

Abstract
Background
Equine influenza virus (EIV) epizootics affect 2·1 million Mongolian horses approximately every 10 years and critically impact economy and nomadic livelihood of Mongolia.
Objectives
An active surveillance program was established in 2011 to monitor influenza viruses circulating among Mongolian horses.
Methods
Nasal swabs were collected from horses in free‐ranging horse herds in Töv, Khentii, and Dundgovi aimags (provinces) from January to September 2011. Real‐time reversetranscriptase–polymerase chain reaction (rRT‐PCR) was used to determine the presence of influenza A virus. Influenza A‐positive specimens were cultured to amplify virus; viral RNA was extracted, and gene segments were amplified and sequenced by Sanger sequencing.
Results
A total of 745 horses were swabbed; most horses were without clinical signs of illness. In July 2011, reports of influenza‐like illnesses emerged among horses in Mongolia's capital, and subsequently, surveillance efforts were adjusted to swab horses associated with the epizootic. Thirty‐four specimens of rRT‐PCR influenza‐positive virus were collected in May, June, August, and September. Three specimens yielded detectable virus. Gene sequence studies suggested that all three isolates were identical H3N8 viruses. Phylogenetic analyses indicated the strain was very similar to other H3N8 EIVs circulating in central Asia between 2007 and 2008.
Conclusions
As large Mongolian equine herds often seem to suffer from EIV epizootics, it seems prudent to continue such routine equine influenza surveillance. Doing so will provide an early warning system, should novel viruses emerge, help in assessing if EIV is crossing over to infect humans and provide data to assess the likely effectiveness of current EIV vaccines.
Keywords: Equine, infectious disease outbreaks, influenza virus, Mongolia, sentinel surveillance
Introduction
Equine influenza virus (EIV) is a highly contagious respiratory disease that infects all equine breeds and odd‐toed ungulates. After an incubation period of 4–10 days, symptoms of running nose, fever, and cough persist for approximately 3–7 days. EIV is known to infect humans1 and dogs.2 A variant EIV H3N8 virus has recently caused widespread epizootics among dogs in the United States.3, 4 EIV strains may have contributed to previous epidemics in humans5, 6 and have potential to play a role in the generation of pandemic viruses.7, 8, 9 In developed countries, the use of EIV vaccine has greatly reduced EIV morbidity. However, EIV vaccine is not often available in developing countries, and among equine species of developing countries, EIV outbreaks and economic impact can be severe.
In recent history, four large EIV enzootics have been documented in Mongolia during the periods 1974–1975, 1983–1984, 1993–1994, and 2007–2008 (Table 1). Estimated attack rates have ranged from 22% to 42%, affecting an estimated total of 2 650 000 horses and causing an estimated 560 000 total deaths. Among the sick horses, death rates ranged from 20% to 30% before vaccines were employed. Fortunately, during the 2007–2008 epizootic, Mongolian government was able to employ 20 000 doses of EIV vaccine and reduced the death rate to 5%. From Chinese and Japanese laboratory studies, the 1974–1975 and 1983–1984 epizootics were likely caused by strains similar to A/equine/Prague/56 (H7N7) (equine‐1) virus10, while the 1993–1994 and 2007–2008 epizootics were due to H3N8 EIV strains.10, 11
Table 1.
Recent equine influenza epizootics in Mongolia. Data are derived from the records compiled from the Department of Veterinary and Animal Breeding, Government of Mongolia
| Years | Virus | Estimated number of infected horses in millions (%) | Estimated number of deaths among infected horses (%) |
|---|---|---|---|
| 1974–1975 | H7N71 | 0.54 (30) of 1·8 | 135 000 (25) |
| 1983–1984 | H7N7 | 0.76 (40) of 1·9 | 225 000 (30) |
| 1993–1994 | H3N8 | 0.891 (42) of 2·1 | 176 000 (20) |
| 2007–2008a | H3N811 | 0.459 (22) of 2·1 | 24 600 (5) |
Equine influenza vaccine used.
In collaboration between Mongolia's Institute of Veterinary Medicine (IVM), the University of Florida (UF), and St. Jude Children's Research Hospital, active EIV surveillance efforts were established in three Mongolian aimags (provinces) in January 2011. Unexpectedly, in July of 2011, Mongolian veterinary officials began to receive reports of Influenza‐like illness (ILIs) among horses near the Mongolian capital, Ulaanbaatar. Within a few weeks of these reports, horse racing enthusiasts and their horses from across Mongolia converged upon Ulaanbaatar to take part in annual horse races as part of the annual national celebration of Nadaam. As owners and their horses returned to their homes, the virus spread to all 21 aimags in Mongolia. The epizootic seemed to subside in September 2011. In total, 74 608 illnesses and 40 deaths were reported among 2·1 million Mongolian horses. In the work presented here, researchers sought to isolate and partially sequence the genome of the virus associated with this most recent epizootic.
Methods
Site selection
Sample collection for the active surveillance study focused on the three aimags in Mongolia with the highest density of horses, Töv, Khentii, and Dundgovi (Figure 1), as recorded by the annual 2011 livestock census produced by the National Statistical Office of Mongolia. In 2011, the number of horses and the ratio of horses to people were 199 837 and 15:1 (Töv); 143 682 and 4·8:1 (Khentii); and 13 664 and 3·8:1 (Dundgovi), respectively. Sums (villages) within the three aimags were selected based on the highest infection rates during the previous 2007–2008 EIV epizootic (one sum in Khentii, six in Töv, and seven in Dundgovi). Horse owners were engaged in these sums and invited to participate in the surveillance program. The surveillance goal was to swab 50 horses in each aimag during each month. A total of 30 herds were followed, including 7 in Töv, 1 in Khentii, and 22 in Dundgovi. Nasal swab would first be collected from horses with signs of influenza‐like illness, and then samples of the remaining otherwise healthy horses were swabbed to reach target sample numbers.
Figure 1.
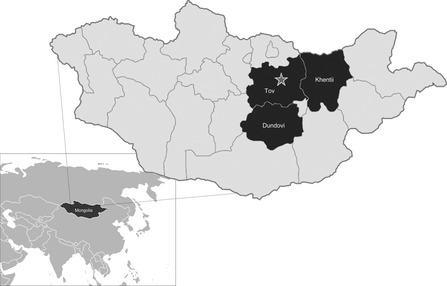
Map of Mongolia indicating the 3 aimags from which horses were sampled. The star is positioned at the location of Mongolia's capital, Ulaanbaatar.
Enrollment
This study was approved by the Department of Veterinary & Animal Breeding, Government of Mongolia. Each month (when roads and weather permitted) researchers from the IVM traveled to each individual horse owner's home. The horse owners gathered the free‐ranging horses and assisted the researchers in collecting the nasal swab specimens normally through use of bridles, ropes, and hand restraining measures. Two nasal swabs were collected from each horse. In large herds, no attempt was made to serially sample specific horses; instead, a number of horses to swab were used as a goal for these specific large herds.
Laboratory methods
Upon collection, each nasal swab was placed into a 2‐ml screwcap test tube containing 1 ml of viral transport medium. Samples were transported to IVM in an insulated cooler with ice packs to maintain a cold chain between 0 and 4°C. At IVM, long‐term storage samples were preserved at −80°C. One swab from each horse was shipped to the UF on dry ice for influenza molecular detection, culture, and sequence studies.
At UF, swabs were screened for generic influenza A using molecular techniques. From a 50‐μl aliquot of swab medium, RNA was extracted using the MagMAX™ viral RNA extraction kit (Ambion, Foster City, CA, USA). Real‐time reverse transcriptase–polymerase chain reaction (rRT‐PCR) was then performed with the iScript™ one‐step rRT‐PCR kit (Bio‐Rad, Hercules, CA, USA) using CDC influenza A primers and probe. Swab specimens (run in duplicate) with Ct values <35 were considered positive; Ct values ≥35 to <40 were classified suspected positives; and specimens Ct values ≥40 were considered negative. Swab samples positive and suspected positive for influenza A were cultured in fertilized eggs or Madin–Darby canine kidney cells and passaged twice to amplify the virus.
RNA from cultured swab isolates was then extracted using the MagMAX™ viral RNA extraction kit, and cDNA was produced using the influenza A universal dodecamer (U12mer) and SuperScript® III Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA) as described.12 PCR was performed with the cDNA using the Platinum® Taq DNA polymerase (Invitrogen) and gene segment‐specific primers for the influenza hemagglutinin (HA), neuraminidase (NA), and matrix (M) genes as described.12 Gene segment‐specific products were isolated with agarose gel electrophoresis and extracted using the QIAquick gel extraction kit (Qiagen, Germantown, MD, USA). Sanger sequencing of HA, NA, and M genes was performed at University of Florida's Interdisciplinary Center for Biotechnology Research using ABI 3130 DNA sequencers.
General BLASTn searches (http://blast.ncbi.nlm.nih.gov/Blast.cgi) of the full‐length HA gene sequences were performed. Representative EIV isolates identified by these analyses were combined with taxa from recent comprehensive phylogenetic analyses to build the final data set.13, 14, 15, 16 Sequence alignments were performed using mafft 5.817 followed by minor manual adjustments in ClustalW.18 The E‐INS‐I alignment strategy was used with the following parameters: scoring matrix (BLOSUM62), gap open penalty (1·53), and offset value (0). The final sequence was trimmed to the first conserved nucleic acid at the 5′ and 3′ end of the HA sequences due to incomplete data available for certain important isolates. The aligned data set was imported into jmodeltest version 0.1.119, and the Akaike information criterion (AIC) was used to select a best‐fit model of evolution for phylogenetic analysis. Phylogenetic trees were constructed using mrbayes v. 3.1.2.20 The Markov chain was run for a maximum of 10 million generations, with a stopping rule implemented so that the analysis would halt when the average deviation of the split frequencies was <0·01. Four independent analyses were conducted, each with 1 cold and 3 heated chains with the default heating parameter (temperature = 0·2). Every 1000 generations were sampled, and the first 25% of MCMC samples discarded as burn‐in.
In addition, for one isolate, all 8 gene segments were amplified and subjected to third‐generation SMRT® DNA sequencing using the PacBio RS system (Pacific Biosciences, Menlo Park, CA, USA) at the University of Florida's Interdisciplinary Center for Biotechnology Research.
Statistical methods
Age of the animal's samples was grouped into three categories (1–6, 7–12, and 13–18 years). Risk factor associations for aimag, age group, and sex with influenza‐positive molecular screening results were studied using binary logistic regression. Final multivariate models were designed using manual backwards elimination and included covariates with P values <0·05. Analyses were performed using sas v9.2 (SAS Institute Inc., Cary, NC, USA).
Results
A total of 745 samples were collected from the three aimags between January and September 2011, with an average of 8 herds tested per month (Table 2). Swab specimens positive for influenza were found in the months of May (13 horses), June (1 horse), August (10 horses), and September (10 horses).
Table 2.
Number of horse swabs with molecular evidence of influenza A virus, and total number of horses from which nasal swabs were collected, by month and aimag, Mongolia, 2011
| Töv | Dundgovi | Khentii | ||||
|---|---|---|---|---|---|---|
| Positive | Total | Positive | Total | Positive | Total | |
| January | 0 | 30 | 0 | 20 | 0 | 0 |
| February | 0 | 30 | 0 | 50 | 0 | 0 |
| March | 0 | 40 | 0 | 25 | 0 | 0 |
| April | 0 | 50 | 0 | 0 | 0 | 0 |
| May | 8 | 50 | 0 | 0 | 5 | 50 |
| June | 0 | 50 | 1 | 50 | 0 | 50 |
| July | 0 | 50 | 0 | 50 | 0 | 0 |
| August | 0 | 50 | 0 | 0 | 10 | 50 |
| September | 0 | 0 | 10 | 50 | 0 | 0 |
| Total | 8 | 350 | 11 | 245 | 15 | 150 |
Generic influenza A qRT‐PCR
Considering all samples collected, 34 (4·6%) of the horse swabs were positive for generic influenza A virus (Table 2). A total of 350 paired horse swab specimens were collected in Töv between January and August 2011. All Töv samples tested were influenza negative except for 8 (2%) samples collected in the month of May. For Khentii, 50 total paired horse swab specimens were collected each month in May, June, and August. Fifteen (10%) of the 150 Khentii samples tested were influenza positive. A total of 245 paired horse nasal swabs were collected in Dundgovi from January to September and saved for the months of April and August. One (2%) of the 50 June samples and 10 (20%) of the 50 September samples screened were positive for influenza A.
The number of positive samples in Khentii was significantly greater compared with Dundgovi (OR = 2·4; 95% CI, 1·1–5·3) and Töv (OR = 4·6; 95% CI, 1·9–11·1), but there was no significant difference between those in Dundgovi and Töv (Table 3). Fifteen (3·9%) of 380 male horses and 19 (5·2%) of 361 female horses screened were influenza A‐positive; however, this difference was not statistically significant. The age of the horses ranged from 1 to 18 years, with a median of 7 years. There was no significant difference between the proportions of horses aged from 1 to 6 years, which tested positive (4·0%), compared with horses aged from 7 to 12 years (3·9%) (P = 0·9). However, horses aged from 13 to 18 years were significantly tested more likely to be influenza A‐positive compared with the horses aged from 1 to 6 years (OR = 3·3; 95% CI, 1·2–8·9). In an adjusted model containing both aimag and age group, horses from Khentii were found more likely to be rRT‐PCR positive compared with horses from Töv (adjusted OR = 4·6; 95% CI, 1·9–11·1). Similarly, the oldest horses were found to have a higher likelihood to screen positive for influenza compared with youngest horses (adjusted OR = 3·3; 95% CI, 1·2–8·9) (Table 1).
Table 3.
Risk factors for positive influenza A by rRT‐PCR among central Mongolian horses, Khentii, Dundgovi, and Töv provinces, Mongolia, 2011
| Variable | N | n a (%) | Unadjusted OR (95% CI) | Adjusted OR (95% CI) |
|---|---|---|---|---|
| Age(years) | ||||
| 13–18 | 65 | 7 (20·6) | 3·5 (1·3–9·4)b | 3·3 (1·2–8·9)b |
| 7–12 | 350 | 16 (47·1) | 1·4 (0·6–3·0) | 1·3 (0·6–3·0) |
| 1–6 | 330 | 11 (32·4) | Reference | Reference |
| Sex | ||||
| Female | 365 | 19 (55·9) | 1·3 (0·7–2·7) | – |
| Male | 380 | 15 (44·1) | Reference | – |
| Aimag (province) | ||||
| Khentii | 150 | 15 (44·1) | 4·8 (2·0–11·5)b | 4·6 (1·9–11·1)b |
| Dundgovi | 245 | 11 (32·4) | 2·0 (0·8–5·1) | 2·1 (0·8–5·2) |
| Töv | 350 | 8 (23·5) | Reference | Reference |
Represents influenza A virus positive samples as determined by rRT‐PCR.
Represents statistically significant results (P < 0·05).
Viral culturing and sequencing studies
Of the 34 rRT‐PCR‐positive swabs, three specimens successfully yielded virus after cultured on embryonated chicken eggs. By sequence studies, all were H3N8 EIVs: A/equine/Mongolia/3/2011 (Töv aimag/May), A/equine/Mongolia/20/2011 (Töv aimag/May), and A/equine/Mongolia/56/2011 (Khentii aimag/May).
Sequence data were acquired representing the HA gene segments for isolates 3, 20, and 56 (GenBank accession numbers JX549062; JX549063 and JX549064, respectively), the M gene segment for isolates 3 and 56 (GenBank accession numbers JX549060 and JX549064, respectively), and NA gene segment for isolate 3 (GenBank accession number JX549061).
Attempts to obtain full genome sequencing of A/equine/Mongolia/3/2011(H3N8) using third‐generation sequencing technology (PacBio RS, Menlo Park, CA, USA) provided full‐length reads for all eight genome segments; however, issues with base call errors and continuity produced sequences that had only 85–90% identity to that obtained by Sanger sequencing, thus highlighting the issues of this particular technology for full viral genome sequence at the time of this study (data not shown).
BLASTn, molecular data set, and phylogenetic analysis
A BLASTn search of the viral HA sequences obtained for the three viruses isolated from this work revealed the highest sequence identity with various H3N8 equine influenza virus isolates circulating in central Asia during 2007 and 2008. In addition, BLASTn searches queried on the M gene and partial NA gene sequences obtained from the viruses isolated in this study also indicated the Mongolian strains were very similar to isolates circulating in central Asia during 2007–2008. Given the similarity in BLASTn search identities among the sequence data for all three gene segments, only the data from the HA gene segment were used in further phylogenetic studies. The final aligned MCP data set, after truncation of the 5′ and 3′ end of the HA sequence to match available sequence coverage among representative EIV sequences available in GenBank, contained 1009 nucleic acid characters encompassing the HA1 coding region (including gaps) for 30 unique taxa inclusive of the three isolates from this work. jModeltest identified the HKY+I model to be the most suitable model for phylogenetic analyses. The results of our phylogenetic analysis of the partial HA genes are in good agreement with recent phylogenetic analyses supporting the recognition of five major H3N8 EIV clades13, 14, 15, 16 (Figure 2). The Bayesian analysis demonstrated with a high level of confidence that the three 2011 H3N8 Mongolian EIV isolates are most closely related to other H3N8 EIV isolates belonging to the Florida clade 2.
Figure 2.
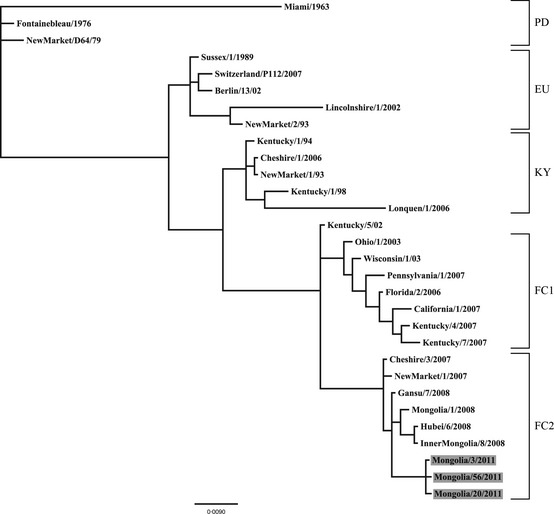
Phylogram depicting the relationship of H 3 N 8 equine influenza virus isolates, including those recovered from the large M ongolian epizootic in 2011 (highlighted in gray). Tree based on the partial hemagglutinin gene sequences (1009 nucleic acid characters of hemagglutinin1 including gaps). All nodes were supported by a posterior probability of >90. Branch lengths are based on the number of inferred substitutions, as indicated by the scale. Major equine influenza clades represented in the phylogram are labeled as follows: PD , pre‐divergent; EU , E uropean; KY , K entucky; FC 1, F lorida clade 1; FC 2, F lorida clade 2.
Discussion
This article describes the results of an EIV surveillance program conducted in Mongolia during 2011. During these surveillance efforts, an outbreak of ILI occurred among horses near the Mongolia capital of Ulaanbaatar in July 2011, with subsequent virus dispersal across the country facilitated by a national horse racing event. Of the 745 paired horse swabs collected in three Mongolia aimags, 34 specimens had evidence of influenza A infection. EIV was isolated from three of those specimens; all three specimens were collected in close temporal proximity to the high incidence of horse ILIs. Sequence analyses of the HA, NA, and M gene segments revealed a similar origin, which suggests that no significant genomic reassortments had occurred between the EIVs circulating among horses in Mongolia.
Our risk analysis identified one unexpected finding, in that horses aged 13–18 years were tested more likely to be influenza A virus positive than horses aged 1–6 years. In areas where EIV is enzootic, older horses are likely at less risk of EIV infection due to acquired immunity from previous exposures. While our systematic sampling approach attempted to collect swabs from a proportionate representation of horses, perhaps sampling bias did exist, as often times when younger horses prized for racing displayed signs of illness, herders did not allow field staff to sample them.
This study faced a key limitation. Transportation of specimens from the rural enrollment sites to IVM and then to the UF was thoughtfully planned and carefully executed; however, factors outside the control of study staff may have led to the degradation of swab samples during transport. While this might have prevented the opportunity to culture and isolate virus, molecular identification of EIV was still possible. Breaks in the cold chain likely explain why the 34 rRT‐PCR‐positive specimens yielded only three viable virus isolates. Efforts to better ensure proper storage and transportation have since been implemented for additional analyses.
In 2011, Mongolia was estimated to have a population of 2·1 million horses, with central Mongolia having the most dense horse populations. Horses are owned by semi‐nomadic family groups and come into frequent contact with horses owned by other family groups. The contact between wide traveling groups of horses provides an ideal setting for EIV transmission. Researchers at the IVM have developed an inactivated trivalent influenza vaccine (using equine H1N1, H3N8, and H7N7 strains). Since 1988, each year approximately 200 000 doses are produced, with only 10% of Mongolian horses vaccinated annually in 7 of Mongolia's 21 aimags. Because Mongolians rely so heavily on their horses for everyday life, the potential for EIV zoonotic infection is high. Monitoring the viruses circulating among the horses provides better insight into its zoonotic capabilities as well as provides novel information needed to keep the animal vaccines current.
While further genomic analyses are in progress, partial genome sequence of the viruses associated with the 2011 Mongolian EIV epizootic appears to be highly similar to a H3N8 EIV that widely circulated in Central Asia and Europe in 2007–2008 and is related to other viruses in Florida clade 2. The World Organization for Animal Health (OIE) has since classified this outbreak as a reoccurrence of virus responsible for the 2008 EIV outbreak in Mongolia. An attempt was made to use third‐generation sequencing to obtain a full flu A genome sequence; however, technical difficulties made interpretation difficult. Future research is warranted to improve this technology and utility for such genomic sequencing efforts.
Acknowledgements
This research was supported by multiple grants: Contract HHSN266200700005C (Dr Richard Webby) from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, USA; R01 AI068803‐ARRA supplement (Dr Gray) from the National Institute of Allergy and Infectious Diseases; and multiple grants (Dr Gray) from the US Department of Defense Armed Forces Health Surveillance Center's Global Emerging Infections Surveillance and Response Program.
Yondon et al (2013) Isolation and characterization of H3N8 equine influenza a virus associated with the 2011 epizootic in Mongolia. Influenza and Other Respiratory Viruses 7(5), 659–665
References
- 1. Alford RH, Kasel JA, Lehrich JR, Knight V. Human responses to experimental infection with influenza A/Equi 2 virus. Am J Epidemiol 1967; 86:185–192. [DOI] [PubMed] [Google Scholar]
- 2. Crispe E, Finlaison DS, Hurt AC, Kirkland PD. Infection of dogs with equine influenza virus: evidence for transmission from horses during the Australian outbreak. Aust Vet J 2011; 89(Suppl. 1):27–28. [DOI] [PubMed] [Google Scholar]
- 3. Crawford PC, Dubovi EJ, Castleman WL et al Transmission of equine influenza virus to dogs. Science [Research Support, Non‐U.S. Gov't]. 2005; 310:482–485. [DOI] [PubMed] [Google Scholar]
- 4. Anderson TC, Bromfield CR, Crawford PC, Dodds WJ, Gibbs EP, Hernandez JA. Serological evidence of H3N8 canine influenza‐like virus circulation in USA dogs prior to 2004. Vet J 2012; 191:312–316. [DOI] [PubMed] [Google Scholar]
- 5. Masurel N, Mulder J. Studies on the content of antibodies for equine influenza viruses in human sera. Bull World Health Organ 1966; 34:885–893. [PMC free article] [PubMed] [Google Scholar]
- 6. Minuse E, McQueen JL, Davenport FM, Francis T Jr. Studies of Antibodies to 1956 and 1963 Equine Influenza Viruses in Horses and Man. J Immunol 1965; 94:563–566. [PubMed] [Google Scholar]
- 7. Gibbs EP, Anderson TC. Equine and canine influenza: a review of current events. Anim Health Res Rev 2010; 11:43–51. [DOI] [PubMed] [Google Scholar]
- 8. Laver WG, Webster RG. Studies on the origin of pandemic influenza. 3. Evidence implicating duck and equine influenza viruses as possible progenitors of the Hong Kong strain of human influenza. Virology 1973; 51:383–391. [DOI] [PubMed] [Google Scholar]
- 9. Morens DM, Taubenberger JK. Historical thoughts on influenza viral ecosystems, or behold a pale horse, dead dogs, failing fowl, and sick swine. Influenza Other Respir Viruses [Historical Article Research Support, N.I.H., Extramural Review] 2010; 4:327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guo Y, Wang M, Zheng GS, Li WK, Kawaoka Y, Webster RG. Seroepidemiological and molecular evidence for the presence of two H3N8 equine influenza viruses in China in 1993–94. J Gen Virol 1995; 76(Pt 8):2009–2014. [DOI] [PubMed] [Google Scholar]
- 11. Motoshima M, Okamatsu M, Asakura S et al Antigenic and genetic analysis of H3N8 influenza viruses isolated from horses in Japan and Mongolia, and imported from Canada and Belgium during 2007‐2010. Arch Virol 2011; 156:1379–1385. [DOI] [PubMed] [Google Scholar]
- 12. Hoffmann E, Stech J, Guan Y, Webster RG, Perez DR. Universal primer set for the full‐length amplification of all influenza A viruses. Arch Virol 2001; 146:2275–2289. [DOI] [PubMed] [Google Scholar]
- 13. Bryant NA, Rash AS, Russell CA et al Antigenic and genetic variations in European and North American equine influenza virus strains (H3N8) isolated from 2006 to 2007. Vet Microbiol 2009; 138:41–52. [DOI] [PubMed] [Google Scholar]
- 14. Daly JM, Lai ACK, Binns MM, Chambers TM, Barrandeguy M, Mumford JA. Antigenic and genetic evolution of equine H3N8 influenza A viruses. J Gen Virol 1996; 77:661–671. [DOI] [PubMed] [Google Scholar]
- 15. Daly JM, MacRae S, Newton JR, Wattrang E, Elton DM. Equine influenza: a review of an unpredictable virus. Vet J 2011; 189:7–14. [DOI] [PubMed] [Google Scholar]
- 16. Murcia PR, Wood JLN, Holmes EC. Genome‐Scale Evolution and Phylodynamics of Equine H3N8 Influenza A Virus. J Virol 2011; 85:5312–5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Katoh K, Kuma K, Toh H, Miyata T. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res 2005; 33:511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thompson JD, Higgins DG, Gibson TJ. CLUSTAL‐W ‐ improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position‐specific gap penalties and weight matrix choice. Nucleic Acids Res 1994; 22:4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Posada D. jModelTest: phylogenetic model averaging. Mol Biol Evol 2008; 25:1253–1256. [DOI] [PubMed] [Google Scholar]
- 20. Huelsenbeck JP, Ronquist F. MRBAYES: bayesian inference of phylogenetic trees. Bioinformatics 2001; 17:754–755. [DOI] [PubMed] [Google Scholar]