadesine appear to be mediated by AMPK.10 To determine whether AMPK mediates the inhibitory effect of acadesine on TF expression, we pretreated HUVEC with AMPK inhibitor compound C before adding acadesine and LPS to the cell medium. Acadesine was able to inhibit TF expression in compound C-treated cells to the same extent as in vehicle (DMSO)-treated cells; this was demonstrated by the 1-step clotting assay for TF activity, real-time reverse-transcription polymerase chain reaction for TF mRNA levels, and Western blots for TF protein levels (Figure 2A–C). As another test of the involvement of AMPK in the inhibitory effect of acadesine on TF expression, we isolated macrophages from mice deficient in α1-AMPK, which is the sole AMPK catalytic unit in vascular cells and leukocytes.8 These mice were obtained from Benoit Viollet Laboratory, Institut Cochin, Paris, France. Compared to wild-type cells, macrophages isolated from α1-AMPK−/−mice exhibited higher levels of TF-mediated clotting, and this difference in activity was observed both with and without LPS treatment (Supplementary Figure IV). Importantly, in wild-type macrophages and α1-AMPK–deficient macrophages, acadesine suppressed LPS-induced TF-mediated clotting to the level seen in nontreated cells. Together, these data indicate that AMPK is not involved in the inhibitory effect of acadesine on LPS-induced TF expression.
Figure 2.

AMPK and A2AR are not involved in the inhibitory effect of acadesine on TF expression. HUVEC were treated with LPS (1 μg/mL), LPS and acadesine (1 mmol/L), or LPS and acadesine in the presence of AMPK inhibitor compound C (10 μmol/L) or A2AR antagonist ZM241385 (5 μmol/L). A and D, TF activity of cell lysate was examined with a 1-stage clotting assay. B and E, Total cellular RNA was extracted and TF mRNA expression analyzed by real-time reverse-transcription polymerase chain reaction. GAPDH was used as normalization control. C and F, TF expression at the protein level was analyzed by Western blotting. Data are shown as means±SEM of 6 independent experiments. *P<0.05. **P<0.01.
Adenosine A2A Receptor Does Not Contribute to the Inhibitory Effect of Acadesine on TF Expression
Acadesine increases adenosine levels to regulate a variety of physiological and pathological functions.20 Adenosine A2A receptor (A2AR) mediates most of the antiinflammatory effects of adenosine.21 To test whether A2AR is involved in the inhibitory effect of acadesine on TF expression, we pretreated HUVEC with the A2AR antagonist ZM241385 before adding acadesine and LPS to the cell medium. Pre-treatment of HUVEC with ZM241385 did not change the inhibitory effect of acadesine on TF expression, as evidenced by the results of the 1-step clotting assay and by TF mRNA and protein levels (Figure 2D-F). We also used macrophages isolated from A2AR−/− mice (obtained from Jiang-Fan Chen Laboratory, Boston University School of Medicine, Boston, Mass) to determine the involvement of A2AR in acadesine-mediated TF suppression. In these A2AR-deficient cells, LPS induced the same level of TF-mediated clotting as it did in wild-type cells, and acadesine suppressed TF activity to the same extent as in wild-type cells (Supplementary Figure V). These data indicate that A2AR does not contribute to the inhibitory effect of acadesine on LPS-induced TF expression.
PI3K/Akt Signaling Is Crucial to the Inhibitory Effect of Acadesine on TF Expression in Endothelial Cells and Monocytes
The PI3K/Akt signaling pathway has been shown to negatively regulate TF expression in vitro and in vivo.1–4 We investigated whether acadesine could activate the PI3K/Akt pathway and affect LPS-induced Akt phosphorylation. Under resting conditions, acadesine increased the levels PI3K activity and Akt phosphorylation in a time-dependent manner in both HUVEC and human monocytes (Figure 3A, B; Supplementary Figure VIA). The PI3K inhibitor wortmannin blocked the activation of Akt by acadesine, indicating that Akt phosphorylation in acadesine-treated cells is attributable to acadesine-mediated PI3K activation. Treatment with LPS initiated minor Akt phosphorylation in HUVEC and monocytes. Acadesine pre-treatment significantly elevated the levels of phospho-Akt in LPS-treated cells (Figure 3C; Supplementary Figure VIB).
Figure 3.

Role of the PI3K/Akt pathway in acadesine-downregulated TF expression in endothelial cells. A-C, HUVEC were treated with either acadesine (1 mmol/L) or LPS (1 μg/mL) and acadesine for different periods and lysed. A, PI3Kα was immunoprecipitated from cell lysates and its activity was assayed using a commercial enzyme-linked immunosorbent assay kit. Akt and phospho-Akt (B, C) were examined by Western blotting. D, Akt, GSK3β, extracellular signal-regulated kinase, and their phosphorylated forms were examined by Western blotting at 30 minutes after treatment with LPS (1 μg/mL), acadesine (1 mmol/L), or LPS and acadesine with or without wortmannin pretreatment (50 nM). E, TF in HUVEC was examined by Western blotting at 4 hours after treatment with LPS (1 μg/mL) or LPS and acadesine (1 mmol/L) with or without wortmannin pretreatment (50 nM) or LY294002 (10 μmol/L).
PI3K/Akt can negatively regulate multiple signaling pathways.22 GSK-3β, a crucial molecule for p65 phosphorylation, is 1 downstream phosphorylation target of Akt.23 Corresponding to the levels of phospho-Akt, acadesine treatment increased the levels of phospho-GSK-3β in HUVEC and monocytes under both resting and LPS-treated conditions (Figure 3D; Supplementary Figure VIC). Extracellular signal-regulated kinase, an important MAPK for TF expression, is the target of the Raf-1 pathway. The latter is regulated by PI3K/Akt signaling.17 In HUVEC treated with LPS, extracellular signal-regulated kinase phosphorylation was almost completely blunted by acadesine (Figure 3D). In human monocytes, the phosphorylation of extracellular signal-regulated kinase and p38 that was induced by LPS was also dramatically inhibited by acadesine (Supplementary Figure VID). To determine the role of PI3K/Akt activation in the inhibitory effect of acadesine on TF expression, HUVEC were pretreated with the PI3K inhibitor wortmannin and LY294002, followed by the addition of LPS and acadesine. Under these conditions, the suppression of TF expression by acadesine was greatly reduced (Figure 3E). Thus, the PI3K/Akt pathway is critically involved in the inhibitory effect of acadesine on TF expression in HUVEC.
The activation of transcription factors NF-κB and AP-1 is necessary for TF expression.14,15,16 We examined whether PI3K/Akt participated in the inhibitory effect of acadesine on the activation of NF-κB and AP-1 in HUVEC and monocytes. Electrophoretic mobility shift assays showed that 60 minutes after treatment with LPS and acadesine, the nuclear levels of AP-1 and NF-κB p50/p65 bound to their oligonucleotides were much lower than those of cells treated with LPS only (Figure 4A–D). Wortmannin, a PI3K inhibitor, reversed the inhibition of AP-1 and NF-κB activation by acadesine (Figure 4A–D). Additionally, electrophoretic mobility shift assays using nuclear extracts from LPS-treated cells also showed that acadesine directly interfered with the binding of transcription factors, especially AP-1, to their oligonucleotides (Supplementary Figure VII).
Figure 4.

Role of the AP-1 and NF-κB pathways in acadesine-downregulated TF expression in endothelial cells. Nuclear protein was extracted from HUVEC or human monocytes at 1 hour after treatment with LPS (1 μg/mL) or LPS and acadesine (1 mmol/L) with or without wortmannin pretreatment (50 nM). F–I, Electrophoretic mobility shift assays were performed using 32P-oligonucleotides corresponding to the AP-1 and NF-κB DNA recognition sequences. Results for AP-1 in HUVEC (A) and monocytes (C), respectively. Results for NF-κB (B, D). The intensity of each AP-1 or NF-κB band was quantified by densitometry and expressed as folds of untreated control cells. Data are shown as means±SEM of 4 independent experiments. *P<0.05.
Acadesine Inhibits TF Expression in Mouse Models of Sepsis, Atherosclerosis, and Thrombosis
In a mouse LPS-induced sepsis model, TF is produced at high levels and is a major contributor to the hypercoagulation state.18 To determine whether acadesine can suppress TF expression in this murine model, we injected acadesine intraperitoneally into C57BL/6J mice (purchased from the Jackson Laboratory, Bar Harbor, Me) 1 hour before LPS administration. LPS induced the expression of TF mRNA in many major organs, including the lung and liver. This expression was dramatically reduced by acadesine treatment (Figure 5A). Consistent with the results for TF mRNA, fibrin levels in the livers of mice treated with LPS and acadesine were much lower than those of mice treated with LPS only (Figure 5B). In immunohistochemistry experiments that used an antibody against fibrin, robust fibrin deposition was found on the endothelium of venules in the livers of LPS-treated mice but not in control mice. Treatment with acadesine almost completely eliminated LPS-induced fibrin deposition (Figure 5C).
Figure 5.
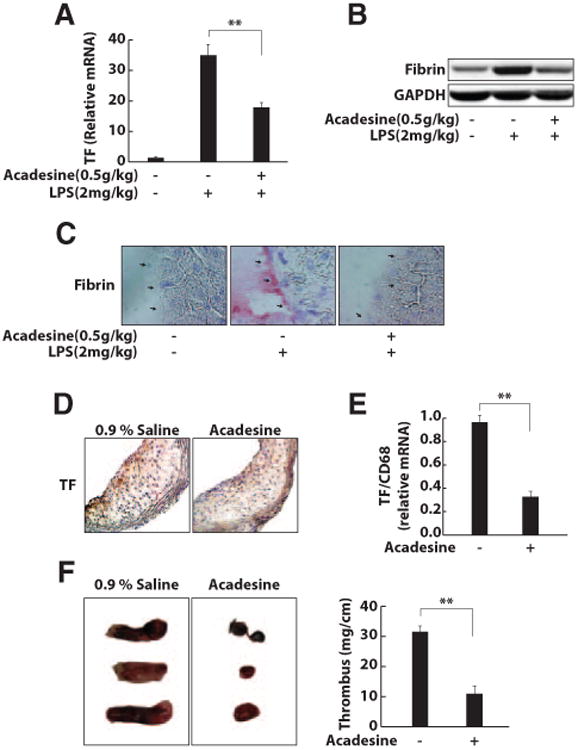
Acadesine inhibits TF production in the mouse models of sepsis, atherosclerosis, and thrombosis. C57 Bl/6J mice (male, 25–30 grams) were injected intraperitoneally with acadesine (500 mg/kg) and LPS (2 mg/kg). At 6 or 24 hours after injections, livers were collected for RNA extraction or processed for Western blots and histochemical analysis. A, TF mRNA expression was analyzed by real-time reverse-transcription polymerase chain reaction. B, Liver lysates were prepared and fibrin expression at the protein level was analyzed by Western blotting. C, Liver sections were immunostained with AP-conjugated antibody against fibrin to detect fibrin on the endothelium of venules (arrows; *P<0.05; ** P<0.01). D and E, Apolipoprotein E−/− mice fed a western diet for 3 months were intraperitoneally injected with vehicle (0.9% saline) or acadesine (500 mg/kg) daily for 5 days. The atherosclerotic arteries were collected and processed for immunostaining and RNA extraction (*P<0.05; **P<0.01). D, The stainings of TF in atherosclerotic lesions of mice treated with the vehicle and acadesine were quantified and compared. E, The levels of TF mRNA were measured with real-time reverse-transcription polymerase chain reaction and compared between 2 groups. F, C57BL/6J mice (male, 25–30 grams) were injected intraperitoneally with vehicle (saline) or acadesine (500 mg/kg) and then underwent a surgical procedure to initiate thrombus formation in the inferior vena cava. The injections were performed daily for 3 days. The thrombosed inferior vena cava was weighed and the length of thrombus was measured. The size of the thrombus was quantified as mg/cm. Three representative thrombosed inferior vena cava from 10 mice in each group and the quantitative data for thrombosed inferior vena cava are shown. *P<0.05. **P<0.01.
Macrophages are a major source of TF in atherosclerotic lesions.24 On sections of atherosclerotic arteries from apolipoprotein E−/− mice fed a western diet for 3 months, strong TF-positive staining (brown) was observed (Figure 5D). Peritoneal injection of acadesine for 5 days did not significantly reduce the number of macrophages in atherosclerotic lesions (data not shown). However, acadesine treatment dramatically decreased TF expression, demonstrated by the weak TF staining on the sections of arteries from mice treated with acadesine (Figure 5D). Additionally, the level of TF mRNA in atherosclerotic arteries of apolipoprotein E−/−mice treated with acadesine was significantly lower than that in arteries from apolipoprotein E−/− mice treated with vehicle (Figure 5E). These results indicate that TF expression by macrophages in atherosclerotic lesions is dramatically inhibited by acadesine treatment.
TF is also important in the formation of deep vein thrombosis.25 Using a mouse model of deep vein thrombosis, we examined whether acadesine can affect thrombus formation. Mice treated with vehicle had large thrombi in their inferior vena cava, whereas mice treated with acadesine had very small thrombi in their ligated inferior vena cava (Figure 5F).
Discussion
Our study demonstrates that acadesine inhibits TF expression in endothelial cells and in monocytes/macrophages, and that this inhibition occurs through the activation of the PI3K/Akt signaling pathway. PI3K/Akt signaling reduces the activity of MAPK, phosphorylates GSK-3/β, and suppresses the AP-1 and NF-κB pathways, thereby inhibiting TF expression. Acadesine is also effective in suppressing the expression of TF in mouse models of sepsis, atherosclerosis, and thrombosis.
Our findings indicate that the PI3K/Akt pathway is a focal point for the inhibitory effect of acadesine on TF expression. The PI3K/Akt pathway is a conserved family of signal transduction enzymes that participate in the regulation of cell proliferation and survival.22 A number of studies have demonstrated that PI3K/Akt is a negative-feedback regulator of TF expression.17,18,26 Acadesine affects this signaling pathway differently in different cells. Acadesine inhibits PI3K/Akt signaling in C6, MCF-7, PC3, CEM, and K562 tumor cells but enhances it in acute lymphoblastic leukemia cells.27,28 Acadesine seems to have no effect on PI3K/Akt in vascular smooth muscle cells.29 However, we found that in endothelial cells and monocytes, the PI3K/Akt pathway is dramatically activated by acadesine under resting and activated conditions. LPS or proinflammatory cytokines, such as tumor necrosis factor-α, also induced Akt phosphorylation in endothelial cells. After incubation with LPS, the levels of phospho-Akt peaked at 30 to 60 minutes, whereas the levels of phospho-MAPK reached the highest level at 15 to 30 minutes (Figure 3D; Supplementary Figure VID). Thus, the delay in PI3K/Akt activation allows proinflammatory cytokines to activate MAPK, thereafter activating NF-κB and AP-117 and inducing TF expression. In contrast, in acadesine-treated cells, Akt phosphorylation was elevated initially and further increased during LPS stimulation. The increased phospho-Akt was able to phosphorylate GSK-3β to prevent it from phosphorylating p65,23 resulting in inhibition of NF-κB activity and NF-κB-dependent TF expression. Furthermore, the increased phospho-Akt was also able to inhibit the phosphorylation of MAPK, thereby suppressing the NF-κB and AP-1 pathways.17 The inhibitory effect of acadesine on TF expression in endothelial cells was reversed by the PI3K inhibitor wortmannin and LY294002, indicating that PI3K/Akt activation is required for the suppression of TF expression by acadesine. LY294002 is more specific than wortmannin for inhibition of PI3K activity. In our study, reversal of the inhibitory effect of acadesine on TF expression by LY294002 was not as great as that by wortmannin, although the difference did not reach statistical significance. This may be attributable to more broad effects on pathways other than PI3K for wortmannin or incomplete inhibition of PI3K activity by LY294002 at the dose used in our assay.
In addition to the inhibitory effect on translocation of transcription factors, acadesine was able to directly interfere with the binding of transcription factors to their specific DNA sequences, indicating 1 more level at which acadesine could inhibit TF expression (Supplementary Figure VII). This observation is consistent with a previous report.30
Our findings also indicate that acadesine-mediated suppression of TF is not associated with its role in AMPK activation. Endogenous AMPK activity is important in maintaining the resting status of cells, and others have suggested that many effects of acadesine, especially its role in the regulation of inflammation, are linked to its activation of AMPK.9–12 We did find that in mouse macrophages lacking α1AMPK, TF activity was increased more than that of wild-type macrophages, even without any stimulation (Supplementary Figure IV). Additionally, acadesine was able to activate AMPK in endothelial cells and macrophages. However, neither the suppression of AMPK activity in human endothelial cells with compound C nor the depletion of α1AMPK in mouse macrophages abrogated the inhibitory effect of acadesine on LPS-induced TF expression. Furthermore, it has been previously reported that acadesine activates endothelial Akt in an AMPK-independent manner.31 In this study, we observed that the inhibitory role of acadesine in TF expression is PI3K/Akt-dependent. These results provide convincing evidence that the observed inhibitory effect of acadesine in TF expression is not related to its role in AMPK activation.
Acadesine increases the bioavailability of adenosine; however, the exact mechanism for this has not been elucidated.32 It is known that after entrance into the intracellular compartment, acadesine is converted by ADK to 5-aminoimidazole-4-carboxamide-1-β4-ribonucleotide. ADK is a crucial adenosine-removing enzyme.33 Thus, it may be that the metabolism of acadesine by ADK interferes with the adenosine-removal function of this enzyme, thus leading to an increase in intracellular adenosine. It is plausible that the suppression of TF expression by acadesine is related to its role in adenosine regulation. Adenosine exerts many of its antiinflammatory effects through the receptor A2AR. In our study, however, blockage or deficiency of A2AR did not influence the inhibitory effect of adenosine on TF expression. This suggests that the mechanism of inhibition is A2AR-independent. Adenosine can activate PI3K/Akt in either an A1AR-dependent or an A1AR-independent manner.34–36 Because HUVEC do not express A1AR, it is likely that our observed activation of PI3K/Akt by acadesine occurs via an adenosine receptor-independent pathway. To mimic the role of acadesine in PI3K/Akt activation and TF expression, we used lentiviral RNAi to knock-down ADK in HUVEC.40 In these cells, PI3K/Akt was activated and LPS-induced TF expression was suppressed (Supplementary Figure VIII). Thus, it is very likely that acadesine suppresses TF expression through its regulation of ADK activity.
Given its inhibitory effect on TF expression, acadesine shows promise as a treatment for diseases associated with pathological thrombosis. Individuals with sepsis have a high level of TF and elevated coagulation states in their blood.37,38 Elevated TF has been found in the local coronary circulation and systemic blood flow of patients with acute coronary syndromes, such as unstable angina and myocardial infarction.39 Deep vein thrombosis is also linked to TF production.39 In our study, acadesine dramatically inhibited TF expression in models of mouse sepsis and deep vein thrombosis, and it also decreased the production of TF by lesional macrophages. Because clinical trials have demonstrated the safety of acadesine for the prevention of perioperative myocardial infarction, its potential extension to other diseases is a realistic option for improving clinical outcomes through the inhibition of TF expression.
Material and Methods
Reagents
Lipopolysaccharide (Escherichia Coli 0111:B4) was from Sigma-Aldrich (St. Louis, MO). Human TNF-α and IL-1β were from R&D systems (Minneapolis, MN). Adenosine receptor A2AR antagonist ZM241385 and compound C were from Tocris (Ellisville, MO) and Calbiochem(Gibbstown, NJ). Rabbit polyclonal antibodies against human tissue factor (for western blotting) and mouse tissue factor (for immunohistochemistry) were kindly provided by Dr. Ronald Bach (VAMC, Minneapolis, MN). Antibodies for western blotting, including Rabbit anti-GAPDH, anti-p38, anti-phospho-p38, anti-extracellular signal-regulated kinase (ERK1/2), anti-phospho-ERK1/2, anti-JNK (JNK), anti-phospho-JNK (p-JNK), anti-Akt, anti-phospho-Akt, anti-GSK3β, and anti-phospho-GSK-3β were purchased from Cell Signaling Technology, Inc. (Danvers, MA). Antibody against Fibrin II beta (Bß 15-42, Clone: T2G1) was from Accurate Chemical & Scientific Corp (Westbury, NY). Prior to immunostaining, this antibody was conjugated with alkaline phosphatase using an AP conjugation kit from KPL, Inc. (Gaithersburg, MD). All AP-conjugated second antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). ECF™ substrate for western blotting was from GE Healthcare (Pittsburgh, PA). The system for isolation of human monocytes was from Miltenyi Biotec (Auburn, CA).
Culture of HUVECs and isolation of human peripheral blood monocytes and mouse peritoneal macrophages
As previously described 1, human endothelial cells were isolated from umbilical cords and cultured. HUVECs at passages 2 or 3 were used for experiments.
We isolated human monocytes by published methods 2. Briefly, fresh human blood was collected from healthy adult volunteers with acid-citrate-dextrose (ACD; 38 mM citric acid, 75 mM trisodium citrate, and 100 mM dextrose; 1:7 ACD final concentration). Whole blood was centrifuged for 15 min. The resultant leukocyte pellet was layered onto the HISTOPAQUE®-1077 (Sigma, 1077-1). The mononuclear cells collected from buffy coat were purified by CD14 microbeads (MACS; Miltenyi Biotec, Auburn, CA), resuspended in RPMI medium with 10% FBS, and incubated at 37°C for 2 h before use.
Mouse peritoneal macrophages were prepared by using a method previously described 3. Two milliliters of 4% thioglycolate medium was injected intraperitoneally into male mice (6 to 8 weeks old, 25–30 g body weight). Three days later, peritoneal macrophages were harvested by peritoneal lavage using sterile HBSS buffer supplemented with 5 mM EDTA. Lavaged macrophages were resuspended in cell grow medium, placed into 6-well plates overnight, then washed to remove non-adherent leukocytes. Macrophages adhering on culture plates were used for experiments.
One-stage clotting TF activity assay
Treated or non-treated cells were lysed. The TF activity of cell lysates was determined by a one-stage clotting assay standardized against recombinant human TF, as previously described 1.
TF activity on the surface of HUVECs
The assay was performed as described 4. HUVECs were cultured in 48-well plates and treated with LPS or/and acadesine. The TF activity on the cell surface of HUVECs was assayed by measuring the enzymatic activity of the TF/Factor VIIa complex. The cells were washed twice and incubated with Tris buffer (0.1 mM NaCl and 0.05 mM Tris, pH 7.3) containing BSA (5 mg/mL) and CaCl2 (4 mM), followed by the addition of human factor VII. After an incubation at 37°C for 30 min, the chromogenic substrate for TF/factor VIIa complex, Spectrozyme fXa (American Diagnostica Inc.,) was added. The reaction was incubated at 22°C for 30 min and stopped with glacial acetic acid. The absorbance was read at 405 nm and compared with those values obtained from a standard curve generated using known amounts of TF.
Real-time RT-PCR
Total RNA from cultured cells or frozen samples was extracted with Trizol reagent (Invitrogen, Carlsbad, CA). cDNA was synthesized with a first-strand cDNA synthesis kit (Fermentas, Glen Burnie, MD). PCR was performed with a Roche LightCycler 2.0 thermal cycler (Roche, Indianapolis, IN) that used SYBR Green as a double-stranded, DNA-specific binding dye. The relative amount of each gene in each sample was estimated by the Δ ΔCT method 5. Primer sequences for TF were as follows: hTF forward, TGACCTCACCGACGAGATTGTGAA; hTF reverse, TCTGAATTGTTGGCTGTCCGAGGT; mTF forward, ACAAGTGCTTCTCGACCACAGACA; and mTF reverse, TTGCTTCATAGGCCCAGGTCACAT. Primer sequences for human TF are able to detect both the membrane-bound, full-length TF and a soluble, alternatively spliced isoform 6.
Western blot analysis
Western blot analyses were performed as described 5. Cells were rinsed twice with ice-cold PBS and lysed in modified RIPA lysis buffer. Samples were separated on 8–10% sodium dodecyl sulfate-polyacrylamide gels, then transferred to polyvinylidene fluoride membranes. Membranes were blocked with 5% non-fat milk in Tris-buffered saline with 0.1% Tween 20 for 1 h at room temperature, then probed overnight at 4°C with primary antibodies against TF, GAPDH, p38, pp-38, p-ERK, ERK, p-Akt, or Akt, followed by incubation with alkaline phosphatase-conjugated second antibody for 1 h at room temperature. Membranes were developed with a chemifluorescence reagent and scanned by Storm 860 (GE, Healthcare, Piscataway, NJ). Band intensities were quantified with the NIH Image J program.
PI3K activity measurement
PI3Kα was immunoprecipitated from cell lysates. PI3Kα activity was assayed using PI3-Kinase Activity ELISA:Pico, a competitive ELISA kit (Echelon Biosciences Inc., Salt Lake City, UT), according to the manufacturer's instructions.
Electrophoretic mobility shift assay (EMSA)
Nuclear protein was extracted with the NucBuster Protein Extraction kit from Novagen (San Diego, CA). The following oligonucleotides were purchased from Santa Cruz Biotechnology: NF-κB consensus (5′-AGTTGAGGGGACTTTCCCAGGC-3′), NF-κB mutant (5′-AGTTGAGGCGACTTTCCCAGGC-3′), SP-1 consensus (5′-ATTCGATCGGGGCGGGGCGAGC-3′), and SP-1 mutant (5′-ATTCGATCGGTTCGGGGCGAGC-3′). Oligonucleotides for the AP-1 consensus site (5′-CTGGGGTGAGTCATCCCTT-3′) and AP-1 mutant control (5′-CTGGGGTGAGTTGTCCCTT-3′) were from Integrated DNA Technologies, Inc. (Coralville, IA). Oligonucleotides were end-labeled with [γ-32P] ATP (Amersham Biosciences) and T4 polynucleotide kinase (Amersham Biosciences). Five micrograms of nuclear extract was loaded per lane (10 μg for the TF- B probe). EMSA was carried out as described 1.
Mice
Wild-type C57BL/6J mice and apolipoprotein E-deficient (apoE−/−) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). A2AR−/− and α1-AMPK−/− mice were produced as described 5 7. A2AR−/− mice were in a C57BL/6J background, whereas α1-AMPK−/− mice had a mixed 129 and C57BL/6J background. For experiments that used α1-AMPK–deficient macrophages, cells were isolated from α1-AMPK−/− mice and from age- and gender-matched littermate α1-AMPK+/+ mice.
Mouse models of endotoxemia, atherosclerosis, and deep vein thrombosis
To initiate the mouse model of endotoxemia, male wild-type mice (8 weeks, 25–30 g body weight) were intraperitoneally injected with LPS (2 mg/kg, E. coli serotype 0111:B4; Sigma Chemical, St Louis, MO) 8. To develop atherosclerotic lesions in arteries, apoE–/– mice were fed a Western diet containing 21% fat, 0.15% cholesterol, and 19.5% casein without sodium cholate for 3 months 9.
To develop deep vein thrombi, male wild-type mice (8 weeks, 25–30 g body weight) underwent general anesthesia. Following a laparotomy, the inferior vena cava (IVC) was ligated with a 6-0 polypropylene suture. Three days later, mice were euthanized, and the thrombosed IVC was collected, weighed, and measured for length. The samples were snap-frozen and stored for histological analysis 10.
Acadesine treatment
For experiments involving a single administration of acadesine (mouse model of endotoxemia), acadesine was dissolved in saline (20 g/L) and injected intraperitoneally without anesthesia at a dose of 500 mg/kg body weight 11, 12. For experiments involving repeated injections of acadesine (mouse models of atherosclerosis and thrombosis), 500 mg/kg of acadesine was dissolved in saline (20 g/L) and injected into mice intraperitoneally once a day for the time indicated (5 d for atherosclerosis model, 3 d for thrombosis model). Saline was injected as a control condition in all experiments.
Statistical analyses
Statistical analyses were performed with Instat software (GraphPad Software). Data are presented as the mean ± SEM. Data were compared with either one-way ANOVA followed by the Bonferroni correction post-hoc test or Student t test to evaluate two-tailed levels of significance. The null hypothesis was rejected at P < 0.05.
Supplementary Material
Supplementary Figure 1. Acadesine suppresses LPS-induced TF activity on the surface of HUVECs.
HUVECs were pretreated with acadesine for 1 h at various concentrations, followed by stimulation with LPS at 1 μg/mL for 4 h. TF activity on the cell surface was assayed by measuring the enzymatic activation by the TF/factor VIIa complex. An activity of 1 is equivalent to 10−15 Mol of TF. Data are shown as means ± SEM of six independent experiments, * P < 0.05, ** P < 0.01.
Supplementary Figure 2. Acadesine suppresses cytokine-induced TF expression in HUVECs. HUVECs were pretreated with acadesine at 1 mM for 1 h, followed by stimulation with TNF-α (10 ng/mL) or IL-1β (10 ng/mL) for 4 h. TF expression at the protein level was analyzed by western blotting. The intensity of the TF bands was quantitated by densitometry, normalized to GAPDH, and expressed as folds of TF expressed by untreated HUVECs. Data are shown as means ± SEM of four independent experiments. The data from cells treated with cytokines and cytokine/acadesine were compared, * P < 0.05, ** P < 0.01.
Supplementary Figure 3. Acadesine suppresses LPS-induced TF expression and activity in murine bEND.3 cells.
Murine bEND.3 endothelial cells were pretreated with acadesine for 1 h at various concentrations, followed by stimulation with LPS at 1 μg/mL for 4 h. TF activity of cell lysates were examined with a one-stage clotting assay (a). Total cellular RNA was extracted, and TF mRNA expression was analyzed by real-time RT-PCR. GAPDH was used as normalization control (b). Data are shown as means ± SEM of six independent experiments, * P < 0.05,** P < 0.01.
Supplementary Figure 4. The effect of acadesine on TF activity in α1AMPK-deficient murine macrophages.
Thioglycollate-elicited peritoneal macrophages were isolated from wild-type or α1AMPK−/− mice, then treated with either LPS (1 μg/mL) or LPS and acadesine (1 mM), followed by the one-stage clotting assay to measure TF activity. Data are shown as means ± SEM of six independent experiments, * P < 0.05, ** P < 0.01.
Supplementary Figure 5. The effect of acadesine on TF activity in A2A R-deficient murine macrophages.
Thioglycollated-elicited peritoneal macrophages were isolated from wild type or A2AR−;/−; mice, then were treated with either LPS (1 μg/mL) or LPS and acadesine (1 mM), followed by a one-stage clotting assay to measure TF activity. Data are shown as means ± SEM of six independent experiments, * P < 0.05, ** P < 0.01.
Supplementary Figure 6. Role of the PI3K/Akt pathway in acadesine-downregulated TF expression in human monocytes.
(a) and (b), Akt and phospho-Akt in human monocytes treated with acadesine (1 mM), LPS (1 μg/mL) or LPS and acadesine for different periods and examined by western blotting. The intensity of the phospho-Akt was quantitated by densitometry and, after normalization to total Akt, expressed as fold difference over untreated monocytes. Data are shown as means ± SEM of six independent experiments. The data from treated cells at different time points were compared with those from untreated monocytes, * P < 0.05. (c), Akt, GSK-3β, and their phosphorylated forms were examined by western blotting at 30 min after treatment with LPS (1 μg/mL), acadesine (1 mM), or LPS and acadesine with or without wortmannin pretreatment (50 nM). Data are shown from one of four independent experiments. (d), At various time points after treatment with LPS (1 μg/mL) or LPS and acadesine (1 mM), monocytes were examined by western blotting for the levels of phospho-ERK1/2, ERK1/2, phospho-JNK, JNK, phospho-p38, and p38. Immunoblots are representative of four independent experiments.
Supplementary Figure 7. Acadesine directly interferes with the binding of transcription factors to their oligonucleotides
Nuclear protein was extracted from HUVECs or human monocytes treated with LPS. EMSA was performed using 32P-oligonucleotides corresponding to the AP-1 and NF-κB DNA recognition sequences and the same amount nuclear protein in the presence of acadesine at different concentrations. The intensities of AP-1 and NF-κB bands were quantified by densitometry and expressed as fold difference over untreated control cells. Data are shown as means ± SEM of four independent experiments, * P > 0.05.
Supplementary Figure 8. PI3K/Akt activation and TF expression in adenosine kinase knockdown HUVECs.
(a), Akt and phospho-Akt in HUVECs transduced with lentiviruses carrying siRNA against adenosine kinase or a scrambled sequence (control). The intensity of phospho-Akt was quantitated by densitometry and, after normalization to total Akt, expressed as the fold change over this value in HUVECs transduced with control virus, * P < 0.05. (b), HUVECs transduced with control or ADK RNAi viruses were examined by western blotting for TF expression at 4 h after treatment with LPS (1 μg/mL) with or without wortmannin pretreatment (50 nM). The intensity of the TF bands was quantitated by densitometry, normalized to GAPDH, and expressed as the fold change over TF expressed by HUVECs without ADK knockdown. Data are shown as means ± SEM of four independent experiments, * P < 0.05.
Acknowledgments
The authors thank Dr Anne Marie Weber-Main for her critical review and editing of manuscript drafts.
Sources of Funding: This work was supported by AHA 0430151N, NIH HL78679, and HL080569 (to Y. Huo).
Footnotes
The online version of this article, along with updated information and services, is located on the World Wide Web at: http://atvb.ahajournals.org/content/30/5/1000
Reprints: Information about reprints can be found online at: http://www.lww.com/reprints
Disclosures: None.
Contributor Information
Weiyu Zhang, Department of Medicine, University of Minnesota, Minneapolis, Minn.
Jianguo Wang, Department of Medicine, University of Minnesota, Minneapolis, Minn.
Huan Wang, Department of Medicine, University of Minnesota, Minneapolis, Minn.
Rong Tang, Department of Medicine, University of Minnesota, Minneapolis, Minn.
John D. Belcher, Department of Medicine, University of Minnesota, Minneapolis, Minn
Benoit Viollet, INSERM U567, CNRS UMR8104, Department of Endocrinology, Metabolism and Cancer, Institut Cochin, Universite Paris 5, Paris, France.
Jian-Guo Geng, Department of Medicine, University of Minnesota, Minneapolis, Minn.
Chunxiang Zhang, Department of Anesthesiology, New Jersey Medical School, University of Medicine and Dentistry of New Jersey, Newark, NJ.
Chaodong Wu, Department of Nutrition and Food Science, Texas A&M University, College Station, Tex.
Arne Slungaard, Department of Medicine, University of Minnesota, Minneapolis, Minn.
Chuhong Zhu, Department of Medicine, University of Minnesota, Minneapolis, Minn; Department of Anatomy, Third Military Medical University, Chongqing, China.
Yuqing Huo, Department of Medicine, University of Minnesota, Minneapolis, Minn.
References
- 1.Mangano DT. Effects of acadesine on myocardial infarction, stroke, and death following surgery. A meta-analysis of the 5 international randomized trials. The Multicenter Study of Perioperative Ischemia (McSPI) Research Group. JAMA. 1997;277:325–332. doi: 10.1001/jama.277.4.325. [DOI] [PubMed] [Google Scholar]
- 2.Mangano DT, Miao Y, Tudor IC, Dietzel C. Post-reperfusion myocardial infarction: long-term survival improvement using adenosine regulation with acadesine. J Am Coll Cardiol. 2006;48:206–214. doi: 10.1016/j.jacc.2006.04.044. [DOI] [PubMed] [Google Scholar]
- 3.Bullough DA, Zhang C, Montag A, Mullane KM, Young MA. Adenosine-mediated inhibition of platelet aggregation by acadesine. A novel antithrombotic mechanism in vitro and in vivo. J ClinInvest. 1994;94:1524–1532. doi: 10.1172/JCI117493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dietrich WD, Miller LP, Prado R, Dewanjee S, Alexis N, Dewanjee MK, Gruber H. Acadesine reduces indium-labeled platelet deposition after photothrombosis of the common carotid artery in rats. Stroke. 1995;26:111–116. doi: 10.1161/01.str.26.1.111. [DOI] [PubMed] [Google Scholar]
- 5.Gruber HE, Hoffer ME, McAllister DR, Laikind PK, Lane TA, Schmid-Schoenbein GW, Engler RL. Increased adenosine concentration in blood from ischemic myocardium by AICA riboside. Effects on flow, granulocytes, and injury. Circulation. 1989;80:1400–1411. doi: 10.1161/01.cir.80.5.1400. [DOI] [PubMed] [Google Scholar]
- 6.Young MA, Henry C, Wong S, Bullough D, Mullane K. Acadesine reduces the frequency of coronary artery reocclusion following rt-PA induced thrombolysis in the dog. Thromb Haemostat. 1995;74:1348–1352. [PubMed] [Google Scholar]
- 7.Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5-aminoimidazole-4-carboxamide ribonucleoside A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem. 1995;229:558–565. doi: 10.1111/j.1432-1033.1995.tb20498.x. [DOI] [PubMed] [Google Scholar]
- 8.Motoshima H, Goldstein BJ, Igata M, Araki E. AMPK and cell proliferation–AMPK as a therapeutic target for atherosclerosis and cancer. J Physiol. 2006;574:63–71. doi: 10.1113/jphysiol.2006.108324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prasad R, Giri S, Nath N, Singh I, Singh AK. 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside attenuates experimental autoimmune encephalomyelitis via modulation of endothelial-monocyte interaction. J Neurosci Res. 2006;84:614–625. doi: 10.1002/jnr.20953. [DOI] [PubMed] [Google Scholar]
- 10.Giri S, Nath N, Smith B, Viollet B, Singh AK, Singh I. 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside inhibits proinflammatory response in glial cells: a possible role of AMP-activated protein kinase. J Neurosci. 2004;24:479–487. doi: 10.1523/JNEUROSCI.4288-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ayasolla KR, Singh AK, Singh I. 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside (AICAR) attenuates the expression of LPS- and Abeta peptide-induced inflammatory mediators in astroglia. J Neuroinflammation. 2005;2:21. doi: 10.1186/1742-2094-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nath N, Giri S, Prasad R, Salem ML, Singh AK, Singh I. 5-aminoimidazole-4-carboxamide ribonucleoside: a novel immunomodulator with therapeutic efficacy in experimental autoimmune encephalomyelitis. J Immunol. 2005;175:566–574. doi: 10.4049/jimmunol.175.1.566. [DOI] [PubMed] [Google Scholar]
- 13.Morrissey JH, Fakhrai H, Edgington TS. Molecular cloning of the cDNA for tissue factor, the cellular receptor for the initiation of the coagulation protease cascade. Cell. 1987;50:129–135. doi: 10.1016/0092-8674(87)90669-6. [DOI] [PubMed] [Google Scholar]
- 14.Bierhaus A, Zhang Y, Deng Y, Mackman N, Quehenberger P, Haase M, Luther T, Muller M, Bohrer H, Greten J, Martin E, Baeuerlei PA, Waldherr R, Kisiel W, Ziegler R, Stern DM, Nawroth PP. Mechanism of the tumor necrosis factor alpha-mediated induction of endothelial tissue factor. J Biol Chem. 1995;270:26419–26432. doi: 10.1074/jbc.270.44.26419. [DOI] [PubMed] [Google Scholar]
- 15.Mackman N. Regulation of tissue factor gene expression in human monocytic and endothelial cells. Haemostasis. 1996;26(Suppl 1):17–19. doi: 10.1159/000217234. [DOI] [PubMed] [Google Scholar]
- 16.Oeth P, Parry GC, Mackman N. Regulation of the tissue factor gene in human monocytic cells. Role of AP-1, NF-kappa B/Rel, and Sp1 proteins in uninduced and lipopolysaccharide-induced expression. Arterioscler Thromb Vasc Biol. 1997;17:365–374. doi: 10.1161/01.atv.17.2.365. [DOI] [PubMed] [Google Scholar]
- 17.Guha M, Mackman N. The phosphatidylinositol 3-kinase-Akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J Biol Chem. 2002;277:32124–32132. doi: 10.1074/jbc.M203298200. [DOI] [PubMed] [Google Scholar]
- 18.Schabbauer G, Tencati M, Pedersen B, Pawlinski R, Mackman N. PI3K-Akt pathway suppresses coagulation and inflammation in endotoxemic mice. Arterioscler Thromb Vasc Biol. 2004;24:1963–1969. doi: 10.1161/01.ATV.0000143096.15099.ce. [DOI] [PubMed] [Google Scholar]
- 19.Wang J, Mahmud SA, Bitterman PB, Huo Y, Slungaard A. Histone deacetylase inhibitors suppress TF-kappaB-dependent agonist-driven tissue factor expression in endothelial cells and monocytes. J Biol Chem. 2007;282:28408–28418. doi: 10.1074/jbc.M703586200. [DOI] [PubMed] [Google Scholar]
- 20.Drew BG, Kingwell BA. Acadesine, an adenosine-regulating agent with the potential for widespread indications. Expert Opin Pharmacother. 2008;9:2137–2144. doi: 10.1517/14656566.9.12.2137. [DOI] [PubMed] [Google Scholar]
- 21.Hasko G, Cronstein BN. Adenosine: an endogenous regulator of innate immunity. Trends Immunol. 2004;25:33–39. doi: 10.1016/j.it.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 22.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 23.Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol. 2005;6:777–784. doi: 10.1038/ni1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang H, Tang R, Zhang W, Amirikian K, Geng Z, Geng J, Hebbel RP, Xia L, Marth JD, Fukuda M, Katoh S, Huo Y. Core2 1-6-N-Glucosaminyltransferase-I Is Crucial for the Formation of Atherosclerotic Lesions in Apolipoprotein E-Deficient Mice. Arterioscler Thromb Vasc Biol. 2009;29:180–187. doi: 10.1161/ATVBAHA.108.170969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li YD, Ye BQ, Zheng SX, Wang JT, Wang JG, Chen M, Liu JG, Pei XH, Wang LJ, Lin ZX, Gupta K, Mackman N, Slungaard A, Key NS, Geng JG. NF-kappaB transcription factor p50 critically regulates tissue factor in deep vein thrombosis. J Biol Chem. 2009;284:4473–4483. doi: 10.1074/jbc.M806010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fukao T, Koyasu S. PI3K and negative regulation of TLR signaling. Trends Immunol. 2003;24:358–363. doi: 10.1016/s1471-4906(03)00139-x. [DOI] [PubMed] [Google Scholar]
- 27.Rattan R, Giri S, Singh AK, Singh I. 5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside inhibits cancer cell proliferation in vitro and in vivo via AMP-activated protein kinase. J Biol Chem. 2005;280:39582–39593. doi: 10.1074/jbc.M507443200. [DOI] [PubMed] [Google Scholar]
- 28.Sengupta TK, Leclerc GM, Hsieh-Kinser TT, Leclerc GJ, Singh I, Barredo JC. Cytotoxic effect of 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside (AICAR) on childhood acute lymphoblastic leukemia (ALL) cells: implication for targeted therapy. Mol Cancer. 2007;6:46. doi: 10.1186/1476-4598-6-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagata D, Takeda R, Sata M, Satonaka H, Suzuki E, Nagano T, Hirata Y. AMP-activated protein kinase inhibits angiotensin II-stimulated vascular smooth muscle cell proliferation. Circulation. 2004;110:444–451. doi: 10.1161/01.CIR.0000136025.96811.76. [DOI] [PubMed] [Google Scholar]
- 30.Kuo CL, Ho FM, Chang MY, Prakash E, Lin WW. Inhibition of lipopolysaccharide-induced inducible nitric oxide synthase and cycloox-ygenase-2 gene expression by 5-aminoimidazole-4-carboxamide riboside is independent of AMP-activated protein kinase. J Cell Biochem. 2008;103:931–940. doi: 10.1002/jcb.21466. [DOI] [PubMed] [Google Scholar]
- 31.Morrow VA, Foufelle F, Connell JM, Petrie JR, Gould GW, Salt IP. Direct activation of AMP-activated protein kinase stimulates nitric-oxide synthesis in human aortic endothelial cells. J Biol Chem. 2003;278:31629–31639. doi: 10.1074/jbc.M212831200. [DOI] [PubMed] [Google Scholar]
- 32.Mullane K. Acadesine: the prototype adenosine regulating agent for reducing myocardial ischaemic injury. Cardiovasc Res. 1993;27:43–47. doi: 10.1093/cvr/27.1.43. [DOI] [PubMed] [Google Scholar]
- 33.Boison D. Adenosine kinase, epilepsy and stroke: mechanisms and therapies. Trends Pharmacol Sci. 2006;27:652–658. doi: 10.1016/j.tips.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 34.Takasuga S, Katada T, Ui M, Hazeki O. Enhancement by adenosine of insulin-induced activation of phosphoinositide 3-kinase and protein kinase B in rat adipocytes. J Biol Chem. 1999;274:19545–19550. doi: 10.1074/jbc.274.28.19545. [DOI] [PubMed] [Google Scholar]
- 35.Shen J, Halenda SP, Sturek M, Wilden PA. Cell-signaling evidence for adenosine stimulation of coronary smooth muscle proliferation via the A1 adenosine receptor. Circ Res. 2005;97:574–582. doi: 10.1161/01.RES.0000181159.83588.4b. [DOI] [PubMed] [Google Scholar]
- 36.Min KJ, Kim JH, Jou I, Joe EH. Adenosine induces hemeoxygenase-1 expression in microglia through the activation of phosphatidylinositol 3-kinase and nuclear factor E2-related factor 2. Glia. 2008;56:1028–1037. doi: 10.1002/glia.20676. [DOI] [PubMed] [Google Scholar]
- 37.Parrillo JE. Pathogenetic mechanisms of septic shock. N Engl J Med. 1993;328:1471–1477. doi: 10.1056/NEJM199305203282008. [DOI] [PubMed] [Google Scholar]
- 38.Riedemann NC, Guo RF, Ward PA. Novel strategies for the treatment of sepsis. Nat Med. 2003;9:517–524. doi: 10.1038/nm0503-517. [DOI] [PubMed] [Google Scholar]
- 39.Steffel J, Luscher TF, Tanner FC. Tissue factor in cardiovascular diseases: molecular mechanisms and clinical implications. Circulation. 2006;113:722–731. doi: 10.1161/CIRCULATIONAHA.105.567297. [DOI] [PubMed] [Google Scholar]
- 40.Ren G, Li T, Lan JQ, Wilz A, Simon RP, Boison D. Lentiviral RNAi-induced downregulation of adenosine kinase in human mesenchymal stem cell grafts: a novel perspective for seizure control. Exp Neurol. 2007;208:26–37. doi: 10.1016/j.expneurol.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1.Wang J, Mahmud SA, Bitterman PB, Huo Y, Slungaard A. Histone deacetylase inhibitors suppress TF-kappaB-dependent agonist-driven tissue factor expression in endothelial cells and monocytes. Journal of Biological Chemistry. 2007;282:28408–28418. doi: 10.1074/jbc.M703586200. [DOI] [PubMed] [Google Scholar]
- 2.An G, Wang H, Tang R, Yago T, McDaniel JM, McGee S, Huo Y, Xia L. P-selectin glycoprotein ligand-1 is highly expressed on Ly-6Chi monocytes and a major determinant for Ly-6Chi monocyte recruitment to sites of atherosclerosis in mice. Circulation. 2008;117:3227–3237. doi: 10.1161/CIRCULATIONAHA.108.771048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang H, Tang R, Zhang W, Amirikian K, Geng Z, Geng J, Hebbel RP, Xia L, Marth JD, Fukuda M, Katoh S, Huo Y. Core2 1-6-N-Glucosaminyltransferase-I Is Crucial for the Formation of Atherosclerotic Lesions in Apolipoprotein E-Deficient Mice. Arterioscler Thromb Vasc Biol. 2009;29:180–7. doi: 10.1161/ATVBAHA.108.170969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim I, Oh JL, Ryu YS, So JN, Sessa WC, Walsh K, Koh GY. Angiopoietin-1 negatively regulates expression and activity of tissue factor in endothelial cells. Faseb J. 2002;16:126–128. doi: 10.1096/fj.01-0556fje. [DOI] [PubMed] [Google Scholar]
- 5.Wang H, Zhang W, Zhu C, Bucher C, Blazar BR, Zhang C, Chen JF, Linden J, Wu C, Huo Y. Inactivation of the adenosine A2A receptor protects apolipoprotein E-deficient mice from atherosclerosis. Arterioscler Thromb Vasc Biol. 2009;29:1046–1052. doi: 10.1161/ATVBAHA.109.188839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eisenreich A, Malz R, Pepke W, Ayral Y, Poller W, Schultheiss HP, Rauch U. Role of the phosphatidylinositol 3-kinase/protein kinase B pathway in regulating alternative splicing of tissue factor mRNA in human endothelial cells. Circ J. 2009;73:1746–1752. doi: 10.1253/circj.cj-99-0225. [DOI] [PubMed] [Google Scholar]
- 7.Jorgensen SB, Viollet B, Andreelli F, Frosig C, Birk JB, Schjerling P, Vaulont S, Richter EA, Wojtaszewski JF. Knockout of the alpha2 but not alpha1 5′-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranosidebut not contraction-induced glucose uptake in skeletal muscle. Journal of Biological Chemistry. 2004;279:1070–1079. doi: 10.1074/jbc.M306205200. [DOI] [PubMed] [Google Scholar]
- 8.Schabbauer G, Tencati M, Pedersen B, Pawlinski R, Mackman N. PI3K-Akt pathway suppresses coagulation and inflammation in endotoxemic mice. Arterioscler Thromb Vasc Biol. 2004;24:1963–1969. doi: 10.1161/01.ATV.0000143096.15099.ce. [DOI] [PubMed] [Google Scholar]
- 9.Huo Y, Zhao L, Hyman MC, Shashkin P, Harry BL, Burcin T, Forlow SB, Stark MA, Smith DF, Clarke S, Srinivasan S, Hedrick CC, Pratico D, Witztum JL, Nadler JL, Funk CD, Ley K. Critical role of macrophage 12/15-lipoxygenase for atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2004;110:2024–2031. doi: 10.1161/01.CIR.0000143628.37680.F6. [DOI] [PubMed] [Google Scholar]
- 10.Li YD, Ye BQ, Zheng SX, Wang JT, Wang JG, Chen M, Liu JG, Pei XH, Wang LJ, Lin ZX, Gupta K, Mackman N, Slungaard A, Key NS, Geng JG. NF-kappaB transcription factor p50 critically regulates tissue factor in deep vein thrombosis. J Biol Chem. 2009;284:4473–4483. doi: 10.1074/jbc.M806010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nath N, Giri S, Prasad R, Salem ML, Singh AK, Singh I. 5-aminoimidazole-4-carboxamide ribonucleoside: a novel immunomodulator with therapeutic efficacy in experimental autoimmune encephalomyelitis. Journal of Immunology. 2005;175:566–574. doi: 10.4049/jimmunol.175.1.566. [DOI] [PubMed] [Google Scholar]
- 12.Prasad R, Giri S, Nath N, Singh I, Singh AK. 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside attenuates experimental autoimmune encephalomyelitis via modulation of endothelial-monocyte interaction. J Neurosci Res. 2006;84:614–625. doi: 10.1002/jnr.20953. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Acadesine suppresses LPS-induced TF activity on the surface of HUVECs.
HUVECs were pretreated with acadesine for 1 h at various concentrations, followed by stimulation with LPS at 1 μg/mL for 4 h. TF activity on the cell surface was assayed by measuring the enzymatic activation by the TF/factor VIIa complex. An activity of 1 is equivalent to 10−15 Mol of TF. Data are shown as means ± SEM of six independent experiments, * P < 0.05, ** P < 0.01.
Supplementary Figure 2. Acadesine suppresses cytokine-induced TF expression in HUVECs. HUVECs were pretreated with acadesine at 1 mM for 1 h, followed by stimulation with TNF-α (10 ng/mL) or IL-1β (10 ng/mL) for 4 h. TF expression at the protein level was analyzed by western blotting. The intensity of the TF bands was quantitated by densitometry, normalized to GAPDH, and expressed as folds of TF expressed by untreated HUVECs. Data are shown as means ± SEM of four independent experiments. The data from cells treated with cytokines and cytokine/acadesine were compared, * P < 0.05, ** P < 0.01.
Supplementary Figure 3. Acadesine suppresses LPS-induced TF expression and activity in murine bEND.3 cells.
Murine bEND.3 endothelial cells were pretreated with acadesine for 1 h at various concentrations, followed by stimulation with LPS at 1 μg/mL for 4 h. TF activity of cell lysates were examined with a one-stage clotting assay (a). Total cellular RNA was extracted, and TF mRNA expression was analyzed by real-time RT-PCR. GAPDH was used as normalization control (b). Data are shown as means ± SEM of six independent experiments, * P < 0.05,** P < 0.01.
Supplementary Figure 4. The effect of acadesine on TF activity in α1AMPK-deficient murine macrophages.
Thioglycollate-elicited peritoneal macrophages were isolated from wild-type or α1AMPK−/− mice, then treated with either LPS (1 μg/mL) or LPS and acadesine (1 mM), followed by the one-stage clotting assay to measure TF activity. Data are shown as means ± SEM of six independent experiments, * P < 0.05, ** P < 0.01.
Supplementary Figure 5. The effect of acadesine on TF activity in A2A R-deficient murine macrophages.
Thioglycollated-elicited peritoneal macrophages were isolated from wild type or A2AR−;/−; mice, then were treated with either LPS (1 μg/mL) or LPS and acadesine (1 mM), followed by a one-stage clotting assay to measure TF activity. Data are shown as means ± SEM of six independent experiments, * P < 0.05, ** P < 0.01.
Supplementary Figure 6. Role of the PI3K/Akt pathway in acadesine-downregulated TF expression in human monocytes.
(a) and (b), Akt and phospho-Akt in human monocytes treated with acadesine (1 mM), LPS (1 μg/mL) or LPS and acadesine for different periods and examined by western blotting. The intensity of the phospho-Akt was quantitated by densitometry and, after normalization to total Akt, expressed as fold difference over untreated monocytes. Data are shown as means ± SEM of six independent experiments. The data from treated cells at different time points were compared with those from untreated monocytes, * P < 0.05. (c), Akt, GSK-3β, and their phosphorylated forms were examined by western blotting at 30 min after treatment with LPS (1 μg/mL), acadesine (1 mM), or LPS and acadesine with or without wortmannin pretreatment (50 nM). Data are shown from one of four independent experiments. (d), At various time points after treatment with LPS (1 μg/mL) or LPS and acadesine (1 mM), monocytes were examined by western blotting for the levels of phospho-ERK1/2, ERK1/2, phospho-JNK, JNK, phospho-p38, and p38. Immunoblots are representative of four independent experiments.
Supplementary Figure 7. Acadesine directly interferes with the binding of transcription factors to their oligonucleotides
Nuclear protein was extracted from HUVECs or human monocytes treated with LPS. EMSA was performed using 32P-oligonucleotides corresponding to the AP-1 and NF-κB DNA recognition sequences and the same amount nuclear protein in the presence of acadesine at different concentrations. The intensities of AP-1 and NF-κB bands were quantified by densitometry and expressed as fold difference over untreated control cells. Data are shown as means ± SEM of four independent experiments, * P > 0.05.
Supplementary Figure 8. PI3K/Akt activation and TF expression in adenosine kinase knockdown HUVECs.
(a), Akt and phospho-Akt in HUVECs transduced with lentiviruses carrying siRNA against adenosine kinase or a scrambled sequence (control). The intensity of phospho-Akt was quantitated by densitometry and, after normalization to total Akt, expressed as the fold change over this value in HUVECs transduced with control virus, * P < 0.05. (b), HUVECs transduced with control or ADK RNAi viruses were examined by western blotting for TF expression at 4 h after treatment with LPS (1 μg/mL) with or without wortmannin pretreatment (50 nM). The intensity of the TF bands was quantitated by densitometry, normalized to GAPDH, and expressed as the fold change over TF expressed by HUVECs without ADK knockdown. Data are shown as means ± SEM of four independent experiments, * P < 0.05.