

Abstract
Two major pathways degrade most cellular proteins in eukaryotic cells: the ubiquitin–proteasome system (UPS), which usually degrades the majority of proteins, and autophagy, primarily responsible for the degradation of most long-lived or aggregated proteins and cellular organelles. Disruption of these processes can contribute to pathology of a variety of diseases. Further, both pathways are critical for the maintenance of several aspects of cellular homeostasis, but, until recently, were thought to be largely distinct. Recent advances in this field, however, now strongly suggest that their activities are carefully orchestrated through several interfacing elements that are presented and discussed in this review.
Keywords: Proteasome, autophagy, ubiquitin, homeostasis
Introduction
For decades, studies have focused on analyzing protein synthesis, particularly in understanding transcriptional and translational signals. However, the other side of protein steady-state, degradation, has been overlooked. Since pioneering studies initiated in the 1940s [1], Two major pathways of degradation have been described for most cellular proteins in eukaryotic cells: 1) the ubiquitin-proteasome system (UPS), responsible for degrading 80-90% of proteins including many regulated, short-lived, abnormal, denatured, or, in general, damaged proteins [2]; and 2) autophagy, which, by contrast, is primarily responsible for the degradation of most long-lived proteins, but also for aggregated proteins as well as cellular organelles (mitochondria, peroxysomes, ribosomes, infectious organisms). These two degradative systems not only degrade proteins into small polypeptides, but help maintain amino acid pools and energy balance, either during acute starvation for the UPS, or in the course of chronic starvation for autophagy. Indeed, because de novo synthesis of amino acids carries high energy costs, recycling of amino acids is important for its contribution to maintenance of cellular homeostasis with less energy expenditure. Additionally, these catabolic pathways constitute essential components of the cellular control of protein quality (PQC), which senses misfolded or damaged proteins, tags them, and, finally, degrades them. Therefore, studying these systems is essential to developing a better understanding of protein homeostasis.
Although the UPS and autophagy mechanisms were primarily thought to be largely distinct catabolic pathways, and were therefore investigated separately, recent advances in understanding the common mechanisms contributing to the UPS regulation and the induction of autophagy have highlighted the relationship between them. As autophagy and the UPS are both critical for the maintenance of aspects of cellular homeostasis (ATP balance, amino acid recycling, PQC), their activities likely need to be carefully orchestrated. Recent evidence has revealed cross-talk mechanisms involving the small ubiquitin ligand molecules and the linking protein p62, as well as cell signaling pathways and transcription factors, and will be discussed in this review to outline the coordination between these systems and its importance to protein homeostasis.
The ubiquitin-proteasome system (UPS)
Ubiquitination-dependent degradation by the proteasomal machinery is involved in the regulation of several processes including maintenance of cellular quality control, transcription, cell cycle progression, DNA repair, receptor-mediated endocytosis, cell stress response, and apoptosis. This mechanism is the predominant mode of targeting proteins for degradation. Before degradation through the proteasomal machinery can occur, substrate proteins designated for degradation must first be tagged with ubiquitin, a highly conserved 76-amino-acid polypeptide.
Ubiquitin and ubiquitin-like tags for degradation
The conjugation system
The small, globular regulatory protein ubiquitin directs other proteins to cellular locations: modification of a target protein by ubiquitin serves as a recognition signal that allows downstream effectors to bind the ubiquitin-modified protein to change its function or fate. Covalent attachment of ubiquitin to the protein substrate occurs through a three-step cascade mechanism (Figure 1): Ubiquitin is first linked to a cysteine residue of an activating enzyme, E1, via its terminal glycine, in an ATP-dependent manner. The activated ubiquitin is then transferred to a conjugating enzyme, E2, also at a cysteine residue, forming an energy-rich thiol ester bond. Finally, ubiquitin is attached to the substrate protein that is specifically bound to one member of the E3 ubiquitin ligase family [3]. In the case of RING (really interesting new gene) finger E3 ligases, the transfer of ubiquitin is direct from E2-ubiquitin to the substrate, even if the presence of E3 is required. In the case of HECT-domain-containing (homologous to the E6AP carboxy terminus) E3 ligases, ubiquitin is first transferred to an intermediate complex, E3-ubiquitin, which, in turn, attaches its ubiquitin moieties to the substrate protein to be degraded [4]. At present, 2 genes are known to encode E1 isoforms, at least 37 genes encode E2, and over 1000 encode E3 ubiquitin ligases in the human genome [5]. Each E1 isoform has a distinct preference for E2, while association of E2 and E3, depending on the cellular context, generates extensive combinatorial complexity.
Figure 1.
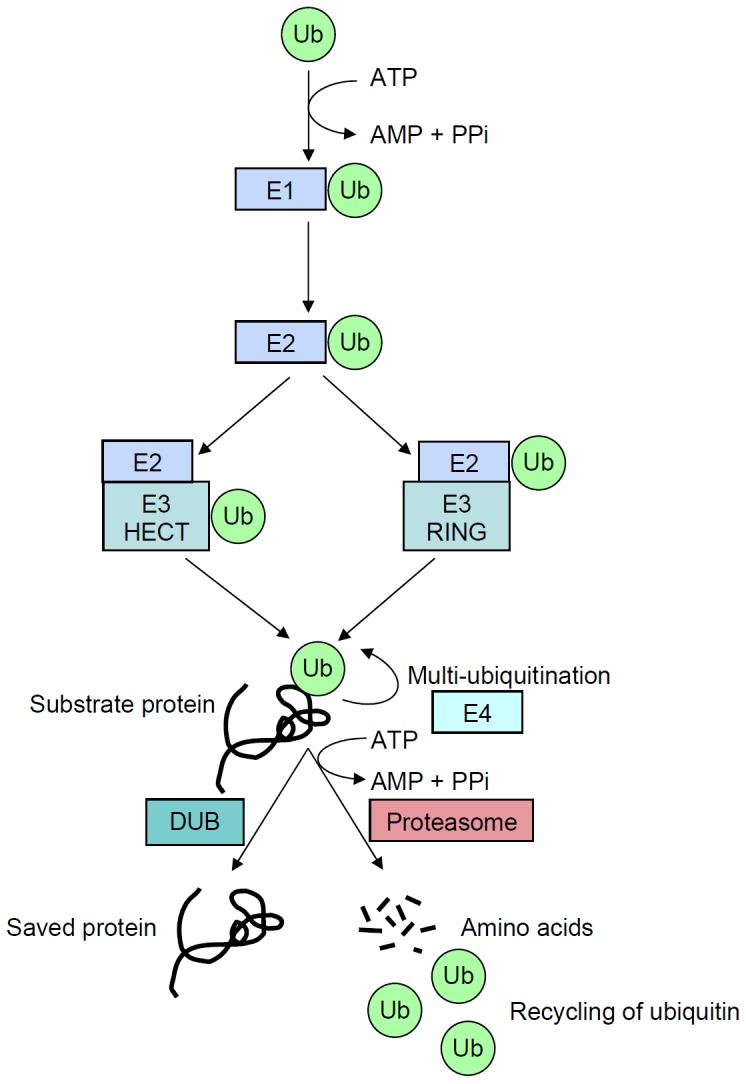
Schematic representation of the ubiquitination process that target proteins for degradation. A ubiquitin molecule (Ub) is first linked by its carboxy-terminal amino acid to an E1-activating enzyme (E1) through a high-energy bond on a cysteine residue, while consuming energy (ATP). Activated ubiquitin is then translocated to the E2-conjugating enzyme (E2). RING E3 ubiquitin ligases (E3 RING) catalyze the transfer of ubiquitin directly from E2 to the substrate, whereas HECT E3 enzymes (E3 HECT) accept activated ubiquitin from E2 before transferring it to the substrate. Following this first step, called monoubiquitination, the process may be repeated by some processive E2/E3 enzymes, or with the help of E4 enzymes and, finally, leads to polyubiquitination. Proteins targeted for degradation into amino acids can however be rescued by deubiquitining enzymes (DUB).
The repertoire of ubiquitin signals
Ubiquitin can attach to substrate proteins in several ways, generating a broad repertoire of signals with different topologies and lengths. These can be diversely recognized by different proteins and lead to various functional outcomes for the tagged substrate protein. Ubiquitin is anchored by an isopeptide bond to the ε-NH2 group of lysine residues in the substrate protein by the C-terminal residue on its last glycine (residue 76), a mechanism called “monoubiquitination”. This signal may be extended by addition of other ubiquitin moieties to the previous one, called “polyubiquitination”. In eukaryotes, most substrates are polyubiquitinated (Figure 2).
Figure 2.
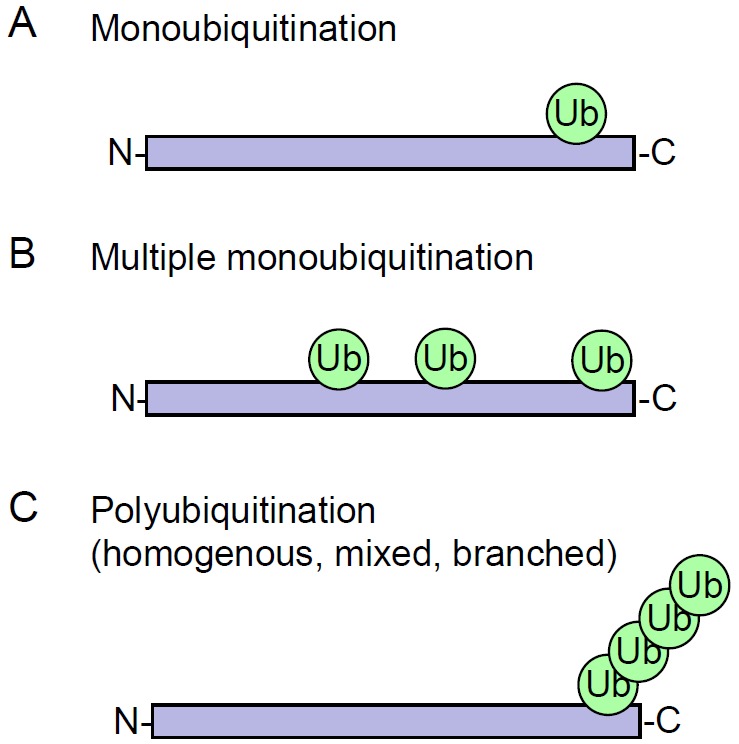
Different types of ubiquitination. Monoubiquitination represents modification of protein by a single ubiquitin (A), or by several single ubiquitins (B). Polyubiquitination (C) corresponds to the successive addition of several ubiquitins on one of their seven lysine residues. All types of ubiquitination involve isopeptide bonds anchoring ubiquitin to either the ε-NH2 group of a lysine in the target substrate, or to the α-NH2 group of its amino-terminal residue.
Polyubiquitination can occur at several sites and in several ways. An additional component of the ubiquitination machinery, an E4 enzyme, is involved in elongation of short ubiquitin chains [6]. In the “classical” pathway, the lysine residue at position 48 of ubiquitin is used, and this pattern is repeated to generate polymers. A chain of four or more moieties is, in general, the minimal requirement for substrate recognition by the 26 S proteasome complex-the major ATP-dependent degradation pathway-although for some proteins monoubiquitination or multiple monoubiquitinations can efficiently target substrates for proteasomal degradation [7]. In fact, seven lysine residues in ubiquitin can be used for chain elongation, at positions 6, 11, 27, 29, 33, 48, and 63. In addition, the N-terminal methionine of ubiquitin can also bind to the C-terminal glycine, generating a linear chain. However, lysine-48 (K48) and lysine-63 (K63) are the most frequent sites for elongation. In contrast to all other positions that can target proteins for degradation, K63 polyubiquitination chains participate in regulation of endocytosis, DNA repair, and protein kinase activity [8]. In addition to homogeneous polyubiquitination, recent evidence describes mixed types of ubiquitin addition using different lysine residues in the same linear chain, or multiple branched chains on several lysine residues on one ubiquitin polypeptide or substrate protein (for details, see review [7]). In fact, the affinity of K48 tetraubiquitin chains for the proteasome is 100-fold higher than for diubiquitin, and does not increase further with longer chains [9]. Monoubiquitination is sufficient for receptor internalization, but evidence suggests that polyubiquitination uses K63 as a chain type for further processing.
Variations in degradation signals
A limited number of short-lived proteins are believed to have their stability governed by their N-terminal amino acid, which provides destabilizing activity upon modification (according to the “N-end” rule). N-end dependent degradation has been shown to have important functions in peptide import, chromosome segregation and meiosis, angiogenesis and cardiovascular development. Several proteins degraded through this pathway have been characterized so far, including RGS4, RGS5 and RGS16 (regulator of G protein signaling), GNG2 (G protein subunit γ2) involved in signal transduction, IAP1 (inhibitor of apoptosis protein 1), and SCC1 (sister chromatid cohesin 1) playing an essential function in cohesion between sister chromatid during DNA replication. Recognition occurs by specific elements called N-recognins, which bind N-terminal sequences of substrates and tag them with ubiquitin for proteasomal degradation (reviewed in [10]). Therefore, ubiquitin can also be conjugated to the α-NH2 group of the N-terminal residue of the substrate.
To add complexity to this process, de-ubiquitinating enzymes can reverse the ubiquitination-degradation process by cleaving ubiquitin residues from both monoubiquitinated substrates and polyubiquitinated proteins. This process allows free ubiquitin regeneration, which is anyway present at limiting concentration in the cells, but also constitutes a “last-chance” proofreading allowing correctly-folded proteins to escape degradation [11].
Other ubiquitin signals
Molecular ubiquitin signals generated by attachment of one or more ubiquitin molecules are recognized by specialized ubiquitin binding domains (UBD) that form transient and noncovalent interactions with monomers or polymers of ubiquitin moieties. So far, around 200 cellular proteins have been recognized to contain one or more UBD [12], including shuttle proteins that transport degradation candidates to the proteasome [R23A (Rad23 in yeast), Dsk2, Ddi1, NBR1] and binding proteins directly participating in the proteasome machinery [Rpn10 (S5a in human), Rpn13] [13].
In addition, several other proteins related to ubiquitin have been identified as mediating protein degradation; these fall into two classes: proteins containing a ubiquitin-like domain (UDP) and ubiquitin-like modifiers (Ubl). UDPs are highly homologous to ubiquitin in amino acid sequence as well as in three-dimensional structure, but they do not form conjugates with proteins. Instead, they serve as adaptors binding to ubiquitin or Ubl proteins. Like UDPs, Ubl proteins are also homologous to ubiquitin in sequence and three-dimensional structure, but, in addition, they possess a glycine residue at their C-terminus that allows them to form conjugates with proteins. Ubl proteins include SUMO (small ubiquitin-like modifier), NEDD8 (neuronal-precursor cell expressed developmentally down-regulated protein) [14], ISG15 (interferon-stimulated gene 15) [15], ATG 8 and 12 (autophagy-related genes in yeast) (see below [16]), FAT10 (F-adjacent transcript-10) [17], and UFM1 (ubiquitin-fold-modifier 1) [18]. The functions of these Ubl proteins are still poorly understood, but they are related to various biological processes including DNA replication, signal transduction, cell cycle control, embryogenesis, cytoskeletal regulation, metabolism, stress response, homeostasis, and mRNA processing.
In summary, ubiquitin is one of the most versatile protein modifiers, producing a wide repertoire of molecular signals. However, the complexity of this first step of tagging proteins to be degraded or regulated is far from being clearly understood and will require further investigation.
The proteasomal machinery
Structure
Degradation of the vast majority of intracellular proteins is performed by the 26 S proteasome. This machinery contains two subcomplexes, the core 20 S proteasome of about 700 kDa and the 19 S regulatory particle of about 900 kDa [19] (Figure 3). The 20 S element forms a barrel-shaped structure with a central cavity of 2-nm diameter. It contains proteases subunits, with two copies of seven different α-subunits and two copies of seven different β-subunits. β1-subunits have caspase-like activity that hydrolyzes the peptide bond after negatively-charged amino acid residues. β2-subunits have trypsin-like activity, which cleaves the peptide bond after positively-charged amino acid residues. β5-subunits are characterized by chymotrypsin-like activity and hydrolyze the peptide bond after large hydrophobic amino acid residues. The size of released peptides varies from 3 to 25 amino acid residues, which are further degraded into single amino acids by peptidases [20].
Figure 3.

Assembly and constitution of the 26 S proteasome. The proteasome is constituted of subcomplexes of 20 S and 19 S core particles. The 20 S is a barrel-shaped structure, which consists of two inner β-rings with proteasic activities, each made up of seven subunits, and of two outer α-rings. The 19 S regulatory particle is composed of approximately 20 different proteins that form a lid and a base. Energy is required for the assembly of the complete 26 S complex (ATP), but also for the unfolding and translocation of the ubiquitinated (green spheres) protein to be degraded (black structure).
In mammals, there also exists an immunoproteasome stimulated by γ-interferon. This cytokine activates the synthesis of alternate β1i, β2i, and β5i subunits incorporated in place of constitutive subunits. The immunoproteasome is believed to generate peptides specifically cleaved for antigen presentation [21].
Each 20 S proteasomal subunit possesses two units of each of the three cleaving activities. To be degraded, a substrate must first be unfolded, then must been translocated into the proteolytic chamber via the narrow gate formed by α residues.
Regulation
The mechanisms regulating 26 S proteasome activity remain poorly understood, but involve numerous proteins that reversibly associate with it: binding of the regulatory 19 S particle, opening of the axial channel into the 20 S core particle, translocation of the protein to be degraded, and association with proteins that modify proteasome-bound ubiquitin chains. The 19 S regulatory particle is the key regulatory component of the 26 S proteasome; it is responsible for 20 S gating, recognition, unfolding, and translocation of ubiquitinated proteins. Attachment of the 19 S particle to the 20 S element controls the gate and helps to avoid degrading a protein in error. Recognition and binding to ubiquitin by 19 S subunits permits selection of the appropriate target protein [22]. A 20 S core particle can exist without a 19 S element and may exert selective degradation, in particular on oxidized proteins. In addition to the 19 S regulatory particle, several other protein complexes, such as PA200, PA28, and PI31, can interact with α-rings of the 20 S core particle, forming alternative isoforms of the proteasome [23-25]. These isoforms regulate ubiquitin-independent substrate degradation by the proteasome, but the physiological functions of such alternative complexes are not well understood.
26 S proteasome activity is ATP-dependent. Indeed, ATP is necessary for 19 S assembly with a 20 S core particle, as well as for substrate unfolding and translocation into the 20 S proteolytic chamber. When cells are subjected to stress factors, and ATP is low, the proteasome system may switch to an ATP-independent mechanism of degradation of proteins, and/or autophagy may be upregulated. Proteasome isoforms involving PA200, PA28, and PI31 are ATP-independent.
Additional regulation of protein degradation occurs directly on the proteasomal complex. In fact, the proteasomal subunit can be subjected to several types of post-translational modifications, including phosphorylation, N-acetylation, N-terminal processing, caspase cleavage, N-myristoylation, O-glycosylation, S-glutathionylation, alkylation, and oxidation (reviewed in [26]). These modifications affect the proteasomal activity, its proteic composition, and have been assumed to control proteasome interaction with proteins and membranes.
Alternative “non-canonical” mechanisms
The well-studied “classical” ubiquitin-dependent proteasomal degradation requires polyubiquitination of the substrate and mobilizes energy from ATP. However, in recent years data have accumulated on alternative mechanisms: ubiquitin-independent, ATP-independent degradation by the 20 S proteasome without a 19 S lid, and processing without degradation. Additionally, ubiquitin-like mechanisms of degradation occur in which another protein plays the role of ubiquitin, or the signal for degradation, called a “degron”, is contained in the sequence of the protein per se. For example, ornithine decarboxylase contains a degradation signal, the “PEST” sequence [27], while α-synuclein and tau proteins involved in Parkinson’s disease [28,29], c-jun [30], calmodulin [31], and oxidized proteins [32] may also be degraded without ubiquitination. Further, natively-disordered or stress-induced unfolded proteins can be degraded without ATP consumption because energy is not required to unfold such proteins, and exposed hydrophobic surfaces resulting from this unfolding replace ubiquitination. In this case, the function of ubiquitin is fulfilled by hydrophobic regions of the unfolded chain. Oxidized proteins can undergo ubiquitin-independent degradation by the 20 S core proteasome in the absence of ubiquitin and ATP, as shown by in vitro experiments [32].
The proteasome can also work by processing a protein, which differs from complete degradation. Processing affects proteins like p105, which is cleaved by a limited endoproteolysis to generate the p50 NF-κB transcription factor [33,34]. Other proteins that undergo processing include YB-1, a DNA/RNA nuclear and cytoplasmic binding protein with multiple protective functions [35], and eIF4G and eIF3a, which are translation initiation factors [36]. Their cleavage by the 20 S proteasome has an inhibitory effect on translation of mRNAs that are dependent on these factors.
Physiopathological alterations in proteasomal efficiency
Though the exact mechanism remains unclear, an age-related decrease in proteasome activity is observed, which weakens the capacity of cells to remove damaged proteins and favors the development of diseases [37]. For example, proteasome activity decreases with advancing age in the cerebral cortex, hippocampus and spinal cord, but not in the cerebellum or brainstem in rats [38]. In addition, a common age-related feature observed in many tissues is accumulation of the ubiquitin-conjugated proteins. Such proteins, tagged for degradation but not efficiently removed, may be harmful to the cells. Several factors may contribute to this situations, including chronic exposure to free radicals, accumulation of genetic errors, or reduced amounts of proteasome synthesis [37]. In addition, the insulin/IGF-1 (insulin-like growth factor type 1), activating a cascade of conserved kinases, inhibits the forkhead transcription factor FoxO (forkhead box O), which coordinates the induction of degradative pathways including the UPS (see third section).
The proteasome ultimately controls almost all intracellular processes, including cell cycle control, transcription, translation, protein quality control (PQC), DNA repair, receptor endocytosis, and cell stress response [39]. It is therefore not surprising that the proteasome system is involved in physiopathological processes resulting in cancer, autoimmune diseases, neurodegenerative disorders, or viral diseases. Many neurodegenerative diseases such as polyglutamine repeat diseases, Parkinson’s disease, Alzheimer’s disease, tauopathies, prion disease, and amyotrophic lateral sclerosis (ALS) share abnormal accumulation of ubiquitinated proteins into aggregates and inclusions as a hallmark feature of the disease pathology. Failure to remove the polyubiquitinated proteins may lead to the accumulation of aggregated proteins. The capacity of the ubiquitin proteasome pathway can be exceeded either by overexpression of substrates or by a decrease in proteasome activity, due to either directly clogging the entrance of other substrates or down-regulation components of the UPS components [40]. In addition, there is a strong evidence demonstrating that proteasome inhibition by pharmacological treatment enhances inclusion formation in cellular models, indicating that dysfunction of the proteasome might be a factor that initiates the formation of inclusions [41]. In these diseases, specific misfolded proteins exert toxicity leading to neuronal cell dysfunction and death, although the exact molecular mechanisms are still poorly understood. Evidence has shown that early protein aggregates may be toxic to neuronal cells [42]. In other cells, toxicity could result from unnecessary interactions of misfolded proteins with normal cellular proteins, disturbing the maturation process and activities of normal cellular proteins, or exhibit toxicity by trapping normal proteins (gain of function).
The cardiomyocyte, similar to the neuron, is a postmitotic cell in which protein degradation may be important in protein quality control. For that reason, impaired or inadequate protein degradation in the heart is associated with and may be a major pathogenic factor for a wide variety of cardiac dysfunctions, while enhanced protein degradation is also implicated in the development of cardiac pathology [43]. In animal models of ischemic cardiopathies, declined peptidase activities accompanied by accumulation of oxidatively modified and ubiquitinated proteins were observed, while several proteasome subunits are subjected to oxidative modifications. In addition, depressed proteasome activities occur in pressure-overload mouse hearts and conversely, genetic deficiency of UPS components causes cardiac pathology (reviewed in [44]).
The importance of protein degradation in carcinogenesis has been exemplified by the successful application of a novel class of cancer therapeutics known as proteasome inhibitors, like Bortezomib which has demonstrated activity towards multiple myeloma, mantle cell lymphoma, prostate cancer and non-small cell lung carcinoma [45]. The mechanisms mediating the antitumor activity of proteasome inhibitors are manifold, including accumulation of p53, p21Cip1 and p27Kip1, upregulation of death receptors and pro-apoptotic members of the Bcl-2 family and induction of endoplasmic reticulum stress [46]. Proteasome inhibitors exhibit preferential cytotoxicity towards cells grown under hypoxic conditions, as is the case in most solid tumors. High levels of proteasome inhibition can be achieved in vivo in the absence of dose-limiting systemic side-effects. In addition, cancer cells usually display higher levels of basal proteasomal activity, rendering the proteasome an attractive target for cancer therapy [47].
Several inhibitors of the proteasome system have been studied; however, proteasomal involvement in many critical cellular processes ultimately makes these inhibitors toxic. Such inhibitors have been used to successfully combat cancer cells [26,48], and the therapeutic use of these reagents is a promising approach to cancer treatment. Few activators of the proteasome have been reported. One of them is the 1-[1-(4-fluorophenyl)-2,5-dimethylpyrrol-3-yl]-2-pyrrolidin-1-ylethanone, which inhibits Usp14, a proteasome-associated de-ubiquitinating enzyme [49].
Conclusion
The rapid, precise, and timely processing of many cellular proteins by the UPS allows tight control of critical cellular functions, including protein quality control, regulation of proliferation, DNA repair, and signal transduction. A functional UPS is also required for cells to cope with various types of stress, including oxidation, exposure to heavy metals, and accumulation of misfolded proteins. Aberrant proteasome activity is directly related to the pathogenesis of many human diseases including cancer, neurodegenerative disorders, viral infection, autoimmune diseases, and aging. The control and proper functioning of this system is therefore critical for the cell.
Autophagy
Autophagy is an intracellular lysosomal (vacuolar) degradation process that is characterized by the formation of double-membrane vesicles, known as autophagosomes, which sequester cytoplasm. Autophagy is involved in cell growth, survival, development, and death, and has been implicated in human physiopathologies such as cancer, neurodegenerative disorders, myopathies, heart and liver diseases, and gastrointestinal disorders [50].
Autophagic pathways
Autophagy occurs in three distinct types, according to the different pathways by which the cargo is delivered to the lysosomes or vacuole: chaperone-mediated autophagy (CMA), microautophagy, and macroautophagy (Figure 4). Additionally, in yeast an autophagy-related constitutive transport system delivers some vacuolar enzymes and is a cytoplasm-to-vacuole targeting (Cvt) pathway [51]. In CMA, which has been characterized in higher eukaryotes but not in yeast, the Hsc70 (heat shock cognate protein of 70 kDa) chaperone protein and co-chaperones BAG1, Hip, Hop, and Hsp40/DNAJB1 first bind to the cytosolic target, by recognizing a specific consensus sequence KFERQ on it (present in 30% of cellular proteins). The chaperone then binds to a specific receptor (LAMP-2A) on the lysosomal membrane [52]. Subsequently, the cargo protein is unfolded and translocated into the lysosome, where it is degraded. In microautophagy, the lysosomal (or vacuolar) membrane is subjected to invagination, protrusion, or septation to incorporate a small part of the cytoplasmic volume into the lysosomal inner space, where its contents are degraded [53]. Macroautophagy (also referred to as “autophagy”) is mediated by a unique organelle, the autophagosome, which encloses a portion of cytoplasm or organelle such as mitochondria, peroxysome, or aggregates, for delivery to the lysosome. Macroautophagy is believed to be the major mode of autophagy, and is the most extensively analyzed [54]. Constitutive autophagy has a housekeeping role and is essential for survival, development, and metabolic regulation. Autophagy is also responsive to stress, and can be activated tenfold by nutrient deprivation under a complex cascade of regulatory signaling pathways, but it can also degrade damaged proteins and organelles, oxidized lipids, and intracellular pathogens. We will present here the main features of macroautophagy (hereafter termed “autophagy”).
Figure 4.
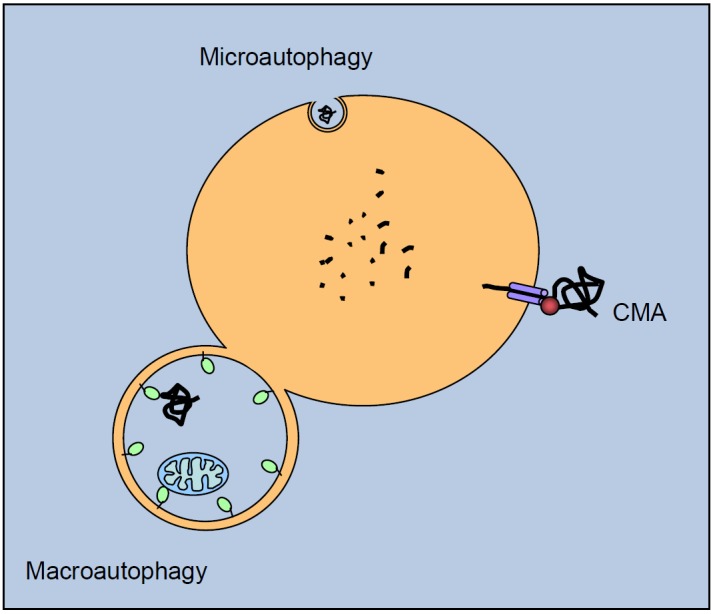
Autophagic pathways. Cytosolic proteins are degraded in the lysosomal lumen (yellow) through three different autophagic mechanisms. In microautophagy, the lysosomal membrane invaginates to engulf a small portion of cytosol with its contents. In chaperone-mediated autophagy (CMA), a targeting motif in the substrate proteins is recognized by a cytosolic chaperone (red sphere) that delivers it to a lysosomal receptor. This receptor multimerizes to form a translocation complex that mediates the translocation of the substrate protein into the lumen of the lysosome. In macroautophagy, a double membrane vesicle sequesters cargo proteins and a whole region of the cytosol, and then fuses with the lysosome for cargo delivering. Once in the lysosomal lumen, proteins as well as other macromolecules are rapidly degraded by multiple enzymes, including cathepsins.
Understanding the molecular mechanisms of autophagy has accelerated since the initial discovery of approximately 35 autophagy-related (Atg) genes from genetic studies in yeast [55]. Mammals have orthologs for most yeast Atg proteins, as well as producing some additional factors specific to higher eukaryotes. The autophagic cascade has been divided into distinct stages: control by cell signaling pathways, nucleation, elongation, and vesicle fusion to lysosomes.
The different steps of the autophagic process
The regulation of autophagy is central to the understanding of its mechanisms. Two main kinase systems control the autophagic pathway: the mTOR–ULK1 and the Beclin1 pathways (Figure 5). mTOR, or target of rapamycin (TOR in nonmammalian species), belongs to the family of phosphoinositide-3-kinase related kinase (PIKKs) [56]. mTOR is so-named because it responds to treatment with rapamycin and other kinase inhibitors that have been widely used to induce autophagy, even under nutrient-rich conditions [57]. mTOR exists in two distinct complexes, complex 1 (mTORC1) and complex 2 (mTORC2); mTORC2 is less sensitive to rapamycin. The two complexes contain several proteins in common (Deptor, GβL, and PRAS40), but other components are specific to mTORC1 (Raptor) or mTORC2 (Rictor, SIN1, and Protor).
Figure 5.
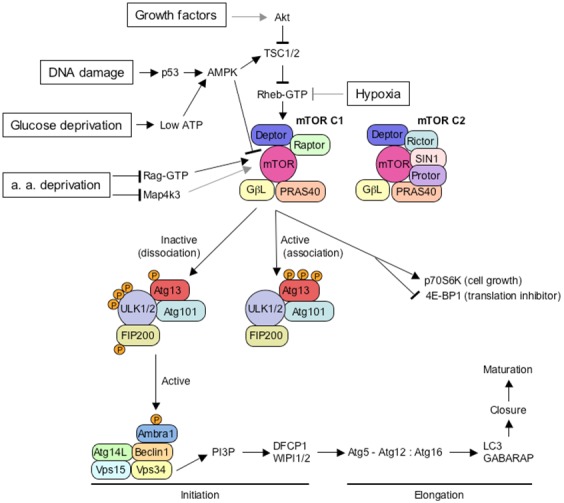
Overview of the major components controlling the initiation step of mammalian autophagy. Several key molecular components participate in the initiation of autophagy. Autophagy inducers such as growth factor, glucose, or amino acid (a. a.) deprivation, DNA damage or hypoxia modulate the inhibitory interaction of mTORC1 with the ULK1/2 complex. Only some of the intermediate molecules are shown in the pathways from autophagy inducers to mTORC1 for the sake of clarity. Grey lines indicate connections between proteins that are not known to be direct. This upper part is condensed and adapted from [56]. The mTORC2 complex is shown only for comparison of its subunit composition with mTORC1, but its function is less well known. When activated by growth factors for example, the mTORC1 complex binds to the ULK1/2 complex and hyperphosphorylates its Atg13 subunit, resulting in the inactivation of the ULK1/2 complex. Starvation, resulting in glucose and amino acid deprivation, low level of growth factor, or stress factors such as DNA damage or hypoxia, results in the inhibition of the mTORC1 complex. As a consequence, the downregulated mTOC1 complex dissociates from the ULK1/2 complex. Subsequent dephosphorylation of the Atg13 subunit allows ULK1/2 activation, with its autophosphorylation, in addition of phosphorylation of Atg13 and FIP200. The activated ULK1/2 complex locates then at the initiating phagophore, where it stimulates the activity of Beclin 1 class III PI3K complex, through phosphorylation of Ambra1, and maybe through other putative interactions. The activated PI3 kinase Vps34 of the Beclin 1 complex produces phosphatidylinositol-3-phosphate (PI3P) at the phagophore membrane that binds to DFCP1 and WIPI 1 and 2 proteins. These proteins allow modifications at the phagophore membrane and subsequent recruitment of the Atg5-Atg12:Atg16 conjugation complex at the phagophore membrane, that in turn allows to anchor LC3, together with its homologous protein, GABARAP. These elongation steps allow subsequent autophagosome growth, closure, and maturation into autolysosomes.
The serine-threonine kinase TOR is central for integrating signaling pathways that regulate cellular homeostasis, by coordinating anabolic and catabolic processes with nutrients, energy and oxygen availability, and growth factor signaling [50]. When activated in the presence of nutrients or growth factors, the mTORC1 complex associates to the ULK1 (or ULK2) complex and hyperphosphorylates its Atg13 subunit, which results in its inactivation and subsequent down-regulation of autophagy. The ULK complex contains the ULK1 or ULK2 kinase, Atg13, FIP200 (focal adhesion kinase-family interacting protein of 200 kDa) and Atg101, an Atg13-binding protein in mammals. Once activated, mTORC1 favors cell growth by promoting translation via the phosphorylation of p70S6K (70 kDa polypeptide 1 ribosomal protein S6 kinase) and of 4E-BP1, an inhibitor of translation initiation, therby inactivating it [58].
Conversely, nutrient or growth factor deprivation downregulates mTORC1, which dissociates from the ULK1 (or ULK2) complex, leading to its subsequent dephosphorylation on specific residues and resulting in its activation. Activated ULK1 (or ULK2) phosphorylates itself and both Atg13 and FIP200 to initiate autophagy. The activated ULK1 (or ULK2) complex localizes at the phagophore during starvation. How ULK1 and ULK2 activate downstream components of the autophagic machinery remains unclear, but ULK1 can phosphorylate Ambra1 (activating molecule of Beclin 1-regulated autophagy 1), a component of the Beclin1 complex associated with the kinase vps34 [59]. The mTOR-ULK1-vps34 pathway can be stimulated by multiple forms of cellular stress in addition to nutrient or growth factor deprivation, including hypoxia, reactive oxygen species, DNA damage, protein aggregates, damaged organelles, or intracellular pathogens. Accordingly, ULK complexes are regulated by multiple pathways including cAMP-dependent protein kinase (AMPK). However, a detailed study of pathways involved in these cellular stresses is complex and beyond the scope of this review [50,56].
Initiation/nucleation
In the initial step of macroautophagy, an isolation membrane forms in the cytoplasm through the activity of specific autophagy effectors (see below, and Figure 6), including LC3 (microtubules-associated protein light chain 3). The nascent membrane, called the “isolation membrane”, wraps around a portion of cytoplasm that may contain soluble proteins, organelles, or aggregates to be degraded. It forms a crescent-shaped structure called the “phagophore” or “omegasome”, which is assembled at the phagophore assembly site (PAS). Sources of the phagophore membranes remain uncertain, but potentially include the endoplasmic reticulum, the golgi complex, endosomes, mitochondria, and the plasma membrane [54,60]. Nucleation and assembly of the initial phagophore are under the control of a complex of class III phosphatidylinositol 3-kinases (PI3K). The core components of this complex include the catalytic PI3K unit Vps34 (vacuolar protein sorting), Vps15, a p150 regulatory kinase, and a positive modulatory unit Beclin 1 (Atg6 in yeast). The activity of this complex is tightly controlled by positive and negative regulators, and determines the level of cellular autophagy. Mammalian cells possess Beclin 1-binding proteins, including positive regulator Atg14L [also known as Barkor (Beclin 1-associated Atg Key regulator)], Bif-1 and UVRAG, and negative regulators, Bcl-2, and Rubicon, characterized by a more transient binding [61]. Phosphatidylinositol-3-phosphate (PtdIns3P) is generated by Vps34 and constitutes an essential membrane component of the elongating phagophore. In mammalian cells, WIPI1 and WIPI2 (WD-repeat protein interacting with phospholipids, orthologous to yeast Atg18), DFCP1 (double FYVE-containing protein), and Alfy (autophagy linked FYVE protein) are recruited by PtdIns3P at the membrane, together with Atg2 (in yeast, but its function in mammalian cells is not known yet [59]). Subsequently, WIPIs induce membrane rearrangements that ultimately facilitate the formation of autophagosomes by an unknown molecular mechanism [62]. Atg9 (mAtg9 or Atg9L1 in mammals) is the only transmembrane Atg protein. mAtg9 traffics between the trans-Golgi network and late endosome in normal cells, but in response to starvation, it localizes to autophagic vacuoles. It is supposed to carry lipids or to serve as platform for recruiting effectors to the phagophore [63].
Figure 6.

Integrated view of mammalian autophagy. Autophagy is initiated (1) by the nucleation of the phagophore, also called isolation membrane. This membrane vesicle then elongates (2) and closes on itself as a double membraned vesicle, the autophagosome. It selectively engulfs proteins (black) or organelles such as mitochondria, as well as non-selectively a portion of the cytosol. The autophagosome usually fuses with endosomes coming from the endocytic pathway (3). The resulting amphisome then fuses with lysosomes to form autolysosomes, in which macromolecules and proteins are degraded in an acidic environment by lysosomal hydrolases. The Beclin1:hVps35:Atg14L complex controls the initiation process, and is regulated by several associated proteins, such as Ambra1, Bif-1, UVRAG, or Bcl-2. The kinase hVps34 allows the synthesis of phosphatidylinositol-3-phosphate (PtdIns3P), which is essential for the phagophore formation. In parallel, Atg9 and WIPI proteins contribute also to the nucleation and elongation of the phagophore. Below, LC3 proteins (Atg8 orthologs) are cleaved by Atg4, and linked to phosphatidylethanolamine (PE) by the Atg7, Atg3, and Atg12-Atg5:Atg16 conjugation complex, to be finally included into the autophagosomal membrane. It plays a crucial function in the elongation process, and in cargo anchoring to the autophagosome inner cavity, with the help of adaptors like p62. The second conjugation complex, Atg12-Atg5:Atg16, is bound to the inner and the outer membrane of the elongating phagophore, and is supposed to play a role in the elongation as well as the closure of the autophagosome.
Elongation
Two ubiquitin-like conjugation systems are involved in the maturation of phagophores into closed autophagosomes during an elongation and a closure stage [16]. In the first conjugation system, the Atg12-Atg5 ubiquitin-like conjugation system promotes elongation and closure of the autophagosome. Atg12 is activated by Atg7 (an E1-like enzyme), is transferred to Atg10 (E2-like enzyme), and is, finally, conjugated with Atg5 [64]. The Atg12-Atg5 conjugate then binds to Atg16L (Atg16 in yeast) in a non-covalent fashion to constitute an E3-like enzyme. The second conjugation system involves the Atg8 proteins. The yeast Atg8 possesses several mammalian orthologs: LC3A, LC3B, LC3C, GABARAP (γ-AminoButyrate acid receptor-associated protein), GABARAPL1 (GABARAP-Like protein 1), GABARAPL2/GATE-16 (Golgi-associated ATPase enhancer of 16 kDa), and GABARAPL3 (a human paralog) [65,66]. LC3B is thought to act as the main Atg8 homolog involved in starvation-induced autophagy [67]. These ubiquitin-like proteins are synthesized as precursors and are essential components of autophagosome formation. LC3 processing involves Atg4, a cysteine protease that cleaves LC3 at its C-terminus, exposing a glycine residue. This form of LC3 is then activated by Atg7 (E1-like enzyme), transferred to Atg3 (E2-like enzyme), and, finally, covalently linked to an amino group of phosphatidylethanolamine (PE), a major membrane phospholipid, by the Atg12-Atg5:Atg16L complex (E3-like enzyme) [68,69]. Atg4 is also able to cleave LC3-PE, thus functioning as a deconjugating enzyme, a reaction that controls the level of active LC3. LC3-PE localizes at both sides of the isolation membrane. Several results suggest that the level of LC3-PE tightly controls the size of autophagosomes [70]. When the autophagosome closes, resulting in a double-membrane vacuole-which is typical, but not exclusive, of autophagosomes-the Atg12-Atg5:Atg16L complex leaves the autophagosome, and LC3-PE molecules associated with the autophagosomal cytosolic surfaces are cleaved from PE by Atg4 and recycled. Recent evidence suggests that, if LC3 members of the LC3 subfamily have a role in elongation of the autophagic membrane, GABARAP proteins act downstream of this step in regulating the size of the autophagosomes, possibly by controlling their closure [66].
Cargo recognition
Although originally considered a non-specific process, autophagy has been more recently found to occur in a selective mode of degradation for targeted defective proteins or damaged organelles. The LC3 family of proteins not only plays an essential function in autophagosome biogenesis, but also in selective autophagy (Figure 7). LC3 family members are able to recruit several adaptor proteins, including p62/SQSTM1 (sequestosome 1), NBR1 (neighbor of BRCA1), and Nix/BNIP3L, as well as other types of proteins, such as calreticulin and clathrin heavy chain [65]. p62 and NBR1 are both selectively degraded by autophagy and are able to act as cargo receptors for degradation of ubiquitinated substrates for autophagy. p62 has been more extensively studied, and contains an LIR (LC3 interacting region, also called LRS, LC3 recognition sequence) domain that binds to LC3 as well as a UBA (ubiquitin-binding adaptor), which allows ubiquitin-conjugated substrate proteins to be incorporated into the autophagosome [41]. p62 interacts preferentially with long K63-linked chains (with more than 7 ubiquitins) [71], but it can also bind short chains linked through both K48 and K63. The LC3 recognition region contains basic residues at its N-terminal α-helix surface, which is involved in an interaction with the acidic cluster of the LRS/LIR domain in p62 [72]. In addition, p62 polymerizes via its N-terminal Phox/Bem1 (PB1) domain, which may be responsible for aggregate formation and accumulation [73]. It can interact with NBR1 through the PB1 domain, but also through the Alfy protein. Though a LIR/LRS in Alfy has not yet been identified, Alfy physically interacts with Atg5 and p62 and functions as a scaffold for recruitment of their cargos to the isolation membrane [74,75]. However, other potential interacting proteins can bind to other surfaces of LC3 proteins, independently of LIR. p62 could also be involved in ubiquitin-independent selective autophagy, as it has recently been found for the superoxide dismutase 1 (SOD1) protein [76].
Figure 7.
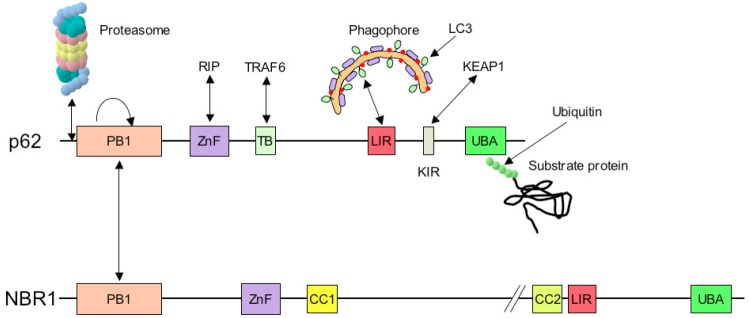
Schematic representation of the structure of p62 and NBR1 adaptors, and of their binding partners. p62 and NBR1 bind preferentially to K63-ubiquitinated proteins with the UBA domain situated at their carboxy-terminus. LC3 molecules, which are situated at the inner membrane of the phagophore, and are involved in the selective autophagic process, recognize p62 through the LIR (LC3-interacting region)/LRS (LC3 recognition sequence). In addition, p62 can bind to the proteasome as a shuttling protein due to the presence of a ubiquitin-like (UBL) domain, situated at its amino-terminal end. Other sites in p62 allow binding of RIP proteins (ZnF: Zinc finger), Traf6 mediator in IL1 or NGF signaling (TB) and Keap1 (KIR). NBR1 posseses two LIR domains and two coiled-coil domains of oligomerization (CC1 and CC2). The common domain Phox and Bem 1 (PB1) can form self-oligomerization and hetero-oligomerization with other proteins containing the PB1 domain such as p62.
NBR1 is a p62-like protein with similar domain architecture to p62. It contains a C-terminal UBA domain interacting with ubiquitin, an LIR motif binding to Atg family members such as LC3, and a PB1 domain allowing interaction with p62 [77]. As NBR1 co-localizes with p62 in ubiquitin-positive aggregates in response to autophagy inhibition, it suggests that p62 and NBR1 cooperate in sequestering and degrading ubiquitinated proteins [78].
Another adaptor, Nix/BNIP3L has been associated exclusively with mitochondrial degradation (mitophagy) during erythroid maturation [79]: mitochondria are progressively lost from the erythroblast until the mature erythrocyte stage. The mitochondrial molecule Nix is responsible for recruiting autophagosomes to depolarized mitochondria and was suggested to be essential for the loss of mitochondrial membrane potential, which would act as a signal to target mitochondria for autophagy. Nix was recently found to bind the mammalian LC3 and GABARAP and thus recruit forming autophagosomes to depolarized mitochondria. As such Nix may represent the closest mammalian homologue of the yeast Atg32, at least in erythroid mitophagy [80].
Fusion
After their formation, autophagosomes can fuse to endocytic compartments that are either early or late endosomes, as well as multivesiculated bodies [59], and are called amphisomes (amphi: both, some: body). Amphisomes fuse with the lysosomal compartment, a step that recruits hydrolases, including cathepsins B, D and L, to contact the cargo transported in autophagosomes [81]. These hydrolases work as endopeptidases under acidic conditions, determined by vacuolar ATPases at the lysosomal membrane. Small peptides and amino acids resulting from that digestion are then released into the cytosol by putative permeases [82]. In S. cerevisiae, Atg22 has recently been identified as a permease that recycles amino acids from the vacuole [83]. These last steps of the autophagic process are of fundamental importance, since any blockade in the autophagosomal flux would result in an accumulation of autophagosomes that would ultimately interrupt the autophagic process. Fusion events between autophagosomes and lysosomes have been studied extensively and indicate that it is a multi-step process regulated through complex molecular machinery that includes ESCRT and HOPS molecular complexes. ESCRT proteins (endosomal sorting complex required for transport) situated at the vesicle membrane may be crucial for the autophagosome-lysosome fusion and may have a role in autophagosome formation [84,85]. HOPS (homotypic fusion and proteins sorting) plays a role in the early stages of docking at the vesicle surface and takes part in the vesicle fusion with SNARE (soluble N-ethylamide-sensitive factor attachment protein receptor) and Rab [86,87]. In fact, fusion between two cellular compartments depends on the canonical cellular fusion machinery, the Rab-SNARE system. SNAREs are membrane-anchored proteins that regulate lipid bilayer fusion, and Rab GTPases associate with membranes and play an important role in tethering and docking vesicles to their target compartment during vesicle fusion [88]. Rab proteins can directly regulate SNARE function during fusion [89,90]. Other proteins such as LAMPs (lysosomal-associated membrane proteins) [91], UVRAG (UV irradiated resistance-associated gene) [92], AAA ATPases [93], and DRAM (damage-regulated autophagy modulator) [94] have also been demonstrated to play a role in the later stage of autophagy in mammals but their precise function remains to be further elucidated.
Non-conventional autophagy
Recent findings suggest that macroautophagy can also occur in the absence of some of the key autophagy proteins, through a non-canonical mechanism: steps involving Beclin1, ULK1, Atg5, and LC3 can thus be bypassed to allow autophagosomes formation, under special condition of stress, such as pro-apoptotic stimuli or nutriment deprivation. The monomeric GTPase Rab9 that is involved in vesicle trafficking between the trans-Golgi network and late endosomes is required in this non-canonical form of autophagy [95].
Physiopathological alterations in autophagy efficiency
Autophagy has been implicated in several diseases, including neurodegenerative, liver, muscle (including heart) diseases, cancer, and aging.
Indeed, reduced macroautophagy activity has been reported in Parkinson’s disease, Huntington’s disease, Alzheimer’s disease, polyglutamine disease, amyotrophic lateral sclerosis and prion diseases. Alteration in autophagy in these disorders spread across problems in autophagosome formation, cargo recognition, autophagosome mobilization toward lysosome fusion or in the degradation of the autophagic cargo once delivered to the lysosomes [41]. The development of neurodegenerative disease in patients with proteinopathies implies that the autophagy may reach a saturation point in which its capacity to degrade the mutant aggregate-prone proteins is exceeded, or that concurrent defects may occur in the autophagy pathway. The benefits of autophagy in improving the pathological conditions caused by the misfolded proteins could be clearly demonstrated in animal models of Alzheimer’s disease, Huntington’s disease or Kennedy’s disease, while mice deficient for autophagy show neurodegeneration in the brain [96,97].
Similar to neurodegenerative diseases, the pathogenesis of myodegenerative diseases may involve either the failure of autophagosomes to fuse with lysosomes or the aggregation of misfolded proteins that exceed the autophagic clearance capacity of the myocyte. Danon disease, a genetic disease characterized by cardiomyopathy, myopathy, and variable mental retardation, results from a mutation in the lysosomal protein LAMP-2 and is associated with extensive accumulation of autophagosomes in the muscles of LAMP-2-deficient mice and patients. Inhibition of other essential components of the autophagic pathways (Atg5 or Atg7) triggers cardiac hypertrophy, left ventricular dilation, and diminished cardiac output, accompanied by increased levels of ubiquitinated proteins and aggregated mitochondria [43]. Conversely, cardiac pathology elicited by multiple stressors, including elevated afterload, chronic ischemia and ischemia/reperfusion, are associated with robust induction of autophagy. However, induction of autophagy can either antagonize disease pathogenesis or contribute to the progression of the disease depending on the context and amplitude of induction [98]. For example, autophagy may be protective in ischemia, when cardiomyocytes need energy. Moreover, rapamycin reduces established cardiac hypertrophy, improves cardiac function in pressure-overloaded rats, and is protective against ischemia/reperfusion injury in cultured mouse cardiomyocytes. By contrast, activation of autophagy is maladaptive in cases of load-stressed hearts, and during the post-ischemic reperfusion phase [99].
The role of autophagy in cancer is complex and highly debated. On one hand, autophagy, as a housekeeping process capable of preventing accumulation of toxic cellular waste, some of which may be carcinogenic, can act as a tumor suppressor. On the other hand, the ability of autophagy to support cell survival in conditions of hypoxia and nutrient deprivation may assist the survival of tumors, as these need to develop strategies to survive in sites where there is poor vasculature or reduced nutrient delivery to the core of a lesion.
In the course of cellular transformation, autophagy may act as a tumor-suppressor pathway. The autophagic machinery can limit DNA damage and chromosomal instability. In addition, many of the signaling pathways leading to tumorigenesis overlap with those regulating autophagy, and a number of autophagy genes such as Beclin 1, ATG5, ATG4c, and Bif-1 also have properties of tumor suppressors in mice. Conversely, the products of several tumor suppressor genes, such as DRAM, PTEN, DAPK, TSC1, and TSC2, positively regulate autophagy (reviewed in [47]). The most frequently mutated in human cancers gene p53, can transactivate genes that induce autophagy, like DRAM and sestrin 1 and 2, though its role in autophagy remains controversial, while other oncogenes such as class I PI3K, Akt/PKB, and Bcl-2, among others, inhibit autophagy. Thus, in general, there is a positive correlation between molecules that induce autophagy and tumor suppression, and between molecules that inhibit autophagy and tumor progression.
On the other hand, macroautophagy serves as a pivotal mechanism to facilitate tumor growth and survival under conditions of nutrient deprivation by liberating free amino acids into the cytosol as a result of self-digestion [46]. Oncogenically transformed cells may have a stronger ability to initiate autophagy and become more dependent on this mechanism to survive, especially when exposed to radiation or chemotherapeutic agents. For example, pharmacological inhibition of autophagy in colorectal tumors causes nutrient deprivation induced cell death, highlighting an essential role of autophagy for the survival of cancer cells [100]. This has led to pharmacologically favorable inhibitors of autophagy, like chloroquine (inhibiting lysosomal acidification) and its derivatives, being tested in clinical trials as sensitizers for radio- and chemotherapy in several malignancies. In addition, a large series of clinically approved and experimental anticancer therapies induce the accumulation of autophagosomes in tumor cell lines in vitro [101]. Altogether, these data suggests that inhibition of autophagy, rather than stimulation of autophagy, might be beneficial in cancer treatment.
Autophagic activity decreases with age, leading to poor response to stress and inefficient clearance of damaged proteins and organelles in cells, which may contribute to the functional deterioration of aging organisms [102]. In particular, increasing evidence suggests that impaired autophagy is important in neuronal dysfunction and death in ageing and age-related disease. The mechanism for the decline in autophagy with aging is unknown but, at least in the rodent liver, is thought to involve alterations both in responses to hormonal regulation of autophagy (e.g., glucagon, insulin) and in the degradation of autophagosomes [103]. It is possible that the accumulation of undigested material inside secondary lysosomes, including lipofuscin, a nondegradable pigmented product and a characteristic feature of aged or postmitotic cells, interferes with the ability of lysosomes to fuse with autophagosomes and degrade their cargo [104]. This situation thus creates a vicious cycle and leads to a progressive defect in autophagosome degradation. However, actively dividing and growing cells can partially eliminate lipofuscin by diluting the pigment in each mitotic cycle. Another explanation to the decrease in autophagy efficacy comes from multiple reports indicating that Atg proteins or other proteins required for autophagy induction, such as Sirtuin 1, have reduced expression in aged tissues and that autophagy diminishes with age [105]. This applies, for instance, to normal human brain aging in which Atg5, Atg7, and Beclin 1 are downregulated. Finally, one interesting feature is that caloric restriction, an inducer of autophagy, extends lifespan of diverse organisms, including yeast, worms, flies, fish and mammals (reviewed in [102]).
In summary, the development of these diseases in patients implies that autophagy may reach a saturation point in which the capacity to degrade aggregate-prone proteins is exceeded. While in degenerative diseases, and in some of cardiopathies a way to specifically stimulate autophagy is sought, in contrast, in anti-cancer therapies, inhibition of autophagy might be beneficial (reviewed in [106]). Moreover, autophagy may also be involved in cell death, liver diseases, and innate and adaptive immunity, and it is therefore likely that more diseases with autophagy associations will be discovered in the future.
Conclusion
Autophagy, a key component of the intracellular protein quality control system, and of the homeostatic pathway, seems to be intricately linked with different adaptive and repair responses to stress. Autophagy can be regulated at least at three levels, upstream signaling pathways, autophagosome formation, and autophagosome maturation and fusion with lysosomes. Many questions remain opened about these mechanisms, that will probably be addressed in the future.
Interactions
The need for energetic homeostasis and protein balance (proteostasis) requires that both degradative systems, the UPS and autophagy, are tightly controlled and coordinated during a cell’s life. The relative contribution of each degradation pathway is different between cell types. Proteasomal degradation is predominant in cultured cells that are not subjected to stress conditions. Depending on the cell type, the relative contribution of the UPS and autophagy may considerably vary: in muscle cells, for example, autophagy can account for 40% of degradation of long-lived proteins [107]. In general, if short-lived proteins are specifically degraded by the ubiquitin-proteasome system [108], long-lived proteins are preferential substrates for autophagy [109]; however, this distinction is only relative [110]. Further, one hypothesis suggests that, in cells in which the UPS is dominant, autophagy may act as a backup system that can help or relieve the burden of the UPS in case of overloading [111]. Ubiquitin-proteasome and autophagy degradation pathways were considered for a long time as independent. However, as seen above, recent results have suggested that ubiquitination can target substrates for degradation via both pathways. In addition, several proteins, such as α-synuclein [112,113], aggregate-prone, or other proteins [110,114], are known to be substrates of both degradative pathways.
At the interface between the UPS and autophagy, the first consideration is the protein to be degraded. This protein either has to be refolded or degraded, and a pathway must be chosen. The decision depends on multiple parameters, including the structure of the protein and the chaperone and co-chaperone molecules that constitute the primary detection device for proteins to be degraded. Additionally, the machinery must consider ubiquitin ligases and ubiquitin structure as a degradative signal (e.g., monoubiquitination, K48, K63). Then, ubiquitin-binding molecules may work as adaptors, shuttling proteins, or proteins linking directly to the proteasome or to the growing phagophore. Another level of cross-talk between the UPS and autophagy involves more general cellular integrative programs, such as the endoplasmic reticulum (ER) stress response, atrophy in muscle cells, or energetic homeostasis.
First level of interface: the protein to be degraded
The protein to be degraded contains in its structure, at least partially, elements that will determine its fate (Figure 8A): 1) The protein’s structure specifies if it is a wild-type or a mutant that may be less stable. Additionally, the protein may contain clusters of hydrophobic amino acids and/or specific signals such as N-terminal amino acids following the N-end rule, or degradation signals or degrons that may determine if the protein belongs to the class of short- or long-lived proteins. 2) The secondary structure may give rise to conformational instability or exposure of hydrophobic regions to the environment. 3) At a higher level of protein structure, only soluble proteins can be degraded by the proteasome, while aggregates cannot, and may even impair its functioning [110]. The candidate protein for degradation thus constitutes the first element at the interface between the two degradative systems, since the triage decision may eventually saturate one system, and modify the working of the other.
Figure 8.

Schematic representation of factors situated at the interface between the ubiquitin-proteasome and the autophagy-lysosome degradative systems. A: Molecular interface between the UPS and autophagy. Misfolded proteins may be degraded preferentially by the UPS or by autophagy, or even by both degradative pathways. The fate of a given protein is dictated first by its altered conformation, and/or by molecular determinants such as exposed hydrophobic amino acids, specific signals (degrons), or N-terminal specific amino acids. These misfolded proteins are then recognized by the front-line detectors constituted of chaperones (Hsp70, Hsc70 or Hsp90) and co-chaperones (Bag1, Bag3, CHIP, HspBP1, HSJ1, etc.) that will attempt to refold it, or will make decisions (triage) about refolding or degrading the substrate protein. For simplicity, the case of Hsp90 has not be treated in this figure (see reference [156] for more details). Co-chaperones molecules can display a ubiquitin-ligase activity (CHIP), and recruit ubiquitin-conjugating enzymes such as Ubc4/5, or facilitate binding to the proteasome (Bag1). On the other hand, the co-chaperone Bag3 will favor K63- (Lysine 63) over K48-polyubiquitination (Lysine 48), and direct the substrate protein to autophagic degradation. The tagged protein is usually recognized then by several types of adaptor proteins that bring them to the proteasome or to autophagic vesicle. Certain adaptor proteins, such as p62, can play different roles for both degradative systems. There are proteins involved in the structure or in regulation of one degradative pathway, and are degraded by the second degradative system, that are thus contributing to the cross-talk between both systems (p53, LC3, or proteasome subunits, see text). At a higher level of integration, the degradative pathways are probably coordinated with more general homeostatic programs, such as response to stress factors (the ER stress response is one example of coordination of signaling pathways directing the activity of both degradative pathways). Another example of integrative program concerns the catabolic atrophy in muscles that involves the coordinating action of the FoxO transcription factors on both pathways. B: Energy interface as a possible balance element between UPS and autophagy activities. The UPS requires energy to assemble the proteasomal complex, activate ubiquitin, and unfold the substrate protein. Although the proteasome structure can be switched to an ATP-free one, and certain proteins can be degraded without needing ubiquitination, low levels of ATP may globally reduce the efficiency of this pathway. Conversely, low level of ATP, amino acids (a.a.), glucids or anabolic hormones (insulin, insulin-like growth ractor (IGF-1)) results in the activation of autophagy. Amino acids resulting from protein degradation may either be used for the synthesis of new proteins, sparing thus energy for their de novo synthesis (except for essential amino acids that have to be brought by nutrition), or may be totally degraded through a catabolic pathway to recover new energy.
Most misfolded soluble proteins would be preferentially degraded by the proteasome, and by autophagy only if the proteasome capacity is exceeded. However, micro- and macroautophagy are both able to degrade soluble proteins, either on a house-keeping basis, or as a bulky process under stress in the case of macroautophagy, as it is the case under amino acid, energy, or growth factor depletion. However, aggregates can only be degraded by autophagy. Notably, this is true even for very large (2 μm) aggregates in cultured cells [73]. This phenomenon may correspond to the process of concentrating aggregated proteins into a single structure at the microtubule organizing center (MTOC). This structure is called the aggresome, which is supposed to facilitate its autophagic degradation [111].
Second level of interface: degradative tags and linking proteins
The second level of cross-talk between the UPS and autophagy depends on the tagging system that marks proteins for degradation, usually with ubiquitin, or ubiquitin-like proteins. The type of ubiquitin structure and polymerization determines the fate of the protein to which it is attached. However, upstream of ubiquitination, misfolded or damaged proteins are first detected by the chaperone/co-chaperone system, which constitutes the front-line of detectors that contribute to cellular homeostasis [115]. Proteins misfolded at the synthesis step, denatured following a stress (thermic shock, oxydation, glycation, etc.), constitutively rapidly degraded for cell signaling, or presenting regions with low levels of structuration, are good candidates for such detection.
Co-chaperone molecules and degradative pathways
Among co-chaperone molecules able to decide the fate of candidate proteins for degradation, CHIP and BAG proteins have been found to be deciding how to direct substrate proteins toward the UPS or the autophagic proteolytic pathways [116]. CHIP (C-terminus of Hsp70-interacting protein) is a co-chaperone with an E3 ubiquitin ligase activity associated to Hsp70. It contains two functional domains and may mediate substrate degradation by both degradative pathways: the tetratricopeptide repeat domain is critical for proteasomal degradation, whereas the U-box domain is sufficient to direct substrates toward the lysosomal degradation pathway [117]. Studies performed with α-synuclein suggest that CHIP acts effectively as a molecular switch between UPS and autophagy degradation pathways [118]. Other co-chaperones known to be involved in switching between the UPS and autophagy are members of the BAG (Bcl-2-associated athanogene) protein family. BAG1 constitutes a link between HSP70 and the proteasome [119,120]. It also interacts with CHIP, and may therefore direct Hsc/Hsp70 substrate to the proteasome. In contrast, BAG3, associated to the sHSP (small heat shock protein) HSPB8 (sHSP20), facilitates the degradation of substrates such as mutated Huntingtin with expanded glutamine repeats via the autophagic process [121]. Moreover, BAG3 interacts with p62 to promote p62-dependent autophagic degradation [120]. The BAG3/BAG1 ratio may therefore regulate autophagy compared to proteasomal pathway, respectively. This ratio has been shown to increase during aging, which indicates that aged cells depend more on autophagy than the UPS machinery to degrade polyubiquitinated proteins [122]. As a result of protein identification by chaperones and co-chaperones, E3 ligases associated to E2-conjugation enzymes are recruited by co-chaperones, or are already present in the co-chaperone molecule, as it is the case for CHIP. This step may be regulated, and one can hypothesize that regulating cofactors will be found that will control the association between co-chaperones and E2/E3 enzymes and to determine precisely which degradative pathway will seal the fate of a specific protein.
The result of this first step of identification by chaperone/co-chaperone complexes is a tag attached to the protein to be degraded, which is usually ubiquitin. The type of ubiquitination may direct the substrate toward one or the other degradative pathway. For example, K48-linked polyubiquitinated chains target the substrate to the proteasome [123], while K63-linked polyubiquitinated chains or monoubiquitinated substrates [124] favor autophagic degradation [125,126]. There also exist substrates that can be degraded by both pathways, like α-synuclein and alpha(1) antitrypsin in the ER [127]. Further studies are needed to clarify these points. In fact, depletion of ubiquitin, which is present as free protein at a limited concentration in cells, by saturating one system of degradation has, therefore, mechanically a strong effect on the second one.
Common adaptor molecules
Following ubiquitination, adaptor proteins associate to ubiquitin or to other degradative tags. These proteins include p62, NBR1 (Figure 7), and, more generally, proteins bearing ubiquitin binding domains (UBD) or ubiquitin-associated domains (UBA). These adaptors are either shuttling proteins, part of the proteasome, or linkers for engulfing autophagic cargo.
P62/SQSTM1 is an adaptor molecule linking ubiquitinated proteins to the autophagic machinery through its ubiquitin-associated (UBA) domain on one side, and directly binds to LC3 and other Atg8 homologs via the LC3 interacting region (LIR) motif on the other side [73]. p62 can also interact with the intrinsic subunits of the proteasome via an N-terminal ubiquitin-like (UBL) domain, therefore shuttling substrates for proteasomal degradation [41,128]. p62 has homologs: NBR1, or Atg32 and Nix for mitophagy in developing erythroid cells.
In fact, these molecules at the interface between the two systems were identified through perturbations in the flux through either pathway that have been reported to affect the activity of the other system. These data indicate cross-talks between the two pathways. Indeed, proteasome inhibitors induce autophagy as a compensatory response [129-131]. The autophagy adaptor protein p62 is also involved in this mechanism. Conversely, genetic inactivation of essential autophagic genes results in the accumulation and aggregation of ubiquitinated proteins [96,97]. However, this converse situation is not symmetrical to the inhibition of the proteasome: long-term autophagy inhibition slows the clearance of short-lived proteasome-specific substrates (like p53) because of elevated levels of p62, which might result in the sequestration of ubiquitinated short-lived substrates [132]. In addition, inhibition of autophagy does not lead to the induction of the proteasome, as a general compensatory mechanism.
A third molecule that may play a role at the interface between the UPS and autophagy is HDAC6 (histone deacetylase 6), and is associated to microtubules through dynein motor molecules. It binds ubiquitin via the C-terminal BUZ domain, thereby allowing retrograde transportation of ubiquitinated proteins toward the microtubule organizing center (MTOC) in the vicinity of the nucleus [129]. Accumulation of aggregated proteins at the aggresome is supposed to help to detoxify cells against oligomers, amyloid fibrils or small aggregates [133]. Proteasomal complexes were found to be more abundant in this region, but there are also reports indicating that aggregates may impair their activity [134]. Moreover, HDAC6 is also essential for the retrograde transport of autophagosomes and lysosomes to MTOC where it may facilitate the autophagic degradation of aggresomes [135]. HDAC6 may therefore favor autophagy upon the proteasome process.
Interaction through common regulatory molecule
Several kinases or transcription factors may be situated at the interface between both degradative pathways. For instance, a regulatory molecule may activate or inhibit one pathway, and may be degraded by the other one. When this second degradative pathway is downregulated, the quantity of regulating molecule is increased, which then affects the first pathway. This is the case for example for p53: in dopaminergic neurons, induction of autophagy following proteasome inhibition may take place via a mechanism requiring p53, which level is increased [136]. p53 is suggested to activate AMPK and inhibit mTOR, and also to induce the transcription of DRAM.
LC3, involved in autophagosomes formation and in cargo selection, has also been shown to be degraded by the proteasome system without absolutely requiring ubiquitination [137]. Further studies are needed to better understand this point. Interestingly, proteasomal subunits were also found to be degraded by lysosomes [138,139].
It may be interesting to further investigate whether other regulatory molecules, such as proteins controling autophagy, like kinases or Atg proteins for example, may be degraded by the proteasome.
Integrative mechanisms
The third level of control is determined by general cellular programs that coordinate the activity of several genes involved in the degradative pathways, through the activity of kinases and transcription factors. So far, only two such programs have been found: the ER stress response [140], and the atrophy program working more specifically in muscle, including cardiac [43], cells. Additionally, the possibility of coordination of the two degradative pathways through the sensing of cellular energy level will be discussed.
The ER stress pathway
One of the main elements that coordinates the activity of the UPS and autophagy is the endoplasmic reticulum (ER). ER is the site for post-translational protein modifications, including folding, oligomerization, glycosylation, and disulfide bond formation. ER stress can be caused by the accumulation of misfolded or premature proteins in the ER lumen or the cytosol [141,142]. In eucaryotic cells, UPS is the main system that degrades misfolded proteins exporting from the ER. The main mechanism of response against ER stress is called unfolded protein response (UPR). In mammalian cells, UPR is mediated by three pathways, PERK, ATF6, and IRE1 [143]. As a consequence of their induction, global protein synthesis will be downregulated, but ER chaperone molecules and proteins involved in degradation pathways will be upregulated. This mechanism helps the UPS because it will lower its burden [144]). However, ER stress, through part of the UPR pathway, can also activate autophagy: the IRE1 pathway is necessary for lipid conjugation of LC3 [145], probably via activation of the JNK pathway, which in particular phosphorylates Bcl-2 and induces Beclin1 activation [130,146]. The PERK/eIF2alpha phosphorylation step is the second essential arm to mediate polyglutamine-induced LC3 conversion [147]. It downregulates general protein synthesis, but permits relative transcription of specific transcription factors such as ATF4. Several studies have highlighted the importance of this transcription factor in the upregulation of autophagy following proteasome inhibition [148,149]. However, signals activating ATF4 are not restricted to the UPR: in response to amino acid deficiency, double-stranded RNA, or heme limitation, in addition to ER stress, this factor stimulates the expression of Atg5, Atg7, and LC3 genes.
FoxO transcription factors
In atrophying muscle cells, FoxO3 coordinately activates protein degradation by both pathways [150,151], and its activation has been found to be essential. FoxO3 activation causes dramatic atrophy of muscles and cultured myotubes via transcription of a set of atrophy-related genes (“atrogenes”) including atrogin-1, a critical ubiquitin ligase involved in proteasomal degradation of proteins [152]. In addition, FoxO3 increases the transcription of many autophagy-related genes, including LC3B, GABARAPL1, ATG12, vps34, ULK2, Atg4B, Bnip3, and Beclin 1. This transcription factor, which is negatively regulated through the insulin/IGF-1/PI3K(I)/Akt pathway, therefore constitutes, at least in muscular cells, a master control element that coordinates the activation of autophagy and the UPS pathways [153].
Level of cellular energy available
The level of cellular energy available, in other words ATP, may also constitute a link between the ubiquitin-proteasome and autophagy (Figure 8B). In fact, when ATP level is high, the degradation process uses more energy, and the ubiquitin-proteasome system is working properly. Refolding of damaged proteins by chaperones and co-chaperones, proteasome assembly, activation of ubiquitin by E1 enzymes, as well as target protein unfolding before entry into the proteasomal cavity for degradation, have an energy cost. While refolding of a certain protein may spare neosynthesis, its complete degradation will spare de novo synthesis of some amino acids. When ATP is low, it may slow down protein refolding as well as proteasomal degradation. However, in some cases, there are alternative approaches to the 19 S subunit of the proteasome that do not need ATP. In addition, degradation of unfolded proteins following a stress, such as oxidative stress, does not consume as much ATP. It would therefore be interesting to measure if a switch in types of proteasomal degradation is triggered in case of low energy. On the other hand, low ATP level results in activating autophagy, in part because of the mechanistic increase of AMP, which activates AMPK, and finally inhibits the mTOR pathway. In addition, mitophagy is the autophagic process that removes damaged mitochondria that are no longer effective in producing energy. It is not clear, however, how high the energy cost of synthesizing autophagic vesicles is. In the case of low resources, autophagy is stimulated, and is probably coupled to catabolic pathways that may also be activated to replenish ATP levels, otherwise the cell would probably be destroyed by apoptosis or necrosis. As a general point of view, it would be interesting to compare the energy cost of each degradative process, UPS and autophagy.
Conclusion
The two synergistic degradation systems in eukaryotic cells, the ubiquitin-proteasome system and autophagy, are cooperative and complementary to maintain cellular homeostasis, in forming a network for critically monitoring and preventing toxicity arising from protein misfolding, and ultimately in dealing with toxic misfolded species [40,46,154,155]. This network includes chaperones, co-chaperones, stress-sensing molecules, aggresomes, UPS, and autophagy. Both degradative pathways can be coactivated to degrade misfolded proteins, but display also compensatory effects when one is dysfunctional.
During the last decades, extensive effort has been invested to understand molecular mechanisms underlying both the UPS and autophagy, and to learn how these two different degradative pathways are integrated as components of cellular catabolism or survival mechanisms. Elucidation of the signaling network bridging these two systems may help to identify novel drug targets for enhancing the cellular sensitivity to degradation inhibitors for anticancer treatments, or to enhance efficiency of misfolded protein degradation to cure degenerative diseases. Further understanding of the molecular mechanisms of communication between UPS and autophagy in response to different stimuli is therefore important to manipulate the pathways for therapeutic goals and will help pharmaceutical development of novel treatment methods for a variety of diseases.
Acknowledgement
This study was supported by University Paris Diderot-Paris 7, CNRS, AFM (French Association against Myopathies). I thank Sheila Cherry, Fresh Eyes Editing, LLC for revision of the English.
References
- 1.Schoenheimer R, Ratner S, Rittenberg D. Studies in protein metabolism: VII. The metabolism of tyrosines. J Biol Chem. 1939;127:333–344. [Google Scholar]
- 2.Rock KL, Gramm C, Rothstein L, Clarck K, Stein R, Dick L, Hwang D, Goldberg AL. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell. 1994;78:761–771. doi: 10.1016/s0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 3.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 4.Ciechanover A. Intracellular protein degradation: From a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Biochim Biophys Acta. 2012;1824:3–13. doi: 10.1016/j.bbapap.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 5.Pickart CM, Eddins MJ. Ubiquitin: structures, functions, mechanisms. Biochim Biophys Acta. 2004;1695:55–72. doi: 10.1016/j.bbamcr.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 6.Koegl M, Hoppe T, Schlenker S, Ulrich HD, Mayer TU, Jentsch S. A novel ubiquitination factor, E4, is involved in multiubiquitin chain assembly. Cell. 1999;96:635–644. doi: 10.1016/s0092-8674(00)80574-7. [DOI] [PubMed] [Google Scholar]
- 7.Kravtsova-Ivantsiv Y, Ciechianover A. Non-canonical ubiquitin-based signals for proteasomal degradation. J Cell Sci. 2012;125:539–548. doi: 10.1242/jcs.093567. [DOI] [PubMed] [Google Scholar]
- 8.Wu CJ, Conze DB, Li T, Srinivasula SM, Ashwell JD. Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-kappaB activation. Nat Cell Biol. 2006;8:398–406. doi: 10.1038/ncb1384. [DOI] [PubMed] [Google Scholar]
- 9.Raasi S, Orlov I, Fleming KG, Pickart CM. Binding of Polyubiquitin Chains to Ubiquitin-associated (UBA) Domains of HHR23A. J Mol Biol. 2004;341:1367–1379. doi: 10.1016/j.jmb.2004.06.057. [DOI] [PubMed] [Google Scholar]
- 10.Sriram SM, Kim BY, Kwon YT. The N-end rule pathway: emerging functions and molecular principles of substrate recognition. Nat Rev Mol Cell Biol. 2011;12:735–747. doi: 10.1038/nrm3217. [DOI] [PubMed] [Google Scholar]
- 11.Kelderon D. De-ubiquitinate to decide your fate. Curr Biol. 1996;6:662–665. doi: 10.1016/s0960-9822(09)00443-6. [DOI] [PubMed] [Google Scholar]
- 12.Dikic I, Wakatsuki S, Walters KJ. Ubiquitin-binding domains - from structures to functions. Nat Rev Mol Cell Biol. 2009;10:659–671. doi: 10.1038/nrm2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hicke L, Schubert HL, Hill CP. Ubiquitin-binding domains. Nat Rev Mol Cell Biol. 2005;6:610–621. doi: 10.1038/nrm1701. [DOI] [PubMed] [Google Scholar]
- 14.Hochstrasser M. There’s the Rab: a novel ubiquitin-like modification linked to cell-cycle regulation. Gene Dev. 1998;12:901–907. doi: 10.1101/gad.12.7.901. [DOI] [PubMed] [Google Scholar]
- 15.Korant BD, Blomstrom DC, Jonak GJ, Knight EJ. Interferon-induced proteins. Purification and characterization of a 15,000 dalton protein from human and bovine cells induced by interferon. J Biol Chem. 1984;259:14835–14839. [PubMed] [Google Scholar]
- 16.Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol. 2001;2:211–216. doi: 10.1038/35056522. [DOI] [PubMed] [Google Scholar]
- 17.Raasi S, Schmidtke G, Groettrup M. The ubiquitin-like protein FAT10 forms covalent conjugates and induces apoptosis. J Biol Chem. 2001;276:35334–35343. doi: 10.1074/jbc.M105139200. [DOI] [PubMed] [Google Scholar]
- 18.Komatsu M, Chiba T, Tatsumi K, iemura S, Tanida I, Okasaki N, Ueno T, Kominami E, Natsume T, Tanaka K. A novel protein-conjugating system for Ufm1, a ubiquitin-fold modifier. EMBO J. 2004;23:1977–1986. doi: 10.1038/sj.emboj.7600205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Groll M, Huber R. Substrate access and processing by the 20S proteasome core particle. Int J Biochem Cell Biol. 2003;35:606–616. doi: 10.1016/s1357-2725(02)00390-4. [DOI] [PubMed] [Google Scholar]
- 20.Tomkinson B, Lindas AC. Tripeptidyl-peptidase II: a multi-purpose peptidase. Int J Biochem Cell Biol. 2005;37:1933–1937. doi: 10.1016/j.biocel.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 21.Griffin TA, Nandi D, Cruz M, Fehling HJ, Kaer LV, Monaco JJ, Colbert RA. Immunoproteasome assembly: cooperative incorporation of interferon gamma (IFN-gamma)-inducible subunits. J Exp Med. 1998;187:97–104. doi: 10.1084/jem.187.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lupas A, Baumeister W, Hofmann K. A repetitive sequence in subunits of the 26S proteasome and 20S cyclosome (anaphase-promoting complex) Trends Biochem Sci. 1997;22:195–196. doi: 10.1016/s0968-0004(97)01058-x. [DOI] [PubMed] [Google Scholar]
- 23.Ustrell V, Hoffman L, Pratt G, Rechsteiner M. PA200, a nuclear proteasome activator involved in DNA repair. EMBO J. 2002;21:3516–3522. doi: 10.1093/emboj/cdf333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma CP, Slaughter CA, DeMartino GN. Identification, purification and characterization of protein activator (PA28) of the 20 S proteasome (macropain) J Biol Chem. 1992;267:10515–10523. [PubMed] [Google Scholar]
- 25.Zaiss DM, Standera S, Kloetzel PM, Sijts AJ. PI31 is a modulator of proteasome formation and antigen presenting. Proc Natl Acad Sci U S A. 2002;99:14344–14349. doi: 10.1073/pnas.212257299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sorokin AV, Kim ER, Ouchinnikov LP. Proteasome system of protein degradation and processing. Biochemistry. 2009;74:1411–1442. doi: 10.1134/s000629790913001x. [DOI] [PubMed] [Google Scholar]
- 27.Rosenberg-Hasson Y, Bercovich Z, Ciechanover A, Kahana C. Degradation of ornithine decarboxylase in mammalian cells is ATP dependent but ubiquitin independent. Eur J Biochem. 1989;185:469–474. doi: 10.1111/j.1432-1033.1989.tb15138.x. [DOI] [PubMed] [Google Scholar]
- 28.Tofaris GK, Layfield R, Spillantini MG. Alpha-synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett. 2001;509:22–26. doi: 10.1016/s0014-5793(01)03115-5. [DOI] [PubMed] [Google Scholar]
- 29.David DC, Layfield R, Serpell L, Narain Y, Goedert M, Spillantini MG. Proteasomal degradation of tau protein. J Neurochem. 2002;83:176–185. doi: 10.1046/j.1471-4159.2002.01137.x. [DOI] [PubMed] [Google Scholar]
- 30.Jariel-Encontre I, Pariat M, Martin F, Carillo S, Salvat C, Piechaczyk M. Ubiquitinylation is not an absolute requirement for degradation of c-jun protein by the 26 S proteasome. J Biol Chem. 1995;270:11623–627. doi: 10.1074/jbc.270.19.11623. [DOI] [PubMed] [Google Scholar]
- 31.Tarcsa E, Szymaska G, Lecker S, O’Connor CM, Goldberg AL. Ca++-free calmodulin and calmodulin damaged by in vitro aging are selectively degraded by 26 S proteasomes without ubiquitination. J Biol Chem. 2002;275:20295–20301. doi: 10.1074/jbc.M001555200. [DOI] [PubMed] [Google Scholar]
- 32.Grune T, Reinheckel T, Davies KJ. Degradation of oxidized proteins in mammalian cells. FASEB J. 1997;11:526–534. [PubMed] [Google Scholar]
- 33.Moorthy AK, Savinova OV, Ho JQ, Wang VY, Vu D, Ghosh G. The 20 S proteasome processes NF-kappaB1 p105 into p50 in a translation-independent manner. EMBO J. 2006;25:1945–1956. doi: 10.1038/sj.emboj.7601081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coux O, Goldberg AL. Enzymes catalysing ubiquitination and proteolytic processing of the p105 precursor of nuclear factor kappaB1. J Biol Chem. 1998;273:8820–8828. doi: 10.1074/jbc.273.15.8820. [DOI] [PubMed] [Google Scholar]
- 35.Sorokin AV, Selyutina AA, Skabkin MA, Guryanov SG, Nazimov IV, Richard C, Th’ing JY, Sorensen PH, Ovchinnikov LP, Evdokinova V. Proteasome-mediated cleavage of the Y-box-binding protein 1 is linked to DNA-damage stress response. EMBO J. 2005;24:3602–3612. doi: 10.1038/sj.emboj.7600830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baugh JM, Pilienpko EV. 20 S proteasome differentially alters translation of different mRNAs via the cleavage of eIF4F and eIF3. Mol Cell. 2004;16:575–586. doi: 10.1016/j.molcel.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 37.Low P. The role of ubiquitin-proteasome system in ageing. Gen Comp Endocrinol. 2001;172:39–43. doi: 10.1016/j.ygcen.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 38.Keller JN, Hanni KB, Markesbery WR. Possible involvement of proteasome inhibition in aging: implications for oxidative stress. Mech Ageing Dev. 2000;113:61–70. doi: 10.1016/s0047-6374(99)00101-3. [DOI] [PubMed] [Google Scholar]
- 39.Konstantinova IM, Tsmokha AS, Mittenberg AG. Role of proteasomes in cellular regulation. Int Rev Cell Mol Biol. 2008;267:59–124. doi: 10.1016/S1937-6448(08)00602-3. [DOI] [PubMed] [Google Scholar]
- 40.Wong E, Cuervo AM. Integration of clearance mechanisms: the proteasome and autophagy. Cold Spring Harb Perspect Biol. 2010;2:a006734. doi: 10.1101/cshperspect.a006734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wooten MW, Hu X, Babu JR, Seibenhener ML, Geetha T, Paine MG, Wooten MC. Signaling, polyubiquitination, trafficking, and inclusions: sequestosome 1/p62’s role in neurodegenerative disease. J Biomed Biotechnol. 2006;2006:62079. doi: 10.1155/JBB/2006/62079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dobson CM. Principles of protein folding, misfolding and aggregation. Semin Cell Dev Biol. 2004;15:3–16. doi: 10.1016/j.semcdb.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 43.Zheng Q, Wang X. Autophagy and the ubiquitin-proteasome system in cardiac dysfunction. Panminerva Med. 2010;52:9–25. [PMC free article] [PubMed] [Google Scholar]
- 44.Portbury AL, Ronnebaum SM, Zungu M, Patterson C, Willis MS. Back to your heart: ubiquitin proteasome system-regulated signal transduction. J Mol Cell Cardiol. 2012;52:526–537. doi: 10.1016/j.yjmcc.2011.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu WK, Cho CH, Lee CW, Wu K, Fan D, Yu J, Sung JJ. Proteasome inhibition: a new therapeutic strategy to cancer treatment. Cancer Lett. 2010;293:15–22. doi: 10.1016/j.canlet.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 46.Wu WK, Sakamoto KM, Milani M, Aldana-Masankgay G, Fan D, Wu K, Lee CW, Cho CH, Yu J, Sung JJ. Macroautophagy modulates cellular response to proteasome inhibitors in cancer therapy. Drug Resist Updat. 2010;13:87–92. doi: 10.1016/j.drup.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 47.Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, Massey DC, Menzies FM, Moreau K, Narayanan U, Renna M, Siddiqi FH, Underwood BR, Winslow AR, Rubinsztein DC. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383–1435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- 48.Ruschak AM, Slassi M, Kay LE, Schimmer AD. Novel proteasome inhibitors to overcome bortezomib resistance. J Natl Cancer Inst. 2011;103:1007–1017. doi: 10.1093/jnci/djr160. [DOI] [PubMed] [Google Scholar]
- 49.Lee BH, Lee MJ, Parks S, Oh DC, Elsasser S, Chen PC, Gartner C, Dimova N, Hanna J, Gygi SP, WIlson SM, King RW, Finley D. Enhancement of proteasome activity by a small-molecule inhibitor Usp14. Nature. 2010;467:179–184. doi: 10.1038/nature09299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2012;40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lynch-Day MA, Klionsky DJ. The Cvt pathway as a model for selective autophagy. FEBS Lett. 2010;584:1359–1366. doi: 10.1016/j.febslet.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kaushik S, Bandyopadhyay U, Sridhar S, Kiffin R, Martinez-Vicente M, Kon M, Orenstein SJ, Wong E, Cuervo AM. Chaperone-mediated autophagy at a glance. J Cell Sci. 2011;124:495–499. doi: 10.1242/jcs.073874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li WW, Li J, Bao JK. Microautophagy: lesser-known self-eating. Cell Mol Life Sci. 2012;69:1125–1136. doi: 10.1007/s00018-011-0865-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–132. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 55.Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol. 2009;10:458–467. doi: 10.1038/nrm2708. [DOI] [PubMed] [Google Scholar]
- 56.Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell. 2010;40:310–322. doi: 10.1016/j.molcel.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hara K, Yonezawa K, Weng QP, Kozlowski MT, Belham C, Avruch J. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J Biol Chem. 1998;273:14484–14494. doi: 10.1074/jbc.273.23.14484. [DOI] [PubMed] [Google Scholar]
- 58.Chen Y, Klionsky DJ. The regulation of autophagy - unanswered questions. J Cell Sci. 2011;124:161–170. doi: 10.1242/jcs.064576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mehrpour M, Esclatine A, Beau I, Codogno P. Overview of macroautophagy regulation in mammalian cells. Cell Res. 2010;20:748–762. doi: 10.1038/cr.2010.82. [DOI] [PubMed] [Google Scholar]
- 60.Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571–580. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Funderburk SF, Wang QJ, Yue Z. The Beclin 1 -VPS34 complex- at the crossroads of autophagy and beyond. Trends Cell Biol. 2010;20:355–362. doi: 10.1016/j.tcb.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mauthe M, Jacob A, Freiberger S, Hentschel K, Stierhof YD, Codogno P, Proikas-Cezanne T. Resveratrol-mediated autophagy requires WIPI-1-regulated LC3 lipidation in the absence of induced phagophore formation. Autophagy. 2011;7:1448–1461. doi: 10.4161/auto.7.12.17802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Longatti A, Tooze SA. Vesicular trafficking and autophagosome formation. Cell Death Differ. 2009;16:956–965. doi: 10.1038/cdd.2009.39. [DOI] [PubMed] [Google Scholar]
- 64.Geng J, Klionsky DJ. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep. 2008;9:859–864. doi: 10.1038/embor.2008.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7:279–296. doi: 10.4161/auto.7.3.14487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. Embo J. 2010;29:1792–1802. doi: 10.1038/emboj.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tanida I, Ueno T, Kominami E. Human light chain 3/MAP1LC3B is cleaved at its carboxyl-terminal Met121 to expose Gly120 for lipidation and targeting to autophagosomal membranes. J Biol Chem. 2004;279:47704–47710. doi: 10.1074/jbc.M407016200. [DOI] [PubMed] [Google Scholar]
- 68.Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, Inagaki F, Ohsumi Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. 2007;282:37298–37302. doi: 10.1074/jbc.C700195200. [DOI] [PubMed] [Google Scholar]
- 69.Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell. 2008;19:2092–2100. doi: 10.1091/mbc.E07-12-1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xie Z, Nair U, Klionsky DJ. Atg8 controls phagophore expansion during autophagosome formation. Mol Biol Cell. 2008;19:3290–3298. doi: 10.1091/mbc.E07-12-1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol. 2004;24:8055–8068. doi: 10.1128/MCB.24.18.8055-8068.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shvets E, Fass E, Scherz-Shouval R, Elazar Z. The N-terminus and Phe52 residue of LC3 recruit p62/SQSTM1 into autophagosomes. J Cell Sci. 2008;121:2685–2695. doi: 10.1242/jcs.026005. [DOI] [PubMed] [Google Scholar]
- 73.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 74.Clausen TH, Lamark T, Isakson P, Finley K, Larsen KB, Brech A, Overvatn A, Stenmark H, Bjorkoy G, Simonsen A, Johansen T. p62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy. 2010;6:330–344. doi: 10.4161/auto.6.3.11226. [DOI] [PubMed] [Google Scholar]
- 75.Filimonenko M, Isakson P, Finley KD, Anderson M, Jeong H, Melia TJ, Bartlett BJ, Myers KM, Birkeland HC, Lamark T, Krainc D, Brech A, Stenmark H, Simonsen A, Yamamoto A, Clausen TH, Finley K, Larsen KB, Overvatn A, Bjorkoy G, Johansen T. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy p62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Mol Cell. 2010;38:265–279. doi: 10.1016/j.molcel.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gal J, Strom AL, Kwinter DM, Kilty R, Zhang J, Shi P, Fu W, Wooten MW, Zhu H. Sequestosome 1/p62 links familial ALS mutant SOD1 to LC3 via an ubiquitin-independent mechanism. J Neurochem. 2009;111:1062–1073. doi: 10.1111/j.1471-4159.2009.06388.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kirkin V, Lamark T, Johansen T, Dikic I. NBR1 cooperates with p62 in selective autophagy of ubiquitinated targets. Autophagy. 2009;5:732–733. doi: 10.4161/auto.5.5.8566. [DOI] [PubMed] [Google Scholar]
- 78.Kirkin V, Lamark T, Sou YS, Bjorkoy G, Nunn JL, Bruun JA, Shvets E, McEwan DG, Clausen TH, Wild P, Bilusic I, Theurillat JP, Overvatn A, Ishii T, Elazar Z, Komatsu M, Dikic I, Johansen T. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell. 2009;33:505–516. doi: 10.1016/j.molcel.2009.01.020. [DOI] [PubMed] [Google Scholar]
- 79.Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009;16:939–946. doi: 10.1038/cdd.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mortensen M, Ferguson DJP, Simon AK. Mitochondrial clearance by autophagy in developing erythrocytes: clearly important, but just how much so? Cell Cycle. 2010;9:1901–1906. doi: 10.4161/cc.9.10.11603. [DOI] [PubMed] [Google Scholar]
- 81.Turk V, Stoka V, Vasiljeva O, Renko M, Sun T, Turk B, Turk D. Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim Biophys Acta. 2012;1824:68–88. doi: 10.1016/j.bbapap.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tong J, Yan X, Yu L. The late stage of autophagy: cellular events and molecular regulation. Protein Cell. 2010;1:907–915. doi: 10.1007/s13238-010-0121-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Teter SA, Eggerton KP, Scott SV, Kim J, Fischer AM, Klionsky DJ. Degradation of lipid vesicles in the yeast vacuole requires function of Cvt17, a putative lipase. J Biol Chem. 2001;276:2083–2087. doi: 10.1074/jbc.C000739200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rusten TE, Filimonenko M, Rodahl LM, Stenmark H, Simonsen A. ESCRTing autophagic clearance of aggregating proteins. Autophagy. 2008;4:233–236. [Google Scholar]
- 85.Raiborg C, Stenmark H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature. 2009;458:445–452. doi: 10.1038/nature07961. [DOI] [PubMed] [Google Scholar]
- 86.Seals DF, Eitzen G, Margolis N, Wickner WT, Price A. A Ypt/Rab effector complex containing the Sec1 homolog Vps33p is required for homotypic vacuole fusion. Proc Natl Acad Sci U S A. 2000;97:9402–9407. doi: 10.1073/pnas.97.17.9402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wurmser AE, Sato TK, Emr SD. New component of the vacuolar class C-Vps complex couples nucleotide exchange on the Ypt7 GTPase to SNARE-dependent docking and fusion. J Cell Biol. 2000;151:551–562. doi: 10.1083/jcb.151.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Atlashkin V, Kreykenbohm V, Eskelinen EL, Wenzel D, Fayyazi A, Fischer von Mollard G. Deletion of the SNARE vti1b in mice results in the loss of a single SNARE partner, syntaxin 8. Mol Cell Biol. 2003;23:5198–5207. doi: 10.1128/MCB.23.15.5198-5207.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gutierrez MG, Munafo DB, Beron W, Colombo MI. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J Cell Sci. 2004;117:2687–2697. doi: 10.1242/jcs.01114. [DOI] [PubMed] [Google Scholar]
- 90.Jager S, Bucci C, Tanida I, Ueno T, Kominami E, Saftig P, Eskelinen EL. Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci. 2004;117:4837–4848. doi: 10.1242/jcs.01370. [DOI] [PubMed] [Google Scholar]
- 91.Eskelinen EL, Tanaka Y, Saftig P. At the acidic edge: emerging functions for lysosomal membrane proteins. Trends Cell Biol. 2003;13:137–145. doi: 10.1016/s0962-8924(03)00005-9. [DOI] [PubMed] [Google Scholar]
- 92.Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, Jung JU. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol. 2006;8:688–699. doi: 10.1038/ncb1426. [DOI] [PubMed] [Google Scholar]
- 93.White SR, Lauring B. AAA+ ATPases: achieving diversity of function with conserved machinery. Traffic. 2007;8:1657–1667. doi: 10.1111/j.1600-0854.2007.00642.x. [DOI] [PubMed] [Google Scholar]
- 94.Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–134. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 95.Codogno P, Mehrpour M, Proikas-Cezanne T. Canonical and non-canonical autophagy: variations on a common theme of self-eating? Nat Rev Mol Cell Biol. 2011;13:7–12. doi: 10.1038/nrm3249. [DOI] [PubMed] [Google Scholar]
- 96.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 97.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 98.Nemchenko A, Chiong M, Turer A, Lavandero S, Hill JA. Autophagy as a therapeutic target in cardiovascular disease. J Mol Cell Cardiol. 2011;51:584–593. doi: 10.1016/j.yjmcc.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 100.Sato K, Tsuchihara K, Fujii S, Sugiyama M, Goya T, Atomi Y, Ueno T, Ochiai A, Esumi H. Autophagy is activated in colorectal cancer cells and contributes to the tolerance to nutrient deprivation. Cancer Res. 2007;67:9677–9684. doi: 10.1158/0008-5472.CAN-07-1462. [DOI] [PubMed] [Google Scholar]
- 101.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 102.Markaki M, Tavernarakis N. The role of autophagy in genetic pathways influencing ageing. Biogerontology. 2011;12:377–386. doi: 10.1007/s10522-011-9324-9. [DOI] [PubMed] [Google Scholar]
- 103.Rajawat YS, Hilioti Z, Bossis I. Aging: central role for autophagy and the lysosomal degradative system. Ageing Res Rev. 2009;8:199–213. doi: 10.1016/j.arr.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 104.Brunk UT, Terman A. Lipofuscin: mechanisms of age-related accumulation and influence on cell function. Free Radic Biol Med. 2002;33:611–619. doi: 10.1016/s0891-5849(02)00959-0. [DOI] [PubMed] [Google Scholar]
- 105.Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146:682–695. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 106.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Clague MJ, Urbe S. Ubiquitin: same molecule, different degradation pathways. Cell. 2010;143:682–685. doi: 10.1016/j.cell.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 108.Hochstrasser M. Ubiquitin, proteasomes, and the regulation of intracellular protein degradation. Curr Opin Cell Biol. 1995;7:215–223. doi: 10.1016/0955-0674(95)80031-x. [DOI] [PubMed] [Google Scholar]
- 109.Yoshimori T. Autophagy: a regulated bulk degradation process inside cells. Biochem Biophys Res Commun. 2004;313:453–458. doi: 10.1016/j.bbrc.2003.07.023. [DOI] [PubMed] [Google Scholar]
- 110.Ding WX, Yin XM. Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy. 2008;4:141–150. doi: 10.4161/auto.5190. [DOI] [PubMed] [Google Scholar]
- 111.Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143:1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–25013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- 113.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 114.Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet. 2002;11:1107–1117. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- 115.Tyedmers J, Mogk A, Bukau B. Cellular strategies for controlling protein aggregation. Nat Rev Mol Cell Biol. 2010;11:777–788. doi: 10.1038/nrm2993. [DOI] [PubMed] [Google Scholar]
- 116.Lanneau D, Wettstein G, Bonniaud P, Garrido C. Heat shock proteins: cell protection through protein triage. ScientificWorldJournal. 2010;10:1543–1552. doi: 10.1100/tsw.2010.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.McDonough H, Patterson C. CHIP: a link between the chaperone and proteasome systems. Cell Stress Chaperones. 2003;8:303–308. doi: 10.1379/1466-1268(2003)008<0303:calbtc>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shin Y, Klucken J, Patterson C, Hyman BT, McLean PJ. The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates alpha-synuclein degradation decisions between proteasomal and lysosomal pathways. J Biol Chem. 2005;280:23727–23734. doi: 10.1074/jbc.M503326200. [DOI] [PubMed] [Google Scholar]
- 119.Sroka K, Voigt A, Deeg S, Reed JC, Schulz JB, Bahr M, Kermer P. BAG1 modulates huntingtin toxicity, aggregation, degradation, and subcellular distribution. J Neurochem. 2009;111:801–807. doi: 10.1111/j.1471-4159.2009.06363.x. [DOI] [PubMed] [Google Scholar]
- 120.Gamerdinger M, Hajieva P, Kaya AM, Wolfrum U, Hartl FU, Behl C. Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. Embo J. 2009;28:889–901. doi: 10.1038/emboj.2009.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Carra S, Seguin SJ, Lambert H, Landry J. HspB8 chaperone activity toward poly(Q)-containing proteins depends on its association with Bag3, a stimulator of macroautophagy. J Biol Chem. 2008;283:1437–1444. doi: 10.1074/jbc.M706304200. [DOI] [PubMed] [Google Scholar]
- 122.Gamerdinger M, Carra S, Behl C. Emerging roles of molecular chaperones and co-chaperones in selective autophagy: focus on BAG proteins. J Mol Med (Berl) 2011;89:1175–1182. doi: 10.1007/s00109-011-0795-6. [DOI] [PubMed] [Google Scholar]
- 123.Baboshina OV, Haas AL. Novel multiubiquitin chain linkages catalyzed by the conjugating enzymes E2EPF and RAD6 are recognized by 26 S proteasome subunit 5. J Biol Chem. 1996;271:2823–2831. doi: 10.1074/jbc.271.5.2823. [DOI] [PubMed] [Google Scholar]
- 124.Kim PK, Hailey DW, Mullen RT, Lippincott-Schwartz J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc Natl Acad Sci U S A. 2008;105:20567–20574. doi: 10.1073/pnas.0810611105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Tan JM, Wong ES, Kirkpatrick DS, Pletnikova O, Ko HS, Tay SP, Ho MW, Troncoso J, Gygi SP, Lee MK, Dawson VL, Dawson TM, Lim KL. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum Mol Genet. 2008;17:431–439. doi: 10.1093/hmg/ddm320. [DOI] [PubMed] [Google Scholar]
- 126.Ikeda F, Crosetto N, Dikic I. What determines the specificity and outcomes of ubiquitin signaling? Cell. 2010;143:677–681. doi: 10.1016/j.cell.2010.10.026. [DOI] [PubMed] [Google Scholar]
- 127.Teckman JH, Perlmutter DH. Retention of mutant alpha(1)-antitrypsin Z in endoplasmic reticulum is associated with an autophagic reponse. Am J Physiol Gastrointest Liver Physiol. 2000 Nov;279:G961–74. doi: 10.1152/ajpgi.2000.279.5.G961. [DOI] [PubMed] [Google Scholar]
- 128.Komatsu M, Ichimura Y. Physiological significance of selective degradation of p62 by autophagy. FEBS Lett. 2010;584:1374–1378. doi: 10.1016/j.febslet.2010.02.017. [DOI] [PubMed] [Google Scholar]
- 129.Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem. 2005;280:40282–40292. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- 130.Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, Yin XM. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol. 2007;171:513–524. doi: 10.2353/ajpath.2007.070188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, Padmanabhan R, Hild M, Berry DL, Garza D, Hubbert CC, Yao TP, Baehrecke EH, Taylor JP. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- 132.Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell. 2009;33:517–527. doi: 10.1016/j.molcel.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–738. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- 134.Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292:1552–1555. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- 135.Watanabe Y, Tanaka M. p62/SQSTM1 in autophagic clearance of a non-ubiquitylated substrate. J Cell Sci. 2011;124:2692–2701. doi: 10.1242/jcs.081232. [DOI] [PubMed] [Google Scholar]
- 136.Du Y, Yang D, Li L, Luo G, Li T, Fan X, Wang Q, Zhang X, Wang Y, Le W. An insight into the mechanistic role of p53-mediated autophagy induction in response to proteasomal inhibition-induced neurotoxicity. Autophagy. 2009;5:663–675. doi: 10.4161/auto.5.5.8377. [DOI] [PubMed] [Google Scholar]
- 137.Gao Z, Gammoh N, Wong PM, Erdjument-Bromage H, Tempst P, Jiang X. Processing of autophagic protein LC3 by the 20S proteasome. Autophagy. 2010;6:126–137. doi: 10.4161/auto.6.1.10928. [DOI] [PubMed] [Google Scholar]
- 138.Cuervo AM, Palmer A, Rivett A, Knecht E. Degradation of proteasomes by lysosomes in rat liver. Eur J Biochem. 1995;227:792–800. doi: 10.1111/j.1432-1033.1995.tb20203.x. [DOI] [PubMed] [Google Scholar]
- 139.Kristensen AR, Schandorff S, Høyer-Hansen M, Nielsen MO, Jäättelä M, Dengjel J, Andersen JS. Ordered organelle degradation during starvation-induced autophagy. Mol Cell Proteomics. 2008;7:2419–2428. doi: 10.1074/mcp.M800184-MCP200. [DOI] [PubMed] [Google Scholar]
- 140.Chen X, Yin XM. Coordination of autophagy and the proteasome in resolving endoplasmic reticulum stress. Vet Pathol. 2011;48:245–253. doi: 10.1177/0300985810385154. [DOI] [PubMed] [Google Scholar]
- 141.Harding HP, Calfon M, Urano F, Novoa I, Ron D. Transcriptional and translational control in the mammalian unfolded protein response. Annu Rev Cell Dev Biol. 2002;18:575–599. doi: 10.1146/annurev.cellbio.18.011402.160624. [DOI] [PubMed] [Google Scholar]
- 142.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 143.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 144.Buchberger A, Bukau B, Sommer T. Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol Cell. 2010;40:238–252. doi: 10.1016/j.molcel.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 145.Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, Shiosaka S, Hammarback JA, Urano F, Imaizumi K. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26:9220–9231. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30:678–688. doi: 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E, Momoi T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007;14:230–239. doi: 10.1038/sj.cdd.4401984. [DOI] [PubMed] [Google Scholar]
- 148.Milani M, Rzymski T, Mellor HR, Pike L, Bottini A, Generali D, Harris AL. The role of ATF4 stabilization and autophagy in resistance of breast cancer cells treated with Bortezomib. Cancer Res. 2009;69:4415–4423. doi: 10.1158/0008-5472.CAN-08-2839. [DOI] [PubMed] [Google Scholar]
- 149.Zhu K, Dunner K Jr, McConkey DJ. Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene. 2010;29:451–462. doi: 10.1038/onc.2009.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, Goldberg AL, Schiaffino S, Sandri M. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–471. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 151.Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, Goldberg AL. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007;6:472–483. doi: 10.1016/j.cmet.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 152.Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117:399–412. doi: 10.1016/s0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zhao J, Brault JJ, Schild A, Goldberg AL. Coordinate activation of autophagy and the proteasome pathway by FoxO transcription factor. Autophagy. 2008;4:378–380. doi: 10.4161/auto.5633. [DOI] [PubMed] [Google Scholar]
- 154.Korolchuk VI, Menzies FM, Rubinsztein DC. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2010;584:1393–1398. doi: 10.1016/j.febslet.2009.12.047. [DOI] [PubMed] [Google Scholar]
- 155.Lamark T, Johansen T. Autophagy: links with the proteasome. Curr Opin Cell Biol. 2010;22:192–198. doi: 10.1016/j.ceb.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 156.Arndt A, Rogon C, Höhfeld J. To be, or not to be - molecular chaperones in protein degradation. Cell Mol Life Sci. 2007;64:2525–2541. doi: 10.1007/s00018-007-7188-6. [DOI] [PMC free article] [PubMed] [Google Scholar]