

Abstract
Background
Prucalopride is a selective, high-affinity, 5-hydroxytryptamine 4 (5-HT4) receptor agonist, which is approved for the symptomatic treatment of chronic constipation in women in whom laxatives fail to provide adequate relief. Women of childbearing potential, many of whom will be using oral contraceptives, comprise a large proportion of patients seeking medical therapy for constipation.
Objective
The aim of this study was to evaluate the effect of prucalopride on the absorption and steady-state pharmacokinetics of oral contraceptives in healthy women.
Methods
Sixteen women (aged 18–45 years) were enrolled in this open-label, two-way crossover trial (ClinicalTrials.gov identifier: NCT01036893) and given two 5-day treatments with a once-daily oral contraceptive (ethinylestradiol 0.035 mg + norethisterone 1 mg), alone and in combination with prucalopride 2 mg once daily. Treatments were separated by a 7 ± 2-day washout period. On days 1 and 5, blood samples were obtained pre-dose and at regular intervals post-dose up to 24 and 48 hours, respectively, to determine ethinylestradiol and norethisterone plasma concentrations. Prucalopride plasma concentrations were determined pre-dose and 3 hours post-dose on days 1 and 5, and 24 hours post-dose on day 6. Safety was assessed.
Results
Thirteen participants completed the study. One participant was thought to be non-compliant on days 3 and/or 4, and was excluded from the day 5 analysis. On days 1 and 5, maximum plasma concentrations of both oral contraceptive constituents were attained in ~1 hour and were unaffected by prucalopride administration. On day 5, steady-state prucalopride and oral contraceptive concentrations had been achieved. Prucalopride did not affect the pharmacokinetics of the oral contraceptives: point estimates for the maximum plasma concentration and area under the plasma concentration–time curve values and their associated 90 % confidence intervals were contained within predefined equivalence limits (80–125 %). Prucalopride was well tolerated, with a safety profile consistent with those observed in previous studies.
Conclusion
Co-administration of prucalopride with an oral contraceptive did not result in any clinically meaningful pharmacokinetic interactions and was well tolerated.
Introduction
The dihydrobenzofuran-carboxamide derivative prucalopride is the first selective, high-affinity, 5-hydroxytryptamine 4 (5-HT4) receptor agonist with potent gastrointestinal prokinetic activity [1, 2]. Prucalopride, at a standard dose of 2 mg once daily, is approved for the symptomatic treatment of chronic constipation in women in whom laxatives fail to provide adequate relief [1]. In three identical pivotal phase III trials in patients with chronic constipation, prucalopride 2 mg once daily for 12 weeks increased the frequency of spontaneous complete bowel movements, improved patient satisfaction with treatment and bowel function, and improved patient perception of constipation severity and constipation-related quality of life [3–5]. In these studies, prucalopride was generally well tolerated, with most adverse events (AEs) being mild to moderate in severity and transient in nature. Across the pivotal trials, the most frequently reported AEs associated with therapy were headache (25 % of patients) and gastrointestinal symptoms (nausea [19 %], diarrhea [12 %], or abdominal pain [12 %]) [3, 4]. AEs occurred predominantly at the start of therapy and usually disappeared within a few days with continued treatment [3, 4].
The prevalence of chronic constipation in the general population is relatively high, with 5–18 % of individuals reporting some form of constipation [6], although the actual numbers may be underestimated because a large proportion do not seek medical attention for their condition [7]. Women, particularly those younger than 50 years, present with constipation more commonly than men (prevalence ratio 2.2:1) [8–10]. Women of childbearing potential, many of whom will be using oral contraceptives, therefore comprise a large proportion of those seeking medical therapy for constipation. It is thus important to understand whether treatments for chronic constipation interact with the pharmacokinetics of oral contraceptives.
Prucalopride has an established pharmacokinetic profile [2]. In summary, the maximum plasma concentration (Cmax) is reached within 2–3 hours of a single 2 mg oral dose. Absolute oral bioavailability is greater than 90 %, and absorption is not influenced by concomitant food intake, which indicates that the drug can be taken with or without meals. Prucalopride undergoes limited metabolism and is largely eliminated unchanged in the urine via passive renal filtration and active secretion. The elimination half-life (t½) of prucalopride is approximately 24–30 hours, supporting once-daily administration.
Compounds that induce cytochrome P450 (CYP) 3A4 (such as estrogen-2-hydroxylase) have been shown to reduce systemic exposure to contraceptive steroids such as ethinylestradiol and norethisterone [11], which carries with it the risks of spotting, breakthrough bleeding, and ultimately contraceptive failure [12]. Currently available data indicate that prucalopride does not act as an inducer of CYP3A4—in vivo studies of prucalopride administered for 1 week or more showed that it did not lower plasma concentrations of erythromycin or R-warfarin (data on file). However, interference with the absorption of oral contraceptives could theoretically result in considerably reduced plasma concentrations, with contraceptive failure as a consequence. In this study of healthy women, we therefore investigated the effects of prucalopride on the pharmacokinetics of the estrogen ethinylestradiol and the progestogen norethisterone, which are the active constituents of several oral contraceptives.
Methods
Study Design
This randomized, open-label, two-way crossover, phase I trial (ClinicalTrials.gov identifier: NCT01036893) was designed to evaluate both the effect of single-dose prucalopride 2 mg (Resolor®;1 prucalopride succinate tablets) on the absorption of ethinylestradiol and norethisterone, and the effect of 5 or 6 days of treatment with prucalopride 2 mg once daily on the steady-state pharmacokinetics of ethinylestradiol and norethisterone in healthy women. The trial was carried out at a single center in Germany (FOCUS Clinical Drug Development GmbH, Neuss, Germany) from December 17, 2009, until February 10, 2010, in accordance with the Declaration of Helsinki and the International Conference on Harmonisation Good Clinical Practice guidelines [13, 14], and was approved by the relevant independent ethics committees. All participants provided written informed consent before screening.
Participants
Eligibility was assessed at a screening visit, which took place within the 4 weeks before the first drug administration. Healthy women (in the age group of 18–45 years) who had regular menstrual cycles of 28 ± 3 days in the previous 6 months were eligible for inclusion in the study if they had a body mass index (BMI) of 18–27 kg/m2; had not smoked in the 6 months before screening; and were using adequate non-hormonal birth control such as the double-barrier method (e.g. a condom and spermicide, a cervical cap and spermicide), were practicing abstinence, or had a partner who was sterile (e.g. had undergone vasectomy).
Individuals were excluded from the study if they had a history or evidence of drug or alcohol abuse; had abnormal electrocardiogram (ECG) intervals or morphology (e.g. QT interval >500 ms or corrected QT interval using Bazett’s formula [QTcB] >470 ms) that were considered to be clinically significant; had a history or evidence of cardiac arrhythmias, bronchospastic disease, or cardiovascular disease; or had a history or evidence of psychiatric, gynecological, hepatic, gastrointestinal, renal, endocrine, neurological, or dermatological disease. Individuals with drug allergies, those who had contraindications for the use of oral contraceptives (e.g. known or suspected active venous thromboembolic disorders, hormone-dependent malignancies, coagulation disorders, menstrual cycle-dependent migraines, lipid metabolism disorders, or hepatic disorders), and those who had used other medications, oral contraceptives, or any hormonal depot device in the 6 months before screening were also excluded. In addition, individuals were excluded if they had participated in an investigational drug study or had donated blood in the 30 days before the first visit, had positive blood test results for human immunodeficiency virus (HIV) or hepatitis B or C at screening, or were pregnant or breastfeeding.
Treatments
In accordance with a two-way randomized crossover study design, participants were given two 5-day treatments (days 1–5 of each crossover phase; Fig. 1) with a once-daily oral contraceptive, once as monotherapy (treatment A) and once with once-daily prucalopride on days 1–6 of the treatment phase (treatment B). The washout period between the two contraceptive treatments was 7 ± 2 days. The stage of the patient’s menstrual cycle was not taken into account in the timings of these treatments. The oral contraceptive Trinovum® (ethinylestradiol 0.035 mg and norethisterone 1 mg; Janssen-Cilag Ltd) was used; prucalopride was administered as 2 mg film-coated tablets containing prucalopride succinate, equivalent to 2.0 mg prucalopride base.
Fig. 1.
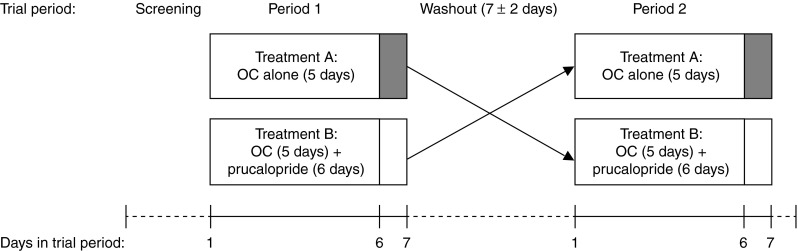
Overview of the trial design. OC oral contraceptive
The oral contraceptive dose was taken at 0800 hours. For the combination treatment, prucalopride was administered immediately before the oral contraceptive. The drugs were taken with a total of 200 mL of non-carbonated water. On days 1 and 5 of each treatment period, the study medication was administered in the clinic following an overnight fast of at least 10 hours, and participants were not permitted to eat or drink until 2 hours after receiving the medication, at which time they received a standard breakfast. On all other days, participants took the study treatments either at the clinic (days 2 and 6) or at home (days 3 and 4) 30 minutes before breakfast. Compliance was assessed by investigator supervision of dosing (except on days 3 and 4) and daily diary entries.
During the study, participants were not permitted to take medication other than the study drugs, with the exception of as-needed paracetamol/acetaminophen (up to a maximum of three 500 mg tablets per day, and no more than 3 g during the study).
Pharmacokinetic Assessments
Serial blood samples for the determination of ethinylestradiol and norethisterone concentrations in plasma were taken on day 1 pre-dose and then at 1, 2, 3, 4, 6, 8, 10, 12, and 24 hours post-dose, and on day 5 pre-dose and then at 1, 2, 3, 4, 6, 8, 10, 12, 24, 36, and 48 hours post-dose. Participants receiving treatment B had serial blood samples collected for the determination of plasma concentrations of prucalopride on days 1 and 5 pre-dose and then at 3 hours post-dose, and on day 6 pre-dose and then at 24 hours post-dose. No pharmacokinetic parameters were calculated for prucalopride.
Assay Validation
Plasma samples were analyzed for prucalopride, ethinylestradiol, and norethisterone, using validated liquid chromatography–tandem mass spectrometry (LC–MS/MS) methods. Assay methods were validated in accordance with the US Food and Drug Administration (FDA) Guidance for Industry: Bioanalytical Method Validation [15] and the Good Laboratory Practice regulations of the Organisation for Economic Co-operation and Development [16]. Validation parameters, including accuracy (expressed as bias), precision (percentage coefficient of variation), recovery, specificity, dilution, and stability were evaluated and amply met the acceptance criteria outlined in the FDA guidance [15].
The method for the determination of prucalopride in human heparin plasma was linear in the range of 0.200–100 ng/mL, with a lower limit of quantification (LLQ) of 0.200 ng/mL. Briefly, prucalopride was extracted from 50 μL plasma by liquid–liquid extraction with tertiary butyl methyl ether under alkaline conditions, using an analog (SSP-002392) as the internal standard. High-performance liquid chromatography–tandem mass spectrometry (HPLC–MS/MS) analysis was carried out with an API-4000 mass spectrometer (AB Sciex, Toronto, ON, Canada) coupled with an Agilent 1100 HPLC system (Agilent, Santa Clara, CA, USA). The mass spectrometer was operating in positive electrospray ionization (ESI) mode, and the chromatographic separation was achieved on a Zorbax Extend-C18 3.5 μm HPLC column, 4.6 × 75 mm, with a mobile-phase gradient.
For ethinylestradiol, the method was linear in the range of 3.00–600 pg/mL, with an LLQ of 3.00 pg/mL, using 500 μL of plasma. Ethinylestradiol and its deuterated internal standard (ethinylestradiol-d4) were extracted from plasma by solid-phase extraction on Isolute C18 (EC) cartridges (Biotage, Uppsala, Sweden). Subsequently, ethinylestradiol was derivatized with dansyl chloride and the derivate was extracted using liquid–liquid extraction with a mixture of tertiary butyl methyl ether and pentane. HPLC–MS/MS analysis was performed using the API-4000 mass spectrometer coupled with the Agilent 1100 HPLC system. The mass spectrometer was operating in positive atmospheric pressure chemical ionization (APCI) mode, and the chromatographic separation was achieved on a Hypersil C8 BDS HPLC column (3.0 μm, 4.6 × 150 mm), with a mobile-phase gradient.
For norethisterone, the method was linear in the range of 0.0500–20.0 ng/mL, with an LLQ of 0.0500 ng/mL, using 500 μL of plasma. In summary, norethisterone and its stable isotope-labeled internal standard (13C2-norethisterone) were extracted from plasma by online solid-phase extraction on HySphere C8 EC-SE cartridges, using a Symbiosis Pharma system (Spark Holland BV, Emmen, The Netherlands), which was preceded by liquid–liquid extraction with a mixture of chloroform and pentane. Chromatographic separation was achieved on a Zorbax XDB-C8 HPLC column (3.5 μm, 75 × 4.6 mm), with a mobile-phase gradient. The API-4000 mass spectrometer was operating in positive APCI mode.
In the current study, each analytical run consisted of a freshly prepared calibration curve, using blank human heparin plasma for all three analytes. Quality control (QC) samples were prepared at three different concentrations (prucalopride: 0.600, 6.00, and 80.0 ng/mL; ethinylestradiol: 9.00, 50.0, and 150 pg/mL; norethisterone: 0.150, 1.00, and 16.0 ng/mL), stored with the study samples, and analyzed in duplicate divided over the analytical run. Run acceptance was performed in accordance with the FDA Guidance for Industry: Bioanalytical Method Validation [15]. In this study, the overall accuracy of the QC samples ranged from −0.4 % to 3.4 % for prucalopride, from 1.1 % to 2.4 % for ethinylestradiol, and from 0.0 % to 0.4 % for norethisterone. The precision ranged from 2.9 % to 4.2 % for prucalopride, from 2.9 % to 8.3 % for ethinylestradiol, and from 1.9 % to 5.8 % for norethisterone. In all methods, no interference was observed at the retention time of the analytes and their internal standards. Moreover, >66 % of 48 re-analyzed plasma samples (for ethinylestradiol and norethisterone) or 12 re-analyzed plasma samples (for prucalopride) showed differences of ≤20 % compared with the original result, therefore demonstrating incurred sample reproducibility for all three analytes.
Pharmacokinetic Analysis
Pharmacokinetic analyses were performed using WinNonlin® software (version 5.20; Pharsight Corporation, Mountain View, CA, USA) and Statistical Analysis System (SAS®) software (version 9.1.3; SAS® Institute Inc., Cary, NC, USA). The following pharmacokinetic parameters were determined on day 1 for norethisterone and ethinylestradiol: Cmax, time to reach Cmax (tmax), and area under the plasma concentration–time curve (AUC) during the first 24-hour dosing interval (AUC24) calculated by linear trapezoidal summation. On day 5, the following parameters were determined: the minimum plasma concentration during a 24-hour dosing interval (Cmin), Cmax, AUC during a 24-hour dosing interval (AUCτ) calculated by linear trapezoidal summation, and t½, defined as 0.693/λ, where λ is the elimination rate constant determined by linear regression of the terminal points of the log-linear plasma concentration–time curve.
Safety Assessments
Safety was assessed by AEs (recorded throughout the study); clinical laboratory measurements (performed at screening, pre-dose on day 1 and day 7 of each treatment period, and at the final visit or discontinuation); physical examinations (at screening, on day 1 of each treatment period, and at the final visit or discontinuation); assessments of vital signs (at screening, pre-dose on day 1, at the end of each treatment period, and at the final visit or discontinuation); and 12-lead ECGs (at screening, on day 1 of each treatment period, and at the final visit or discontinuation). A blood sample for serology testing (HIV and hepatitis B and C) was obtained at screening, and samples for hematology and coagulation tests were obtained at screening, on days 1 and 7 of each treatment period, and at the final visit or discontinuation. Drug and alcohol screening (at screening and on day −1 of each treatment period) and pregnancy testing (at screening, on day −1 of each treatment period, and at the final visit or discontinuation) were also performed.
Statistical Analysis
No formal sample size calculation was performed. A sample size of 16 participants (in order to have at least 12 individuals completing the trial) with a crossover design was considered sufficient to determine relevant changes in the plasma concentrations of ethinylestradiol and norethisterone.
Descriptive statistics were calculated for the plasma concentrations of norethisterone and ethinylestradiol at each sampling time and for the derived pharmacokinetic parameters. Mixed effects modeling (with the participant as the random effect and with the sequence, period, and treatment as fixed effects) was used to compare natural log-transformed Cmax and AUC24 values between treatments on day 1, and to compare natural log-transformed Cmin, Cmax, and AUCτ values, and untransformed t½ values between treatments on day 5. Using the mean squared error from the model, 90 % confidence intervals (CIs) were calculated for the treatment difference (B − A) of the log-transformed bioavailability parameters Cmax and AUC24 on day 1, and Cmin, Cmax, and AUCτ values on day 5. Classical 90 % CIs for the ratios of treatment B (oral contraceptive plus prucalopride) to treatment A (oral contraceptive alone) were then obtained by exponentiation and expressed as percentages. The absence of an interaction was declared if the 90 % CIs were contained within the range of 80–125 %. The non-parametric Koch procedure was used to compare tmax values on day 1 and day 5 between treatments. A non-parametric 90 % CI for the treatment difference (B − A) was calculated using a distribution-free procedure adapted to two-way crossover designs. Descriptive statistics were calculated for the prucalopride plasma concentrations at each sampling time. Safety data were analyzed descriptively and comprised data from all participants who had taken study medication, including those not included in the pharmacokinetic analysis.
Results
Participants
Sixteen individuals were enrolled in the study, all of whom were Caucasian women. Their mean age was 35.5 years (range 19–44 years), their mean body weight was 65.9 kg (range 51–80 kg), their mean height was 169 cm (range 163–180 cm), and their mean BMI was 23.0 kg/m2 (range 19.0–27.0 kg/m2). At screening, all participants had a regular menstrual cycle and there were no abnormal findings.
Three participants discontinued the trial, all of whom were withdrawn because of AEs. These AEs (vomiting, dental pulpitis, and headache; all moderate in intensity) occurred with oral contraceptive plus prucalopride (treatment B) in the group that received this treatment combination first. These three participants therefore did not receive oral contraceptive alone. In total, 14 individuals reported protocol deviations, of which only two were major (prohibited concomitant medications used: ibuprofen 150 mg and metoclopramide 8 mg); both participants with major protocol deviations were among those who were withdrawn because of AEs. On day 5 of the oral contraceptive plus prucalopride period, one participant had pre-dose concentrations of prucalopride, ethinylestradiol, and norethisterone that were much lower than would be theoretically expected and much lower than the pre-dose concentrations measured on other days of the same treatment period in this participant. On day 3 this individual had reported nausea and vomiting, and on days 3 and 4 she had not reported intake of trial medication in her participant diary (although later she stated that she had taken the trial medication). After supervised drug intake on day 5, drug absorption appeared normal (as evidenced by the ethinylestradiol and norethisterone profiles on day 5, and the day 6 prucalopride pre-dose and 24-hour post-dose concentrations), which strongly suggests that this individual did not take the study medication on days 3 and/or 4. Therefore, statistical comparison of the day 5 pharmacokinetic parameters was also performed on a subset of 12 participants, excluding this suspected non-compliant participant.
Ethinylestradiol Pharmacokinetics
On day 1, Cmax was reached at a median time of 1 hour after dosing with both treatments (Fig. 2 and Table 1). There were no statistically significant differences in Cmax, tmax, or AUC24 between treatments (oral contraceptive vs. oral contraceptive plus prucalopride; Table 1). The geometric mean treatment ratios for Cmax and AUC24 were 110.37 % and 95.52 %, respectively, and the associated 90 % CIs were within the predefined equivalence limits of 80–125 % (Table 1).
Fig. 2.

Mean ethinylestradiol plasma concentration–time profiles on day 1 and day 5 (n = 13). OC oral contraceptive
Table 1.
Pharmacokinetic parameters and summary of the equivalence analysis for ethinylestradiol
| Parameter | Treatment A | Treatment B | OC + prucalopride versus OC alone | ||
|---|---|---|---|---|---|
| OC alonea | OC + prucalopridea | PE (%) | 90 % CI | p value | |
| Day 1 (n = 13) | |||||
| tmax (h) | 1.0 [1.0–2.0] | 1.0 [1.0–2.0] | 0.00 | −0.50, 0.00 | 0.4224 |
| Cmax (pg/mL) | 90.5 ± 21.8 | 103 ± 32.0 | 110.37 | 99.74, 122.13 | 0.1079 |
| AUC24 (pg·h/mL) | 727 ± 156 | 720 ± 204 | 95.52 | 90.70, 100.61 | 0.1409 |
| Day 5 (n = 13)b | |||||
| tmax (h) | 1.0 [1.0–3.0] | 1.0 [1.0–3.0] | −0.50 | −1.00, 0.00 | 0.0644 |
| Cmin (pg/mL) | 18.6 ± 7.4 | 17.8 ± 8.1 | 83.00 | 65.43, 105.29 | 0.1872 |
| Cmax (pg/mL) | 130 ± 34 | 123 ± 27 | 96.07 | 89.37, 103.28 | 0.3412 |
| AUCτ (pg·h/mL) | 1,153 ± 323 | 1,090 ± 296 | 92.54 | 85.07, 100.66 | 0.1260 |
| t½ (h) | 17.1 ± 2.4 | 15.0 ± 3.2 | – | – | 0.0154 |
| Day 5 (n = 12)b | |||||
| tmax (h) | 1.0 [1.0–3.0] | 1.0 [1.0–3.0] | −0.25 | −0.50, 0.00 | 0.1530 |
| Cmin (pg/mL) | 19.4 ± 7.0 | 19.3 ± 6.3 | 97.10 | 86.83, 108.59 | 0.6438 |
| Cmax (pg/mL) | 132 ± 35 | 126 ± 27 | 99.12 | 92.80, 105.88 | 0.8140 |
| AUCτ (pg·h/mL) | 1,135 ± 331 | 1,119 ± 288 | 97.65 | 93.36, 102.14 | 0.3605 |
| t½ (h) | 17.4 ± 2.2 | 15.3 ± 3.1 | – | – | 0.0305 |
aValues are expressed as means ± standard deviations, except for tmax, for which median [range] values are given
bResults are based on all data (n = 13) and on n = 12 after exclusion of one participant because circumstantial evidence indicated that her medication was not taken on days 3 and/or 4
AUC τ area under the plasma concentration–time curve during a 24-hour dosing interval, AUC 24 area under the plasma concentration–time curve during the first 24-hour dosing interval, CI confidence interval, C max maximum plasma concentration, C min minimum plasma concentration, OC oral contraceptive, PE point estimate of the geometric mean treatment ratio, t ½ elimination half-life, t max time to reach Cmax
Ethinylestradiol steady state was reached on day 5, with similar plasma concentrations of ethinylestradiol observed before and 24 hours after administration of oral contraceptive alone (20.7 ± 8.1 pg/mL and 20.5 ± 6.7 pg/mL, respectively) and oral contraceptive plus prucalopride (18.5 ± 8.5 pg/mL and 19.2 ± 6.7 pg/mL, respectively) [Fig. 2]. On day 5, Cmax was reached at a median time of 1 hour after dosing and there were no statistically significant differences in tmax, Cmin, Cmax, or AUCτ between treatments (Table 1). There was a statistically significant difference in t½, but this difference was considered too small to be clinically meaningful. The geometric mean treatment ratios for Cmax and AUCτ were 96.07 % and 92.54 %, respectively, and the associated 90 % CIs were within the predefined equivalence limits of 80–125 % (Table 1). The lower limit of the 90 % CI was well below 80 % for Cmin when all participants were included in the analysis, but fell within the predefined equivalence limits when the data from the suspected non-compliant participant were omitted (Table 1).
Norethisterone Pharmacokinetics
On day 1, Cmax was reached at a median time of 1 hour after administration (Fig. 3 and Table 2); there were no statistically significant differences in Cmax, tmax, or AUC24 between treatments (Table 2). The geometric mean treatment ratio for Cmax was 94.14 %, and the associated 90 % CI was within the predefined equivalence limits (Table 2). The geometric mean treatment ratio for AUC24 was 90.29 %, and the lower limit of the 90 % CI (79.12 %) was very slightly below the pre-set lower limit of 80 % (Table 2). However, this difference was considered too small to be clinically relevant.
Fig. 3.

Mean norethisterone plasma concentration–time profiles on day 1 and day 5 (n = 13). OC oral contraceptive
Table 2.
Pharmacokinetic parameters and summary of the equivalence analysis for norethisterone
| Parameter | Treatment A | Treatment B | OC + prucalopride versus OC alone | ||
|---|---|---|---|---|---|
| OC alonea | OC + prucalopridea | PE (%) | 90 % CI | p value | |
| Day 1 (n = 13) | |||||
| tmax (h) | 1.0 [1.0–2.0] | 1.0 [1.0–2.0] | 0.00 | −0.03, 0.00 | 0.3210 |
| Cmax (ng/mL) | 12.6 ± 5.0 | 12.4 ± 4.4 | 94.14 | 81.02, 109.37 | 0.4845 |
| AUC24 (ng·h/mL) | 61.1 ± 30.7 | 58.2 ± 26.2 | 90.29 | 79.12, 103.02 | 0.1918 |
| Day 5 (n = 13)b | |||||
| tmax (h) | 1.0 [1.0–2.0] | 1.0 [1.0–2.0] | 0.00 | 0.00, 0.00 | 0.7261 |
| Cmin (ng/mL) | 0.93 ± 0.45 | 0.92 ± 0.50 | 73.92 | 49.05, 111.39 | 0.2125 |
| Cmax (ng/mL) | 17.1 ± 4.6 | 17.0 ± 4.7 | 98.07 | 88.37, 108.84 | 0.7434 |
| AUCτ (ng·h/mL) | 105 ± 39 | 98.9 ± 33.7 | 91.36 | 82.58, 101.09 | 0.1370 |
| t½ (h) | 10.2 ± 2.0 | 9.8 ± 1.8 | – | – | 0.1858 |
| Day 5 (n = 12)b | |||||
| tmax (h) | 1.0 [1.0–2.0] | 1.0 [1.0–2.0] | 0.00 | −0.50, 0.00 | 0.6000 |
| Cmin (ng/mL) | 0.97 ± 0.45 | 1.00 ± 0.44 | 97.94 | 84.37, 113.70 | 0.8059 |
| Cmax (ng/mL) | 17.0 ± 4.8 | 17.1 ± 4.9 | 99.00 | 88.02, 111.35 | 0.8801 |
| AUCτ (ng·h/mL) | 100 ± 37 | 100 ± 35 | 96.04 | 88.28, 104.47 | 0.4045 |
| t½ (h) | 10.3 ± 2.0 | 9.9 ± 1.9 | – | – | 0.1637 |
aValues are expressed as means ± standard deviations, except for tmax, for which median [range] values are given
bResults are based on all data (n = 13) and on n = 12 after exclusion of one participant because circumstantial evidence indicated that her medication was not taken on days 3 and/or 4
AUC τ area under the plasma concentration–time curve during a 24-hour dosing interval, AUC 24 area under the plasma concentration–time curve during the first 24-hour dosing interval, CI confidence interval, C max maximum plasma concentration, C min minimum plasma concentration, OC oral contraceptive, PE point estimate of the geometric mean treatment ratio, t ½ elimination half-life, t max time to reach Cmax
Norethisterone steady state was reached on day 5, with plasma concentrations of norethisterone being similar before and 24 hours after administration of oral contraceptive alone (0.97 ± 0.47 ng/mL and 1.13 ± 0.51 ng/mL, respectively) and oral contraceptive plus prucalopride (0.92 ± 0.51 ng/mL and 1.11 ± 0.48 ng/mL, respectively) [Fig. 3]. On day 5, Cmax was reached at a median time of 1 hour after dosing. There were no statistically significant differences in tmax, Cmin, Cmax, AUCτ, or t½ between treatments (Table 2). The geometric mean treatment ratios for Cmax and AUCτ were 98.07 % and 91.36 %, respectively, and the associated 90 % CIs were within the predefined equivalence limits of 80–125 % for Cmax and AUCτ (Table 2). For Cmin, the geometric mean treatment ratio and the lower limit of the 90 % CI were below 80 % when all participants were included in the analysis. However, these parameters fell within the predefined equivalence limits when the data from the suspected non-compliant participant were omitted (Table 2).
Prucalopride Pharmacokinetics
On day 1, the mean near-peak (3-hour) concentration of prucalopride was 4.56 ± 0.87 ng/mL. On day 5, prucalopride steady state was reached, with similar plasma concentrations pre-dose on days 5 and 6 and at 24 hours post-dose on day 6 (3.00 ± 1.16 ng/mL, 3.20 ± 0.84 ng/mL, and 3.13 ± 0.58 ng/mL, respectively). On day 5, the mean near-peak (3-hour) steady-state plasma concentration of prucalopride was 8.18 ± 1.64 ng/mL.
Prucalopride Safety and Tolerability
No unexpected safety findings for prucalopride were identified on administration with ethinylestradiol and norethisterone. No deaths or serious or severe treatment-emergent AEs were reported. Treatment-emergent AEs were more common in participants receiving prucalopride plus oral contraceptive (39 events, n = 15 [93.8 %]) than in those receiving oral contraceptive alone (4 events, n = 4 [30.8 %]). The most common AEs that occurred with oral contraceptive plus prucalopride were headache (11 events, n = 11 [68.8 %]), nausea (11 events, n = 10 [62.5 %]), vomiting (7 events, n = 6 [37.5 %]), and diarrhea (4 events, n = 4 [25.0 %]). All cases of headache and diarrhea started on day 1, as did eight of the cases of nausea. Five participants experienced vomiting on day 1 (one of whom also experienced vomiting on day 7), and a sixth participant reported vomiting on day 3. Three of the episodes of vomiting occurred 1.5–3.5 hours post-dose, while the other four episodes occurred 9–21 hours post-dose. The four AEs that were reported in participants receiving oral contraceptive alone were all moderate cases of headache, three of which occurred on day 1 and one that occurred on day 8. One episode of palpitations was reported, but this did not result in drug discontinuation and was not associated with other serious cardiovascular events. No clinically relevant abnormalities or trends were observed in the laboratory data, vital signs, ECGs, or physical examinations.
Discussion
Prucalopride was developed for the treatment of chronic constipation, which tends to be more common in women than in men. A high proportion of patients taking prucalopride are therefore also likely to be taking oral contraceptives. Several oral contraceptives (including ethinylestradiol and norethisterone) are metabolized by CYP3A4, induction of which can reduce exposure to the components of the oral contraceptives and risk contraceptive failure. Although there is no indication that prucalopride has CYP3A4-inducing properties, and it has a very low potential for enzyme inhibition, the pharmacodynamic properties of prucalopride may theoretically lead to reduced absorption of concomitantly used drugs. However, the findings of the current study indicate that prucalopride has no clinically relevant effects on the pharmacokinetics of either ethinylestradiol or norethisterone.
Single-dose prucalopride had no effect on the rate or extent of ethinylestradiol and norethisterone absorption, despite a number of participants reporting diarrhea on day 1 of treatment. Thus, the faster transit associated with diarrhea and the known prokinetic effects of prucalopride appear not to have been associated with any clinically relevant effects in terms of drug absorption. This suggests that the absorption of oral contraceptives is unaffected by the changes in transit time evoked by prucalopride, and points to the limited importance of enterohepatic circulation (with possible second-pass absorption as a consequence) in the absorption of oral steroids in humans [17]. In addition, prucalopride did not affect the pharmacokinetics of ethinylestradiol and norethisterone once steady-state concentrations of prucalopride and oral contraceptive were achieved, indicating that there was no metabolic interaction of prucalopride with the oral contraceptive constituents.
Prucalopride was well tolerated, and the safety profile was consistent with observations from previous studies of prucalopride in adult populations [3, 18, 19]. The most frequent treatment-emergent AEs were gastrointestinal symptoms (nausea, vomiting, and diarrhea), which were predominantly limited to day 1 of drug administration. These findings are in agreement with those of previous studies, in which diarrhea and nausea were more frequently reported with prucalopride treatment than with placebo, with most cases occurring during the first 1–2 days of treatment [3, 4]. Importantly, the present study was performed in healthy volunteers who were not constipated, which might have been an influencing factor in the occurrence of gastrointestinal-related AEs due to the potent gastrointestinal prokinetic activity of prucalopride. Nonetheless, these events did not affect the pharmacokinetics of the oral contraceptive. In particular, the vomiting did not occur at a time that would affect absorption of the oral contraceptive. However, as with all drugs, if vomiting were to occur very soon after oral contraceptive administration, then full absorption of the drug(s) could not be guaranteed.
Consistent with the high affinity and selectivity of prucalopride for 5-HT4 receptors [20, 21], there were no clinically relevant changes in vital signs or ECG parameters, and no significant cardiovascular AEs were observed.
This is the first study to look at the interaction between prucalopride and oral contraceptives. However, a number of limitations should be noted. First, the findings are applicable only to the oral contraceptives evaluated in the study, and may not be generalizable to other oral contraceptives. A second potential limitation is that women with a BMI greater than 27 kg/m2 were excluded from the study, and therefore the findings may not be applicable to obese women.
Conclusion
Administering prucalopride with an oral contraceptive containing ethinylestradiol and norethisterone is not associated with any clinically meaningful drug–drug interactions or safety concerns. These findings are important because oral contraceptive therapy often combines the estrogen ethinylestradiol and the progestogen norethisterone, and these constituents are likely to be among concomitant medications used by women taking prucalopride.
Acknowledgments
The authors thank Dr Andreas Schrödter (of FOCUS Clinical Drug Development GmbH) for his invaluable assistance in performing the study, and Matthias Gurniak (of FOCUS Clinical Drug Development GmbH) for additional operational support. This clinical research was funded by the sponsor, Shire-Movetis NV. Under the direction of the authors, Tom Potter and Catherine Hill (employees of Oxford PharmaGenesis™ Ltd [Oxford, UK] and PharmaGenesis™ London [London, UK]) provided writing assistance for this publication. Editorial assistance in formatting, proofreading, copy editing, and fact checking was also provided by Oxford PharmaGenesis™ Ltd. Slavka Baronikova and David Pierce from Shire-Movetis NV reviewed and edited the manuscript for scientific accuracy. Shire-Movetis NV provided funding to Oxford PharmaGenesis™ Ltd for support in writing and editing this manuscript. Although the sponsor was involved in the design, collection, analysis, interpretation, and fact checking of information, the content of this manuscript, the ultimate interpretation, and the decision to submit it for publication in Drugs in R&D was made by the authors independently. The authors confirm that the data presented provide an accurate representation of the study results.
Author Contributions
Vera Van de Velde and Lieve Vandeplassche were involved in the conception of the study and interpretation of the data. Mieke Hoppenbrouwers was involved in conception, analysis, and interpretation. Mark Boterman was involved in laboratory testing and analysis of the data. Jannie Ausma was responsible for coordinating the study and was also involved in the conception, analysis, and interpretation of the data. All authors were involved throughout the development of the manuscript.
Conflict of Interest Disclosures
Vera Van de Velde has received consultancy fees from Shire-Movetis NV. Mark Boterman’s institution (Analytisch Biochemisch Laboratorium BV) received a grant from Shire-Movetis NV for analysis of the study samples. Lieve Vandeplassche, Mieke Hoppenbrouwers, and Jannie Ausma are employees of Shire-Movetis NV and hold stock/stock options in Shire. The authors have no other conflicts of interest that are directly relevant to the content of this article.
Footnotes
Resolor® is a CTM registered trademark of Shire-Movetis NV.
References
- 1.European Medicines Agency. Resolor (prucalopride): summary of product characteristics. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/001012/WC500053998.pdf. Accessed 26 March 2012.
- 2.Frampton JE. Prucalopride. Drugs. 2009;69(17):2463–2476. doi: 10.2165/11204000-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 3.Camilleri M, Kerstens R, Rykx A, et al. A placebo-controlled trial of prucalopride for severe chronic constipation. N Engl J Med. 2008;358(22):2344–2354. doi: 10.1056/NEJMoa0800670. [DOI] [PubMed] [Google Scholar]
- 4.Quigley EM, Vandeplassche L, Kerstens R, et al. Clinical trial: the efficacy, impact on quality of life, and safety and tolerability of prucalopride in severe chronic constipation: a 12-week, randomized, double-blind, placebo-controlled study. Aliment Pharmacol Ther. 2009;29(3):315–328. doi: 10.1111/j.1365-2036.2008.03884.x. [DOI] [PubMed] [Google Scholar]
- 5.Tack J, van Outryve M, Beyens G, et al. Prucalopride (Resolor) in the treatment of severe chronic constipation in patients dissatisfied with laxatives. Gut. 2009;58(3):357–365. doi: 10.1136/gut.2008.162404. [DOI] [PubMed] [Google Scholar]
- 6.Wald A, Scarpignato C, Mueller-Lissner S, et al. A multinational survey of prevalence and patterns of laxative use among adults with self-defined constipation. Aliment Pharmacol Ther. 2008;28(7):917–930. doi: 10.1111/j.1365-2036.2008.03806.x. [DOI] [PubMed] [Google Scholar]
- 7.Tack J, Muller-Lissner S, Stanghellini V, et al. Diagnosis and treatment of chronic constipation: a European perspective. Neurogastroenterol Motil. 2011;23(8):697–710. doi: 10.1111/j.1365-2982.2011.01709.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pare P, Ferrazzi S, Thompson WG, et al. An epidemiological survey of constipation in Canada: definitions, rates, demographics, and predictors of health care seeking. Am J Gastroenterol. 2001;96(11):3130–3137. doi: 10.1111/j.1572-0241.2001.05259.x. [DOI] [PubMed] [Google Scholar]
- 9.Higgins PD, Johanson JF. Epidemiology of constipation in North America: a systematic review. Am J Gastroenterol. 2004;99(4):750–759. doi: 10.1111/j.1572-0241.2004.04114.x. [DOI] [PubMed] [Google Scholar]
- 10.Choung RS, Locke GR, 3rd, Schleck CD, et al. Cumulative incidence of chronic constipation: a population-based study 1988–2003. Aliment Pharmacol Ther. 2007;26(11–12):1521–1528. doi: 10.1111/j.1365-2036.2007.03540.x. [DOI] [PubMed] [Google Scholar]
- 11.Barditch-Crovo P, Trapnell CB, Ette E, et al. The effects of rifampin and rifabutin on the pharmacokinetics and pharmacodynamics of a combination oral contraceptive. Clin Pharmacol Ther. 1999;65(4):428–438. doi: 10.1016/S0009-9236(99)70138-4. [DOI] [PubMed] [Google Scholar]
- 12.Bolt HM. Interactions between clinically used drugs and oral contraceptives. Environ Health Perspect. 1994;102(Suppl 9):35–38. doi: 10.1289/ehp.94102s935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.European Medicines Agency. ICH harmonised tripartite guidelines for good clinical practice, 1996. http://www.emea.europa.eu/pdfs/human/ich/013595en.pdf. Accessed 11 June 2012.
- 14.World Medical Association. Declaration of Helsinki: ethical principles for medical research involving human subjects. http://www.wma.net/en/30publications/10policies/b3/. Accessed 12 August 2009.
- 15.US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Veterinary Medicine (CVM). Guidance for industry: bioanalytical method validation, 2001. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm070107.pdf. Accessed 14 December 2011.
- 16.Organisation for Economic Co-operation and Development. OECD principles of good laboratory practice (GLP). http://www.oecd.org/document/63/0,3746,en_2649_34377_2346175_1_1_1_1,00.html. Accessed 13 April 2012.
- 17.Hanker JP. Gastrointestinal disease and oral contraception. Am J Obstet Gynecol. 1990;163(6 Pt 2):2204–2207. doi: 10.1016/0002-9378(90)90562-L. [DOI] [PubMed] [Google Scholar]
- 18.Camilleri M, Van Outryve MJ, Beyens G, et al. Clinical trial: the efficacy of open-label prucalopride treatment in patients with chronic constipation: follow-up of patients from the pivotal studies. Aliment Pharmacol Ther. 2010;32(9):1113–1123. doi: 10.1111/j.1365-2036.2010.04455.x. [DOI] [PubMed] [Google Scholar]
- 19.Quigley EM, Tack J, Kerstens R, et al. The efficacy and safety of oral prucalopride in female patients with chronic constipation who had failed laxative therapy (EMA-authorised population) is similar to that of the ITT population in the initial pivotal trials: pooled data analysis. Gastroenterology. 2012;142(Suppl I):S820–S821. [Google Scholar]
- 20.De Maeyer JH, Aerssens J, Verhasselt P, et al. Alternative splicing and exon duplication generates 10 unique porcine 5-HT 4 receptor splice variants including a functional homofusion variant. Physiol Genomics. 2008;34(1):22–33. doi: 10.1152/physiolgenomics.00038.2008. [DOI] [PubMed] [Google Scholar]
- 21.De Maeyer JH, Prins NH, Schuurkes JA, et al. Differential effects of 5-hydroxytryptamine4 receptor agonists at gastric versus cardiac receptors: an operational framework to explain and quantify organ-specific behavior. J Pharmacol Exp Ther. 2006;317(3):955–964. doi: 10.1124/jpet.106.101329. [DOI] [PubMed] [Google Scholar]