

Abstract
Fibrosis, the hallmark of systemic sclerosis (SSc), is characterized by persistent fibroblast activation triggered by transforming growth factor-β (TGF-β). Since the acetyltransferase p300 plays a key role in fibrosis and its availability governs the intensity of fibrotic responses, we investigated p300 expression in SSc and the molecular basis of its regulation. We found that expression of p300 was markedly elevated in SSc skin biopsies, and was induced by TGF-β in explanted normal skin fibroblasts. Stimulation of p300 by TGF-β was independent of Smads, and involved the early-immediate transcription factor Egr-1, a key regulator of profibrotic TGF-β signaling. Indeed, Egr-1 was both sufficient and necessary for p300 regulation in vitro and in vivo. Increased p300 accumulation in TGF-β-treated fibroblasts was associated with histone hyperacetylation, whereas p300 depletion, or selective pharmacological blockade of its acetyltransferase activity, attenuated TGF-β-induced responses. Moreover, TGF-β enhanced both p300 recruitment and in vivo histone H4 acetylation at the COL1A2 locus. These findings implicate p300-mediated histone acetylation as a fundamental epigenetic mechanism in fibrogenesis, and place Egr-1 upstream in TGF-β-driven stimulation of p300 gene expression. The results establish a firm link between fibrosis with aberrant p300 expression and epigenetic activity to our knowledge previously unreported. Targeted disruption of p300-mediated histone acetylation might therefore represent a viable anti-fibrotic strategy.
Keywords: Acetyltransferase p300, TGF-β, fibroblast, systemic sclerosis, fibrosis, EGR-1, epigenetics
INTRODUCTION
The ubiquitous nuclear phosphoprotein p300 is a component of the chromatin remodeling and transcriptional complex that modulates the expression of genes involved in cell cycle regulation, apoptosis, growth and development (Goodman and Smolik, 2000). The histone acetyltransferase (HAT) domain enables p300 to catalyze histone acetylation and augment activator-dependent transcription (Kraus and Kadonaga, 1998; Vo and Goodman, 2001). In addition to chromatin histone, p300 can also catalyze acetylation of early response gene-1 (Egr-1) and other transcription factors (Ghosh and Varga, 2007). While p300 is essential for normal embryogenesis, altered expression or function is implicated in the Rubinstein-Taybi syndrome, cancer, neurodegenerative diseases and other pathologies (Giles et al., 1998; Iyer et al., 2004; Janknecht, 2002; Petrij et al., 1995; Roelfsema et al., 2005; Saha and Pahan, 2006). Recent studies reveal important roles for p300 in the regulation of extracellular matrix (ECM) homeostasis, myofibroblast transformation and epithelial-mesenchymal transition (Ghosh and Varga, 2007; Kaimori et al., 2010; Lim et al., 2012; Yokomizo et al., 2011).
We have shown previously that the HAT activity of p300 and its interaction with activated Smads are essential for transforming growth factor -β (TGF-β)-induced profibrotic signaling (Ghosh et al., 2004; Ghosh et al., 2009; Ghosh et al., 2001; Ghosh et al., 2000). Moreover, levels of p300 are limiting in normal fibroblasts, and ectopic expression of p300 dramatically enhanced the magnitude of TGF-β responses, whereas ribozyme-mediated knockdown of cellular p300 resulted in selective abrogation of the stimulation of collagen gene expression. Levels of p300 were significantly elevated in explanted SSc fibroblasts (Bhattacharyya et al., 2005), and in skin from mice with chronic GVHD-associated skin fibrosis, suggesting that p300 might have an important role in the pathogenesis of SSc.
Surprisingly, the regulation of p300 expression and activity under physiological and pathological conditions have received scant attention. The present studies demonstrate that p300 expression was significantly elevated in SSc skin biopsies, and was markedly stimulated by TGF-β in explanted normal fibroblasts via a Smad-independent pathway. Increased p300 abundance in TGF-β-stimulated fibroblasts, as well as in cells conditionally overexpressing p300, was associated with p300 accumulation on target gene promoters, histone H4 acetylation and elevated Type I collagen transcriptional activity. These results establish an important mechanistic link between TGF-β signaling, excessive collagen gene expression and fibrosis that is mediated through Egr-1 induction of p300 and histone hyperacetylation at the collagen gene promoter. Precise delineation of the molecular events underlying fibrogenesis and the role of p300 in TGF-β-inducible transcriptional responses may accelerate the development of targeted therapies to control the progression of fibrosis in SSc.
RESULTS
Elevated levels of p300 in SSc
We have previously shown that p300 levels were elevated in explanted SSc fibroblasts (Bhattacharyya et al., 2005). We confirmed this finding in additional fibroblasts from patients with dSSc (Fig. 1a). Next we evaluated biopsies of lesional skin from ten patients with SSc and of dorsal forearms from five healthy controls by immunohistochemistry. The results showed markedly increased p300 immunostaining in SSc biopsies (Fig. 1b). In the SSc lesional dermis, p300 was localized largely to the nucleus in fibroblast-like cells. Staining with isotype control antibodies indicated the specificity of anti-p300 antibodies. The intensity of cellular p300 staining in diffuse cutaneous SSc biopsies showed a modest positive correlation with the modified Rodnan skin score (slope=0.23, R2=0.29).
Figure 1. Enhanced expression of p300 in SSc fibroblasts and skin biopsies.
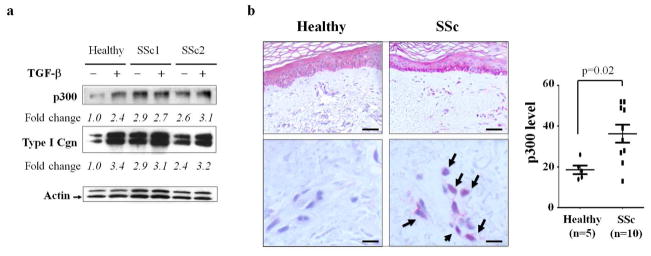
a. Fibroblasts explanted from patients with dSSc and a healthy adult control were incubated with TGF-β for 24 h. Whole cell lysates were analyzed by Western blotting followed by densitometry. The results show relative fold changes normalized with actin. Arrow indicates actin bands. b. Skin biopsies from ten SSc patients and five healthy controls were stained with anti-p300 antibodies or isotype control. Left panels, representative images of randomly selected microscopic fields (upper panels, bar = 80 μm; lower panels, bar = 20μm). Arrows indicate immunopositive dermal cells with the nuclear staining. Right panel, dot-plot of p300 staining score in healthy control and SSc patients. The intensity of p300 staining in each slide was quantified as described in Methods.
TGF-β stimulates p300 gene expression
In view of the increased expression of p300 in SSc skin biopsies, and its pivotal role in mediating TGF-β-induced profibrotic responses, we investigated the regulation of p300 expression by TGF-β. The results of semi-quantitative PCR, Western blot and immunofluorescence analysis revealed that in normal fibroblasts, TGF-β enhanced the levels of p300 (Fig. 2a–b). Stimulation could be detected as early as 6 h, with a maximal >3-fold increase at 48 h. Remarkably, in contrast to p300, levels of CBP, a p300 paralogue with comparable histone acetyltransferase and coactivator function, remained unaltered in TGF-β-treated fibroblasts (data not shown). Northern analysis revealed that stimulation in p300 mRNA expression by TGF-β was concentration-dependent, with a maximal >3-fold increase (Fig. 2c).
Figure 2. TGF-β augments the levels of p300.

a–d. Confluent dermal fibroblasts were incubated with TGF-β2 (12.5 ng/ml unless otherwise indicated). a. At the end of the experiments, whole cell lysates and RNA were harvested and examined by Western blot or semi-quantitative RT-PCR. Upper panels, Western blot; lower panels, semi-quantitative RT-PCR. Representative images. b. Fibroblasts were stained with DAPI (nuclei: blue) or anti-p300 antibodies (green), and viewed under a fluorescent microscope. Original magnification 400X. Representative images. c. Following 10 h incubation, total RNA was isolated and subjected to Northern analysis. d. Fibroblasts were incubated with TGF-β2 for 24 h. Actinomycin D was added for a further 6 or 24 h, and mRNA levels were analyzed by RT-PCR. Lower panel, levels of p300 mRNA relative to actin. e. NIH3T3 fibroblasts were transfected with pGL3-p300-luc, then incubated in serum-free media with TGF-β2 or in media with 20% FBS. Whole cell lysates were assayed for luciferase activities. The results represent the means ± SEM. of triplicate determinations. *p<0.05
A series of experiments were performed in neonatal and adult skin fibroblasts to address the mechanisms underlying p300 stimulation. To examine changes in the stability of p300 mRNA transcripts, fibroblasts were incubated with TGF-β in the presence or absence of actinomycin D (5 μg/ml), followed by determination of mRNA levels at the indicated intervals. The results of RT-PCR showed that p300 mRNA level declined at a comparable rate in the absence or presence of TGF-β, with an estimated half-life of 6–9 h (Fig. 2d). To evaluate the effect of TGF-β on p300 gene transcription, confluent NIH3T3 fibroblasts were transiently transfected with p300-luc reporter constructs. Following 24 h of incubation in serum-free media containing TGF-β, cultures were harvested and luciferase activities were determined. The results indicated that TGF-β induced a >2-fold stimulation of luciferase activity driven by a p300 promoter (Fig. 2e). Addition of serum strongly stimulated p300 promoter activity in transfected fibroblasts, consistent with previous reports in prostate cancer cells (Yu et al., 2004). Together, these results indicate that p300 gene expression is stimulated by TGF-β via transcriptional stimulation.
Stimulation of p300 is Smad-independent
Pretreatment of the cultures with a selective inhibitor of AKL5-mediated Smad2/3 phosphorylation (SB431542) did not block the stimulation of p300 induced by TGF-β, while significantly reducing Smad2 phosphorylation (80% reduction in lane 6 compared to lane 2) (Fig. 3a). In sharp contrast, the ERK1/2 inhibitor PD98059 completely prevented the stimulation (Fig. 3a). Consistent with these results, TGF-β stimulated p300 mRNA levels with a comparable magnitude in Smad3 null and wildtype MEFs (90±18% in wildtype MEFs and 86±3% in Smad3−/− MEFs, p=n.s., data not shown). Moreover, transient transfection assays showed that the ERK1/2 inhibitor, but not the ALK5 inhibitor, abrogated the stimulation of p300 promoter activity (Fig. 3b). Identical Smad-independent TGF-β stimulation of p300 expression was seen in human adult skin fibroblasts incubated with TGF-β (Fig. 3c).
Figure 3. p300 stimulation by TGF-β is mediated via ERK1/2.

a. Confluent fibroblasts were preincubated with SB431542 (left panel) or PD98059 (right panel) followed by TGF-β2 (12.5 ng/ml) for 24 h. Whole cell lysates were examined by immunoblot analysis. b. NIH3T3 fibroblasts transfected with p300-luc were preincubated with SB431542 or PD98059, followed by TGF-β for 24 h. Whole cell lysates were assayed for their luciferase activities. Open bars, untreated fibroblasts; closed bars, TGF-β-treated fibroblasts. The results normalized with protein concentration represent the means ± S.D. of triplicate determinations. * p<0.05. c. Confluent normal adult dermal fibroblasts were preincubated with SB431542 or U0126 followed by TGF-β2 for 24 h. The real-time qPCR results, normalized with GAPDH, represent the means ± SD of triplicate determines from the representative experiment. *, p<0.05.
Stimulation of p300 is mediated by Egr-1
We demonstrated previously that expression of Egr-1 was stimulated by TGF-β in a variety of cell types in a rapid and transient manner and requiring ERK1/2, but not Smads (Bhattacharyya et al., 2011). Moreover, Egr-1 was both necessary and sufficient for TGF-β stimulation of collagen gene expression. Inspection of the human p300 gene promoter revealed the presence of multiple conserved Egr-1 recognition sites (data not shown), suggesting a potential role for Egr-1 in regulation of p300. To investigate this possibility directly, Egr-1 was ectopically expressed in normal fibroblasts with/without TGF-β. The results from multiple transfection assays demonstrated that Egr-1 by itself was capable of enhancing p300 expression, with a magnitude of stimulation comparable to that induced by TGF-β (Fig. 4a). To define the role of DNA-binding activity of Egr-1 in mediating transactivation, we used a mutant p300 promoter construct harboring base changes disrupting all six Egr-1 binding sites (Yu et al., 2004). Transient transfection of this Egr-1-unresponsive mutant p300 promoter revealed reduced basal transcriptional activity, and importantly, complete abrogation of the TGF-β stimulatory response (Fig. 4b). Next, to directly examine the role of Egr-1 in p300 regulation, we used fibroblasts lacking Egr-1. For this purpose, MEFs from Egr-1−/− mice and Egr-1+/+ wildtype littermates in parallel were grown to confluence and then incubated with TGF-β. Western analysis revealed that absence of cellular Egr-1 was associated with decreased levels of p300, and a substantial loss of the TGF-β stimulatory response (Fig. 4c). The role of Egr-1 in TGF-β-induced p300 expression was next examined in vivo. For this purpose, Egr-1−/− mice and wildtype littermates were injected in parallel with TGF-β. Analysis of mRNA directly isolated from the injected skin revealed that in the absence of Egr-1, TGF-β failed to induce p300 expression in vivo (Fig. 4d). Consistent results at the level of protein expression were obtained by immunofluorescence (Fig. 4e). The results from these in vitro and in vivo experiments collectively establish the indispensible role of Egr-1 in the regulation of p300 by TGF-β.
Figure 4. Cellular Egr-1 is required for the stimulation of p300 by TGF-β.

a. Fibroblasts were transfected with Egr-1 or empty vector and harvested following a 24 h incubation with TGF-β. Whole cell lysates were subjected to Western blotting. Arrow indicates actin bands. b. Fibroblasts were cotransfected with p300-luc or mutant p300-luc (Δp300-luc) along with Egr-1, and incubated with TGF-β2 for 24 h. Whole cell lysates were assayed for luciferase activities. Open bars, untreated fibroblasts; closed bars, TGF-β-treated fibroblasts. The results represent the means ± S.D. of triplicate determinations, normalized with protein concentration. *p<0.05. c. Embryonic fibroblasts from Egr-1−/− mice or wildtype littermates were incubated with TGF-β (12.5 ng/ml) for 2 to 24 h. Whole cell lysates were examined by Western analysis. Arrow indicates actin bands. d, e. Skin was harvested from control (PBS)- and TGF-β-treated wildtype and Egr-1−/− mice. d. RNA extracted from the skin of control (PBS)- and TGF-β-treated wildtype and Egr-1−/− mice was examined by RT-PCR. e. Tissue was processed for immunofluorescence using anti-p300 antibodies. Original magnifications, 400X.
TGF-β induces p300-dependent histone H4 hyperacetylation
Maximal stimulation of collagen synthesis by TGF-β depends on the acetyltransferase activity of p300 (Ghosh et al., 2000). In light of its importance, we sought to investigate the role of p300-mediated histone H4 acetylation in fibrotic responses elicited by TGF-β. Confluent fibroblasts were incubated with TGF-β for 24 h, and total cellular histones were acid-extracted. Western analysis showed that TGF-β induced a significant increase in acetylated histone H4, whereas levels of total histone H4 remained unaltered (Fig. 5a). The role of p300 in TGF-β-induced histone hyperacetylation was further evaluated by complementary gain-of-function and loss-of-function experiments. Ad-Tet infection of fibroblasts stably expressing inducible p300 resulted in markedly increased accumulation of p300 in these cells, which was associated with significantly enhanced histone H4 acetylation (Fig. 5b). To further examine the contribution of p300 in the TGF-β response, endogenous p300 was knocked down in normal dermal fibroblasts using p300-specific ribozymes. The results of Western analysis demonstrated that while TGF-β treatment resulted in increased histone H4 acetylation in control fibroblasts, ribozyme-mediated depletion of cellular p300 substantially abrogated the response (Fig. 5c). The effects of TGF-β on the recruitment of p300 to the COL1A2 promoter, and associated locus-specific histone acetylation in vivo, were examined next. The results of ChIP assays revealed markedly elevated p300 accumulation and histone H4 acetylation at the Smad binding region of COL1A2 promoter in fibroblasts incubated with TGF-β (Fig. 6a).
Figure 5. TGF-β induces p300-mediated histone H4 hyperacetylation.

a. Fibroblasts were incubated with TGF-β2 (12.5 ng/ml) for 24 h. Total histones were extracted and analyzed by Western blotting followed by densitometry. Lower panel, quantitation. The results show levels of AcH4 histone normalized to total H4 histone (mean ± S.D) from three independent experiments(lower panel). *p<0.05. b. Rat-12 fibroblasts stably expressing conditional p300 were infected with Ad-β-gal or Ad-Tet to induce p300 expression, and whole cell lysates were examined by Western analysis. Arrow indicates actin bands. c. Foreskin fibroblasts transfected with p300-specific ribozyme or inactive ribozyme were incubated with TGF-β2 (12.5 ng/ml) for 48 h. Whole cell lysates were analyzed by Western blot.
Figure 6. TGF-β induces p300 recruitment and HAT-dependent fibrotic responses.

a. Fibroblasts were incubated with TGF-β2 (12.5 ng/ml) for 60 min, and whole cell lysates were subjected to ChIP assays using antibodies to p300 or acetylated histone H4. Immunoprecipitated chromatins were reverse cross-linked and purified DNA was subjected to PCR (left panels) and real-time qPCR (right panels) using primers to amplify Smad binding region of Col1A2 promoter. The results represent the means ± S.D. of triplicate determinations. *p<0.05. b. Adult skin fibroblasts were pretreated with anacardic acid (ANA) followed by incubation with TGF-β for 24 h. Results of real-time qPCR shown as means ± SEM. *p<0.05. c. Illustration of the TGF-β-Egr-1-p300-chromatin remodeling-fibrotic response relay system. In mesenchymal cells, TGF-β stimulates Egr-1 expression, which activates transcription of both COL1A2 and p300. In turn, p300 causes COL1A2 promoter chromatin histone acetylation, facilitating transcriptional activation by Smad2/3. Elevated levels of p300 enhance Smad2/3-dependent transcriptional activity. EBS, Egr binding site; SBE, Smad binding element.
To directly establish the functional significance of the acetyltransferase activity of p300 in the fibrotic responses elicited by TGF-β, we used anacardic acid, a selective pharmacological inhibitor of histone acetyltransferase activity (Eliseeva et al., 2007). Confluent cultures were pretreated with anacardic acid, followed by incubation with TGF-β. Pharmacological inhibition of HAT activity dramatically abrogated the stimulation of collagen expression induced by TGF-β (Fig. 6b). Taken together, these results indicate that enhanced p300 expression and HAT activity in TGF-β-stimulated fibroblasts resulted in increased histone H4 acetylation that was associated with, and directly responsible for, up-regulation of fibrotic gene expression in these cells.
DISCUSSION
Previous studies have identified an essential role for p300 acetyltransferase and its interaction with Smads in TGF-β-induced profibrotic responses (Czuwara-Ladykowska et al., 2002; Ghosh et al., 2004; Ghosh et al., 2009; Ghosh et al., 2001; Ghosh et al., 2000). Despite its critical role in modulating profibrotic responses elicited by TGF-β and other mediators, the regulation of p300 expression and activity, and the mechanisms underlying their derangement in fibrotic diseases, have received scant attention to date.
The present results reveal that levels of p300 are significantly elevated in lesional skin biopsies from patients with SSc. Moreover, the expression of p300 in explanted normal fibroblasts was markedly up-regulated by TGF-β. Stimulation involved the canonical Smad-independent ERK1/2 -mediated up-regulation of the early immediate transcription factor Egr-1, which then directly stimulated p300 gene transcription. Treatment of the fibroblasts with TGF-β also resulted in enhanced histone H4 hyperacetylation concomitant with increased p300 accumulation at the COL1A2 promoter. Moreover, pharmacological blockade of HAT activity using anacardic acid abrogated the stimulation of collagen gene expression elicited by TGF-β. Taken together with the previous demonstration that augmented p300 expression in fibroblasts sensitized them to the profibrotic effects of TGF-β, the present results provide previously unreported insight into the mechanism of fibrosis in SSc, and indicate a fundamental role of acetyltransferase p300-mediated histone modifications in mediating this process.
Elevated p300 levels were seen in the skin from mice with bone-marrow transplantation-induced chronic GVHD (Bhattacharyya et al., 2005). Similar up-regulation of p300 was also observed in the skin from mice with bleomycin-induced scleroderma (Wu M, Bhattacharrya S, Varga J; unpublished results). The expression of p300 is elevated in explanted SSc skin fibroblasts (Bhattacharyya et al., 2005). Other studies linking fibrosis and p300 showed that the activated SSc fibroblast phenotype was attributed to ligand-independent constitutive association of p300 with Smad2/3 and consequent transcriptional activation of target genes (Ihn et al., 2006). The mechanism accounting for the increased accumulation of p300 in the lesional SSc skin, and its persistence in explanted SSc fibroblasts, remain unknown. Of note, elevated p300 expression has been reported in fibrotic kidneys from patients with glomerulonephritis (Kassimatis et al., 2006), as well as in a mouse model of lung fibrosis (Liu et al., 2004). The levels of p300 are elevated in various tumors (Li et al., 2011a; Li et al., 2011b). Moreover, the relative tissue expression of p300 in these cancers correlates with tumor aggressiveness and survival, suggesting a potential role for p300 as a biomarker of disease severity. Importantly, selective inhibition of p300 acetyltransferase activity with a small molecule blocked profibrotic TGF-β responses in normal fibroblasts in the present studies, and was shown to be reduced the invasiveness of prostate cancer cells displaying elevated p300 (Santer et al., 2011). Taken together, these observations suggest that p300 plays a fundamental role in progression of fibrosis and cancer, and determining its levels may serve as disease biomarkers in these conditions.
Since TGF-β can stimulate collagen gene expression via both canonical and Smad-independent signaling pathways (Ghosh, 2002; Varga, 2002), we investigated the role of the TGF-β receptor kinase and activation of downstream Smads in p300 regulation. Surprisingly, we found that an ERK1/2 kinase inhibitor, but not an ALK5 inhibitor, blocked the stimulation of p300 by TGF-β. The early-immediate transcription factor Egr-1 plays an indispensible role in fibrogenesis, and its expression is constitutively elevated in SSc biopsies (Bhattacharyya et al., 2011). Egr-1 has been implicated in regulation of p300 expression in prostate cancer cells (Yu et al., 2004). In these cells, incubation with serum triggered a potent up-regulation of p300. We have shown previously that in normal skin fibroblasts, Egr-1 was rapidly and transiently induced by TGF-β via a Smad-independent pathway (Bhattacharyya et al., 2011; Chen et al., 2006). Egr-1 stimulated collagen transcription by binding to collagen promoter (Chen et al., 2006). Here we found that Egr-1 directly augmented p300 gene expression and further enhanced the TGF-β-induced stimulation. In contrast, TGF-β failed to stimulate p300 transcription from a promoter with Egr-1 binding sites disrupted. Based on these findings, we propose a complex signaling relay network. Since levels of Egr-1, p300 and collagen are all significantly elevated in lesional SSc tissues, the fibrogenic TGF-β → Egr-1 → p300 → HAT activity and chromatin remodeling → target gene transcription relay pathway is likely to contribute to the progression of fibrosis in a concerted way (Fig. 6c).
Taken together, these results demonstrate that p300, a ubiquitous transcriptional coactivator and epigenetic regulator that is required for optimal Smad-dependent profibrotic responses, is itself a target of TGF-β signaling. Lesional tissue levels of p300 are significantly elevated in mouse models of scleroderma and pulmonary fibrosis, as well as in SSc skin biopsies and explanted SSc skin fibroblasts. Induction of p300 by TGF-β is dependent on Egr-1, which directly binds to regulatory DNA elements in the p300 gene and stimulates its transcription. Augmented abundance of cellular p300 is associated with enhanced intensity of profibrotic TGF-β signaling due to increased histone H4 hyperacetylation and Smad-dependent transcription. Because the cellular availability of p300 governs the intensity of TGF-β responses, Egr-1-mediated up-regulation of p300 expression and epigenetic activity is expected to augment the stimulation of collagen and other fibrotic genes, and thus plays a vital role in the pathogenesis of fibrosis. Small molecules are available to selectively disrupt the histone acetylransefrase activity of p300. Inducing competition for limiting amounts of cellular p300 using agents that activate the AMP kinase pathways, cyclic AMP or PPAR-γ might represent alternate “drug repurposing” approaches for repressing p300 function. Therefore, blocking the stimulation of p300 expression or activity might represent innovative therapeutic strategies to control TGF-β-mediated fibrogenesis.
MATERIALS AND METHODS
Human subjects
Skin biopsies were obtained from the dorsal forearm of five healthy adults, and the lesional skin of ten SSc patients (clinical characteristics in Supplemental table 1). Fibroblasts were explanted from biopsies of lesional skin from patients with dSSc and a healthy adult and studied at early passage. All skin biopsies were performed with written informed consent and in compliance with the Institutional (Boston University and Northwestern University) Review Board for Human Studies, in adherence to the Declaration of Helsinki Principles.
Cell cultures
Primary cultures of dermal fibroblasts were established by explantation from neonatal foreskin or adult skin (Chen et al., 1999). Mouse embryonic fibroblasts explanted from Smad3−/− or wild-type embryos were maintained in Dulbecco’s modified Eagle medium. Culture and infection of Rat-12 cells harboring Tet-inducible p300 were described previously (Baluchamy et al., 2003). At confluence, cultures were incubated with indicated concentrations of TGF-β2 (Genzyme, Framingham, MA), the ALK5 inhibitor SB431542 (Glaxo SmithKline, King of Prussia, PA), the ERK1/2 kinase inhibitor PD98059 or U0126 (Sigma, St. Louis, MO), or actinomycin D (Cell Signaling, Beverly, MA). To reach maximum induction of fibrotic responses, 12.5 ng/ml TGF-β was used in selected experiments. The histone acetyltransferase inhibitor anacardic acid (EMD Millipore, Billerica, MA) was added to the cultures 60 min prior to TGF-β.
Plasmids, transient transfection and reporter assays
The pGL3 p300-luc, pGL3 p300-luc-abcdef, PCB-pCMV-Egr-1 and p300-specific ribozyme constructs were from Dr Adamson (Yu et al., 2004). Confluent foreskin fibroblasts were transiently cotransfected with reporter and expression vectors or empty vectors using Superfect reagent (Qiagen, Valencia, CA), and incubated with TGF-β2 (12.5 ng/ml) for up to 48 h. Transfection with pRLTK-luc was used as an internal control.
Histone extraction and chromatin immunoprecipitation assays
Confluent cultures of fibroblasts were incubated with TGF-β (12.5 ng/ml). Twenty-four h later, cultures were harvested and histones were extracted as described (Ghosh et al., 2007). Chromatin immunoprecipitation (ChIP) assays were performed using the EZ magna ChIP Assay Kit (Upstate/Millipore, Billerica, MA) as described (Ghosh et al., 2009). PCR amplification of the captured DNA sequences was performed using primers complementary to COL1A2 sequences flanking the Smad binding element.
Western analysis
Following incubation with TGF-β for indicated periods, adult human dermal fibroblasts, foreskin fibroblasts or rat fibroblasts were harvested and whole cell lysates were prepared using M-PER lysis buffer (Pierce, Rockford, IL). Equal aliquots of total proteins or acid-extracted histones were electrophoresed on 4–20% Tris-Glycine gradient gels (BIORAD, Hercules, CA), followed by immunoblot analysis using primary antibodies against phospho-ERK1 (Cell Signaling), p300 and actin (Santa Cruz), total histone H4 or acetylated histone H4 (Ac-H4) (Upstate/Millipore). Blots were washed with TBST buffer, followed by incubation with appropriate HRP-conjugated secondary antibodies, and antigen-antibody complexes were visualized by chemiluminescence (ECL Reagent, Amersham BioSciences, Piscataway, NJ). To quantify the bands obtained via Western blot, developed films were scanned in grayscale and analyzed by ImageJ software (http://rsb.info.nih.gov/ij/).
Immunohistochemistry and immunofluorescence
Sections of human skin biopsies were deparaffinized, rehydrated and blocked for 30 min with 3% bovine serum albumin in Tris-buffered saline (pH 7.4), and then incubated overnight with anti-p300 antibody (Santa Cruz). Slides were then incubated for 30 min with a biotinylated donkey anti-rabbit secondary antibody followed by streptavidin-linked alkaline-phosphatase (Jackson Immuno Research, West Grove, PA), or with alkaline phosphatase-conjugated anti-mouse/anti-rabbit antibody (DakoCytomation, Carpinteria, CA) for 30 min and detected with Fast Red (DakoCytomation). Quantification was performed by two independent observers, and the intensity of staining was recorded for at least 200 individual fibroblasts using a scale of 0–3 (0=no visible staining, 1=faint staining observable, 2= moderate staining, 3=strong staining). The average intensity of fibroblast staining was calculated based on images from random fields throughout the dermis of the whole tissue section.
To examine the role of Egr-1 in the regulation of p300 expression in vivo, six- to eight-week-old Egr-1 knockout mice in the C57BL/6 background (Wu et al., 2009) and wildtype littermates were used under protocols institutionally approved by the Northwestern University Animal Care and Use Committee. Mice were injected with TGF-β (250 ng) or PBS. After 48 h, mice were sacrificed, 4 μm sections from dorsal skin were deparaffinized, rehydrated, and immersed in TBS-T buffer, and treated with target retrieval solution (DAKO). Sections were incubated with anti-p300 antibody (Santa Cruz), and bound antibodies were detected using Alexa®488 conjugated donkey-anti-rabbit antibody (Invitrogen). Rabbit control IgG was used as negative controls. Sections were counterstained with 4′-6-diamidino-2-phenylindole (DAPI) for nuclei. Following washing, slides were examined under a Zeiss UV Meta 510 confocal microscope (Carl Zeiss, Jena, Germany).
Northern analysis, RT-PCR and Real-time qPCR
Total RNA was isolated from confluent fibroblasts using TRIZOL reagent (Life Technologies, Grand Island, NY), and examined by Northern analysis or RT-PCR (Bhattacharyya et al., 2005). Reverse transcription were performed using SuperScript First-strand synthesis system (Invitrogen) according to the manufacturer’s protocol. Primers (Sigma Genosys, Woodlands, TX) for PCR were as following: p300, sense 5′-CTTTACCGTCAGGATCCAG-3′, antisense 5′-AGTATTTGTATACCCGT ATG-3′; actin, sense 5′-ATCTGGCACCACACCTTCTACAATGAG CTGCG-3′, antisense 5′-CGTCATACTCCTGCTTGTGATCCACATCTGC-3′. For real-time qPCR, reactions were performed on ABI-Prism 7300 sequence detection PCR machine (Applied Biosystem, Forster City, CA) according to the manufacturer’s protocol. The primer sequences are available upon request. Relative mRNA expression levels were normalized with GAPDH mRNA levels and determined by calculating ΔΔCt.
Statistical analysis
The data are presented as the means ± S.D or means ± SEM. Statistical differences between experimental and control groups were determined by analysis of variance. Values of p<0.05 by Student t-test were considered significant.
Supplementary Material
Acknowledgments
We are grateful to Eileen Adamson (Burnham Institute, CA) for helpful suggestions and for providing plasmids. This work was supported by grants from the National Institutes of Health (AR42309 and CA74403) and the Scleroderma Foundation.
Footnotes
CONFLICT OF INTREST
The authors state no conflict of interest.
References
- Baluchamy S, Rajabi HN, Thimmapaya R, Navaraj A, Thimmapaya B. Repression of c-Myc and inhibition of G1 exit in cells conditionally overexpressing p300 that is not dependent on its histone acetyltransferase activity. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:9524–9. doi: 10.1073/pnas.1633700100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya S, Ghosh AK, Pannu J, Mori Y, Takagawa S, Chen G, et al. Fibroblast expression of the coactivator p300 governs the intensity of profibrotic response to transforming growth factor beta. Arthritis and rheumatism. 2005;52:1248–58. doi: 10.1002/art.20996. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S, Sargent JL, Du P, Lin S, Tourtellotte WG, Takehara K, et al. Egr-1 induces a profibrotic injury/repair gene program associated with systemic sclerosis. PloS one. 2011;6:e23082. doi: 10.1371/journal.pone.0023082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SJ, Ning H, Ishida W, Sodin-Semrl S, Takagawa S, Mori Y, et al. The early-immediate gene EGR-1 is induced by transforming growth factor-beta and mediates stimulation of collagen gene expression. The Journal of biological chemistry. 2006;281:21183–97. doi: 10.1074/jbc.M603270200. [DOI] [PubMed] [Google Scholar]
- Chen SJ, Yuan W, Mori Y, Levenson A, Trojanowska M, Varga J. Stimulation of type I collagen transcription in human skin fibroblasts by TGF-beta: involvement of Smad 3. The Journal of investigative dermatology. 1999;112:49–57. doi: 10.1046/j.1523-1747.1999.00477.x. [DOI] [PubMed] [Google Scholar]
- Czuwara-Ladykowska J, Sementchenko VI, Watson DK, Trojanowska M. Ets1 is an effector of the transforming growth factor beta (TGF-beta) signaling pathway and an antagonist of the profibrotic effects of TGF-beta. The Journal of biological chemistry. 2002;277:20399–408. doi: 10.1074/jbc.M200206200. [DOI] [PubMed] [Google Scholar]
- Eliseeva ED, Valkov V, Jung M, Jung MO. Characterization of novel inhibitors of histone acetyltransferases. Molecular cancer therapeutics. 2007;6:2391–8. doi: 10.1158/1535-7163.MCT-07-0159. [DOI] [PubMed] [Google Scholar]
- Ghosh AK. Factors involved in the regulation of type I collagen gene expression: implication in fibrosis. Exp Biol Med (Maywood) 2002;227:301–14. doi: 10.1177/153537020222700502. [DOI] [PubMed] [Google Scholar]
- Ghosh AK, Bhattacharyya S, Varga J. The tumor suppressor p53 abrogates Smad-dependent collagen gene induction in mesenchymal cells. The Journal of biological chemistry. 2004;279:47455–63. doi: 10.1074/jbc.M403477200. [DOI] [PubMed] [Google Scholar]
- Ghosh AK, Bhattacharyya S, Wei J, Kim S, Barak Y, Mori Y, et al. Peroxisome proliferator-activated receptor-gamma abrogates Smad-dependent collagen stimulation by targeting the p300 transcriptional coactivator. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2009;23:2968–77. doi: 10.1096/fj.08-128736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AK, Mori Y, Dowling E, Varga J. Trichostatin A blocks TGF-beta-induced collagen gene expression in skin fibroblasts: involvement of Sp1. Biochemical and biophysical research communications. 2007;354:420–6. doi: 10.1016/j.bbrc.2006.12.204. [DOI] [PubMed] [Google Scholar]
- Ghosh AK, Varga J. The transcriptional coactivator and acetyltransferase p300 in fibroblast biology and fibrosis. Journal of cellular physiology. 2007;213:663–71. doi: 10.1002/jcp.21162. [DOI] [PubMed] [Google Scholar]
- Ghosh AK, Yuan W, Mori Y, Chen S, Varga J. Antagonistic regulation of type I collagen gene expression by interferon-gamma and transforming growth factor-beta. Integration at the level of p300/CBP transcriptional coactivators. The Journal of biological chemistry. 2001;276:11041–8. doi: 10.1074/jbc.M004709200. [DOI] [PubMed] [Google Scholar]
- Ghosh AK, Yuan W, Mori Y, Varga J. Smad-dependent stimulation of type I collagen gene expression in human skin fibroblasts by TGF-beta involves functional cooperation with p300/CBP transcriptional coactivators. Oncogene. 2000;19:3546–55. doi: 10.1038/sj.onc.1203693. [DOI] [PubMed] [Google Scholar]
- Giles RH, Peters DJ, Breuning MH. Conjunction dysfunction: CBP/p300 in human disease. Trends in genetics: TIG. 1998;14:178–83. doi: 10.1016/s0168-9525(98)01438-3. [DOI] [PubMed] [Google Scholar]
- Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes & development. 2000;14:1553–77. [PubMed] [Google Scholar]
- Ihn H, Yamane K, Asano Y, Jinnin M, Tamaki K. Constitutively phosphorylated Smad3 interacts with Sp1 and p300 in scleroderma fibroblasts. Rheumatology (Oxford) 2006;45:157–65. doi: 10.1093/rheumatology/kei124. [DOI] [PubMed] [Google Scholar]
- Iyer NG, Ozdag H, Caldas C. p300/CBP and cancer. Oncogene. 2004;23:4225–31. doi: 10.1038/sj.onc.1207118. [DOI] [PubMed] [Google Scholar]
- Janknecht R. The versatile functions of the transcriptional coactivators p300 and CBP and their roles in disease. Histology and histopathology. 2002;17:657–68. doi: 10.14670/HH-17.657. [DOI] [PubMed] [Google Scholar]
- Kaimori A, Potter JJ, Choti M, Ding Z, Mezey E, Koteish AA. Histone deacetylase inhibition suppresses the transforming growth factor beta1-induced epithelial-to-mesenchymal transition in hepatocytes. Hepatology. 2010;52:1033–45. doi: 10.1002/hep.23765. [DOI] [PubMed] [Google Scholar]
- Kassimatis TI, Giannopoulou I, Koumoundourou D, Theodorakopoulou E, Varakis I, Nakopoulou L. Immunohistochemical evaluation of phosphorylated SMAD2/SMAD3 and the co-activator P300 in human glomerulonephritis: correlation with renal injury. Journal of cellular and molecular medicine. 2006;10:908–21. doi: 10.1111/j.1582-4934.2006.tb00534.x. [DOI] [PubMed] [Google Scholar]
- Kraus WL, Kadonaga JT. p300 and estrogen receptor cooperatively activate transcription via differential enhancement of initiation and reinitiation. Genes & development. 1998;12:331–42. doi: 10.1101/gad.12.3.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Luo RZ, Chen JW, Cao Y, Lu JB, He JH, et al. High expression of transcriptional coactivator p300 correlates with aggressive features and poor prognosis of hepatocellular carcinoma. Journal of translational medicine. 2011a;9:5. doi: 10.1186/1479-5876-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Yang HX, Luo RZ, Zhang Y, Li M, Wang X, et al. High expression of p300 has an unfavorable impact on survival in resectable esophageal squamous cell carcinoma. The Annals of thoracic surgery. 2011b;91:1531–8. doi: 10.1016/j.athoracsur.2010.12.012. [DOI] [PubMed] [Google Scholar]
- Lim JY, Oh MA, Kim WH, Sohn HY, Park SI. AMP-activated protein kinase inhibits TGF-beta-induced fibrogenic responses of hepatic stellate cells by targeting transcriptional coactivator p300. Journal of cellular physiology. 2012;227:1081–9. doi: 10.1002/jcp.22824. [DOI] [PubMed] [Google Scholar]
- Liu T, Dhanasekaran SM, Jin H, Hu B, Tomlins SA, Chinnaiyan AM, et al. FIZZ1 stimulation of myofibroblast differentiation. The American journal of pathology. 2004;164:1315–26. doi: 10.1016/S0002-9440(10)63218-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrij F, Giles RH, Dauwerse HG, Saris JJ, Hennekam RC, Masuno M, et al. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature. 1995;376:348–51. doi: 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]
- Roelfsema JH, White SJ, Ariyurek Y, Bartholdi D, Niedrist D, Papadia F, et al. Genetic heterogeneity in Rubinstein-Taybi syndrome: mutations in both the CBP and EP300 genes cause disease. American journal of human genetics. 2005;76:572–80. doi: 10.1086/429130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha RN, Pahan K. HATs and HDACs in neurodegeneration: a tale of disconcerted acetylation homeostasis. Cell death and differentiation. 2006;13:539–50. doi: 10.1038/sj.cdd.4401769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santer FR, Hoschele PP, Oh SJ, Erb HH, Bouchal J, Cavarretta IT, et al. Inhibition of the acetyltransferases p300 and CBP reveals a targetable function for p300 in the survival and invasion pathways of prostate cancer cell lines. Molecular cancer therapeutics. 2011;10:1644–55. doi: 10.1158/1535-7163.MCT-11-0182. [DOI] [PubMed] [Google Scholar]
- Varga J. Scleroderma and Smads: dysfunctional Smad family dynamics culminating in fibrosis. Arthritis and rheumatism. 2002;46:1703–13. doi: 10.1002/art.10413. [DOI] [PubMed] [Google Scholar]
- Vo N, Goodman RH. CREB-binding protein and p300 in transcriptional regulation. The Journal of biological chemistry. 2001;276:13505–8. doi: 10.1074/jbc.R000025200. [DOI] [PubMed] [Google Scholar]
- Wu M, Melichian DS, Chang E, Warner-Blankenship M, Ghosh AK, Varga J. Rosiglitazone abrogates bleomycin-induced scleroderma and blocks profibrotic responses through peroxisome proliferator-activated receptor-gamma. The American journal of pathology. 2009;174:519–33. doi: 10.2353/ajpath.2009.080574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokomizo C, Yamaguchi K, Itoh Y, Nishimura T, Umemura A, Minami M, et al. High expression of p300 in HCC predicts shortened overall survival in association with enhanced epithelial mesenchymal transition of HCC cells. Cancer letters. 2011;310:140–7. doi: 10.1016/j.canlet.2011.06.030. [DOI] [PubMed] [Google Scholar]
- Yu J, de Belle I, Liang H, Adamson ED. Coactivating factors p300 and CBP are transcriptionally crossregulated by Egr1 in prostate cells, leading to divergent responses. Molecular cell. 2004;15:83–94. doi: 10.1016/j.molcel.2004.06.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.