

Abstract
An exciting approach to tumor delivery is encapsulation of the drug in self-assembled polymer-peptide nanoparticles. The objective of this work was to synthesize a conjugate of low molecular weight polylactide (LMW PLA) and V6K2 peptide, and investigate self-assembly, drug release kinetics, cell uptake and toxicity, drug pharmacokinetics, and tumor cell invasion with Doxorubicin (DOX) or paclitaxel (PTX). The results for PLA-V6K2 self-assembled NPs were compared with those of polyethylene glycol stabilized PLA (PLA-EG) NPs. The size of PLA-V6K2 and PLA-EG NPs were 100±20 and 130±50 nm, respectively, with polydispersity index of 1.04 and 1.14. The encapsulation efficiency of DOX in PLA-V6K2 and PLA-EG NPs was 44±9% and 55±5%, respectively, and that of PTX was >90 for both NP types. The release of DOX and PTX from PLA-V6K2 was slower than that of PLA-EG and the release rate was relatively constant with time. Based on molecular dynamic simulation, the less hydrophobic DOX was distributed in the lactide core as well as the peptide shell while the hydrophobic PTX was localized mainly to the lactide core. PLA-V6K2 NPs had significantly higher cell uptake by 4T1 mouse breast carcinoma cells compared to PLA-EG NPs, which was attributed to the electrostatic interactions between the peptide and negatively charged moieties on the cell membrane. PLA-V6K2 NPs showed no toxicity to marrow stromal cells. DOX loaded PLA-V6K2 NPs showed higher toxicity to 4T1 cells and the DNA damage response and apoptosis was delayed compared to the free DOX. DOX or PTX encapsulated in PLA-V6K2 NPs significantly reduced invasion of 4T1 cells compared to those cells treated with the drug in PLA-EG NPs. Invasion of 4T1 cells treated with DOX in PLA-V6K2 and PLA-EG NPs was 5±1% and 30±5%, respectively, and that of PTX was 11±2% and 40±7%. The AUC of DOX in PLA-V6K2 NPs was 67% and 21% higher than those of free DOX and PLA-EG NPs, respectively. DOX loaded PLA-V6K2 NPs injected in C3HeB/FeJ mice inoculated with MTCL syngeneic breast cancer cells displayed higher tumor toxicity than PLA-EG NPs and lower host toxicity than the free DOX. Cationic PLA-V6K2 NPs with higher tumor toxicity than the PLA-EG NPs are potentially useful in chemotherapy.
Keywords: Self-assembling peptide, polymer conjugation, hybrid nanoparticle, cell uptake, drug pharmacokinetics, tumor toxicity
1. Introduction
Natural and synthetic nanoparticles (NPs) are widely used in drug delivery, imaging, detection of pathogens, and separation of biomolecules because they are very effective in overcoming biological barriers with their small size and large surface area [1]. For example, NPs that can overcome the short half-life and low solubility of chemotherapeutic drugs in physiological medium and selectively pass through the leaky tumor vasculature are very useful in cancer therapy [2, 3]. Furthermore, NPs grafted with multifunctional ligands can enhance drug targeting by specific interaction with cell surface receptors [4, 5]. Variety of platforms including liposomes [6], lipid-based NPs [7], drug-conjugated micelles [8], and polymer-based NPs [9, 10] are used for tumor delivery. Among these, polymeric NPs provide enormous flexibility in designing drug delivery systems with a wide range of size distribution, encapsulation efficiency, drug loading, circulation time, dissolution rate, and drug release kinetics, and cell uptake [11, 12]. Polylactide (PLA) and copolymers with glycolide are widely used to synthesize biodegradable NPs [13]. Although chemotherapeutic agents are stabilized in the PLA matrix against dissolution by hydrophobic interaction and drug release kinetics is controlled by matrix degradation [3], surface active agents are used to prevent NPs aggregation, reduce the initial burst release, or improve the cell uptake of NPs [14–17]. These active agents include amphiphilic PEG-based copolymers [18, 19], biomolecules such as vitamin E [16], peptides [20, 21], and proteins [22]. These modifiers not only stabilize NPs against aggregation and improve cell uptake, but they also affect the drug safety profile including immunogenicity and systemic toxicity [23].
An exciting approach to tumor delivery is the use of polymer-peptide hybrids that self-assemble to form NPs. PLA macromers provide a stable matrix for encapsulation and sustained release of chemotherapeutic agents while peptides derived from the proteins of intracellular vesicles self-assemble to produce NPs with enhanced cell uptake. In addition, peptide mimics have lower toxicity while maintaining their functionality. For example, the peptide mimic of the trans-activating transcriptional activator (Tat) of HIV-1 penetrates the cell membrane with efficiency comparable to that of Tat peptide but with less toxicity [24]. The VVVVVVKK (V6K2) peptide, derived from the intracellular proteins, self-assemble into vesicles with a narrow size distribution [25, 26] and the positively charged groups of the peptide interact with negatively charged moieties on the cell membrane to enhance NPs penetration and uptake [17, 27]. Matrices produced from low molecular weight (LMW, <10K) PLA macromers display higher drug bioavailability and encapsulation efficiency [16, 19].
The objective of this work was to synthesize hybrid LMW PLA-V6K2 conjugates and investigate self-assembly, drug release kinetics and pharmacokinetics, cell uptake, and tumor toxicity using Doxorubicin (DOX) or paclitaxel (PTX) as the chemotherapy drug. The hypothesis was that the conjugate of LMW PLA and V6K2 self-assembles into NPs with improved cell uptake and tumor toxicity, compared with polyethylene glycol stabilized PLA (PLA-EG) NPs. We demonstrate that the hydrophobic interactions between the lactide groups of LMW PLA as well as specific interactions between the amino acids of V6K2 peptide guide the process of self-assembly to form NPs with the PLA serving as a matrix for encapsulation of DOX or PTX and amino acid residues of the peptide. Dissipative particle dynamics (DPD) method was used to simulate the NPs self-assembly and predict the distribution of drug molecules in the NPs [28, 29].
2. Materials and methods
2.1. Materials
L-lactide (LA, >99.5% purity) monomer was obtained from Ortec (Easley, SC). Calcium hydride, ninhydrin reagent, tetrahydrofuran (THF), trimethylsilane (TMS), poly(ethylene glycol) (PEG, nominal molecular weight 3.4 kDa), triethylamine (TEA), tin (II) 2-ethylhexanoate (TOC), dimethylsulfoxide (DMSO), piperidine and Doxorubicin hydrochloride (DOX) were purchased from Sigma-Aldrich (St. Louis, MO). Paclitaxel was obtained from LC Laboratories (Woburn, MA). PEG was dried by azeotropic distillation from toluene. Acryloyl chloride (AC) and fumaryl chloride (FC) were obtained from Sigma-Aldrich and redistilled prior to use. Diethylene glycol (DEG) initiator (>99% purity) was purchased from Fisher (Pittsburgh, PA). The protected amino acids and Rink Amide NovaGel resin were purchased from EMD Biosciences (San Diego, CA). N,N-Dimethylformamide (DMF), methylene chloride (DCM), N,N-diisopropylcarbodiimide (DIC), triisopropylsilane (TIPS), N,N-dimethylaminopyridine (DMAP), hydroxybenzotriazole (HOBt) and trifluoroacetic acid (TFA) were received from Acros Organics (Pittsburgh, PA). Diethyl ether and hexane were obtained from VWR (Bristol, CT). DCM was dried by distillation over calcium hydride. Spectro/Por dialysis tube (molecular weight cutoff 3.5 kDa) was purchased from Spectrum Laboratories (Rancho Dominquez, CA).
2.2. Macromer synthesis
LMW PLA was synthesized by ring opening polymerization of L-lactide (LA) monomer with DEG as the initiator and TOC as the polymerization catalyst as described [30]. In a typical reaction, 90 g LA monomer dried under vacuum at 40°C for 12 h was placed in a three-neck reaction flask equipped with an overhead stirrer under a stream of nitrogen. Under constant stirring, temperature was gradually increased to 130°C in an oil bath to melt the monomer. After melting, 5.0 mL DEG and 5.5 mL TOC were added to the flask with stirring and the reaction continued for 12 h. After the reaction, unreacted monomers and initiators were removed under vacuum (<1 mm Hg) at 130°C for at least 6 h. The product was dissolved in DCM, precipitated in ice cold ether to remove the high molecular weight PLA fraction. The ether was removed by rotary evaporation and the polymer was re-dissolved in DCM and precipitated twice in hexane. The fractionated LMW PLA was dried under vacuum (<5 mmHg and 40°C) to remove any residual solvent and stored at −20°C. Next, acrylate-terminated PLA (Ac-PLA) was synthesized by condensation polymerization of PLA with acryloyl chloride as described [31]. Similarly, PLA-EG was synthesized by the reaction of LMW PLA and PEG with FC as the linker [32, 33]. In a typical reaction, 2 g PLA and 18 g PEG, dried by azeotropic distillation with toluene, were dissolved in 150 mL DCM under dry nitrogen atmosphere. After cooling to 5°C, 0.6 mL FC and 1.55 mL TEA each dissolved in DCM were added dropwise to the reaction with stirring. The reaction continued for 6 h on ice followed by 12 h under ambient conditions. After the reaction, solvent was removed and residue was dissolved in 100 mL anhydrous ethyl acetate to precipitate and remove the by-product triethylamine hydrochloride. The product was dissolved in DCM, precipitated twice in hexane and cold ether, and dried in vacuum.
2.3. Peptide synthesis and conjugation
The amino acid sequence V6K2 and its mutant sequence (V2K)2V2 were synthesized on Rink Amide NovaGel resin (0.62 mmol active sites/g) using solid phase synthesis as described [30, 34]. The Fmoc-protected amino acid derivative (1 equiv) and HOBt (2 equiv) were dissolved in dry DMF (3 mL) and DIC (1.1 equiv) was added to the mixture. Next, 0.2 mL of 0.05 M DMAP was added and the mixture was shaken for 4–6 h at 30°C in an orbital shaker. The coupling reaction was repeated until a negative result was obtained for the presence of unreacted amines using the Kaiser reagent [35]. Then, the resin was treated with 20% piperidine in DMF for Fmoc deprotection. The subsequent amino acids were coupled using the same procedure. Next, a cysteine amino acid was coupled to the valine end of the peptide for conjugation to LMW PLA by Michael addition reaction. After coupling and deprotecting the last amino acid, the resin was treated with 95% TFA/ 2.5% TIPS/ 2.5% water for 2 h to cleave the peptide from the resin. The peptide was precipitated in cold ether and dried. The dried product was purified by HPLC and characterized by mass spectrometry.
The peptide was conjugated to the macromer by Michael addition between the cysteine’s sulfhydryl group on the peptide and the acrylate group on the polymer to produce the CV6K2 peptide-conjugated macromer. In brief, the peptide was dissolved in sodium borate buffer (pH 8.5) and added to PLAA solution in DMF (2:1 peptide:macromer molar ratio). The mixture was placed in an orbital shaker at 30°C for 12 h. The solution was then dialyzed against DI water and lyophilized to obtain the dry conjugated product. The peptide-conjugated macromer is henceforth referred to as PLA-V6K2. A similar procedure was used to graft the mutant C(V2K)2V2 peptide to PLAA macromer.
2.4. Characterization of the macromer, peptide, and conjugate
The chemical structure of the synthesized macromers was characterized by a Varian Mercury-300 1H NMR (Varian, Palo Alto, CA) at ambient conditions. The sample was dissolved in deuterated DMF at a concentration of 5 mg/mL. The synthesized peptide was purified by HPLC on a 250×10 mm, 10 μm Xterra® Prep RP18 column (Waters, Milford, MA) with a flow rate of 2 mL/min using a gradient 5–95% MeCN in 0.1% aqueous TFA at the detection wavelength of 214 nm as described [33]. Mass spectrometric studies were performed with a Fannigan 4500 electrospray ionization mass spectrometer (ESI-MS; Thermo Electron, Waltham, MA) as described [33]. The molecular weight distribution of the synthesized macromers was characterized by gel-permeation chromatography (GPC). Measurements were carried out with a Waters 717 Plus Autosampler GPC system (Waters, Milford, MA) connected to a model 616 HPLC pump, model 600S controller, and model 410 refractive index detector. The columns consisted of a styragel HT guard column (7.8 mm × 300 mm, Waters) in series with a styragel HR 4E column (7.8 mm × 300 mm, Waters) heated to 37°C in a column heater. The Empower software was used for data analysis and determination of number ( ) and weight ( ) average molecular weights and polydispersity index (PI). The sample (20 μL) with a concentration of 10 mg/mL in THF was eluted with degassed THF at a flow rate of 1 mL/min. Monodisperse polystyrene standards (Waters) with PI<1.1 were used to construct the calibration curve.
2.5. Macromer self-assembly and NPs characterization
The polymer-peptide conjugate, dissolved in DMF:DMSO (1:4) solvent mixture, was self-assembled into NPs by dialysis against PBS. Briefly, 50 mg macromer in 10 mL DMF:DMSO mixture was loaded in a dialysis tube and dialyzed against PBS. The solution was dialyzed for 24 h with a change of buffer every 4 h. A similar procedure was used to produce the control PLA-EG NPs. The size distribution of NPs was measured by light scattering with a NICOMP Submicron Particle Sizer (Autodilute Model 370, NICOMP Particle Sizing Systems, Santa Barbara, CA). 500 μL of the diluted suspension was placed in the instrument cell holder and the scattered light was measured. The scattered intensity was inverted to size distribution by inverse Laplace transform using the CW370 software (NICOMP Particle Sizing Systems). X-ray photoelectron spectroscopy (XPS) was used to determine the surface chemical composition of the NPs. XPS spectra were collected under ultra-high vacuum (6.7×10−7 Pa) with a resolution of 0.5 eV using a hemispherical analyzer on a Kratos Axis Ultra DLD XPS (Shimadzu) with a monochromatic Al Kα X-ray source. TEM was used to investigate the morphology of the NPs. A drop of the NPs’ suspension was placed on a TEM grid and allowed to dry. The dried NPs were stained with 5% uranyl acetate and observed with TEM at an accelerating voltage of 200 keV.
2.6. Degradation of NPs, drug loading efficiency, and release kinetics
Degradation of the NPs was characterized by measuring their particle size and mass loss with incubation time as described [36]. After self-assembly, the NPs suspension was centrifuged at 15,000 rpm for 10 min, the supernatant was decanted to remove the unassembled macromers, and the NPs were resuspended in PBS. For degradation experiments, 50 mg NPs suspended in 1 mL PBS was incubated at 37°C until complete degradation (no mass remaining or NPs not detectable by light scattering). Next, samples were freeze-dried and mass of the dried powder was measured. The fraction of mass remaining was determined by dividing the dried mass at time t by the initial mass at time zero. At each time pint, NPs mean diameter was measured by light scattering.
DOX and PTX were used for determination of encapsulation efficiency and release kinetics of the NPs. DOX or PTX loaded NPs were prepared by the addition of 6 wt% DOX or PTX based on the macromer weight, to the dialysis solution. For determination of DOX encapsulation efficiency, the suspension was centrifuged at 15,000 rpm for 10 min and the supernatant was removed. DOX concentration in the supernatant was measured with a plate reader (Synergy HT, Bio-Tek, Winooski, VT) at excitation and emission wavelengths of 485 and 570 nm, respectively. The measured intensities were correlated to concentration using a calibration curve constructed with solutions of known concentration in the linear range of the detector. For determination of PTX encapsulation efficiency, 2 mg of the NPs was dissolved in 200 μL of DMSO and 3 mL of acetonitrile–water mixture (50:50 v/v) was added. After 2 h, the suspension was centrifuged at 15,000 rpm for 10 min and the supernatant was transferred to HPLC vials for determination of PTX concentration. The drug concentration was determined by isocratic reverse-phase HPLC (Waters system, Milford, MA) using a 250×10 mm, 10 μm Xterra® Prep RP18 column (Waters) at a flow rate of 2 mL/min using 50:50 v/v acetonitrile/water mixture as the mobile phase. A photodiode array detector (model 996, Waters) was used for detection of PTX at the wavelength of 227 nm. The elution time of PTX was 16 min. The measured intensities were correlated to concentrations using a calibration curve constructed with solutions of known PTX concentration ranging 0.65–65 μg/mL (in the linear range of the detector) [37]. The encapsulated amount of DOX or PTX was divided by the initial amount to determine encapsulation efficiency. For determination of release kinetics, DOX or PTX loaded NPs were placed in 15 mL tubes and incubated with 10 mL PBS (pH 7.4). At each time interval, the suspension was centrifuged at 15,000 rpm for 10 min and the supernatant was collected for analysis. Next, the NPs were resuspended in 10 mL fresh PBS and incubated until the next time interval. At each time point, the amount of released DOX or PTX in the supernatant was measured by the plate reader or HPLC, respectively. The released amount was divided by the initial encapsulated amount to determine the fraction of drug released. To investigate the effect of medium [38], DOX encapsulated PLA-V6K2 NPs were incubated in 100% fetal bovine serum (FBS) and the release kinetics was compared with that incubated in PBS.
2.7. Nanoparticle uptake
For cell uptake experiments, 4T1 murine breast carcinoma cells (Scripps Research Institute, La Jolla, CA) were seeded at a density of 5×104 cells/cm2 in 24-well plates and cultured in RPMI-1640 media containing 1 mM sodium pyruvate, 2 mM L-glutamine, 4500 mg/L glucose, 10 mM HEPES, and 1500 mg/L sodium bicarbonate (ATCC, Manasses, VA) [30, 39]. Next, the media was replaced with RPMI-1640 media containing 2 mg/mL FITC-loaded NPs (2% by weight FITC in NPs; excitation and emission wavelength of 485 and 520 nm, respectively) and incubated. At each time point, the culture media was removed; cells were washed three times with PBS to remove the unattached NPs and divided into two groups. Cells in the first group were fixed with paraformaldehyde in PBS at 37°C for 20 min. After fixation, the cells were permeabilized using PBS containing 0.1% Triton X-100 and 100 mM glycine for 30 min and blocked in 1.5% bovine serum albumin/ 0.5% glycine in PBS for 1 h at ambient conditions. For visualization of the cell cytoskeleton, cells were incubated with 200 μL of 6.6 μM Alexa FluorR 594 phalloidin solution (1:200 dilution; excitation and emission wavelength of 581 and 609 nm, respectively) for 30 min at ambient conditions to stain the actin filaments. Then, the cell nuclei were stained with DAPI (excitation and emission wavelength of 358 and 461 nm, respectively) at 1:5000 dilution. Samples were imaged with an inverted fluorescent microscope (Nikon Eclipse Ti-ε, Nikon, Melville, NY). The cells in the second group were collected and FITC fluorescent intensity was analyzed with a plate reader (Synergy HT) at excitation and emission wavelength of 495 and 520 nm, respectively. The measured fluorescent intensities were correlated to FITC concentration using a calibration curve constructed from solutions with known FITC concentration. Encapsulation of FITC in the NPs did not affect its fluorescent intensity as the NPs had negligible fluorescence.
2.8. In vitro cytotoxicity of DOX loaded NPs
Marrow stromal cells (MSCs) isolated from the bone marrow of young adult Wistar rats were used to assess the toxicity of PLA-V6K2 NPs with non-cancerous cells. MSCs were isolated under a protocol approved by the Institutional Animal Care and Use Committee of the University of South Carolina as described [32]. After isolation and removal of hematopoietic and other unattached cells, the cells were detached from the flasks with 0.05% trypsin-0.53 mM EDTA and used for cytotoxicity studies. The seeded MSCs (2.5x104 cells/cm2) were cultured with 0.17, 0.68, or 1.36 mg NP/mL for up to 3 days. At each time point, the fraction of viable cells was measured by MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay. Briefly, after removal of the medium, cells were incubated in 100 μl of serum free medium with 1 mg/mL MTT for 3 h at 37°C. Next, the purple crystals that accumulated within the viable cells were dissolved by 100 μl DMSO and the color intensity was measured by a plate reader (Synergy HT) at the wavelength of 590 nm. The fraction of viable cells was determined by dividing the measured intensity to those cultures treated with PBS only. 4T1 mouse breast tumor cells were used to assess the toxicity of PLA-V6K2 NPs as a function of DOX concentration. 4T1 cells were incubated in medium with DOX or DOX-loaded NPs for 3 days. After incubation, the fraction of viable cells was measured by MTT assay as described above. To investigate the effect of DOX encapsulation on DNA damage response and apoptosis, MTCL mouse breast carcinoma cells (Dorn Research Institute, Columbia, SC [40]) and 4T1 cells were incubated for 2 h with PBS (control), DOX, PLA-V6K2 NPs, or DOX in PLA-V6K2 NPs (1 μM DOX). After removing the conditioned medium and washing with PBS, the cells were incubated in regular medium (RPMI-1640 with 10% FBS) for zero, 4, and 22 h. Next, the cells were washed with PBS and the total protein content was extracted by NP40 buffer (0.5% NP40, 150mM NaCl, 50mM Tris-Cl pH7.4, protease inhibitors). The extracted proteins were separated by standard SDS-PAGE, transferred to nitrocellulose membranes, and detected by specific antibodies. Antibodies against γ-H2AX and cleaved PARP were from Cell signaling Technology (Danvers, MA) and those against Cyclin B1 and GAPDH were from Santa Cruz Biotechnology (Santa Cruz, CA). The western blots were quantified by Image J software (National Institutes of Health, Bethesda, MD).
2.9. Pharmacokinetics of DOX encapsulated PLA-V6K2 NPs
DOX encapsulated in PLA-V6K2, PLA-EG NPs, or free DOX in PBS was injected in the tail vein of C3HeB/FeJ mice (Jackson Laboratory, Bar Harbor, ME; 6 mg/kg body weight). At each time point, 50 μl of blood was collected by retro-orbital bleeding. To avoid frequent bleeding, 5 mice from each group were bled alternatively. The blood samples were centrifuged at 10,000g for 5 min and the supernatants (sera) were collected. The Dox in the sera was extracted with DMSO and quantified by HPLC as described in section 2.6. Near-infrared imaging was used to determine the relative amount of the NPs in the animal organs [37]. The near-infrared dye IRDye 800RS Carboxylate (LI-COR Biosciences), with peak absorption at 786 nm, was loaded in the NPs. 500 μL of the NPs suspension was injected in the tail vein of the C3HeB/FeJ mice. After 2 h, the animal was sacrificed, the organs were removed, and the relative amount of NPs in each organ was imaged by infrared imaging. The organs were scanned in two infrared channels simultaneously (700 and 800 nm) to normalize the intensities (700 nm for normalization).
2.10. Tumor cell migration and invasion
Migration experiments were done in transwell inserts as described [41]. 4T1 cells were seeded on the upper chamber of 24-well Costar® Transwell® plates (Corning, NY) at a density of 1.5×104 cells/well. Invasion experiments were carried out with Matrigel™ in transwells as the invading matrix [42, 43]. 50 μL Matrigel™ (basement membrane matrix, BD Biosciences, Franklin Lakes, NJ) was placed in the upper chamber of the plate and incubated for 30 min at 37°C to gel. After gelation, 4T1 cells were seeded on the Matrigel™ at the same cell density. After cell attachment, media was supplemented with DOX-loaded or PTX-loaded NPs (6 wt%) and incubated for 24 h to allow migration of tumor cells through the transwell membrane (8 μm pore size). After 24 h, cells on the upper side of the membrane were removed with a cell scraper (Corning) and the migrated cells on the lower membrane side were fixed with methanol. DOX or PTX directly added to the medium and NPs without DOX or PTX were used as positive and negative controls, respectively. The fixed cells on transwell membranes were stained with Eosin-Y and Azure-B dyes (IMEB, San Marcos, CA) according to manufacturer’s instructions. The membranes were allowed to dry for 12 h, mounted on microscope slides (bottom side up), and protected with coverslips. Samples were imaged with an inverted fluorescent microscope (Nikon Eclipse Ti-ε) and cells in randomly selected areas on the membrane surface (1.2 mm2 surface area) were counted. The cell count was divided by that in the absence of DOX or PTX in the culture medium to obtain percent cell migration or invasion.
2.11. In vivo cytotoxicity
DOX encapsulated in PLA-V6K2 and PLA-EG NPs was injected in the tail vein of C3HeB/FeJ mice inoculated with MTCL mouse breast carcinoma cells, and tumor volume (related to tumor toxicity) and body weight (related to host toxicity) were monitored for 9 days. Mice were kept under 12-h light and 12-h dark schedule. When mice were at 13–15 weeks of age, experiments were performed in compliance with the NIH and institutional guidelines for care and use of laboratory animals. Cultured MTCL cells were trypsinized and resuspended in serum-free RPMI 1640 medium to the concentration of 2x105 viable cells/mL. 100 μL of the cell suspension was injected subcutaneously in the back of mice. These tumors became measurable after 1 week of inoculation. The tumor size and body weight were measured daily at the same time of the day. After 3 measurements, mice were randomly divided into 4 groups with 10–15 mice in each group. Mice received PBS, PLA-V6K2 NPs, PLA-EG NPs, Dox, Dox in PLA-V6K2 NPs (Dox+PLA-V6K2), or Dox in PLA-EG NPs (Dox+PLA-EG) by tail vein injection (100 μL injection volume and 6 mg Dox/kg body weight). Tumor size and body weight were measured for additional 9 days before the animals were euthanized.
2.12. Statistical analysis
Data expressed as means ± standard deviation. All experiments, except for pharmacokinetics and in vivo toxicity experiments were done in triplicate. Significant differences between groups were evaluated using a two-way ANOVA with replication test followed by a two-tailed Student’s t-test. A value of p<0.05 was considered statistically significant.
3. Simulation method
The self-assembly of PLA-V6K2 and PLA-EG macromers into NPs and the distribution of DOX or PTX in the NPs were simulated via Dissipative Particle Dynamics (DPD) approach as described previously [28]. Figure 1 shows the molecular structure and different bead types on PLA-V6K2 macromer. The bead types with equal mass and volume in the simulation volume were L (lactide repeating unit), EO (ethylene oxide), Ac (acrylate group), P (peptide backbone), C (cystine side chain), V (valine side chain), K (lysine side chain) and W (three water molecules). Each DOX or PTX molecule was coarse grained into three bead types [44, 45] and represented in red and pink colors in the results section, respectively. In DPD, each bead represents a soft particle interacting with the other beads via a soft pair-wise force function given by [46, 47]:
| (1) |
Figure 1.
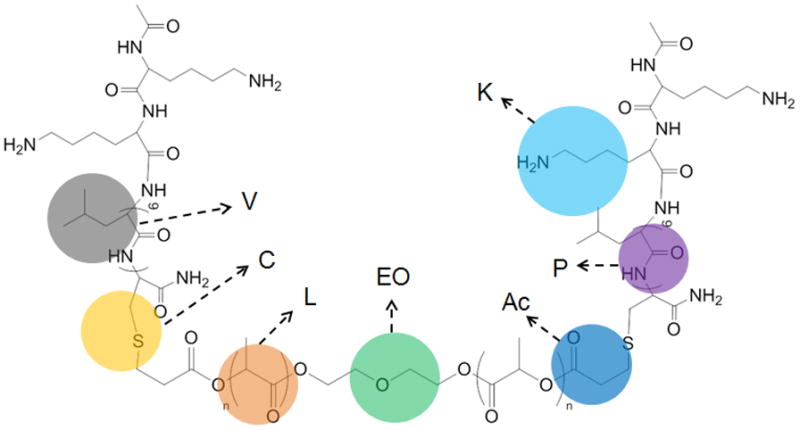
Coarse-grained representation of the PLA-V6K2 conjugate. The brown, green, dark blue, purple, gray, light blue and yellow colors represent lactide repeating unit, ethylene oxide unit, acrylate group, peptide backbone, valine, lysine and cystine side chains, respectively.
Where f is the total force and and are the conservative, dissipative, random and spring components of the force, respectively. For charged beads, a columbic term was added to the simulation force function. The force functions and were calculated as previously described [28]. was calculated from the pair-wise interaction parameter αij which was determined using the DPD length scale (rc) of 6.85 Ǻ and the Flory-Huggins pair-wise interaction parameter between beads i and j, χij, using the following equation [29, 48]:
| (2) |
The interaction parameters which are related to the solubility parameters between beads i and j were calculated via atomistic molecular dynamics simulation (Forcite and Amorphous Cell modules, Materials Studio v5.5, Accelrys) [49] using the COMPASS force field, which is an ab initio force field optimized for condensed-phase systems [50]. The position and velocity of the beads at each time point were determined by solving the equation of motion using the force function (equation 1) [46]. All DPD simulations were performed in a 25×25×25 rc simulation box with 3D periodic boundary conditions with at least 1x105 time steps and dimensionless time step of 0.04. The Mesocite module of the Materials Studio (v5.5) [49] was used to perform the DPD calculations.
4. Results and discussion
4.1. Characterization of the macromer, peptide, and conjugate
, and polydispersity index (PI) of Ac-PLA were 5.31 kDa, 6.52 kDa, and 1.23, respectively. The calculated molecular weight of the peptide was 1013 Da. In the ESI-MS spectrum, mass numbers (m/z) 1014 and 1036 corresponded to the monovalent hydrogen cation [(M+H)+] and monovalent sodium cation [(M+Na)+] of the peptide, respectively (Figure 2a). , and PI of PLA-V6K2 were 5.81 kDa, 11.25 kDa, and 1.9, respectively. NMR results for Ac-PLA and PLA-V6K2 macromers are shown in Figure 2b. The chemical shifts with peak positions at 1.12, 1.62 and 4.66 ppm were assigned to the methyl and methine hydrogens of the peptide, respectively. The chemical shifts with peak positions at 1.75 and 5.46 ppm were assigned to the methyl and methine hydrogens of the lactide in PLA, respectively. The chemical shift with peak position at 3.64 ppm was assigned to the methylene hydrogens of DEG in PLA. The chemical shift with peak position at 5.21ppm was assigned to the amine hydrogens of the lysine residue in the peptide. The presence of chemical shifts with peak positions at 6.22–6.65 ppm was attributed to the vinyl hydrogens of Ac-PLA. The ratio of the peaks at 6.22–6.65 ppm (hydrogens of the acrylate group) and 5.46 ppm (hydrogens of the lactide groups) was related to acrylate/LA molar ratio in Ac-PLA macromer and PLA-V6K2 conjugate. The number of peptides in the conjugate was determined using the acrylate/LA molar ratio before and after conjugation. The average number of peptides per conjugate was 1.2 indicating that there was on average one peptide conjugated to each macromer.
Figure 2.
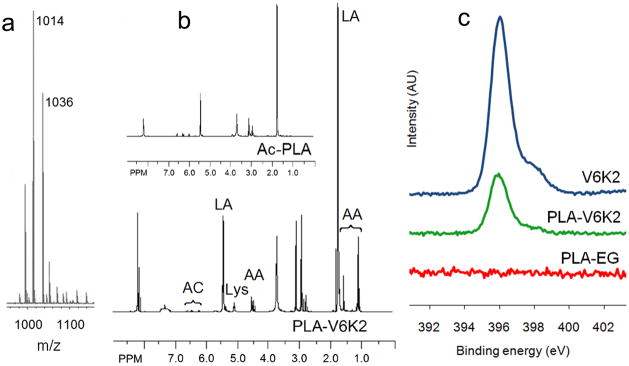
(a) ESI-MS spectrum of cysteine-terminated V6K2 peptide; (b) NMR spectra of Ac-PLA macromer and PLA-V6K2 conjugate. The marked peaks are for hydrogens of the peptide main chain (AA), lactide (LA), lysine (Lys), and acrylate group (AC); (c) XPS spectra of cysteine-terminated V6K2, PLA-EG, and PLA-V6K2 NPs in the 392–402 eV region, corresponding to the binding energy of nitrogen 1s electrons.
4.2. Nanoparticle characterization
XPS spectra of V6K2, PLA-EG, and PLA-V6K2 NPs in the 392–402 eV region, corresponding to the binding energy of nitrogen 1s electrons, are shown in Figure 2c. PLA-EG NPs, due to the absence of nitrogen in the macromer, did not have a peak in this region while V6K2 NPs had a strong nitrogen peak centered at 396 eV. A similar strong peak was observed in the spectra of PLA-V6K2 NPs. Considering the relatively high penetration depth of the incident beam in polymers (7.5 nm) [51], the data demonstrates significant presence of the peptide on the NPs surface. PLA-V6K2 NPs had a spherical shape as shown in the TEM micrograph of Figure 3. Size distribution of the NPs is shown in Figure 4. The number-average diameter of the NPs, the breadth of the distribution corresponding to ±1 SD from the mean, and PI are given in Table 1. Control groups included V6K2 NPs, PLA-EG NPs, and PLA macromer conjugated with a mutant sequence of V6K2 peptide [PLA-(V2K)2V2]. V6K2 NPs without conjugation to PLA had the lowest average size and narrow distribution (35±14 nm; PI=1.01), close to the previously reported size [26]. In contrast, PLA-EG NPs (130±50 nm; PI=1.14) and NPs conjugated with the mutant (V2K)2V2 peptide [PLA-(V2K)2V2; 150±80 nm; PI=1.28] had much higher average size and wider distribution compared to PLA-V6K2 (100±20 nm; PI=1.04). A blend of Ac-PLA macromer with cysteine-terminated V6K2 peptide formed aggregates with ~1μm average size (PLA/V6K2; 850±350 nm; PI=1.17). The slightly lower size of PLA-V6K2 compared to PLA-EG NPs was attributed to the long range ionic interactions between the amino acids. Furthermore, the presence of hydrophobic valine side chains surrounding the PLA hydrophobic core, packed the peptide segments on the NPs surface leading to a higher coverage of the hydrophobic core and lower NP/water interfacial energy in PLA-V6K2 NPs. The pore cutoff size of most tumor tissues is 200–400 nm and NPs with <200 nm size are selectively taken up by tumor-associated cells [52, 53].
Figure 3.

TEM micrograph of the uranyl acetate stained PLA-V6K2 NPs.
Figure 4.
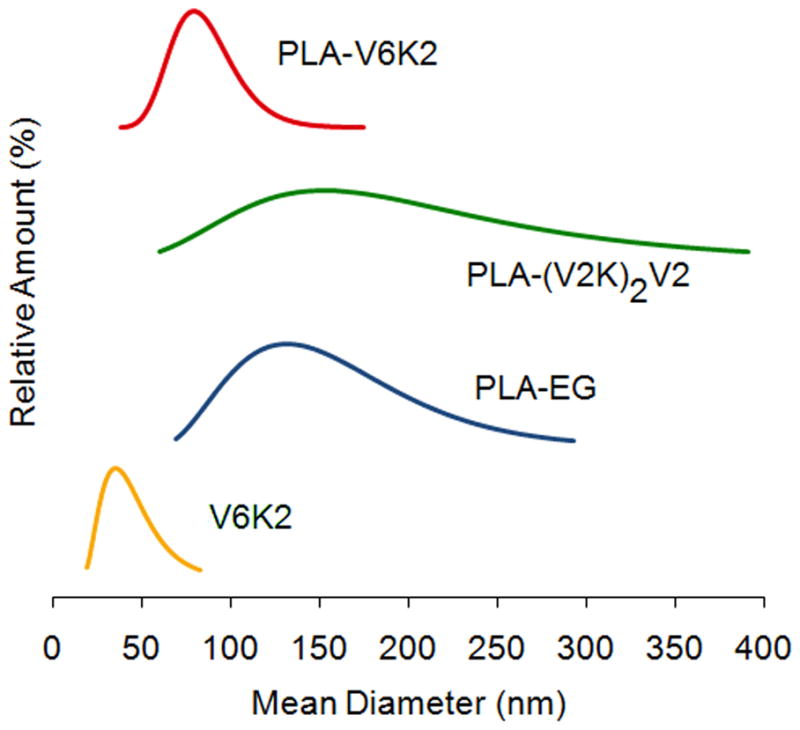
Size distribution of V6K2, PLA-EG, PLA-(V2K)2V2, and PLA-V6K2 NPs.
Table 1.
Diameter and distribution of the synthesized NPs.
| NPs | Diameter (nm) | SD (nm) | PI (nm) |
|---|---|---|---|
| V6K2 | 35 | 4 | 1.01 |
| PLA-EG | 130 | 50 | 1.14 |
| PLA-(V2K)2V2 | 150 | 80 | 1.28 |
| PLA-V6K2 | 100 | 20 | 1.04 |
4.3. Nanoparticle release characteristics and degradation
The hydrophobic PLA core provided a degradable matrix for encapsulation and sustained release of therapeutic agents. The release kinetics of DOX or PTX from PLA-V6K2 and PLA-EG NPs are shown in Figure 5a. The encapsulation efficiency of DOX in PLA-V6K2 and PLA-EG NPs was 44±9% and 55±5%, respectively. The encapsulation efficiency of the more hydrophobic PTX in both NPs was >90%. According to DPD simulation results (see Figure 6), DOX partitioned in the PLA core as well as the V6 shell of the NPs, resulting in lower encapsulation efficiency compared to PTX [54–56]. The release of DOX from PLA-EG NPs was faster than PLA-V6K2 NPs. The burst release for PLA-V6K2 in the first 12 h was <10% followed by a relatively constant release rate for 35 days and 54% of the encapsulated DOX was released after 10 days. For PLA-EG NPs, the release in the first 12 h was <10% but 80% of the encapsulated DOX was released in 10 days. 100% of the DOX was released from PLA-EG and PLA-V6K2 NPs after 28 and 35 days respectively. The release of PTX from PLA-V6K2 NPs was initially slower than PLA-EG NPs. The released amount of PTX from PLA-V6K2 and PLA-EG NPs after 10 days was 46% and 74%, respectively. However, 100% of the PTX was released from both NPs after 28 days.
Figure 5.
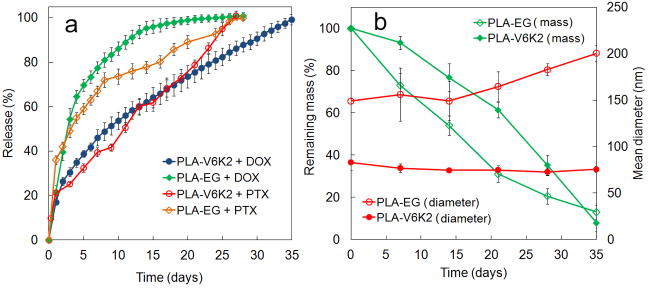
(a) Release profiles of DOX or PTX encapsulated in PLA-V6K2 and PLA-EG NPs incubated in PBS; the release profile of DOX encapsulated in PLA-V6K2 NPs and incubated in 100% FBS (open blue data points) is compared with that incubated in PBS (full blue data points); (b) the effect of incubation time on degradation kinetics (green) and diameter (red) of PLA-V6K2 (full symbols) and PLA-EG (open symbols) NPs; Error bars correspond to the mean ±1 SD for n=3.
Figure 6.
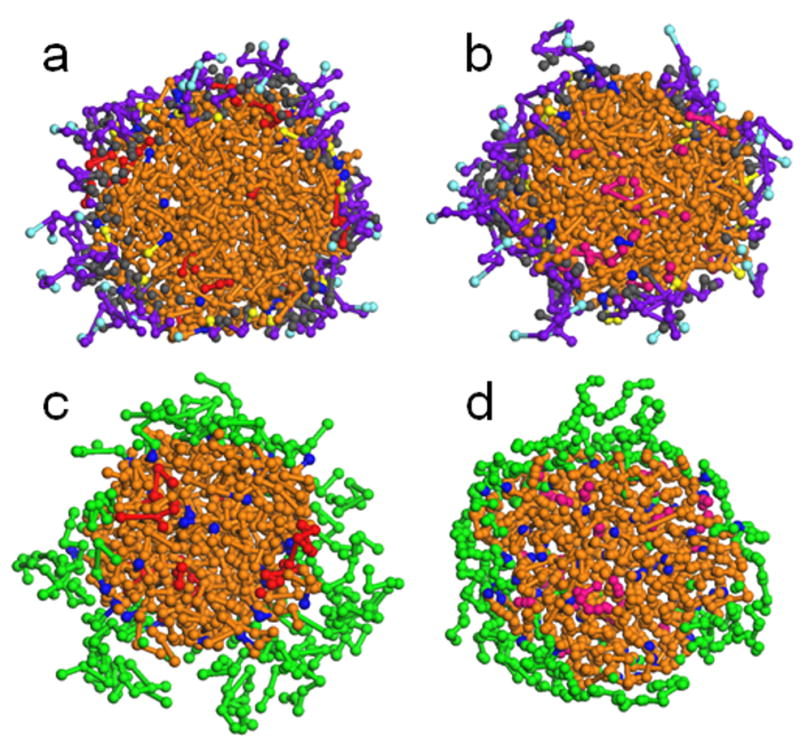
DPD simulations of the cross-section of self-assembled PLA-V6K2 and PLA-EG NPs in the presence of DOX or PTX. Images (a) and (b) show DOX and PTX encapsulation in PLA-V6K2 NPs, respectively, while images (c) and (d) show DOX and PTX encapsulation in PLA-EG NPs. L, EO, Ac, P, V, K and C beads are shown in brown, green, dark blue, purple, gray, light blue and yellow, respectively. DOX and PTX beads are shown in red and pink colors, respectively.
DPD simulation of the cross-sections for the self-assembly of PLA-V6K2 and PLA-EG NPs in the presence of DOX or PTX are shown in Figure 6. Images (a) and (b) show DOX and PTX encapsulation in PLA-V6K2 NPs, respectively, while images (c) and (d) show DOX and PTX encapsulation in PLA-EG NPs. L, EO, Ac, P, V, K and C beads are shown in brown, green, dark blue, purple, gray, light blue and yellow, respectively. DOX and PTX beads are shown in red and pink colors, respectively. All simulations were carried out in aqueous medium. The hydrophobic L beads aggregated and formed the central part of the NPs. The hydrophilic P and K beads in PLA-V6K2 NPs and EO beads in PLA-EG NPs shielded the core from the aqueous phase and formed the shell of the NPs. In PLA-V6K2 NPs, the hydrophobic interactions between V and L beads overcame the extensional free energy of the peptide leading to a higher coverage of the core surface compared to that of PLA-EG NPs. The distribution of DOX and PTX drugs in PLA-V6K2 and PLA-EG NPs is clearly shown in the cross-sections. DOX in PLA-V6K2 and PLA-EG NPs (see images (a) and (c) in Figure 6) was distributed in both the lactide core and peptide shell, explaining its lower encapsulation efficiency. PTX in both PLA-V6K2 and PLA-EG NPs (see images (b) and (d) in Figure 6) was localized to the hydrophobic core and at the proximity of L beads, explaining its high encapsulation efficiency. The slower release of DOX or PTX in PLA-V6K2, compared to PLA-EG NPs, was attributed to the dense coverage of the NPs with the peptide, which had attractive interactions with the drug and delayed the release upon degradation of the lactide core.
PTX has low solubility in water (<0.1 mg/mL) but its solubility can be enhanced by the addition of amphiphilic polymers to the aqueous medium [57]. For example, amphiphilic block copolymers of PEG as the micelle shell and poly(2-(4-vinylbenzyloxy)-N,N-diethylnicotinamide) as the micelle core effectively provided solubilization and stabilization for PTX [58]. DOX has a relatively higher solubility in aqueous medium compared to PTX and aggregation is reported for DOX encapsulated in egg phosphatidylcholine (EPC)/cholesterol liposomes [59]. The drug solubility in the NPs depends on the relative interaction energy between the drug molecules, drug with water, PLA, PEG, and amino acids with water, drug with PLA and PEG, and drug with the amino acids in PLA-V6K2. Based on the simulation results, 62 ±4% and 86 ±3% of the encapsulated DOX was localized at the PLA-PEG and PLA-V6K2 interface, respectively, those fractions were 25 ±3% and 30 ±4% for PTX. Results indicate that the PLA-V6K2 interface had a stabilizing effect on DOX encapsulation while PTX was mainly stabilized in the PLA core. The simulation results were analyzed for the fractional size distribution of drug aggregates. Based on simulation results, DOX underwent aggregation in PLA-EG NPs with the highest fraction of 3-mers. However, DOX did not seem to form aggregates in PLA-V6K2 NPs. PTX did not seem to aggregate in PLA-EG or PLA-V6K2 NPs. The aggregation of DOX in PLA-EG stemmed from the domination of DOX-DOX energetic interaction compared to the interactions with the matrix. The results are consistent with the reported DOX aggregation in EPC/cholesterol liposomes [55].
It is well-established that the incubation medium can dramatically affect the degradation and drug release from the NPs [38]. To investigate the effect of medium, the DOX release from PLA-V6K2 NPs suspended in PBS (full blue circles) is compared with that in 100% fetal bovine serum (FBS, open blue circles) in Figure 5a. In PBS, 90% of the DOX was released in 28 days while the same fraction of DOX was released in only 5 days in FBS. In addition to that, the acidic environment of the lysosomes can accelerate NPs degradation as they cross the cell membrane. Furthermore, intracellular enzymes can enhance NPs degradation and drug release. These results demonstrate that the DOX release kinetics may be significantly faster in vivo within the tumor environment.
DOX and PTX release profiles in Figure 5a for PLA-V6K2 and PLA-EG NPs followed the NPs’ mass loss shown in Figure 5b. Unlike high molecular weight PLA in which mass loss follows hydrolytic degradation of the matrix below a critical molecular weight [60, 61], PLA-V6K2 NPs lost mass concurrent with degradation [36]. Degradation of PLA-V6K2 and PLA-EG NPs was relatively constant and lasted for 5 weeks, with PLA-EG having slightly slower degradation compared to PLA-V6K2. Mass loss results suggest that erosion as well as diffusion contributed to the drug release. The average size of PLA-V6K2 and PLA-EG NPs, shown in Figure 5b, did not change appreciably with incubation, consistent with previous results for short-chain triblock poly(ε-caprolactone)-b-polyethylene glycol-b-poly(ε-caprolactone) (PCL-PEG-PCL) micelles [62]. It is well-established that sulfhydryl group of cysteine residues in peptides and proteins react to form disulfide crosslinks in the presence of disulfide isomerase enzyme, other cysteine residues, or electron-accepting reagents such as oxygen [2]. The unexpected persistence of the NPs with degradation and the absence of any change in their size (see Figure 5b) can be explained by disulfide bond formation in PLA-V6K2 NPs [63].
4.4. Tumor cell uptake of nanoparticles
The fluorescent images in Figure 7 compare the uptake of FITC-loaded PLA-V6K2 (left panel) and PLA-EG (right panel) NPs by 4T1 breast tumor cells after 2, 12 and 24 h. The blue, red, and green colors in the images show the cell nuclei, cytoskeletal actin, and FITC-labeled NPs. The areas in yellow are the overlap of cytoskeletal red and green FITC-labeled NPs. The inset in the 24 h images with 2X higher magnification show with more clarity that the NPs are positioned in to the cell cytoplasm around the nuclei. When the green fluorescent image of the NPs is much stronger than the red actin, the green intensity dominates. The PLA-V6K2 NPs in the left inset image around the cells are green color while the PLA-EG NPs in the right image are yellow. These color variations indicate that the fluorescent intensity of PLA-V6K2 NPs (inset in the left image) was significantly higher than that of red cytoskeletal actin and the fluorescent intensity of PLA-EG NPs (inset in the right image) was comparable to that of cytoskeletal actin. Therefore, the PLA-V6K2 NPs had higher uptake than the PLA-EG NPs after 24 h incubation with 4T1 cells.
Figure 7.

Fluorescent images of FITC-labeled PLA-V6K2 (left column) and PLA-EG (right column) NPs incubated with 4T1 breast tumor cells after 2, 12 and 24 h. The cell nuclei and cytoskeleton were stained with DAPI (blue) and phalloidin (red), respectively; the insets in the 24 h images show 2X higher magnification of the stained cells and NPs. The FITC-loaded NPs can be seen as bright green or yellow (overlap of red and green) dots.
The fraction of PLA-V6K2 and PLA-EG NPs taken up by 4T1 tumor cells with incubation time is shown in Figure 8a. Percent uptake of PLA-V6K2 and PLA-EG NPs by 4T1 cells increased from 22±1% to 49±1% and from 19±1% to 32±1%, respectively, with increasing incubation time from 2h to 24 h. Overall, the uptake of PLA-V6K2 NPs was significantly higher than that of PLA-EG NPs (p-value=0.005). DPD simulation results in Figure 6 demonstrate that the lysine side chains of PLA-V6K2 are positioned on the NPs surface facing water. NPs uptake is affected by many factors including particle size and composition, surface polarity and charge, and cell density [64, 65]. NPs with size <100 nm are taken up by clathrin-mediated endocytosis and macropinocytosis while those >100 nm size are taken up by phagocytosis [66, 67]. Since the size of NPs ranged between 50–200 nm, both pathways contributed to the uptake. The higher uptake of PLA-V6K2 NPs was related to electrostatic interactions between the positively-charged lysine groups of the NPs and negatively-charged cell surface moieties such as heparan sulfates, sialic acids and phospholipid head groups [68]. Electrostatic interactions at the NP-cell interface have been shown to reduce the free energy barrier for uptake leading to direct translocation of NPs to the cytoplasm [69]. For example, short arginine/lysine sequences like YGRKKRRQRRR from the TAT protein transduction domain of HIV virus [70] have the ability to disrupt and penetrate cell membrane, allowing direct delivery of a cargo molecule to the cytoplasm by macropinocytosis [70, 71].
Figure 8.

(a) The effect of incubation time on percent uptake of PLA-V6K2 and PLA-EG NPs by 4T1 breast tumor cells; (b) the effect of PLA-V6K2 NPs (without DOX) on viability of non-cancerous MSCs with incubation time; (c) the effect of free DOX and DOX in PLA-V6K2 NPs after 3 days of incubation on percent viability (normalized to untreated cells) of 4T1 tumor cells as a function of DOX concentration; (d) DOX concentration in the blood circulation with time after tail vein injection of free DOX, DOX in PLA-V6K2 NPs, and DOX in PLA-EG NPs (6 mg DOX/kg mice) in C3HeB/FeJ mice. In (a-c), error bars correspond to the mean ±1 SD for n=3 and in (d), error bars correspond to mean ±1 SD for n=5. Cell viability was measured by MTT assay.
4.5. Cytotoxicity of PLA-V6K2 nanoparticle and circulation time
The cytotoxicity of PLA-V6K2 NPs (with no drug) to normal cells was evaluated with MSCs and the results are shown in Figure 8b. There was no significant change in cell viability as the NP concentration in the medium was increased from 0.17 to 1.36 mg/mL and incubation time was increased from 2 to 72 h. MSC cell viability was >90% for all incubation times and NP concentrations. Electrostatic interaction between cationic NPs and the negatively-charged cell surface may increase endocytosis across the cell membrane. In retrospect, the electrostatic interaction can also affect the integrity of the cell membrane and cause undesired cytotoxicity. Goodman et al. investigated the toxicity of functionalized gold NPs and observed that cationic NPs, due to the initial electrostatic interaction, are moderately toxic to fibroblasts and red blood cells [72]. Bexiga et al. showed evidence for an apoptotic pathway in human brain astrocytoma cells with respect to toxicity of cationic polystyrene NPs with activation of caspases, increase in reactive oxygen species (ROS), and damage to mitochondria [73]. Chompoosor et al. reported that the toxicity of cationic gold NPs on HeLa cells depended on ligand hydrophobicity with more hydrophobic ligands having higher ROS, leading to increased NPs degradation and less DNA damage [74]. Basarkar et al. observed that cationic PLGA NPs surface-modified with cetyltrimethyl ammonium bromide had no toxicity to human embryonic kidney cells [75]. Cationic PCL-pluronic-PCL NPs showed lower cytotoxicity in several cancer cell lines compared to lipofectamine [76]. Tomita et al. measured cytotoxicity and inflammatory response of anionic and cationic n-butylcyanoacrylate (NCBA) NPs with murine macrophages [77]. Both anionic and cationic NCBA NPs showed no cytotoxicity at low concentrations (<10 μg/mL). Interestingly, at higher concentrations cationic NCBA NPs exhibited lower cytotoxicity in macrophages compared to anionic NPs, indicating that cationic NPs may have less interaction with macrophages and inflammatory cells than anionic NPs, leading to a longer circulation. Adsorption of serum proteins to the surface of cationic polysaccharide NPs partially screens the positive charges, leading to lower toxicity [78]. The low toxicity of cationic NPs may also be related to their higher rate of exocytosis as well as endocytosis. Zhang et al. reported that toxicity of cationic silica NPs depended on differentiation state of the bronchial epithelial cells with cells that are differentiated showing higher toxicity than the undifferentiated cells [79]. Since undifferentiated cells have high rate of exocytosis, Zhang results are consistent with those of Merhi [78] with cationic polysaccharide NPs and Lu [80] with cationic albumin-conjugated PEGylated NPs. Niu et al. observed enhanced cellular uptake of cationic Paclitaxel NPs by NIH-3T3 and MDR-3T3 (active p-glycoprotein) cells compared to liposomal Paclitaxel and Taxol [81]. Taken together, the previous results indicate that cytotoxicity of cationic NPs strongly depends on differentiation state of the cell, adsorption of serum proteins, and P-glycoprotein efflux. Cationic NPs exhibit higher cell uptake than neutral or anionic NPs and lower interaction with inflammatory cells. The low toxicity of PLA-V6K2 NPs cultured with MSCs (see Figure 8b) is consistent with the premise that cytotoxicity of cationic NPs is dependent on the cell differentiation state and rate of exocytosis [78, 79, 81].
The effect of DOX encapsulation in PLA-V6K2 NPs on cytotoxicity was evaluated by incubation with 4T1 cells for 3 days and the results are shown in Figure 8c as a function of DOX concentration. Similar to the MSC viability results in Figure 8b, the drug-free PLA-V6K2 NPs did not show any toxicity to 4T1 cells after incubation for 3 days. At low drug concentrations (<1 μM), encapsulation did not have a noticeable effect on cytotoxicity. However, as the DOX concentration was increased from 1 to 6 μM, the encapsulated DOX had significantly higher toxicity to 4T1 cells than the free drug (p-value=0.013). The higher toxicity of DOX in PLA-V6K2 NPs to 4T1 cells in Figure 8c compared to free DOX is consistent with the high endocytosis of cationic NPs [78].
The investigate the effect of DOX encapsulation on DNA damage response and apoptosis, MTCL and 4T1 cells were incubated for 2 h with PBS (control), DOX, PLA-V6K2 NPs, and DOX-loaded PLA-V6K2 NPs (1 μM DOX). After removing the conditioned medium, the cells were incubated in regular medium (RPMI-1640 with 10% FBS) for zero, 4, and 22 h and the response was determined by the expression of γ-H2AX DNA damage marker, cleaved PARP apoptosis marker, and Cyclin B1 cell cycle arrest marker. The γ–H2AX binds to the broken ends of the double strand DNA and induces DNA damage response [82]. Dox induced DNA damage leads to cell cycle arrest and apoptosis. The G2/M cell cycle arrest by DOX results in the accumulation of Cyclin B1 [83] while the cleavage of caspases and PARP triggers apoptosis [84]. The electrophoretic bands of γ-H2AX, cleaved PARP, and Cyclin B1 after zero, 4, and 22 h of incubation for all groups (PBS, NP, DOX, NP-DOX) are shown in Figures 9a and 9b, respectively. The bands of the 4T1 groups for γ-H2AX and cleaved PARP were quantified and the amounts are shown with time in Figures 9c and 9d, respectively. Free DOX induced DNA damage instantly at time zero while the effect of NP-DOX was delayed for both cell types (see Figures 9a-c). The amount of γ-H2AX in the free DOX group decreased after 22 h due to the repair of damaged DNA while that of NP-DOX was delayed and began to increase after 22 h. Both DOX and NP-DOX caused cell cycle arrest after 22 h (see Figures a and b for Cyclin B1) with the amount of Cyclin B1 slightly higher in the NP-DOX group. In MTCL cells, free DOX induced apoptosis earlier after 4 h (see Figure 9a) but the amount of cleaved PARP was higher in NP-DOX group after 22 h in both cell lines (see Figure 9a-b). Together, the results in Figures 8c and 9 indicate that NP-DOX was efficiently taken up by the tumor cells but the DNA damage response was delayed due to DOX encapsulation. In addition, the sustained release of DOX at lower dose in normal tissues allows the cell to repair the damage, thus diminishing DOX side effects. This is supported by the in vivo results in Figure 11 in which the animals treated with DOX loaded PLA-V6K2 NPs had lower body weight loss than those treated with free DOX.
Figure 9.
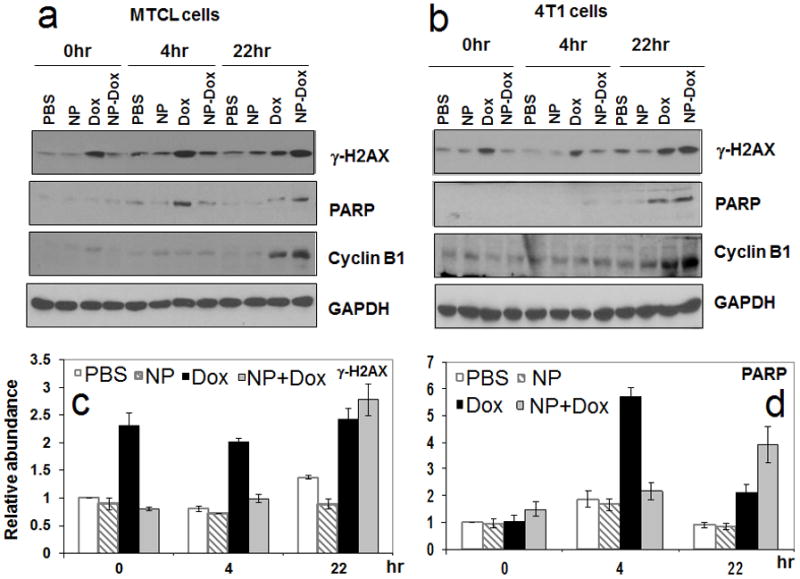
The effect of DOX encapsulation in PLA-V6K2 NPs on DNA damage and apoptosis of MTCL (a) and 4T1 (b) mouse breast cancer cells; Groups included PBS, PLA-V6K2 NPs, free DOX (1 μM), and DOX in PLA-V6K2 NPs (1 μM); tumor cells were incubated with drug-loaded NPs for 2 h; Next, the medium was replaced with drug-free medium and incubated for zero, 4, and 22 h. The response was determined by the expression of γ-H2AX DNA damage marker, cleaved PARP apoptosis marker, and Cyclin B1 cell cycle arrest marker); quantified intensities of γ-H2AX (c) and cleaved PARP (d) markers; GAPDH was used as the internal control and the amount of the markers in PBS-treated cells at time zero was set as one.
Figure 11.
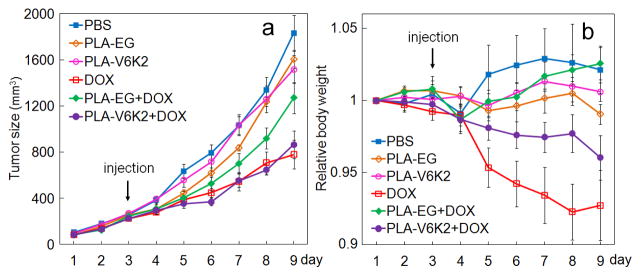
Tumor volume (a) and the change in body weight (b) with time after tail vein injection of DOX loaded NPs (on day 3). Groups included PBS (control, blue), PLA-EG NPs (control, brown), PLA-V6K2 NPs (control, pink), direct injection of DOX (red), DOX in PLA-EG NPs (green), and DOX in PLA-V6K2 NPs (purple). Mice were inoculated with MTCL mouse breast cancer cells by subcutaneous injection in the back of the animal. The injected dose was 6 mg Dox/kg body weight.
The effect of encapsulation on circulation time was determined in C3HeB/FeJ mice. Figure 8d shows DOX concentration in the blood with time after injection of free DOX, DOX in PLA-EG and DOX in PLA-V6K2 in the mice circulation. For all time points up to 72 h, the drug concentration in blood of DOX-loaded NPs was significantly higher than free DOX (p-values <0.0001 for PLA-V6K2 and PLA-EG). Furthermore, for all time points, the drug concentration in blood of DOX-loaded PLA-V6K2 NPs was significantly higher than that of PLA-EG NPs (p-value <0.0001). For example, after 24 h the DOX blood concentration for PLA-V6K2 NPs, PLA-EG NPs, and free DOX was 72, 55, and 26 ng/mL, respectively; after 48 h the concentration dropped to 45, 31, and 17 ng/mL. The area under the concentration-time curve (AUC) for free Dox, DOX in PLA-EG and DOX in PLA-V6K2 NPs was 3400, 4700, and 5700 ng/mL.h, respectively, consistent with the previously reported AUC for free DOX [85]. Encapsulation of DOX in PLA-EG and PLA-V6K2 NPs increased AUC by 38% and 67%, respectively. Assuming a minimum effective concentration of 50 ng/nmL [85], the circulation time of DOX in PLA-V6K2 NPs, PLA-EG NPs, and free DOX was 42, 26, and 12 h, respectively. The higher circulation time of PLA-V6K2 NPs may be related to the lower interaction of cationic NPs with macrophages and inflammatory cells [77]. PLA-V6K2 NPs were loaded with a near-infrared dye and the distribution of the NPs in different organs was imaged by near-infrared imaging. The NPs accumulated mainly in the kidney and liver with little accumulation in the lung and spleen.
4.6. Tumor cell migration and invasion of DOX-loaded nanoparticles
The effect of DOX or PTX loading in PLA-V6K2 NPs (shown in brown) on migration and invasion of 4T1 cells is compared with PLA-EG NPs (shown in blue) in Figures 10a-d. Figures 10a and 10b show the effect of DOX loading on cell migration and invasion, respectively. Figures 10c and 10d show the effect of PTX loading on cell migration and invasion, respectively. The ordinate in the figure is the number of migrated cells divided by that in the control group (migrated/invaded cells in the absence of drug and NPs). Columns 1 through 3 correspond to cell migration/invasion in culture medium supplemented with unloaded NPs (labeled “NP”), DOX/PTX without encapsulation in NPs (5 μM directly added to media, labeled “DOX” or “PTX”), and DOX/PTX loaded in NPs (labeled “NP+DOX” or “NP+PTX”), respectively. NPs without DOX or PTX had little effect on migration and invasion of tumor cells. DOX added directly to the medium (shown in green) reduced cell migration and invasion to 68% and 30%, respectively, as the drug acted intracellularly to trigger mitochondrial and DNA damage [86, 87], leading to cell cycle arrest and impaired cell migration and proliferation. DOX encapsulation in NPs significantly slowed tumor cell migration and invasion compared to DOX or PTX group (shown by one star). DOX encapsulated in PLA-V6K2 NPs had lower average cell migration compared to PLA-EG NPs but the difference was not statistically significant. However, DOX encapsulated in PLA-V6K2 NPs significantly reduced cell invasion compared to DOX (indicated by two stars with p<0.0002). For example, direct addition of DOX reduced cell invasion to 30±10% while DOX in PLA-V6K2 NPs reduced cell invasion to 5±1%. PTX added directly to the medium decreased cell migration and invasion to 65% and 48% of the control, respectively. Cell migration and invasion decreased to 45% and 40% in the presence of PTX loaded PLA-EG NPs, which was not significantly different from PTX directly added to the media. PTX encapsulated in PLA-V6K2 NPs decreased migration and invasion to 12% and 11%, respectively, which was significantly lower than the NP or PTX groups. Based on these results PLA-V6K2 NPs, due to higher rate of endocytosis, were more effective in delivering DOX or PTX intracellularly compared with PLA-EG NPs.
Figure 10.

The effect of DOX (a, b) and PTX (c, d) loaded in PLA-V6K2 (brown) and PLA-EG (blue) NPs on (a, c) migration and (b, d) invasion of 4T1 cells. The ordinate is the number of invaded cells divided by that in the absence of DOX or PTX and NPs. Columns 1 through 3 are the percent cell migration or invasion for culture media supplemented with unloaded PLA NPs, DOX or PTX (5 μM directly added to media) and DOX or PTX in NPs (PLA-EG and PLA-V6K2), respectively. One star indicates statistically significant difference between the test group and control (medium without DOX and NPs); two stars indicate statistically significant difference between the test group and DOX or PTX directly added to the culture medium. Error bars correspond to the mean ±1 SD for n=3.
4.7. In vivo toxicity
C3HeB/FeJ mice were inoculated with MTCL syngeneic breast cancer cells. When the tumor volume reached 400 mm3, animals were injected with PLA-V6K2 or PLA-EG NPs, DOX, and DOX encapsulated in PLA-V6K2 or PLA-EG NPs. Animals injected with PBS were used as control. Figures 11a and 11b show the tumor size and the change in body weight with time for all groups, respectively. PBS and the NP groups (without DOX) had the highest tumor size after 9 days followed by DOX-loaded PLA-EG group. DOX directly injected and DOX-loaded PLA-V6K2 NPs displayed the lowest tumor size after 9 days. According to Figure 11b, DOX had the highest decrease in body weight followed by DOX-loaded PLA- V6K2 NPs. PLA-V6K2 and PLA-EG NPs (without DOX) displayed no change in body weight while PBS and DOX in PLA-EG NPs showed a slight increase in body weight. The in vivo data in Figure 11 demonstrates that PLA-V6K2 NPs displayed higher tumor and host toxicity while PLA-EG NPs showed lower tumor and host toxicity. In other words, PLA-V6K2 NPs were more effective in delivering DOX to the tumor cells while PLE-EG NPs were more effective in shielding normal tissues from the effect of DOX.
PEGylation is used extensively to increase the residence time of NPs in the circulation [88]. PEGylation increases the fraction of NPs that are taken up by the tumor tissue, thus reducing the host toxicity. However, PEGylation can adversely affect cell uptake and tumor toxicity [89]. Conjugation of peptides to the surface of PEGylated NPs has been shown to improve tumor toxicity of chemotherapy drugs. For example, conjugation of cylic RGD peptide to PEG-PLA-Paclitaxel NPs significantly reduced tumor growth in glioblastoma-bearing nude mice, compared with those NPs without cyclic RGD conjugation [90]. In that work, the reduction in tumor growth was explained by specific interaction of the cyclic RGD conjugated NPs with αvβ3 surface receptors on glioblastoma cells as well as non-specific interaction with ionic moieties on the cell membrane. As another example, PLA-PEG NPs functionalized with penetratin, a cell penetrating peptide, showed higher uptake in the brain (the target tissue) and lower accumulation in non-target tissues [91]. Our work, for the first time compares cell uptake, tumor and host toxicity of PLA NPs stabilized with a self-assembling cationic peptide with that stabilized by PEG. Results demonstrate that DOX in PEG stabilized PLA NPs dramatically reduced host toxicity while DOX in V6K2 stabilized PLA NPs significantly increased tumor cell uptake and tumor toxicity of the drug. These results indicate that methods that can activate the removal of PEG from the NPs surface in the tumor tissue and uncover the underlying functional groups that interact with cell surface moieties can increase tumor toxicity of the encapsulated drugs while maintaining low host toxicity.
5. Conclusions
In this work, we investigated the drug release kinetics, cell uptake, pharmacokinetics, tumor cell migration and invasion, and tumor and host toxicity of DOX encapsulated in hybrid NPs made of low molecular weight PLA and self-assembling V6K2 peptide. PLA-V6K2 NPs had spherical shape and number average size of 100 nm. DOX or PTX encapsulated in PLA-V6K2 NPs displayed constant release rate kinetics concurrent with the degradation of NPs. Molecular dynamic simulation of the NPs demonstrated that the less hydrophobic DOX was distributed in the lactide core as well as the peptide shell while the hydrophobic PTX was localized to the lactide core. PLA-V6K2 NPs had significantly higher tumor cell uptake compared to PLA NPs stabilized with PEG (PLA-EG), which was attributed to the electrostatic interactions between the lysine groups of the peptide and negatively charged moieties on the cell surface. PLA-V6K2 NPs (with no drug) showed no toxicity to non-cancerous marrow stromal cells and 4T1 breast carcinoma cells. DOX loaded PLA-V6K2 NPs showed higher toxicity to 4T1 cells with delayed DNA damage response and apoptosis compared to free DOX. With respect to the effect of encapsulation on pharmacokinetics, DOX encapsulated in PLA-V6K2 and PLA-EG NPs increased AUC by 67% and 38%, respectively, compared to free DOX. The invasion of 4T1 tumor cells incubated with DOX or PTX in PLA-V6K2 NPs was significantly lower than that of direct treatment with the drug or the drug in PLA-EG NPs. DOX encapsulated PLA-V6K2 NPs injected in C3HeB/FeJ mice inoculated with MTCL syngeneic breast carcinoma cells displayed tumor toxicity similar to that of direct injection of DOX but significantly lower host toxicity. Furthermore, DOX in PLA-V6K2 NPs showed higher tumor toxicity than in PLA-EG NPs. Taken together, results demonstrate that PLA-V6K2 have stronger electrostatic interactions with the cell membrane, leading to higher cell uptake, lower tumor cell invasion of DOX loaded NPs, higher tumor toxicity, with a host toxicity higher than PLA-EG NPs but lower than free DOX. The findings of this work are potentially useful for designing more effective tumor delivery systems that can activate the cleavage of PEG from the NPs surface in the tumor tissue to uncover the underlying functional groups for stronger interaction with the tumor cell surface leading to higher rate of exocytosis and higher tumor toxicity.
Acknowledgments
This work was supported by grants from the National Science Foundation under Grant Numbers CBET0756394, CBET0931998, and DMR1049381, and the National Institutes of Health under Grant No. DE19180. E. Jabbari thanks Dr. Ralph A. Reisfeld (The Scripps Research Institute, La Jolla, CA) for providing the 4T1 murine breast carcinoma cells. Authors thank Dr. Soumitra Ghoshroy (Electron Microscopy Center, University of South Carolina) for his assistance with TEM. Authors would like to thank Mr. Ryan Cassaro and Ms. Sowjanya Kadali for their assistance with peptide synthesis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jabbari E. Targeted delivery with peptidomimetic conjugated self-assembled nanoparticles. Pharm Res. 2009;26(3):612–630. doi: 10.1007/s11095-008-9802-1. [DOI] [PubMed] [Google Scholar]
- 2.Owens DE, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;307(1):93–102. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 3.Upadhyay KK, Agrawal HG, Upadhyay C, Schatz C, Le Meins JF, Misra A, Lecommandoux S. Role of block copolymer nanoconstructs in cancer therapy. Crit Rev Ther Drug. 2009;26(2):157–205. doi: 10.1615/critrevtherdrugcarriersyst.v26.i2.20. [DOI] [PubMed] [Google Scholar]
- 4.Rossi EA, Sharkey RM, McBride W, Karacay H, Zeng L, Hansen HJ, Goldenberg DM, Chang CH. Development of new multivalent-bispecific agents for pretargeting tumor localization and therapy. Clin Cancer Res. 2003;9(10):3886S–3896S. [PubMed] [Google Scholar]
- 5.Pirollo KF, Chang EH. Does a targeting ligand influence nanoparticle tumor localization or uptake? Trends Biotechnol. 2008;26(10):552–558. doi: 10.1016/j.tibtech.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 6.Buyens K, De Smedt SC, Braeckmans K, Demeester J, Peeters L, van Grunsven LA, du Jeu XD, Sawant R, Torchilin V, Farkasova K, Ogris M, Sanders NN. Liposome based systems for systemic siRNA delivery: Stability in blood sets the requirements for optimal carrier design. J Contr Rel. 2012;158(3):362–370. doi: 10.1016/j.jconrel.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 7.Battaglia L, Gallarate M. Lipid nanoparticles: state of the art, new preparation methods and challenges in drug delivery. Exp Opin Drug Deliv. 2012;9(5):497–508. doi: 10.1517/17425247.2012.673278. [DOI] [PubMed] [Google Scholar]
- 8.Liu ZH, Wang YT, Zhang N. Micelle-like nanoassemblies based on polymer-drug conjugates as an emerging platform for drug delivery. Exp Opin Drug Deliv. 2012;9(7):805–822. doi: 10.1517/17425247.2012.689284. [DOI] [PubMed] [Google Scholar]
- 9.Ma WX, Chen MS, Kaushal S, McElroy M, Zhang Y, Ozkan C, Bouvet M, Kruse C, Grotjahn D, Ichim T, Minev B. PLGA nanoparticle-mediated delivery of tumor antigenic peptides elicits effective immune responses. Int J Nanomed. 2012;7:1475–1487. doi: 10.2147/IJN.S29506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parveen S, Sahoo SK. Long circulating chitosan/PEG blended PLGA nanoparticle for tumor drug delivery. Eur J Pharmacol. 2011;670(2–3):372–383. doi: 10.1016/j.ejphar.2011.09.023. [DOI] [PubMed] [Google Scholar]
- 11.Gupta M, Agrawal GP, Vyas SP. Polymeric nanomedicines as promising vehicle for solid tumor therapy and targeting. Curr Mol Med. 2012 [PubMed] [Google Scholar]
- 12.Deepa G, Kumar NA, Pillai JJ, Kumar GS. Polymer Nanoparticles - A novel strategy for administration of paclitaxel in cancer chemotherapy. Curr Med Chem. 2012 [PubMed] [Google Scholar]
- 13.Nishiyama N, Kataoka K. Current state, achievements, and future prospects of polymeric micelles as nanocarriers for drug and gene delivery. Pharmacol Therapeut. 2006;112(3):630–648. doi: 10.1016/j.pharmthera.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 14.Avgoustakis K, Beletsi A, Panagi Z, Klepetsanis P, Karydas AG, Ithakissios DS. PLGA-mPEG nanoparticles of cisplatin: in vitro nanoparticle degradation, in vitro drug release and in vivo drug residence in blood properties. J Contr Rel. 2002;79(1–3):123–135. doi: 10.1016/s0168-3659(01)00530-2. [DOI] [PubMed] [Google Scholar]
- 15.Manchanda R, Fernandez-Fernandez A, Nagesetti A, McGoron AJ. Preparation and characterization of a polymeric (PLGA) nanoparticulate drug delivery system with simultaneous incorporation of chemotherapeutic and thermo-optical agents. Colloids Surf B: Biointerf. 2010;75(1):260–267. doi: 10.1016/j.colsurfb.2009.08.043. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Z, Feng S-S. Nanoparticles of poly(lactide)/vitamin E TPGS copolymer for cancer chemotherapy: Synthesis, formulation, characterization and in vitro drug release. Biomaterials. 2006;27(2):262–270. doi: 10.1016/j.biomaterials.2005.05.104. [DOI] [PubMed] [Google Scholar]
- 17.Musyanovych A, Dausend J, Dass M, Walther P, Mailander V, Landfester K. Criteria impacting the cellular uptake of nanoparticles: A study emphasizing polymer type and surfactant effects. Acta Biomater. 2011;7(12):4160–4168. doi: 10.1016/j.actbio.2011.07.033. [DOI] [PubMed] [Google Scholar]
- 18.Li M, Panagi Z, Avgoustakis K, Reineke J. Physiologically based pharmacokinetic modeling of PLGA nanoparticles with varied mPEG content. Int J Nanomedicine. 2012;7:1345–1356. doi: 10.2147/IJN.S23758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park J, Fong PM, Lu J, Russell KS, Booth CJ, Saltzman WM, Fahmy TM. PEGylated PLGA nanoparticles for the improved delivery of doxorubicin. Nanomed Nanotechnol Biol Med. 2009;5(4):410–418. doi: 10.1016/j.nano.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu P, Qin L, Wang Q, Sun Y, Zhu M, Shen M, Duan Y. cRGD-functionalized mPEG-PLGA-PLL nanoparticles for imaging and therapy of breast cancer. Biomaterials. 2012;33(28):6739–6747. doi: 10.1016/j.biomaterials.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 21.Mercado AE, Yang X, He X, Jabbari E. Effect of grafting BMP2-derived peptide to nanoparticles on osteogenic and vasculogenic expression of stromal cells. J Tissue Eng Regen Med. 2012 doi: 10.1002/term.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ge J, Neofytou E, Lei J, Beygui RE, Zare RN. Protein-Polymer Hybrid Nanoparticles for Drug Delivery. Small. 2012;8(23):3573–3578. doi: 10.1002/smll.201200889. [DOI] [PubMed] [Google Scholar]
- 23.Clift MJ, Rothen-Rutishauser B, Brown DM, Duffin R, Donaldson K, Proudfoot L, Guy K, Stone V. The impact of different nanoparticle surface chemistry and size on uptake and toxicity in a murine macrophage cell line. Toxicol Appl Pharmacol. 2008;232(3):418–427. doi: 10.1016/j.taap.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 24.Niu Y, Bai G, Wu H, Wang RE, Qiao Q, Padhee S, Buzzeo R, Cao C, Cai J. Cellular translocation of a gamma-AApeptide mimetic of Tat peptide. Mol Pharm. 2012;9(5):1529–1534. doi: 10.1021/mp300070w. [DOI] [PubMed] [Google Scholar]
- 25.Santoso S, Hwang W, Hartman H, Zhang S. Self-assembly of surfactant-like peptides with variable glycine tails to form nanotubes and nanovesicles. Nano Lett. 2002;2(7):687–691. [Google Scholar]
- 26.von Maltzahn G, Vauthey S, Santoso S, Zhang SU. Positively charged surfactant-like peptides self-assemble into nanostructures. Langmuir. 2003;19(10):4332–4337. [Google Scholar]
- 27.Hillaireau H, Couvreur P. Nanocarriers’ entry into the cell: relevance to drug delivery. Cell Mol Life Sci. 2009;66(17):2873–2896. doi: 10.1007/s00018-009-0053-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moeinzadeh S, Jabbari E. Mesoscale simulation of the effect of a lactide segment on the nanostructure of star poly(ethylene glycol-co-lactide)-acrylate macromonomers in aqueous solution. J Phys Chem B. 2012;116(5):1536–1543. doi: 10.1021/jp211056p. [DOI] [PubMed] [Google Scholar]
- 29.Groot RD, Warren PB. Dissipative particle dynamics: Bridging the gap between atomistic and mesoscopic simulation. J Chem Phys. 1997;107(11):4423–4435. [Google Scholar]
- 30.Jabbari E, He X. Synthesis and characterization of bioresorbable in situ crosslinkable ultra low molecular weight poly(lactide) macromer. J Mater Sci Mater Med. 2008;19(1):311–318. doi: 10.1007/s10856-006-0020-2. [DOI] [PubMed] [Google Scholar]
- 31.Jabbari E, He X, Valarmathi MT, Sarvestani AS, Xu W. Material properties and bone marrow stromal cells response to in situ crosslinkable RGD-functionlized lactide-co-glycolide scaffolds. J Biomed Mater Res A. 2009;89(1):124–137. doi: 10.1002/jbm.a.31936. [DOI] [PubMed] [Google Scholar]
- 32.He X, Jabbari E. Material properties and cytocompatibility of injectable MMP degradable poly(lactide ethylene oxide fumarate) hydrogel as a carrier for marrow stromal cells. Biomacromolecules. 2007;8(3):780–792. doi: 10.1021/bm060671a. [DOI] [PubMed] [Google Scholar]
- 33.He X, Ma J, Jabbari E. Effect of grafting RGD and BMP-2 protein-derived peptides to a hydrogel substrate on osteogenic differentiation of marrow stromal cells. Langmuir. 2008;24(21):12508–12516. doi: 10.1021/la802447v. [DOI] [PubMed] [Google Scholar]
- 34.He X, Jabbari E. Solid-phase synthesis of reactive peptide crosslinker by selective deprotection. Prot Pept Lett. 2006;13:515–518. doi: 10.2174/092986606777790610. [DOI] [PubMed] [Google Scholar]
- 35.Kaiser E, Bossinger CD, Colescott RL, Olsen DB. Color test for terminal prolyl residues in the solid-phase synthesis of peptides. Anal Chim Acta. 1980;118(1):149–151. [Google Scholar]
- 36.Mercado AE, He X, Xu W, Jabbari E. The release characteristics of a model protein from self-assembled succinimide-terminated poly(lactide-co-glycolide ethylene oxide fumarate) nanoparticles. Nanotechnology. 2008;19:12. doi: 10.1088/0957-4484/19/32/325609. [DOI] [PubMed] [Google Scholar]
- 37.He X, Ma J, Mercado AE, Xu WJ, Jabbari E. Cytotoxicity of Paclitaxel in biodegradable self-assembled core-shell poly(lactide-co-glycolide ethylene oxide fumarate) nanoparticles. Pharm Res. 2008;25(7):1552–1562. doi: 10.1007/s11095-007-9513-z. [DOI] [PubMed] [Google Scholar]
- 38.Faisant N, Akiki J, Siepmann F, Benoit JP, Siepmann J. Effects of the type of release medium on drug release from PLIGA-based microparticles: Experiment and theory. Int J Pharmaceut. 2006;314(2):189–197. doi: 10.1016/j.ijpharm.2005.07.030. [DOI] [PubMed] [Google Scholar]
- 39.Huang M, Ma Z, Khor E, Lim L-Y. Uptake of FITC-chitosan nanoparticles by a549 cells. Pharm Res. 2002;19(10):1488–1494. doi: 10.1023/a:1020404615898. [DOI] [PubMed] [Google Scholar]
- 40.Yang X, Wood PA, Oh EY, Du-Quiton J, Ansell CM, Hrushesky WJM. Down regulation of circadian clock gene Period 2 accelerates breast cancer growth by altering its daily growth rhythm. Breast Cancer Res Treat. 2009;117(2):423–431. doi: 10.1007/s10549-008-0133-z. [DOI] [PubMed] [Google Scholar]
- 41.He X, Ma J, Jabbari E. Migration of marrow stromal cells in response to sustained release of stromal-derived factor-1[alpha] from poly(lactide ethylene oxide fumarate) hydrogels. Int J Pharmaceut. 2010;390(2):107–116. doi: 10.1016/j.ijpharm.2009.12.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Elstner A, Holtkamp N, Deimling Av. Involvement of Hif-1 in desferrioxamine-induced invasion of glioblastoma cells. Clin Exp Metastasis. 2007;24:57–66. doi: 10.1007/s10585-007-9057-y. [DOI] [PubMed] [Google Scholar]
- 43.Shorts-Cary L, Xu M, Ertel J, Kleinschmidt-Demasters BK, Lillehei K, Matsuoka I, Nielsen-Preiss S, Wierman ME. Bone morphogenetic protein and retinoic acid- inducible neural specific protein-3 is expressed in gonadotrope cell pituitary adenomas and induces proliferation, migration, and invasion. Endocrinology. 2007;148(3):967–975. doi: 10.1210/en.2006-0905. [DOI] [PubMed] [Google Scholar]
- 44.Guo XD, Zhang LJ, Wu ZM, Qian Y. Dissipative particle dynamics studies on microstructure of ph-sensitive micelles for sustained drug delivery. Macromolecules. 2010;43(18):7839–7844. [Google Scholar]
- 45.Guo XD, Tan JPK, Kim SH, Zhang LJ, Zhang Y, Hedrick JL, Yang YY, Qian Y. Computational studies on self-assembled paclitaxel structures: Templates for hierarchical block copolymer assemblies and sustained drug release. Biomaterials. 2009;30(33):6556–6563. doi: 10.1016/j.biomaterials.2009.08.022. [DOI] [PubMed] [Google Scholar]
- 46.Espanol P, Warren P. Statistical-mechanics of dissipative particle dynamics. Europhys Lett. 1995;30(4):191–196. [Google Scholar]
- 47.Hoogerbrugge PJ, Koelman JMVA. Simulating microscopic hydrodynamic phenomena with dissipative particle dynamics. Europhys Lett. 1992;19(3):155–160. [Google Scholar]
- 48.Groot RD, Rabone KL. Mesoscopic simulation of cell membrane damage, morphology change and rupture by nonionic surfactants. Biophys J. 2001;81(2):725–736. doi: 10.1016/S0006-3495(01)75737-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Materials Studio, V 5.5. Accelrys; San Diego: 2011. [Google Scholar]
- 50.Sun H. COMPASS: An ab initio force-field optimized for condensed-phase applications - Overview with details on alkane and benzene compounds. J Phys Chem B. 1998;102(38):7338–7364. [Google Scholar]
- 51.Russell TP. The characterization of polymer interfaces. Ann Rev Mater Sci. 1991;21:249–268. [Google Scholar]
- 52.Hobbs SK, Monsky WL, Yuan F, Roberts WG, Griffith L, Torchilin VP, Jain RK. Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc Natl Acad Sci. 1998;95:4607–4612. doi: 10.1073/pnas.95.8.4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Unezaki S, Maruyama K, Hosoda J, Nagae I, Koyanagi Y, Nakata M, Ishida O, Iwatsuru M, Tsuchiya S. Direct measurement of the extravasation of polyethyleneglycol-coated liposomes into solid tumor tissue by in vivo fluorescence microscopy. Int J Pharm. 1996;144:11–17. [Google Scholar]
- 54.Ahmed F, Pakunlu RI, Brannan A, Bates F, Minko T, Discher DE. Biodegradable polymersomes loaded with both paclitaxel and doxorubicin permeate and shrink tumors, inducing apoptosis in proportion to accumulated drug. J Contr Rel. 2006;116:150–158. doi: 10.1016/j.jconrel.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 55.Lin R, Ng LS, Wang C-H. In vitro study of anticancer drug doxorubicin in PLGA-based microparticles. Biomaterials. 2005;26:4476–4485. doi: 10.1016/j.biomaterials.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 56.Xu X, Chen X, Wang Z, Jing X. Ultrafine PEG–PLA fibers loaded with both paclitaxel and doxorubicin hydrochloride and their in vitro cytotoxicity. Eur J Pharm Biopharm. 2009;72:18–25. doi: 10.1016/j.ejpb.2008.10.015. [DOI] [PubMed] [Google Scholar]
- 57.Yang T, Cui FD, Choi MK, Cho JW, Chung SJ, Shim CK, Kim DD. Enhanced solubility and stability of PEGylated liposomal paclitaxel: In vitro and in vivo evaluation. Int J Pharmaceut. 2007;338(1–2):317–326. doi: 10.1016/j.ijpharm.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 58.Lee SC, Huh KM, Lee J, Cho YW, Galinsky RE, Park K. Hydrotropic polymeric micelles for enhanced paclitaxel solubility: In vitro and in vivo characterization. Biomacromolecules. 2007;8(1):202–208. doi: 10.1021/bm060307b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li XG, Hirsh DJ, Cabral-Lilly D, Zirkel A, Gruner SM, Janoff AS, Perkins WR. Doxorubicin physical state in solution and inside liposomes loaded via a pH gradient. Biochimica Et Biophysica Acta-Biomembranes. 1998;1415(1):23–40. doi: 10.1016/s0005-2736(98)00175-8. [DOI] [PubMed] [Google Scholar]
- 60.Crow BB, Borneman AF, Hawkins DL, Smith GM, Nelson KD. Evaluation of in vitro drug release, pH change, and molecular weight degradation of poly(L-lactic acid) and poly(D, L-lactide-co-glycolide) fibers. Tissue Eng. 2005;11(7–8):1077–1084. doi: 10.1089/ten.2005.11.1077. [DOI] [PubMed] [Google Scholar]
- 61.Messaritaki A, Black SJ, van der Walle CF, Rigby SP. NMR and confocal microscopy studies of the mechanisms of burst drug release from PLGA microspheres. J Cont Rel. 2005;108(2–3):271–281. doi: 10.1016/j.jconrel.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 62.Hu Y, Zhang L, Cao Y, Ge H, Jiang X, Yang C. Degradation behavior of poly(ε-caprolactone)-b-poly(ethylene glycol)-b-poly(ε-caprolactone) micelles in aqueous solution. Biomacromolecules. 2004;5:1756–1762. doi: 10.1021/bm049845j. [DOI] [PubMed] [Google Scholar]
- 63.Bulaj G. Formation of disulfide bonds in proteins and peptides. Biotechnol Adv. 2005;23(1):87–92. doi: 10.1016/j.biotechadv.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 64.Oyewumi MO, Mumper RJ. Influence of formulation parameters on gadolinium entrapment and tumor cell uptake using folate-coated nanoparticles. Int J Pharm. 2003;251:85–97. doi: 10.1016/s0378-5173(02)00587-2. [DOI] [PubMed] [Google Scholar]
- 65.Oyewumi MO, Yokel RA, Jay M, Coakley T, Mumper RJ. Comparison of cell uptake, biodistribution and tumor retention of folate-coated and PEG-coated gadolinium nanoparticles in tumorbearing mice. J Contr Rel. 2004;95:613–626. doi: 10.1016/j.jconrel.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 66.Chithrani BD, Ghazani AA, Chan WC. Determining the size and shape dependence of gold nanoparticle uptake into mammalian cells. Nano Lett. 2006;6:662–668. doi: 10.1021/nl052396o. [DOI] [PubMed] [Google Scholar]
- 67.O’Brien ME, Wigler N, Inbar M, et al. Reduced cardiotoxicity and comparable efficacy in a Phase III trial of pegylated liposomal doxorubicinHCl (CAELYX/Doxil) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Ann Oncol. 2004;15:440–449. doi: 10.1093/annonc/mdh097. [DOI] [PubMed] [Google Scholar]
- 68.Nakase I, Tadokoro A, Kawabata N, Takeuchi T, Katoh H, Hiramoto K, Negishi M, Nomizu M, Sugiura Y, Futaki S. Interaction of arginine-rich peptides with membrane-associated proteoglycans is crucial for induction of actin organization and macropinocytosis. Biochemistry. 2007;46:492–501. doi: 10.1021/bi0612824. [DOI] [PubMed] [Google Scholar]
- 69.Brooks H, Lebleu B, Vives E. Tat peptide-mediated cellular delivery: back to basics. Adv Drug Deliv Rev. 2005;57:559–577. doi: 10.1016/j.addr.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 70.Hansen M, Kilk K, Langel U. Predicting cell-penetrating peptides. Adv Drug Deliv Rev. 2008;60(4–5):572–579. doi: 10.1016/j.addr.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 71.Rothbard JB, Jessop TC, Lewis RS, Murray BA, Wender PA. Role of membrane potential and hydrogen bonding in the mechanismof translocation of guanidinium-rich peptides into cells. J Am Chem Soc. 2004;126:9506–9507. doi: 10.1021/ja0482536. [DOI] [PubMed] [Google Scholar]
- 72.Goodman CM, McCusker CD, Yilmaz T, Rotello VM. Toxicity of gold nanoparticles functionalized with cationic and anionic side chains. Bioconj Chem. 2004;15(4):897–900. doi: 10.1021/bc049951i. [DOI] [PubMed] [Google Scholar]
- 73.Bexiga MG, Varela JA, Wang FJ, Fenaroli F, Salvati A, Lynch I, Simpson JC, Dawson KA. Cationic nanoparticles induce caspase 3-, 7- and 9-mediated cytotoxicity in a human astrocytoma cell line. Nanotoxicology. 2011;5(4):557–567. doi: 10.3109/17435390.2010.539713. [DOI] [PubMed] [Google Scholar]
- 74.Chompoosor A, Saha K, Ghosh PS, Macarthy DJ, Miranda OR, Zhu ZJ, Arcaro KF, Rotello VM. The role of surface functionality on acute cytotoxicity, ros generation and dna damage by cationic gold nanoparticles. Small. 2010;6(20):2246–2249. doi: 10.1002/smll.201000463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Basarkar A, Devineni D, Palaniappan R, Singh J. Preparation, characterization, cytotoxicity and transfection efficiency of poly(DL-lactide-co-glycolide) and poly(DL-lactic acid) cationic nanoparticles for controlled delivery of plasmid DNA. Int J Pharmaceut. 2007;343(1–2):247–254. doi: 10.1016/j.ijpharm.2007.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhao J, Gou ML, Dai M, Li XY, Cao M, Huang MJ, Wen YJ, Kan B, Qian ZY, Wei YQ. Preparation, characterization, and in vitro cytotoxicity study of cationic PCL-Pluronic-PCL (PCFC) nanoparticles for gene delivery. J Biomed Mater Res A. 2009;90A(2):506–513. doi: 10.1002/jbm.a.31950. [DOI] [PubMed] [Google Scholar]
- 77.Tomita Y, Rikimaru-Kaneko A, Hashiguchi K, Shirotake S. Effect of anionic and cationic n-butylcyanoacrylate nanoparticles on NO and cytokine production in Raw cells. Immunopharmacol Immunotoxicol. 2011;33(4):730–737. doi: 10.3109/08923973.2011.565345. [DOI] [PubMed] [Google Scholar]
- 78.Merhi M, Dombu CY, Brient A, Chang J, Platel A, Le Curieux F, Marzin D, Nesslany F, Betbeder D. Study of serum interaction with a cationic nanoparticle: Implications for in vitro endocytosis, cytotoxicity and genotoxicity. Int J Pharmaceut. 2012;423(1):37–44. doi: 10.1016/j.ijpharm.2011.07.014. [DOI] [PubMed] [Google Scholar]
- 79.Zhang HY, Xia T, Meng H, Xue M, George S, Ji ZX, Wang X, Liu R, Wang MY, France B, Rallo R, Damoiseaux R, Cohen Y, Bradley KA, Zink JI, Nel AE. Differential expression of syndecan-1 mediates cationic nanoparticle toxicity in undifferentiated versus differentiated normal human bronchial epithelial cells. ACS Nano. 2011;5(4):2756–2769. doi: 10.1021/nn200328m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lu W, Tan YZ, Hu KL, Jiang XG. Cationic albumin conjugated pegylated nanoparticle with its transcytosis ability and little toxicity against blood-brain barrier. Int J Pharmaceut. 2005;295(1–2):247–260. doi: 10.1016/j.ijpharm.2005.01.043. [DOI] [PubMed] [Google Scholar]
- 81.Niu GQ, Castro CH, Nguyen N, Sullivan SM, Hughes JA. In vitro cytotoxic activity of cationic paclitaxel nanoparticles on MDR-3T3 cells. J Drug Targeting. 2010;18(6):468–476. doi: 10.3109/10611860903508804. [DOI] [PubMed] [Google Scholar]
- 82.Foster ER, Downs JA. Histone H2A phosphorylation in DNA double-strand break repair. FEBS J. 2005;272(13):3231–3240. doi: 10.1111/j.1742-4658.2005.04741.x. [DOI] [PubMed] [Google Scholar]
- 83.Perdiguero E, Nebreda AR. Regulation of Cdc25C activity during the meiotic G(2)/M transition. Cell Cycle. 2004;3(6):733–737. [PubMed] [Google Scholar]
- 84.Riedl SJ, Salvesen GS. The apoptosome: signalling platform of cell death. Nature Rev Mol Cell Biol. 2007;8(5):405–413. doi: 10.1038/nrm2153. [DOI] [PubMed] [Google Scholar]
- 85.Twelves CJ, Dobbs NA, Aldhous M, Harper PG, Rubens RD, Richards MA. Comparative pharmacokinetics of doxorubicin given by 3 different schedules with equal dose intensity in patients with breast-cancer. Cancer Chemother Pharmacol. 1991;28(4):302–307. doi: 10.1007/BF00685539. [DOI] [PubMed] [Google Scholar]
- 86.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 87.Guano F, Pourquier P, Tinelli S, Binaschi M, Bigioni M, Animati F, Manzini S, Zunino F, Kohlhagen G, Pommier Y, Capranico G. Topoisomerase poisoning activity of novel disaccharide anthracyclines. Mol Pharmacol. 1999;56:77–84. doi: 10.1124/mol.56.1.77. [DOI] [PubMed] [Google Scholar]
- 88.Ayen WY, Kumar N. In vivo evaluation of doxorubicin-loaded (peg)(3)-pla nanopolymersomes (polydoxsome) using dmba-induced mammary carcinoma rat model and comparison with marketed LipoDox. Pharm Res. 2012;29(9):2522–2533. doi: 10.1007/s11095-012-0783-8. [DOI] [PubMed] [Google Scholar]
- 89.Amoozgar Z, Yeo Y. Recent advances in stealth coating of nanoparticle drug delivery systems. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2012;4(2):219–233. doi: 10.1002/wnan.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhan C, Gu B, Xie C, Li J, Liu Y, Lu W. Cyclic RGD conjugated poly(ethylene glycol)-co-poly(lactic acid) micelle enhances paclitaxel anti-glioblastoma effect. J Contr Rel. 2010;143(1):136–142. doi: 10.1016/j.jconrel.2009.12.020. [DOI] [PubMed] [Google Scholar]
- 91.Xia H, Gao X, Gu G, Liu Z, Hu Q, Tu Y, Song Q, Yao L, Pang Z, Jiang X, Chen J, Chen H. Penetratin-functionalized PEG-PLA nanoparticles for brain drug delivery. Int J Pharm. 2012;436(1–2):840–850. doi: 10.1016/j.ijpharm.2012.07.029. [DOI] [PubMed] [Google Scholar]