

Abstract
Depolarization of presynaptic terminals that arises from activation of presynaptic ionotropic receptors, or somatic depolarization, can enhance neurotransmitter release; however, the molecular mechanisms mediating this plasticity are not known. Here we investigate the mechanism of this enhancement at the calyx of Held synapse, in which presynaptic glycine receptors depolarize presynaptic terminals, elevate resting calcium levels, and potentiate release. Using knock-out mice of the calcium-sensitive PKC isoforms (PKCCa), we find that enhancement of evoked but not spontaneous synaptic transmission by glycine is mediated primarily by PKCCa. Measurements of calcium at the calyx of Held indicate that deficits in synaptic modulation in PKCCa knock-out mice occur downstream of presynaptic calcium increases. Glycine enhances synaptic transmission primarily by increasing the effective size of the pool of readily releasable vesicles. Our results reveal that PKCCa can enhance evoked neurotransmitter release in response to calcium increases caused by small presynaptic depolarizations.
Introduction
Like G-protein-coupled receptors (Wu and Saggau, 1997; Blackmer et al., 2001; Schmitz et al., 2001), presynaptic ionotropic receptors modulate synaptic transmission (Kerchner et al., 2001; Turecek and Trussell, 2001; Engelman and MacDermott, 2004). The latter form of synaptic regulation can involve small depolarizations of presynaptic boutons that enhance neurotransmitter release by increasing presynaptic calcium (Turecek and Trussell, 2001; Awatramani et al., 2005; Christie et al., 2011). In this way, synaptic enhancement by ionotropic receptors is similar to synaptic enhancement produced by subthreshold somatic depolarizations at cortical, hippocampal, and cerebellar synapses (Glitsch and Marty, 1999; Alle and Geiger, 2006; Shu et al., 2006; Yu et al., 2010; Christie et al., 2011). In general, it is not understood how presynaptic depolarizations enhance neurotransmitter release.
Glycinergic enhancement of evoked and spontaneous release at the calyx of Held synapse is a well-studied form of plasticity that involves a small presynaptic depolarization arising from activation of presynaptic ionotropic receptors (Turecek and Trussell, 2001; Balakrishnan et al., 2009). Glycine, which is released by interneurons in the medial nucleus of the trapezoid body (Turecek and Trussell, 2001), activates presynaptic ionotropic receptors that depolarize presynaptic terminals as a result of the relatively high chloride reversal potential at this synapse (−50 mV) (Price and Trussell, 2006; Huang and Trussell, 2008; Kim and Trussell, 2009). This depolarization is sufficient to open a small fraction of P-type voltage-gated calcium channels, elevating presynaptic calcium and, ultimately, increasing the amplitude of evoked EPSCs and miniature EPSC (mEPSC) frequency (Turecek and Trussell, 2001; Trussell, 2002; Awatramani et al., 2005; Kim and Trussell, 2009). This form of enhancement can be mimicked by direct depolarization of the presynaptic bouton (Awatramani et al., 2005), suggesting that it can provide general insight into enhancement arising from both presynaptic ionotropic receptor activation and depolarization of presynaptic boutons conveyed from the soma. The molecular mechanisms that respond to elevated calcium to produce such synaptic enhancement are not known. The increases in presynaptic calcium associated with glycine-induced enhancement are too small [tens to hundreds of nanomolar (Turecek and Trussell, 2001; Awatramani et al., 2005)] to effectively activate synaptotagmin, the low-affinity calcium sensor that mediates vesicle fusion (Schneggenburger and Neher, 2005; Südhof and Rothman, 2009). This suggests that an unidentified calcium sensor provides the link between presynaptic calcium elevations induced by glycine and enhanced vesicle fusion.
The calcium-sensitive PKC isoforms (PKCCa), PKCα and PKCβ, are good candidates to mediate glycine-induced enhancement. They are present at the calyx of Held, and they respond to modest calcium increases after tetanic stimulation [posttetanic potentiation (PTP)] to enhance evoked synaptic responses (Fioravante et al., 2011). Because PTP and glycine-induced synaptic enhancement both depend on small increases in presynaptic calcium, we hypothesize that PKCα and PKCβ also mediate glycine-induced enhancement.
Here we test the involvement of PKCα and PKCβ in synaptic enhancement mediated by glycine at the calyx of Held synapse. By using PKCα and PKCβ knock-out (ko) mice and PKCαβ double knock-out (dko) mice, we find that most of the glycine-induced enhancement of evoked synaptic responses is mediated by PKCα and PKCβ but that these isoforms do not mediate the increase in mEPSC frequency after glycine application. Presynaptic calcium signaling is unaltered in dko mice compared with wild-type animals, indicating that the effect of PKCCa is downstream of Ca2+ influx. Blocking glycine receptors with strychnine, however, prevents normal Ca2+ increases in wild-type mice in response to glycine application. We find that glycine enhances synaptic transmission primarily by increasing the effective size of the pool of readily releasable vesicles. Thus, by depolarizing the calyx of Held, glycine opens presynaptic calcium channels to activate PKCα and PKCβ and thus increases neurotransmitter release. Moreover, these findings establish that PKCα and PKCβ can play central roles in increasing synaptic strength after small presynaptic depolarizations.
Materials and Methods
Animals.
PKCα and PKCβ single ko animals and PKCαβ dko mice were generated as described previously (Leitges et al., 1996, 2002). All animal handling and procedures were completed in accordance with the guidelines of the Harvard Medical Area Standing Committee on Animals.
Brain slices and electrophysiology.
Postnatal day P11–P14 mice of either sex were deeply anesthetized with isoflurane, and transverse 190-μm-thick brainstem slices containing the medial nucleus of the trapezoid body (MNTB) region were cut using a vibratome slicer (VT1000S; Leica). Brains were dissected and cut at 4°C in slicing solution containing the following (in mm): 125 NaCl, 25 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 0.1 CaCl2, 3 MgCl2, 25 glucose, 3 myo-inositol, 2 Na-pyruvate, and 0.4 ascorbic acid, pH 7.4 (continuously bubbled with 95% O2/5% CO2). Slices were incubated at 32°C for 30 min in a solution consisting of the following (in mm): 125 NaCl, 25 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 2 CaCl2, 1 MgCl2, 25 glucose, 3 myo-inositol, 2 Na-pyruvate, and 0.4 ascorbic acid, pH 7.4 (continuously bubbled with 95% O2/5% CO2). After incubation, slices were transferred to a recording chamber at room temperature. All recordings were made using an upright microscope (Olympus) with a 60×, 0.9 NA objective.
Slices were perfused at 2–3 ml/min during the recording with a standard perfusion solution that contained 25 μm bicuculline and 2 μm CGP 55845 [(2S)-3-[(15)-1-(2,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl)(phenylmethyl)-phosphinic acid] to block transmission mediated by GABAA and GABAB receptors, respectively. Whole-cell postsynaptic patch-clamp recordings were made from visually identified MNTB principle cells using glass pipettes with 2–3 MΩ resistance, filled with internal solution containing the following (in mm): 135 CsF, 5 CsCl, 5 EGTA, 10 HEPES, 2 QX314 [N-(2,6-dimethylphenylcarbomoylmethyl)triethylammonium chloride], and 1 DIDS (4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid disodium salt hydrate) (300 mOsm) at pH 7.4 with CsOH. Whole-cell recordings were compensated electronically by 70% and voltage clamped to −60 mV.
EPSCs were evoked by stimulating presynaptic axons with a custom-made bipolar stimulating electrode placed midway between the midline of the brainstem and the medial border of the MNTB. Multiclamp 700A and 700B (Molecular Devices) amplifiers were used. All recordings were digitized at 20 kHz with an ITC-18 analog-to-digital converter (InstruTECH) using custom macros (written by M. A. Xu-Friedman, State University of New York at Buffalo, Buffalo, NY) in Igor Pro (Wavemetrics) and filtered at 8 kHz. The access resistance and leak current were monitored, and experiments were rejected if either of these parameters changed significantly.
Glycine-induced changes in EPSC and the probability of release (p) were measured by first recording the baseline synaptic strength with paired stimuli (20 ms interval) repeated every 20 s for 3–5 min followed by wash-in of 1 mm glycine with or without 1 μm strychnine. Changes in mEPSCs were measured by recording spontaneous activity for 10 s before each paired-pulse stimuli. The size of the readily releasable pool (RRP) and p were estimated via two methods. First, RRP resulting from a train of stimuli (RRPtrain) elicited at 100 Hz, 0.4 s was obtained by plotting the cumulative EPSC as a function of the stimulus number (Schneggenburger et al., 1999); back-extrapolation to the y-axis using a linear fit of the last 15 points yielded the ∑EPSC0. The fraction of the RRPtrain released by the first stimulus (p) was calculated by dividing the first EPSC by the ∑EPSC0. The second method, referred to as the RRPEQ method (Elmqvist and Quastel, 1965), involved plotting the EPSC amplitude as a function of the cumulative EPSC for the synaptic response obtained with each 100 Hz, 0.4 s train. Extrapolation to the x-axis using a linear fit of the first seven points in the plot yielded the ∑EPSC0 as an estimate of the RRP, as described previously by Elmqvist and Quastel (1965).
Presynaptic calcium measurements.
Presynaptic calyceal terminals were loaded for 5 min with 0.025–0.1% Alexa Fluor 594 dextran and 0.5% Calcium Green-1 dextran (10 kDa; Invitrogen) and incubated at 32°C for 30 min before imaging with a two-photon microscope as described previously (Beierlein et al., 2004; Fioravante et al., 2011). Fluorescence signals from calyces were converted to calcium by determining the Fmax/Fmin ratio (4.8) in a blank pipette and by determining Fmax using high-frequency stimulation as described previously (Maravall et al., 2000). Calyces that exhibited bright green fluorescence at rest were found to be unhealthy for additional study because they either had elevated resting calcium levels or were overloaded with calcium indicator.
Data analyses.
All data analyses were performed using macros written in Igor Pro (Wavemetrics). Glycine-induced synaptic changes were calculated as the ratio of EPSC amplitude 3–5 min after wash-in of 1 mm glycine over the average baseline. mEPSCs were detected as described previously (Fioravante et al., 2011). Statistical analyses were performed using one-way ANOVA tests for multiple group comparisons followed by Tukey's post hoc analysis. Student's paired t tests were used for pairwise comparisons. Level of significance was set at p < 0.05.
Results
Our ultimate goal was to use ko mice to study the mechanism of glycine-induced enhancement at the calyx of Held, but previously this form of synaptic enhancement had only been described in rats (Turecek and Trussell, 2001). We therefore examined the effect of glycine on transmission in the mouse and found that it enhanced transmission at calyx of Held synapses, as shown in a representative experiment (Fig. 1A). In this experiment, glycine increased the EPSC by ∼1.5-fold, and this enhancement reversed when glycine was washed out of the slice. We found that glycine enhances synaptic transmission by 40 ± 4% (n = 23) in wild-type mice. Thus, in both rats and mice, glycine enhances synaptic transmission at calyx of Held synapses.
Figure 1.

Glycine-induced presynaptic enhancement partially occludes PTP. A, A representative experiment showing that the bath application of glycine reversibly enhances synaptic strength at the calyx of Held synapse in mice. Top traces depict the average baseline EPSC response (control), potentiated responses in glycine, and responses after washout of glycine. B, C, Experiments were performed to assess whether the enhancement produced by glycine interacted with the enhancement of synaptic strength produced by tetanic stimulation (PTP). The presynaptic calyceal terminal was stimulated at low frequency (0.2 Hz) to monitor synaptic strength, and the effect of tetanic stimulation (4 s, 100 Hz; inverted triangles) was assessed in control conditions and in the presence of glycine. B, Left, The time course of synaptic amplitudes is the average of eight experiments performed in 2 mm [Ca]e. EPSCs were normalized to the average EPSC recorded in 2 mm [Ca]e before tetanic stimulation. B, Middle, The synaptic enhancement produced by tetanic stimulation in the left is shown for control conditions and in the presence of glycine. EPSCs are normalized to the average EPSC amplitude recorded before tetanic stimulation. B, Right, The effect of glycine on PTP is summarized for eight individual experiments (open circles) and the average for each group (filled circles). C, Similar experiments to B were performed but in 1.5 mm external calcium to decrease the initial release probability by ∼50%, thereby allowing us to assess whether glycine reduced the amplitude of PTP by saturating the probability of release. In C, left, EPSCs are normalized to the EPSC amplitude in 2 mm [Ca]e. Results shown are the average of six cells. *p < 0.05, paired t test. Errors bars are SEM.
Previous studies have shown that calcium acts presynaptically to enhance release in both glycine-induced enhancement and PTP (Turecek and Trussell, 2001; Fioravante et al., 2011), raising the possibility that they could be mediated by the same calcium-dependent target. If these forms of enhancement share a mechanism, they should partially occlude each other. We determined whether this is the case by measuring the amount of PTP induced by tetanic stimulation (4 s, 100 Hz) under control conditions and in the presence of glycine. In control conditions, there was a 92 ± 11% (n = 8) increase in the EPSC amplitude after tetanic stimulation. Glycine not only potentiated the synaptic response but also reduced PTP to 54 ± 13% (n = 8) (Fig. 1B). The reduction of the magnitude of PTP by glycine is consistent with the two forms of enhancement sharing a mechanism leading to occlusion, but saturation of release could also contribute to a reduction in PTP in the presence of glycine. We therefore repeated these experiments in 1.5 mm external calcium (Fig. 1C), which decreases the initial EPSC by ∼50% and thereby greatly reduces the possibility that release is saturated. We found that, under these conditions, glycine significantly reduced the magnitude of PTP from 121 ± 19 to 53 ± 11% (n = 6; Fig. 1C, middle). The finding that glycine-induced potentiation partially occludes PTP, even for conditions under which release is not saturated, supports the hypothesis that glycine-induced plasticity and PTP act through similar presynaptic mechanisms.
The involvement of PKCαβ in glycine-induced enhancement
The observation that glycine partially occludes PTP, combined with our previous finding that calcium-dependent PKC isoforms PKCα and PKCβ play a crucial role in PTP, suggested the hypothesis that PKCα and PKCβ could also mediate glycine-induced enhancement. We tested this hypothesis by studying wild-type and PKCαβ dko mice. Application of glycine enhanced evoked synaptic transmission in wild-type mice, and this enhancement was blocked by the ionotropic glycine receptor antagonist strychnine (Fig. 2A). We found that this form of synaptic enhancement was significantly reduced in PKCαβ dko mice (p < 0.002) (Fig. 2B). The extent of glycine-induced enhancement was variable for all groups (Fig. 2C). On average, glycine enhanced synaptic strength by 40 ± 4% in wild-type mice (n = 23), by 25 ± 7% in PKCα ko mice (n = 8), by 25 ± 6% in PKCβ ko mice (n = 11), and by 10 ± 7% (n = 13) in PKCαβ dko mice (Fig. 2D). These results suggest that PKCα and PKCβ together mediate the bulk of glycine-dependent enhancement of evoked transmission at the calyx of Held and that PKCα and PKCβ make comparable contributions.
Figure 2.

Calcium-dependent isoforms of PKC are necessary for glycine-induced presynaptic potentiation. A, B, From representative experiments, top traces show the average baseline EPSC response (left), potentiated response in glycine (middle), and response with strychnine application (right). Bottom, Plots of normalized EPSC amplitudes as a function of time. At t = 0 s, glycine wash-in was initiated, eliciting potentiated EPSC amplitudes in wild-type (wt) controls (A) but not littermate PKCαβ dko animals (B). C, Cumulative histograms of normalized amplitudes of the potentiated response in glycine for wild-type (black), PKCα (green), PKCβ (red), and dko (purple) calyces. D, Percentage ± SEM EPSC enhancement in glycine for wild-type and PKC ko groups. EPSCs were normalized to baseline before determining the amount of synaptic enhancement. *p < 0.05.
Previous studies found that the glycine-induced increase in mEPSC frequency is prevented by blocking voltage-gated calcium channels, suggesting that it is a consequence of elevated presynaptic calcium entry leading to small increases in basal calcium levels (Turecek and Trussell, 2001). Although increases in evoked transmission and mEPSC frequency are both produced by elevated presynaptic calcium levels, it was not known whether these two forms of synaptic enhancement were produced by the same molecular mechanism. We therefore tested whether, in addition to enhancing evoked release, calcium-dependent PKCs mediate increases in mEPSC frequency. We found that glycine produced a significant increase in mEPSC frequency in wild-type mice (Fig. 3A) and had a similar effect in PKCαβ dko mice (Fig. 3B). Glycine increased mEPSC frequencies to similar extents in wild-type (2.9 ± 0.4-fold increase, n = 21), PKCα (2.8 ± 0.4-fold increase, n = 8), PKCβ (2.7 ± 0.6-fold increase, n = 10), and PKCαβ dko (2.8 ± 0.5-fold increase, n = 13) mice (Fig. 3C, left). The increases in mEPSC frequency were not significantly different among groups (p = 0.99), and glycine did not significantly alter the amplitudes of mEPSCs for all groups (Fig. 3C, right, p = 0.85).
Figure 3.
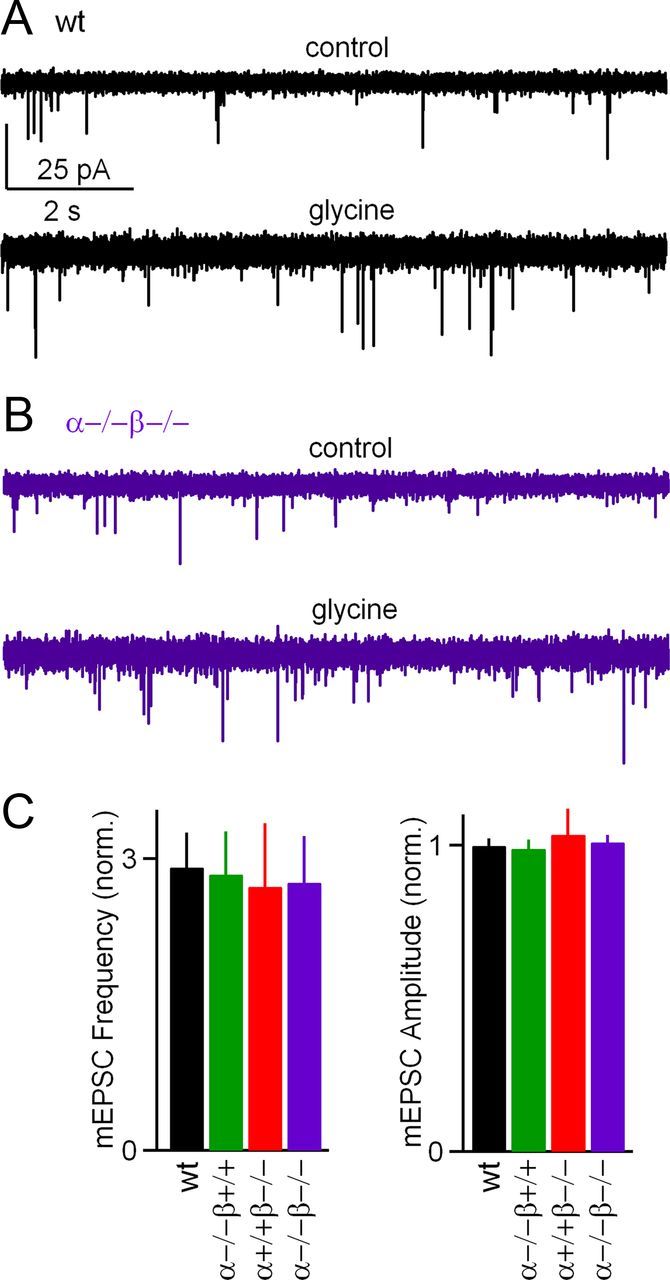
PKCα and PKCβ are not required for glycine-induced increases in spontaneous transmission. A, B, Representative traces of spontaneous transmission before (control; top) and after (bottom) glycine application from wild-type (wt; black) and PKCα/β dko (purple) slices. C, Average mEPSC frequencies and mEPSC amplitudes recorded in the presence of glycine, normalized to the values measured before the application of glycine.
The lack of an effect of PKCCa deletion on mEPSC enhancement by glycine is important in two ways. First, it suggests that different molecular mechanisms mediate glycine-induced enhancement of evoked and spontaneous release. Second, it shows that, in PKCαβ dko mice, glycine still activates presynaptic glycine receptors and depolarizes the terminals sufficiently to open voltage-gated calcium channels and enhance spontaneous synaptic transmission, suggesting that presynaptic glycine signaling is intact in dko animals.
Presynaptic calcium signaling
We explicitly tested whether glycine signaling is intact in PKCαβ dko mice by measuring the effects of glycine on basal calcium levels and stimulus-evoked calcium signals. We used dextran-conjugated calcium indicators and two-photon microscopy to measure presynaptic calcium influx and basal calcium concentrations (Fioravante et al., 2011). In wild-type mice, glycine elevated basal presynaptic calcium at the calyx of Held by 56 ± 13 nm (Fig. 4A, left, B, C), and these calcium increases were prevented by strychnine (4.5 ± 3.8 nm) (Fig. 4A, middle, B, C). This was shown previously for rats (Turecek and Trussell, 2001). We also found that calcium increases evoked by single stimuli ([Ca]1) were not significantly altered by glycine in wild-type mice ([Ca]1 increased by 4.4 ± 2.5 nm, n = 7, p = 0.22; Fig. 4A, left, C, right) and in wild-type mice in the presence of strychnine [Ca]1 increased by 3.1 ± 3.7 nm (n = 4, p = 0.21; Fig. 4A, middle, C, right). If calcium signals were the same in wild-type and dko mice, it would indicate that glycine receptors are functional in the presynaptic terminals of dko mice, and, when activated, they continue to depolarize the calyx and elevate calcium; if not, it would suggest that PKCαβ act upstream of presynaptic calcium signaling. We found that, in PKCαβ dko mice, glycine elevated basal calcium levels to the same degree as in wild-type mice (53 ± 7 nm; n = 8; p = 0.51; Fig. 4A, right, B, C) and that [Ca]1 was not significantly altered by glycine (an increase of 4.2 ± 4.1 nm, n = 8; p = 0.27; Fig. 4A, right, C, right). The consistent lack of an effect of glycine on [Ca]1 indicates that glycine does not significantly alter stimulus-evoked calcium entry. These findings indicate that glycine elevates basal calcium in dko mice and suggest that the reduced ability of glycine to enhance evoked transmission must occur downstream of the elevation in presynaptic calcium concentration.
Figure 4.

Glycine elevates presynaptic calcium levels to a similar extent in wild-type (wt) and PKCα/β dko animals. The effects of glycine on the resting calcium concentration and on calcium transients after single stimuli at the calyx of Held were measured for wild-type mice (black), wild-type mice in the presence of the glycine receptor antagonist strychnine (blue), and PKCα/β dko (purple) mice. Representative calcium signals from individual experiments (A), the average time courses of glycine on the resting presynaptic calcium concentration (B), and the average effect of glycine on the resting calcium concentration and the amplitude of presynaptic calcium signals evoked by single stimuli (C) are shown. *p < 0.05.
Mechanisms of glycine-induced enhancement
To determine whether changes in p contribute to the glycine-induced enhancement observed under our experimental conditions, we examined the effects of glycine on paired-pulse ratio (PPR = EPSC2/EPSC1) (Fig. 5). These experiments were conducted in the presence of cyclothiazide (CTZ) and kynurenate (Kyn) to minimize AMPA receptor desensitization and saturation, respectively. Glycine did not significantly alter PPR in wild-type mice (PPRglycine/PPRcontrol = 0.96 ± 0.03, n = 13, p = 0.44; Fig. 5, black), suggesting that glycine did not change p. We tested the sensitivity of PPR to changes in p by assessing the effect of elevating external calcium from 2 to 3 mm. This manipulation produced a synaptic enhancement similar in amplitude to glycine and significantly decreased PPR (PPR3Ca/PPRcontrol = 0.85 ± 0.01, n = 6, p < 0.05; Fig. 5, red). Glycine did not alter PPR in dko mice (PPRglycine/PPRcontrol = 0.97 ± 0.02, n = 15, p = 0.87; Fig. 5, purple) and in wild-type mice in the presence of strychnine (PPRglycine/PPRcontrol = 0.99 ± 0.03, n = 4, p = 0.98; Fig. 5, blue). These findings suggest that increases in p do not make large contributions to glycine-induced enhancement in mice.
Figure 5.

Glycine-induced synaptic potentiation is not accompanied by an increase in PPR. A, Representative traces from the four groups show superimposed EPSCs measured in two different conditions, with traces normalized to the first EPSCs. B, The effects of the indicated experimental manipulations on PPR are summarized by plotting the ratio of the PPR measured in the indicated condition to the PPR measured under control conditions. wt, Wild type. *p < 0.05.
If synaptic enhancement is not mediated by increases in p, then the effective size of the RRP must be increased. We therefore measured the effective pool size by using high-frequency trains (100 Hz, 0.4 s) to evoke a series of EPSCs in the presence of CTZ and Kyn. We calculated the cumulative EPSC and determined the effective pool size (RRPtrain) by fitting the last 15 points and back-extrapolating to the y-axis to correct for replenishment of the RRP (Fig. 6A). In this method, the probability of release in response to the first stimulus was determined by ptrain =EPSC/RRPtrain (Schneggenburger et al., 1999). In wild-type mice, glycine increased the EPSC size (57 ± 10%, n = 13) primarily by increasing RRPtrain (increased by 44 ± 8%) and only slightly increasing ptrain (increased by 5.8 ± 3.5%) (Fig. 6B, black). We compared the effects of glycine on synaptic transmission with the effects of raising the external calcium concentration from 2 to 3 mm and found that increasing the external calcium concentration elevated synaptic strength by 48 ± 7% (n = 6), primarily by increasing ptrain (39 ± 6%), with only a small contribution from the effective pool size (13 ± 8%) (Fig. 6B, red). The increases in synaptic strength (10 ± 3%, n = 17) and RRPtrain (8 ± 3%) by glycine were essentially eliminated in PKCαβ dko mice (Fig. 6B, purple) and in the presence of strychnine (6 ± 5% increase in EPSC; 3.8 ± 0.8% increase in RRPtrain) (Fig. 6B, blue).
Figure 6.

Glycine-induced synaptic potentiation occurs primarily by increases in the effective pool size. Calyces were stimulated with a 100 Hz, 40 s pulse train in the presence of CTZ and Kyn, and two methods were used to determine the mechanism by which glycine enhances synaptic transmission. A, In the first approach, the cumulative EPSC was plotted against stimulus number, in which back-extrapolation using a linear fit of the last 15 points to the y-intercept yielded the RRPtrain as an estimate of the effective pool size. B, This method was used to determine the effect of the indicated manipulations on EPSC amplitude, RRPtrain and ptrain. Representative experiments are shown above, and summary data are shown below for the indicated conditions. C, The same experiments were reanalyzed using an approach introduced by Elmqvist and Quastel (1965). In this approach, the reduction in the amplitude of the EPSC is attributed to depletion and is used to estimate RRPEQ. D, Representative experiments are shown above, and summary data are shown below for the indicated conditions. Paired t tests were used to compare the EPSC, RRPtrain, ptrain, RRPEQ, and pEQ amplitudes in control and in the presence of either glycine or elevated external calcium, as indicated (*p < 0.05, **p < 0.01, ***p < 0.001). All other values were not statistically significant. wt, Wild type.
We further tested the contributions of different mechanisms to glycine-mediated enhancement by using an alternate method to estimate effective pool size and p that was introduced by Elmqvist and Quastel (1965) (Fig. 6C). In this approach, the reduction in the amplitudes of EPSCs during a stimulus train is attributed to vesicle depletion, and the decrease in amplitude of the first few EPSCs in the train is used to estimate the effective pool size (RRPEQ). Although this approach and that described above both rely on the use of stimulus trains, the Elmqvist and Quastel method uses the initial EPSCs in the train and provides an upper-bound estimate of the effective pool size because it ignores the contribution of replenishment of the RRP from reserve pools during the first few EPSCs to the measured RRP size. In contrast, the cumulative method used previously relies on the synaptic responses measured late in the train and provides a lower-bound estimate of the effective pool size because it assumes a maximal steady-state replenishment rate throughout the train and probably overestimates recovery at the beginning of the train (see Discussion). As expected, the estimates of pool size with the Elmqvist and Quastel method are consistently higher than those obtained with the cumulative method. According to the Elmqvist and Quastel method, glycine enhances synaptic transmission in wild-type mice primarily by increasing RRPEQ (35 ± 8%), although small increases in pEQ (8.2 ± 2.1%) also contribute (Fig. 6D, black). Increases in external calcium enhance synaptic strength exclusively by increasing pEQ (47 ± 9%) rather than increasing RRPEQ (1.3 ± 2.2%; Fig. 6D, red). Finally, only very small increases in EPSC size (10 ± 3%), RRPEQ (8 ± 3%), and pEQ (3.3 ± 2.5%) were observed in PKCαβ dko mice (Fig. 6D, purple) and in wild-type mice treated with strychnine (EPSC, 6 ± 5%; RRPEQ, 4 ± 1%; pEQ, 6 ± 7%; Fig. 6D, blue). Thus, we find that glycine-induced increases in EPSC size are produced primarily by an increase in the effective pool size.
The use of a one-way ANOVA for wild-type, PKCαβ dko, and strychnine-treated wild-type mice revealed that the glycine-induced increase in EPSC amplitude (p < 0.01), RRPtrain (p < 0.01), and RRPEQ (p < 0.01) were all reduced in dko mice compared with control. Similarly, the enhancement of EPSC amplitude (p < 0.05), RRPtrain (p < 0.01), and RRPEQ (p < 0.01) were all reduced in strychnine-treated wild-type mice compared with control mice. There was no significant difference between ptrain and pEQ among groups and no significant difference in any synaptic properties among PKCαβ dko and strychnine-treated wild-type mice.
Previous studies of glycine-induced enhancement performed in rats reported that glycine decreases the magnitude of paired-pulse plasticity (Turecek and Trussell, 2001), suggesting that glycine increases p. Moreover, it was found that glycine does not alter the overall size of the pool of vesicles released by a large presynaptic voltage step (Turecek and Trussell, 2001). These findings suggested that, in rats, increases in p make a prominent contribution to glycine-induced enhancement. We therefore reexamined the mechanism of glycine-induced enhancement in rats and found that glycine decreased PPR (Fig. 7A), which is consistent with glycine increasing p in rats. The use of trains to determine the contribution of effective RRP and p revealed that glycine increases p but produces larger increases in effective RRP (Fig. 7B–D). This was found to be the case for both the cumulative EPSC method and the Elmqvist and Quastel method. Thus, for both rats and mice, two distinct methods of measuring pool size and p support the conclusion that glycine enhances transmission primarily by increasing the effective pool size rather than by increasing p, although in rat there is also an appreciable contribution from increases in p.
Figure 7.

Glycine-induced synaptic potentiation in rats primarily reflects an increase in the effective pool size, but increases in the probability of release also contribute. Experiments were conducted in the presence of CTZ and Kyn to assess the mechanism of glycine-induced enhancement in rats (P11–P14), in which previous studies have suggested that increases in p contribute to glycine-induced enhancement. Glycine led to an average synaptic enhancement of 45 ± 4% (n = 6). A, Comparison of pairs of EPSCs recorded in control conditions and in the presence of glycine (the EPSCs in glycine were normalized to the amplitude of the first EPSC in control) showed that glycine decreased the PPR. Experiments with stimulus trains (40 pulses, 100 Hz) were used to determine the contributions of p and the effective RRP to glycine-induced enhancement using the cumulative EPSC method (B) and the Elmqvist and Quastel method (C). D, The effects of glycine on synaptic properties are summarized (n = 6, ±SEM). In these experiments, glycine decreased PPR in rats by 12 ± 3%. The use of trains to determine the contribution of effective RRP and p revealed that glycine produced small increases in p (ptrain, 15 ± 4%; pEQ, 17 ± 4%; n = 6) but produced larger increases in effective RRP (RRPtrain, 26 ± 2%; RRPEQ, 24 ± 3%; n = 6). Thus, in rats, glycine increases p in addition to increasing the effective pool size, but as in mice, increased neurotransmitter release is mainly attributable to an increase in the effective pool size.
Discussion
Our primary finding is that glycine-induced enhancement of evoked synaptic currents is significantly reduced in mice lacking PKCα and PKCβ, indicating that these calcium-dependent PKC isoforms mediate the bulk of this form of enhancement at the calyx of Held synapse. In contrast, calcium-dependent PKCs are not required for glycine-induced increases in mEPSC frequency, indicating that spontaneous and evoked release are differentially regulated. Activated calcium-dependent PKCs enhance transmitter release primarily by increasing the effective pool size, with only a modest increase in the probability of vesicle release.
Glycine-induced enhancement is mediated primarily by PKCα and PKCβ
Previous studies of glycine-induced enhancement have shown that the opening of chloride channels, and activation of persistent sodium channels, depolarizes the calyx of Held sufficiently to activate a small fraction of P-type calcium channels (Turecek and Trussell, 2001; Price and Trussell, 2006; Huang and Trussell, 2008). Although glycine receptor activation drives chloride efflux, the observation that glycine-induced synaptic enhancement can be mimicked by small sustained presynaptic depolarizations suggests that changes in presynaptic chloride levels are not important in this form of plasticity (Turecek and Trussell, 2001). Previous studies have shown, however, that introducing the calcium chelator EGTA into the calyx of Held abolished synaptic enhancement caused by glycine, indicating that calcium increases are crucial to this form of plasticity (Turecek and Trussell, 2001).
Our findings indicate that PKCα and PKCβ are required to allow glycine-induced presynaptic calcium increases to enhance evoked neurotransmitter release. Based on our observation that glycine continues to elevate presynaptic calcium levels and increase mEPSC frequency at the calyx of Held synapse in PKCαβ dko mice, we conclude that PKCα and PKCβ act downstream from increases in calcium entry.
The involvement of calcium-dependent PKC isoforms in glycine-induced synaptic potentiation raises the possibility that these isoforms are directly activated by elevated presynaptic calcium levels to produce synaptic enhancement. Although the calcium-binding affinities for PKCα and PKCβ [∼40 μm (Kohout et al., 2002)] are much higher than the average increase in presynaptic calcium levels evoked by glycine (50–100 nm), other intracellular factors can raise the binding affinity of PKCs for calcium to 0.1–5 μm or less (Corbalán-García et al., 1999; Kohout et al., 2002; Corbin et al., 2007; Guerrero-Valero et al., 2007), a range that would allow PKCα and PKCβ to respond to the calcium increases produced by glycine. Another possibility is that PKCα and PKCβ are located sufficiently close to P-type calcium channels such that, during calcium channel opening, they are exposed to high local calcium increases within a microdomain that are much higher than the average presynaptic calcium levels.
The mechanism of synaptic enhancement
Several lines of evidence suggest that glycine increases synaptic transmission primarily by increasing the effective size of the RRP of vesicles. First, despite the fact that the application of glycine and increasing extracellular calcium [Ca]e to 3 mm produced the same synaptic enhancement, glycine did not affect paired-pulse plasticity whereas alterations in external calcium decreased PPR in a manner that is consistent with an increase in p. The lack of an effect of glycine on paired-pulse plasticity suggests that glycine does not alter the initial p.
Moreover, plots of the cumulative EPSC as a function of stimulus number revealed that glycine increased the effective size of the RRP. Both the synaptic enhancement and the increase in the effective pool size were eliminated when ionotropic glycine receptors were blocked pharmacologically and in PKCαβ dko mice. There are challenges associated with measuring effective pool size and p. For example, determining the effective pool size using the cumulative EPSC is complicated by replenishment of the pool during stimulation (Schneggenburger et al., 1999) (Fig. 6). Extrapolation corrects for replenishment during the train but assumes that the rate of replenishment is constant throughout the train. This cumulative EPSC method provides a lower-bound estimate of the effective pool size because the assumed rate of replenishment is calculated from the steady-state rate of replenishment at the end of the train, which is likely higher than the initial rate of replenishment.
Finally, we also estimated the effective pool size and p with a method introduced by Elmqvist and Quastel (1965). With this approach, the progressive decrease in the amplitude of EPSCs during a stimulus train is attributed to vesicle depletion, and the decreases in amplitude of the initial EPSCs in the train can be used to estimate the effective pool size. Because this approach assumes that there is no replenishment, it serves as an upper-bound estimate of the effective pool size. As expected, the estimates of pool size with this approach were consistently higher than those obtained with the cumulative method. Nonetheless, this approach also indicated that glycine increased synaptic strength primarily by increasing the effective pool size and that synaptic enhancement and the increase in effective pool size were eliminated in dko mice, whereas increasing [Ca]e increased synaptic strength primarily by increasing p. These results suggest that, in mice, glycine increases EPSC size primarily by increasing the effective size of the RRP.
A comparison of PTP and glycine-induced enhancement
Glycine-induced enhancement and PTP share many features. Both forms of plasticity are mediated by an increase in presynaptic calcium levels (Turecek and Trussell, 2001; Kim et al., 2005; Korogod et al., 2007) and by two PKCCa, PKCα and PKCβ (Fioravante et al., 2011) (Fig. 1). In addition, increases in mEPSC frequency after either tetanic stimulation (Habets and Borst, 2005; Kim et al., 2005; Korogod et al., 2007) or the application of glycine were mediated by calcium increases (Zucker and Lara-Estrella, 1983; Bao et al., 1997; Kim et al., 2005) but did not involve PKCα and PKCβ (Fioravante et al., 2011). Thus, in both cases, spontaneous vesicle fusion and evoked release are differentially regulated, and other calcium-sensitive proteins must mediate enhancement of mEPSC frequency (Groffen et al., 2010; Pang et al., 2011; Yao et al., 2011). In addition, PKCα and PKCβ enhance neurotransmitter release primarily by increasing the effective size of the RRP for both forms of synaptic enhancement (Fioravante et al., 2011) (Fig. 6). It is likely that, once activated, PKCα and PKCβ phosphorylate the same target, possibly Munc 18-1 (Wierda et al., 2007), to produce this increase in effective RRP size.
There are also several interesting differences between PTP and glycine-induced enhancement. Notably, PTP tetanic stimulation increases mEPSC size by 10–20%, and this contributes to synaptic enhancement (He et al., 2009; Fioravante et al., 2011), but in glycine-induced enhancement, no such increase in mEPSC size is observed. This suggests that the high calcium levels evoked by tetanic stimulation are needed to increase mEPSC size. In addition, PKCα and PKCβ are differentially effective in these two phenomena. PKCβ makes a much larger contribution to PTP than PKCα (Fioravante et al., 2011), whereas PKCα and PKCβ make comparable contributions to glycine-induced enhancement. The different calcium signals that induce these forms of plasticity may underlie the differential contributions of these isoforms. PTP is induced by a presynaptic calcium signal that decays more rapidly than PTP (Regehr et al., 1994; Brager et al., 2003; Beierlein et al., 2007; Fioravante et al., 2011). At the calyx of Held, tetanic stimulation increases presynaptic calcium by several hundred nanomolar, and it decays to resting levels with a time constant of ∼22 s compared with synaptic enhancement that decays with a time constant of ∼45 s (Fioravante et al., 2011). For glycine-induced enhancement, the opening of voltage-gated calcium channels leads to a sustained elevation of residual calcium signal by ∼50 nm and larger local calcium increases near open calcium channels. Together, these findings indicate that, despite being similar in many ways, PKCα and PKCβ can make differential contributions to synaptic plasticity.
Implications for related forms of synaptic plasticity
Related forms of synaptic plasticity have been described at a number of synapses. For example, somatic depolarization can lead to depolarization of presynaptic boutons that can increase neurotransmitter release. This has been described at cortical (Sjöström et al., 2003; Corlew et al., 2007; Christie and Jahr, 2009), hippocampal (Vogt and Regehr, 2001), and cerebellar (Glitsch and Marty, 1999) synapses. These forms of plasticity are similar to glycine-mediated modulation, which can be effectively mimicked with small steady presynaptic depolarizations (Awatramani et al., 2005). The similarity of these forms of plasticity to glycine-induced modulation raises the possibility that calcium-dependent PKCs could also contribute to synaptic plasticity arising from somatic depolarization.
Footnotes
This work was supported by National Institutes of Health Grant R37 NS032405 (W.G.R.) and a Howard Hughes Medical Institute Medical Research Fellow grant (Y.C.). We thank Miki Antal, Lindsey Glickfeld, Court Hull, Todd Pressler, and Skyler Jackman for comments on a previous version of this manuscript. We thank Kimberly McDaniels for help with genotyping.
References
- Alle H, Geiger JR. Combined analog and action potential coding in hippocampal mossy fibers. Science. 2006;311:1290–1293. doi: 10.1126/science.1119055. [DOI] [PubMed] [Google Scholar]
- Awatramani GB, Price GD, Trussell LO. Modulation of transmitter release by presynaptic resting potential and background calcium levels. Neuron. 2005;48:109–121. doi: 10.1016/j.neuron.2005.08.038. [DOI] [PubMed] [Google Scholar]
- Balakrishnan V, Kuo SP, Roberts PD, Trussell LO. Slow glycinergic transmission mediated by transmitter pooling. Nat Neurosci. 2009;12:286–294. doi: 10.1038/nn.2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao JX, Kandel ER, Hawkins RD. Involvement of pre- and postsynaptic mechanisms in posttetanic potentiation at Aplysia synapses. Science. 1997;275:969–973. doi: 10.1126/science.275.5302.969. [DOI] [PubMed] [Google Scholar]
- Beierlein M, Gee KR, Martin VV, Regehr WG. Presynaptic calcium measurements at physiological temperatures using a new class of dextran-conjugated indicators. J Neurophysiol. 2004;92:591–599. doi: 10.1152/jn.00057.2004. [DOI] [PubMed] [Google Scholar]
- Beierlein M, Fioravante D, Regehr WG. Differential expression of posttetanic potentiation and retrograde signaling mediate target-dependent short-term synaptic plasticity. Neuron. 2007;54:949–959. doi: 10.1016/j.neuron.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackmer T, Larsen EC, Takahashi M, Martin TF, Alford S, Hamm HE. G protein βγ subunit-mediated presynaptic inhibition: regulation of exocytotic fusion downstream of Ca2+ entry. Science. 2001;292:293–297. doi: 10.1126/science.1058803. [DOI] [PubMed] [Google Scholar]
- Brager DH, Cai X, Thompson SM. Activity-dependent activation of presynaptic protein kinase C mediates post-tetanic potentiation. Nat Neurosci. 2003;6:551–552. doi: 10.1038/nn1067. [DOI] [PubMed] [Google Scholar]
- Christie JM, Jahr CE. Selective expression of ligand-gated ion channels in L5 pyramidal cell axons. J Neurosci. 2009;29:11441–11450. doi: 10.1523/JNEUROSCI.2387-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie JM, Chiu DN, Jahr CE. Ca2+-dependent enhancement of release by subthreshold somatic depolarization. Nat Neurosci. 2011;14:62–68. doi: 10.1038/nn.2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbalán-García S, Rodríguez-Alfaro JA, Gómez-Fernández JC. Determination of the calcium-binding sites of the C2 domain of protein kinase Calpha that are critical for its translocation to the plasma membrane. Biochem J. 1999;337:513–521. [PMC free article] [PubMed] [Google Scholar]
- Corbin JA, Evans JH, Landgraf KE, Falke JJ. Mechanism of specific membrane targeting by C2 domains: localized pools of target lipids enhance Ca2+ affinity. Biochemistry. 2007;46:4322–4336. doi: 10.1021/bi062140c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corlew R, Wang Y, Ghermazien H, Erisir A, Philpot BD. Developmental switch in the contribution of presynaptic and postsynaptic NMDA receptors to long-term depression. J Neurosci. 2007;27:9835–9845. doi: 10.1523/JNEUROSCI.5494-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmqvist D, Quastel DM. A quantitative study of end-plate potentials in isolated human muscle. J Physiol. 1965;178:505–529. doi: 10.1113/jphysiol.1965.sp007639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman HS, MacDermott AB. Presynaptic ionotropic receptors and control of transmitter release. Nat Rev Neurosci. 2004;5:135–145. doi: 10.1038/nrn1297. [DOI] [PubMed] [Google Scholar]
- Fioravante D, Chu Y, Myoga MH, Leitges M, Regehr WG. Calcium-dependent isoforms of protein kinase C mediate posttetanic potentiation at the calyx of Held. Neuron. 2011;70:1005–1019. doi: 10.1016/j.neuron.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glitsch M, Marty A. Presynaptic effects of NMDA in cerebellar Purkinje cells and interneurons. J Neurosci. 1999;19:511–519. doi: 10.1523/JNEUROSCI.19-02-00511.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groffen AJ, Martens S, Díez Arazola R, Cornelisse LN, Lozovaya N, de Jong AP, Goriounova NA, Habets RL, Takai Y, Borst JG, Brose N, McMahon HT, Verhage M. Doc2b is a high-affinity Ca2+ sensor for spontaneous neurotransmitter release. Science. 2010;327:1614–1618. doi: 10.1126/science.1183765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero-Valero M, Marín-Vicente C, Gómez-Fernández JC, Corbalán-García S. The C2 domains of classical PKCs are specific PtdIns(4,5)P2-sensing domains with different affinities for membrane binding. J Mol Biol. 2007;371:608–621. doi: 10.1016/j.jmb.2007.05.086. [DOI] [PubMed] [Google Scholar]
- Habets RL, Borst JG. Post-tetanic potentiation in the rat calyx of Held synapse. J Physiol. 2005;564:173–187. doi: 10.1113/jphysiol.2004.079160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Xue L, Xu J, McNeil BD, Bai L, Melicoff E, Adachi R, Wu LG. Compound vesicle fusion increases quantal size and potentiates synaptic transmission. Nature. 2009;459:93–97. doi: 10.1038/nature07860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Trussell LO. Control of presynaptic function by a persistent Na+ current. Neuron. 2008;60:975–979. doi: 10.1016/j.neuron.2008.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerchner GA, Wang GD, Qiu CS, Huettner JE, Zhuo M. Direct presynaptic regulation of GABA/glycine release by kainate receptors in the dorsal horn: an ionotropic mechanism. Neuron. 2001;32:477–488. doi: 10.1016/s0896-6273(01)00479-2. [DOI] [PubMed] [Google Scholar]
- Kim MH, Korogod N, Schneggenburger R, Ho WK, Lee SH. Interplay between Na+/Ca2+ exchangers and mitochondria in Ca2+ clearance at the calyx of Held. J Neurosci. 2005;25:6057–6065. doi: 10.1523/JNEUROSCI.0454-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Trussell LO. Negative shift in the glycine reversal potential mediated by a Ca2+- and pH-dependent mechanism in interneurons. J Neurosci. 2009;29:11495–11510. doi: 10.1523/JNEUROSCI.1086-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohout SC, Corbalán-García S, Torrecillas A, Goméz-Fernandéz JC, Falke JJ. C2 domains of protein kinase C isoforms alpha, beta, and gamma: activation parameters and calcium stoichiometries of the membrane-bound state. Biochemistry. 2002;41:11411–11424. doi: 10.1021/bi026041k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korogod N, Lou X, Schneggenburger R. Posttetanic potentiation critically depends on an enhanced Ca2+ sensitivity of vesicle fusion mediated by presynaptic PKC. Proc Natl Acad Sci U S A. 2007;104:15923–15928. doi: 10.1073/pnas.0704603104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitges M, Schmedt C, Guinamard R, Davoust J, Schaal S, Stabel S, Tarakhovsky A. Immunodeficiency in protein kinase c β-deficient mice. Science. 1996;273:788–791. doi: 10.1126/science.273.5276.788. [DOI] [PubMed] [Google Scholar]
- Leitges M, Plomann M, Standaert ML, Bandyopadhyay G, Sajan MP, Kanoh Y, Farese RV, Letiges M. Knockout of PKCα enhances insulin signaling through PI3K. Mol Endocrinol. 2002;16:847–858. doi: 10.1210/mend.16.4.0809. [DOI] [PubMed] [Google Scholar]
- Maravall M, Mainen ZF, Sabatini BL, Svoboda K. Estimating intracellular calcium concentrations and buffering without wavelength ratioing. Biophys J. 2000;78:2655–2667. doi: 10.1016/S0006-3495(00)76809-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, Bacaj T, Yang X, Zhou P, Xu W, Südhof TC. Doc2 supports spontaneous synaptic transmission by a Ca2+-independent mechanism. Neuron. 2011;70:244–251. doi: 10.1016/j.neuron.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price GD, Trussell LO. Estimate of the chloride concentration in a central glutamatergic terminal: a gramicidin perforated-patch study on the calyx of Held. J Neurosci. 2006;26:11432–11436. doi: 10.1523/JNEUROSCI.1660-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regehr WG, Delaney KR, Tank DW. The role of presynaptic calcium in short-term enhancement at the hippocampal mossy fiber synapse. J Neurosci. 1994;14:523–537. doi: 10.1523/JNEUROSCI.14-02-00523.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz D, Mellor J, Frerking M, Nicoll RA. Presynaptic kainate receptors at hippocampal mossy fiber synapses. Proc Natl Acad Sci U S A. 2001;98:11003–11008. doi: 10.1073/pnas.191351498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneggenburger R, Neher E. Presynaptic calcium and control of vesicle fusion. Curr Opin Neurobiol. 2005;15:266–274. doi: 10.1016/j.conb.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Meyer AC, Neher E. Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron. 1999;23:399–409. doi: 10.1016/s0896-6273(00)80789-8. [DOI] [PubMed] [Google Scholar]
- Shu Y, Hasenstaub A, Duque A, Yu Y, McCormick DA. Modulation of intracortical synaptic potentials by presynaptic somatic membrane potential. Nature. 2006;441:761–765. doi: 10.1038/nature04720. [DOI] [PubMed] [Google Scholar]
- Sjöström PJ, Turrigiano GG, Nelson SB. Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron. 2003;39:641–654. doi: 10.1016/s0896-6273(03)00476-8. [DOI] [PubMed] [Google Scholar]
- Südhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trussell LO. Modulation of transmitter release at giant synapses of the auditory system. Curr Opin Neurobiol. 2002;12:400–404. doi: 10.1016/s0959-4388(02)00335-5. [DOI] [PubMed] [Google Scholar]
- Turecek R, Trussell LO. Presynaptic glycine receptors enhance transmitter release at a mammalian central synapse. Nature. 2001;411:587–590. doi: 10.1038/35079084. [DOI] [PubMed] [Google Scholar]
- Vogt KE, Regehr WG. Cholinergic modulation of excitatory synaptic transmission in the CA3 area of the hippocampus. J Neurosci. 2001;21:75–83. doi: 10.1523/JNEUROSCI.21-01-00075.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierda KD, Toonen RF, de Wit H, Brussaard AB, Verhage M. Interdependence of PKC-dependent and PKC-independent pathways for presynaptic plasticity. Neuron. 2007;54:275–290. doi: 10.1016/j.neuron.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Wu LG, Saggau P. Presynaptic inhibition of elicited neurotransmitter release. Trends Neurosci. 1997;20:204–212. doi: 10.1016/s0166-2236(96)01015-6. [DOI] [PubMed] [Google Scholar]
- Yao J, Gaffaney JD, Kwon SE, Chapman ER. Doc2 is a Ca2+ sensor required for asynchronous neurotransmitter release. Cell. 2011;147:666–677. doi: 10.1016/j.cell.2011.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Maureira C, Liu X, McCormick D. P/Q and N channels control baseline and spike-triggered calcium levels in neocortical axons and synaptic boutons. J Neurosci. 2010;30:11858–11869. doi: 10.1523/JNEUROSCI.2651-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Lara-Estrella LO. Post-tetanic decay of evoked and spontaneous transmitter release and a residual-calcium model of synaptic facilitation at crayfish neuromuscular junctions. J Gen Physiol. 1983;81:355–372. doi: 10.1085/jgp.81.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]