

Abstract
The sporadic forms of cancer and Alzheimer's disease (AD) are both age-related metabolic disorders. However, the molecular mechanisms underlying the two diseases are distinct: cancer is described by essentially limitless replicative potential, whereas neuronal death is a key feature of AD. Studies of the origin of both diseases indicate that their sporadic forms are the result of metabolic dysregulation, and a compensatory increase in energy transduction that is inversely related. In cancer, the compensatory metabolic effect is the upregulation of glycolysis—the Warburg effect; in AD, a bioenergetic model based on the interaction between astrocytes and neurons indicates that the compensatory metabolic alteration is the upregulation of oxidative phosphorylation—an inverse Warburg effect. These two modes of metabolic alteration could contribute to an inverse relation between the incidence of the two diseases. We invoke this bioenergetic mechanism to furnish a molecular basis for an epidemiological observation, namely the incidence of sporadic forms of cancer and AD is inversely related. We furthermore exploit the molecular mechanisms underlying the diseases to propose common therapeutic strategies for cancer and AD based on metabolic intervention.
Keywords: Warburg effect, inverse Warburg effect, glycolysis, oxidative phosphorylation, metabolic stability
1. Introduction
The vast majority of cancers and neurodegenerative disorders such as Alzheimer's disease (AD) in the general population are sporadic in nature. The monogenic forms of cancer and AD are relatively rare, less dependent on age and have proved genetic factors. The sporadic forms of the diseases are strongly age-related. The incidence of cancer increases exponentially with age, and it abates with age at very advanced ages. The incidence of AD also increases exponentially with age. In view of the age-dependence of these diseases and the dramatic increase in life expectancy during the last 25 years, there has been a tremendous increase in the number of individuals affected with both diseases. Cancer and AD represent increasingly common pathologies with significant effects on human health and welfare [1].
The sporadic forms of cancer and AD are multi-step, age-related processes that implicate two distinct classes of metabolic alterations in cells. The metabolic reprogramming in cancer is increased glycolysis, the Warburg effect [2–4]. Cancer is also associated with tissue invasion and metastasis, leading to a condition defined by essentially limitless replicative potential [5].
The sequence of biochemical events that characterizes AD are alterations that transform normal healthy neurons into degenerate cells. The metabolic reprogramming, as hypothesized in Demetrius & Simon [6], is the upregulation of oxidative phosphorylation (OxPhos) in impaired neurons. A key associated change is the propagation of the disease state to neighbouring cells, resulting in a condition of progressive neuronal death [7,8].
Limitless replicative potential and apoptotic cell death can be viewed as the opposite ends of a demographic spectrum, and hence one might predict an inverse relationship between the two diseases. Epidemiological studies are consistent with this argument. Statistical models [9–11] were used to evaluate associations between prevalent dementia and the risk of future cancer hospitalization, and correspondences between prevalent cancer and subsequent dementia. These studies indicate that prevalent AD was longitudinally related to a reduced risk of cancer, whereas a history of cancer was associated with a reduced risk of AD. These investigations thus point to an inverse correlation between the risk of developing cancer and the risk of developing dementia.
Elucidating the origins of this inverse correlation would have practical applications, because the understanding of the common processes underlying these diseases may help to guide a more fundamental approach towards therapeutic strategies.
Current efforts to understand the epidemiological findings have been reviewed by Plun-Favreau et al. [12] and Driver et al. [11]. The article by Plun-Favreau et al. [12] focused on certain common genetic factors implicated in both diseases. However, the monogenic forms of cancer and AD are very rare. Driver et al. [11] have argued for the plausibility of a link between sporadic forms of the two diseases on the grounds that they share several genes and biological pathways. Replicative immortality and apoptotic cell death, the demographic hallmarks of cancer and AD, respectively, are presumed to be the result of signalling along these common pathways.
The rationale for the link between these two families of diseases that we now propose is based on the fundamental premise that the sporadic forms of cancer and AD are age-related metabolic disorders. Accordingly, these diseases will share many common underlying gene regulatory, biochemical and metabolic characteristics. The link between the two diseases derives from the claim that age-related metabolic disorders can be explained in terms of the metabolic stability theory of ageing [13]. The implications of this theory for the origin of cancer were proposed in Demetrius et al. [14] and for the origin of AD in Demetrius & Simon [6].
The concept of metabolic stability, which is the mainstay of the molecular model of the ageing process, refers to the rate at which critical metabolites in cellular regulatory networks return to their steady-state values after a random perturbation.
Ageing, according to the metabolic stability theory, results from the gradual decline in the homeostatic capacity of cells and tissues, once the state of reproductive maturity is attained. The primary cause of this decline is the increase in the random covalent alterations and conformal modifications of the bioorganic molecules, DNA, RNA and proteins. These molecular changes will generate an increase in molecular disorder and a loss of molecular fidelity [13,15]. This loss will be cumulative, and will ultimately exceed the capacity of cellular repair mechanisms. The subsequent imbalance between loss and repair will result in the impairment of energy transduction, and concomitantly, metabolic dysregulation.
The metabolic stability theory entails that the sporadic forms of cancer and AD are the result of the following suite of dynamic processes that cells have adopted in order to mitigate the deleterious effects of metabolic dysregulation.
— Metabolic alteration: a compensatory increase in glycolysis (in cancer cells) and OxPhos (in neurons) in order to meet the energetic demands owing to metabolic dysregulation.
— Natural selection: competition for substrates between normal cells using normal modes of energy production, and the variant cells, defined by compensatory increases in energy production.
— Propagation: the spread of metabolic abnormalities in the case of AD, and tissue invasion of the variant cell types, in the case of cancer, owing to their selective advantage in the cellular micro-environment.
OxPhos and glycolysis are the two main modes of energy production in cells. The model for the origin of cancer [14] was based on the discovery by Warburg [2] that most normal cells completely catabolize glucose by OxPhos, whereas most cancer cells primarily metabolize glucose by glycolysis—the Warburg effect. The proliferation of cancer cells and their limitless replicative potential were explained in terms of the selective advantage of cancer cells in micro-environments in which resources are subject to large variation in abundance.
The model for the origin of AD [6] was based on the premise that, owing to mild age-related mitochondrial impairment, healthy neurons in aged individuals will have diminished efficiency of mitochondrial energy metabolism. With normal levels of OxPhos, this restricted metabolism will be sufficient to maintain the housekeeping activities of the neurons, but insufficient to maintain normal signalling between neurons. The model predicts that owing to the inherently weak activity of the glycolytic enzyme 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase-3 (PFKFB3) in neurons, upregulation of OxPhos activity—the inverse Warburg effect—will be the predominant compensatory mechanism invoked to furnish sufficient energy for processes such as cell–cell signalling. Neuronal degeneration and neuronal loss, the characteristic hallmark of AD, was explained in terms of the selective advantage of neurons with upregulated OxPhos in a micro-environment in which the energy substrate for OxPhos—lactate generated by astrocytes—is constant and limited.
The inverse roles that the enzyme PFKFB3, the master regulator of glycolysis, plays in the upregulation of glycolysis in cancer cells and the upregulation of OxPhos activity in neurons raise the possibility that upregulation of this glycolytic enzyme may increase the risk of cancer but may be protective against AD.
This paper is organized as follows. We first give an account of the two modes of energy production used in cells. The selection principles that underlie the dynamic processes leading to cancer and AD are briefly reviewed and then applied to the study of the origin of cancer and AD. This analysis is proposed to account for the inverse relation between the prevalence of cancer and AD. Therapeutic strategies are then described in the light of this inverse relation.
2. Energy metabolism in cells
Adenosine triphosphate (ATP) is the main energy currency in living organisms: it sustains the biosynthesis of cellular components, and the transport of ions and organic chemical gradients against concentration gradients. A stable amount of energy must be continuously available as it is essential for maintaining the viability of the cell. The energy is derived from two classes of bioenergetic processes, substrate phosphorylation, namely glycolysis, which typically contributes 12 per cent of the total energy, and OxPhos, which provides about 88 per cent [4].
2.1. Substrate-level phosphorylation
Glycolysis is the anaerobic breakdown of glucose to two molecules of lactic acid. The breakdown proceeds by a mechanism coupled to the formation of ATP from adenosine diphosphate (ADP) and phosphate. The ATP so formed conserves a large fraction of the energy of the original substrate-level molecules. Glucose, the primary energy source, and lactate, the end product, are transported through the cell membrane. They are both non-phosphorylated compounds unlike the intermediate of the glycolytic sequences that are unable to penetrate through the cell membrane. All the enzymes that catalyse the sequence of reactions in the glycolytic pathway are aggregated into a single labile complex in the cytosol. Hence, the glycolytic enzymes occur in relatively stable multi-enzyme complexes, with metabolites passing from one active site to the next without exchanging with the bulk cytoplasm [16].
The metabolic rate, that is, the rate of energy production owing to the conversion of glucose to the end product lactate, will be determined by the mean reaction rate of the enzymes that are involved in the glycolytic activity. This quantity, denoted k, is given by
| 2.1 |
Here, E is the activation free energy, R the gas constant, T the absolute temperature and c a numerical constant.
2.2. Oxidative phosphorylation
OxPhos (or respiration) is the enzymatic oxidation of substrate molecules by molecular oxygen. The enzyme systems that catalyse respiration and the conservation of energy as ATP do not occur as multi-enzyme complexes located in the cytoplasm. The enzymes are fixed in geometric-specific arrays in the mitochondria [16].
The energy for OxPhos is derived from the exergonic electron transport chain reactions that generate a proton gradient across the inner mitochondrial membrane. Protons move exergonically down their electrochemical gradient from the outer to the inner aspects of the membrane. This process yields the energy to drive the endergonic protons of the phosphorylation reaction to generate ATP from ADP.
The metabolic rate, that is the rate of energy production by OxPhos activity, is determined by the proton motive force Δp, which is given by
| 2.2 |
Here, Δψ denotes the electrochemical gradient, ΔpH is the pH difference and c is a numerical constant.
2.3. The energetics of fermentation and respiration
Respiration is a more efficient mode of energy production than fermentation. Anaerobic cells obtain energy from the conversion of glucose to lactate. This metabolite is then extruded as waste. In aerobic organisms, lactate does not leave the cell, and it is oxidized to CO2 and H2O. Accordingly, the maximum energy available under fermentation is about 52 000 cal per mole of glucose. In respiration, the maximum energy available is 686 000 cal per mole. In view of this difference, anaerobic cells must use much more glucose per unit of weight to accomplish the same amount of cellular work as an aerobic cell. The metabolic rate that is the rate of energy production will also be greater in respiratory than in fermentative systems [17].
Empirical studies have shown that the metabolic rate is allometrically related to an organism's body size [18,19]. Quantum metabolism, a mathematical model of energy transduction based on molecular processes in cells, provides explicit expressions for metabolic rate as a function of body size in the case of uni-cells, plants and animals [20,21].
The metabolic rate 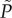 is given by
is given by
| 2.3 |
The quantity d, 1 < d< ∞, is a dimensionality parameter: it describes the number of degrees of freedom of the enzymes localized in the energy-transducing organelles. The quantity W represents body size.
The allometric coefficient α will depend on whether the mode of energy transduction is by glycolytic activity, or by OxPhos. The value of α for glycolysis is a function of the reaction rate of the glycolytic enzymes, as expressed by equation (2.1). In the case of OxPhos activity, the function α will depend on the proton motive force which is given by equation (2.2).
The characterization of the metabolic rate given by equation (2.3) indicates that changes in cellular metabolic rate can be realized by altering the enzymatic reaction rate, as in glycolysis, or regulating the proton conductance of the biomembrane, as in OxPhos.
3. Competition and natural selection
Evolution by natural selection is a two-step process. At the first step, new variation is produced. Here, chance is the dominant factor. At the second step, the metabolic activity of the new variant is evaluated through competition with the resident population. The variants which are most efficient in acquiring resources and transforming these resources into cellular work will have the best chance of surviving and reproducing successfully.
Analytical and empirical studies of competition between incumbent and the variant types in a population now show that natural selection is a stochastic process contingent on the magnitude of resource abundance and its diversity, and predicted by the robustness of the population, that is, the rate at which the population returns to its steady-state condition after a random perturbation in the individual birth and death rate. Robustness is correlated with the statistical parameter evolutionary entropy, a statistical parameter that can be measured by the diversity of the pathways of energy flow within the system [22].
Evolutionary entropy is a generic concept that characterizes the rate of energy production in populations of metabolizing entities: cells, higher organisms, ecosystems. In cells, low entropy describes a system in which there is a low diversity of pathways of energy flow within the system. A low entropic network is described by a restricted number of metabolic pathways that participate in energy transduction. A high entropic system corresponds to a network where metabolic activity is governed by a large number of enzymes catalysing reactions in a diverse class of pathways.
The entropic selection principle, the cornerstone of the model, pertains to competition between an incumbent population of large size and a variant population of relatively small size. These two populations will necessarily have different entropies, and hence different capacities for converting the resources into metabolic activity. The entropic selection principle asserts: ‘the outcome of competition between incumbent and variant types will be predicted by the difference in entropy, and contingent on the resource abundance and the resource diversity’.
The principle can be qualitatively described by the following two propositions [21,22]:
A(1) When resources are diverse and constant in abundance, variants with increased entropy will have a selective advantage, and increase in frequency.
A(2) When resources are singular with large variation in abundance, variants with decreased entropy will have a selective advantage, and increase in frequency.
Consequently, when resource is diverse and abundance constant, high entropic populations will replace low entropic types. However, when resource is singular and subject to large variations in abundance, low entropic types will displace their high entropic relatives.
3.1. Competition between respirators and fermentators
The criteria described by A(1) and A(2) can also be expressed in terms of the bioenergetic parameter metabolic rate, the rate of energy production. We showed [23] that evolutionary entropy, denoted S, is analytically related to the metabolic rate. The relation between the two quantities is given by
| 3.1 |
Here, P denotes the metabolic rate, and a is a numerical constant. The relation given by equation (3.1) entails that the criteria for success in competition can be expressed in terms of the mechanisms of energy production—respiration (OxPhos) and fermentation (glycolysis). We note that cells that use respiratory metabolism are not only more efficient than cells that use fermentatory metabolism they also have a greater metabolic rate.
We can appeal to the entropic selection principle and equation (3.1) to reformulate the criteria for competitive success in terms of conditions invoking metabolic properties, OxPhos and glycolytic activity. These criteria can be qualitatively annotated as follows:
B(1) When resources are diverse and constant, cells using OxPhos activity will have a selective advantage and hence increase in frequency.
B(2) When the resource is singular and varies in abundance, cells using glycolysis will have a selective advantage and consequently increase in frequency.
4. Energy metabolism and ageing
All cells need a constant production of energy to maintain their genetically programmed function. The energy can be stored in the form of ATP and is released during the hydrolysis of the terminal phosphate bond: the standard energy of hydrolysis is tightly regulated in all cells. The random loss of molecular fidelity which is characteristic of the ageing process will result in deviations in the efficiency of ATP production and levels of ATP. Any disturbance in ATP energy levels will compromise cell viability.
The constancy of ATP production in cells is maintained by a highly integrated metabolic network consisting of enzymes and substrates that transform the energy of nutrients into the chemical energy of ATP which is then used for cellular maintenance.
4.1. Thermodynamic entropy, evolutionary entropy and ageing
The capacity of the metabolic network to convert the free energy of the nutrients into chemical energy that can be used to generate cellular biosynthesis and maintain cell viability will depend on two main conditions.
— The spatial configuration of the enzymes which are localized in the cytosol and the mitochondria.
— The temporal organization of the enzymes and substrate that constitute the metabolic network.
The spatial configuration can be analytically characterized in terms of the statistical measure thermodynamic entropy S. This quantity describes the extent to which energy is dispersed or spread out among the different molecular components of the cell. Thermodynamic entropy can be characterized in terms of the geometrical organization of the enzymes in the cellular matrix. A system in which the enzymes occupy fixed position in the cellular matrix has low entropy. By contrast, a system where the enzymes are free to move about in the cytosol and the mitochondria has high entropy.
In a system with low thermodynamic entropy, the transformation from substrate to products will be achieved with relatively high efficiency and minimum dissipation of heat. The change dS in entropy induced by a change dQ in heat energy, as a result of random perturbations in the molecular dynamics of the enzymatic reactions will be given by the Clausius relation
Here, T is the absolute temperature.
The temporal organization of the metabolic network can be analytically described in terms of the statistical parameter evolutionary entropy. This quantity describes the rate at which energy is appropriated from the external environment and transduced into cellular work [22].
A system with low evolutionary entropy has few links between the various metabolites in the network. In addition, the kinetic activity that characterizes the various links is weak. High entropy networks have a large number of links with correspondingly strong activity between the links.
In metabolic-replicating systems, a change 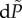 in the metabolic rate due to a change in the activity of the enzymes will induce a change
in the metabolic rate due to a change in the activity of the enzymes will induce a change 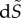 in evolutionary entropy given by [23]
in evolutionary entropy given by [23]
Here, 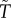 is the mean cycle time of the metabolic reactions.
is the mean cycle time of the metabolic reactions.
An increase in the thermodynamic entropy of the molecular aggregates within cells of living organisms and a concomitant decrease in evolutionary entropy of the biochemical network that regulates the metabolic processes are two bioenergetic signatures of the ageing process [13].
At the molecular level, the common denominator of these signatures is the intrinsic instability of biological molecules whose folded structure is highly sensitive to random molecular changes and a resultant impairment of molecular fidelity. Such impairment can lead to covalent modifications, conformational alterations and mitochondrial DNA damage. The impact of these modifications will be a change in the spatial configuration of the enzymes in the cellular organelles, and hence an increase in the spread and distribution of energy throughout the various microscopic energy modes of the system—an increase in thermodynamic entropy.
Cells are metabolic systems. They maintain their viability by appropriating energy from the external environment and converting this energy into osmotic, biosynthetic and mechanical work. Now, an increase in thermodynamic entropy will ultimately lead to a decrease in the efficiency of the cell to appropriate resources from the external environment and to transform these resources into cellular work. This impairment in efficiency entails a diminished energy production rate and a concomitant decrease in evolutionary entropy.
Evolutionary entropy is positively correlated with metabolic stability, the rate at which the system returns to its steady-state condition after a random perturbation in the kinetic variables that describe the metabolic network. Weak stability implies a high vulnerability to exogenous age-related perturbations, a condition that may result in mitochondrial abnormalities and metabolic dysregulation.
Cancer and AD are the results of strategies of metabolic alteration adopted by the cells to compensate for the age-dependent diminished energy production, and the decrease in evolutionary entropy which this impairment in energy production induces.
5. Cancer and Alzheimer's: metabolic dysregulation
The rationale for the link between cancer and AD is based on the premise that the sporadic forms of both diseases are age-related and can be explained in terms of the metabolic stability theory of ageing. Consequently, the disorders will share a common mitochondrial pathophysiology, namely metabolic dysregulation.
5.1. Cancer and the Warburg effect
Normal cells use OxPhos predominantly for ATP production. Our model for the origin of cancer contends that the metabolic alteration which results from the dysregulation in energy production is characterized by a compensatory increase in glycolysis. The system will then consist of a resident population of normal cells using predominantly OxPhos, and a small population of variants with predominantly glycolytic activity.
The process of natural selection now involves competition between normal cells with intact mitochondria and OxPhos activity, and variant cells with impaired mitochondria and upregulated glycolysis.
Normal cells and cancer cells use both glucose and glutamine as substrates to generate energy. Both cell types also require oxygen. However, the fluctuating oxygen tension, for example, due to inconsistent haemodynamics, entails that the oncologic micro-environment in which competition between normal and cancer cells occurs will be described by substrates that undergo large variation in abundance.
The selection principle enunciated in the preceding sections implies that the variant cells, characterized by an upregulated glycolysis, will have a selective advantage, and hence they will increase in frequency. Sustained glycolysis in the micro-environment will result in the induction of oncogene expression and certain growth abnormalities. These effects will be mitigated by various cellular repair mechanisms resulting in a quasi-stable state consisting of normal cells and cancer cells.
The model proposed in Demetrius et al. [14] contends that various factors, such as metabolic changes in the micro-environment, can cause a disruption in this quasi-stable state, leading to invasion and metastasis, and the expression of the main hallmarks of cancer: self-sufficiency in growth signals, insensitivity to anti-growth signals and limitless replicative potential.
5.2. Alzheimer's disease and an inverse Warburg effect
Brain energy metabolism involves both astrocytes and neurons [24]. The astrocytes are involved in biochemical support of endothelial cells that propagate intracellular  waves over long distances in response to stimuli. These cells perform many functions that include regulation of ion concentration in the extracellular space and modulation of synaptic transmission [25]. Neurons are electrically excitable and maintain voltage gradients across their membranes by ion pumps. Neurons also process and transmit information by chemical signals.
waves over long distances in response to stimuli. These cells perform many functions that include regulation of ion concentration in the extracellular space and modulation of synaptic transmission [25]. Neurons are electrically excitable and maintain voltage gradients across their membranes by ion pumps. Neurons also process and transmit information by chemical signals.
In astrocytes, the predominant mode of energy production is by glycolysis. Glucose is metabolized anaerobically to lactate, and the lactate is released into the extracellular milieu. In neurons, by contrast, the predominant mode of energy production is by OxPhos. The lactate generated from glucose by the astrocytes now constitutes an additional energy source for neurons [26].
The enzyme PFKFB3, a master regulator of glycolysis, is weakly active only in neurons. In neurons, PFKFB3 is degraded constantly [27]. Consequently, neurons are unable to upregulate glycolytic activity to compensate for diminished ATP production.
5.2.1. Metabolic alteration and natural selection
The model for the origin of sporadic forms of AD [6] postulates that AD is a consequence of compensatory alterations in brain energy metabolism that occur in response to reduced energy production in impaired neurons. A central assumption of the model is the upregulation of OxPhos activity in impaired neurons to compensate for the diminished efficiency of energy production due to age-related metabolic dysregulation.
At first glance, this may seem to contradict a body of evidence indicating reduced brain energy metabolism in AD. However, most data on diminished energy metabolism fail to distinguish between neurons and astrocytes, a distinction that is critical to the inverse Warburg effect hypothesis. Furthermore, many such studies focus on late stage AD and also fail to consider the heterogeneity in the stage of degeneration of individual neurons. Interestingly, studies by Zhu et al. [28] have found that an early feature of susceptible neurons in AD is an increase in mtDNA and in levels of cytochrome oxidase-1 protein. The upregulation of OxPhos activity which such increases entail may lead to increased oxidative stress, resulting in further mitochondrial dysregulation, and ultimately in neuronal death, an idea consistent with the increased mitochondrial degradation products reported by Zhu et al. [28].
The idea that neurons at early stages of the disease process may undergo an upregulation of mitochondrial energy metabolism that is not sustainable was reviewed recently in Swerdlow [29]. This concept is consistent with the hypothesis that the upregulation of OxPhos activity is an upstream regulator of the biochemical and histopathological changes associated with AD.
The inverse Warburg hypothesis contends that, as a result of this metabolic reprogramming in the neuronal population, the brain will now consist of two classes of neurons.
— Healthy neurons (type 1). These cells are described by normal OxPhos activity.
— Impaired neurons (type 2). These cells are described by an upregulated OxPhos activity.
These two types of neurons, the healthy cells, type (1), and the impaired cells, type (2), will now compete for the additional energy resource, the lactate produced by the astrocytes.
The model contends that the age-related random loss of molecular fidelity will also cause impairment in the astrocytes. With ageing, these cells will be subject to a diminished glycolytic activity. Consequently, the lactate generated by astrocytes and extruded in the extracellular milieu will have a limited abundance. By appealing to the metabolic selection principle, we predict that, in view of the limited resource constraint, the type (2) neurons, that is the neurons with an increased OxPhos activity, will have selective advantage and consequently higher viability.
Although many factors influence the rate of production of reactive oxygen species (ROS), increased OxPhos activity tends to result in an increase in the production of ROS. ROS are small diffusible molecules that are generated as by-products of OxPhos. These molecules have dual effects. At appropriate levels, they act as second messengers in signal transduction, and hence they serve an important physiological function. Excessive concentration of ROS, however, can cause damage to proteins, lipids and DNA. This condition, however, will be attenuated by the activity of anti-oxidant enzymes that will regulate the activity of ROS, thus resulting in a quasi-stable state consisting of normal neurons, described by normal levels of OxPhos activity, and impaired neurons described by upregulated levels of OxPhos. This quasi-stable state is characteristic of the neuronal population achieved during normal ageing.
The argument advanced in Demetrius & Simon [6] contends that this quasi-stable state can be disrupted by environmental factors or metabolic changes that increase the intensity of selection for the type (2) neurons. Under this condition, the protection given by anti-oxidant enzymes will be overwhelmed and the impairment in the type (2) neurons will be propagated to neighbouring neurons.
This process will now be described by a collapse in metabolic regulation and the invasion of damaged neuronal cells to other regions of the brain. Synaptic loss or dysfunction will now result in a physiological downregulation of OxPhos activity in neurons: physiological downregulation, we argue, entails a transition from healthy to pathological ageing. This shift will lead to neuronal degeneration and ultimately neuronal loss; the major demographic hallmark of AD.
5.2.2. Inverse Warburg effect and the amyloid cascade hypothesis
Our model for the origin of AD postulates age-related changes in metabolism as the primary cause of the disease. According to this model, the shift towards AD is characterized by two critical changes in metabolism. The primary and most critical metabolic reprogramming event is the early upregulation of OxPhos activity as a compensatory mechanism to maintain cellular energy levels in the face of diminished energy metabolism. A consequence of this upregulation of OxPhos activity and natural selection, the system will evolve to a state of dynamical equilibrium involving a large population of healthy neurons and a smaller population of impaired neurons—a signature of healthy ageing.
The second metabolic reprogramming event is the downregulation of OxPhos activity as the state of dynamic equilibrium collapses. This metabolic shift defines the transition from healthy to pathological ageing, a condition that leads to the various hallmarks of AD, namely the production of beta-amyloid and the proliferation of plaques.
The model specifies that the biochemical and histopathological changes will unfold according to the following sequence:
— Oxidative stress: a consequence of the upregulation of OxPhos activity.
— Increased levels of beta-amyloid: a result that derives, in part, from oxidative stress.
— Neuronal loss: a consequence of the integrated action of several factors, namely downregulation of OxPhos, overproduction of beta-amyloid and synaptic dysregulation.
Now, epidemiological studies of AD distinguish between two classes of AD individuals: those with an autosomal dominant inheritance—an early onset group; and those without—the late-onset group. Individuals with autosomal dominant AD are rare. The late-onset group is 20 times more common.
Studies of the early onset AD have led to the amyloid cascade hypothesis. According to this model, the primary event in AD neurodegeneration is the production of the beta-amyloid derivative of the amyloid precursor protein (APP) [30,31].
Beta-amyloid, a small peptide, forms long, insoluble amyloid fibrils that accumulate in spherical microscopic deposits known as senile plaques. It is claimed that synaptic dysfunction and neuronal loss derive from the toxic moieties of beta-amyloid that are small and soluble enough to diffuse readily throughout the brain parenchyma and cause injury and loss. The toxicity of the soluble moieties of the peptide and the spread of the disease through diffusion have been adduced in various therapeutic strategies to arrest the spread of the disease.
Jhamandas & MacTavish [32] and Jhamandas et al. [33], for example, have discovered that the amylin receptor can block certain electrophysiological effects of beta-amyloid. Accordingly, they proposed the receptor as a target for mitigating the neurotoxic effects of the peptide.
Recent investigations, however, indicate that therapeutics based on inhibiting beta-amyloid toxicity or targeting the metabolic pathways involved in beta-amyloid generation are generally ineffective in clinical trials [34,35], although the reasons for the failure of these trials and their relevance to the amyloid cascade hypothesis are controversial. This therapeutic limitation underscores the complex, multifaceted aspect of the disease.
The metabolic reprogramming model for the origin of AD we propose, contends that although late-onset and early onset AD have similar clinical and histopathological signatures, their aetiologies are distinct. Early onset AD, we postulate, is primarily a genetic disease—a disorder owing to a defect in the APP gene and the subsequent overproduction of beta-amyloid. Late-onset AD is primarily a metabolic disease—a disorder due to mitochondrial dysregulation that is a derivative of the ageing process. The primary event in late-onset AD is metabolic alteration—the upregulation of OxPhos activity in impaired mitochondria. This model makes a series of predictions that we now delineate, together with their empirical support.
(1) Oxidative damage is an early stage event that precedes the overproduction of beta-amyloid. This prediction is a consequence of our proposition that the upregulation of OxPhos activity in dysfunctional neurons is the primary event in the transition towards AD, and that increased OxPhos in impaired neurons may lead to oxidative stress.
Empirical support for this prediction derives from studies by Nunomura et al. [36]. These studies indicate that oxidative damage to RNA, proteins and lipids is an early event that precedes the overproduction and aggregation of beta-amyloid. These authors also show that the level of oxidative stress decreases with the duration of the disease and with increased pathophysiology. This decrease in oxidative stress is consistent with the subsequent downregulation of OxPhos activity that we predict will occur during the late stages of the diseases.
(2) Beta-amyloid toxicity and glycolytic activity are negatively correlated. According to the inverse Warburg effect, upregulation of OxPhos activity may result in increased oxidative stress and overproduction and toxicity of beta-amyloid. As a corollary, we can infer that cell lines described by enhanced glycolytic activity will have reduced amyloid toxicity.
Recent studies by Newington et al. [37] are consistent with these predictions. These authors have used the Warburg effect as a generic term to describe the upregulation of aerobic glycolysis in cell lines. Newington et al. [37] showed that nerve cell lines that are resistant to beta-amyloid break down glucose using aerobic glycolysis.
(3) Brain regions show different vulnerability to neurodegeneration. The model predicts that neurons with high energy demands will be more vulnerable to neuronal loss. This prediction issues from the upregulation in OxPhos activity that we argue should characterize neurons in brain regions with a high energy demand.
Empirical support for this prediction is furnished by the systematic study of variation in neuronal vulnerability reported in Mattson & Magnus [38]. These authors report that the prefrontal cortex, which has greater energy demands than the cerebellum, is also more vulnerable to neurodegeneration. It is also reported that the entorhinal cortex surpasses the hippocampus in terms of the complexity of the synaptic architecture and its vulnerability to neurodegeneration.
5.3. Cancer and Alzheimer's disease: a comparison
The sporadic forms of cancer and AD are both age-related diseases. Both diseases have their origin in the random loss in molecular fidelity that characterizes the ageing process. In the context of this model, localized, non-metastatic forms of tumours and neuronal degeneration are predictable results of normal ageing. Normal ageing is characterized by metabolic dysregulation and alterations in metabolic processing. Competition for the available substrates in the cellular micro-environment will result in the emergence of certain quasi-stable states consisting of normal and metabolically impaired cells. In tumours, the metabolically impaired cells will be characterized by increased glycolytic activity. In neuronal systems, the metabolically impaired cells will be defined by increased OxPhos activity.
The glycolytic enzyme PFKFB3 is a critical indicator of the alteration in metabolic processing in both systems. In cancer cells, metabolic dysregulation results in increased glycolysis and the upregulation of PFKFB3. In neuronal systems, the inherent downregulation of PFKFB3 entails that metabolic compensation will be characterized by an increased OxPhos activity.
The metabolic alterations that characterize cancer and AD indicate that the diseases have an inverse relation. The models predict that an increased glycolysis will result in a high risk for the cancer phenotype. An increased OxPhos activity in neurons will result in a high risk of neuronal degeneration and the expression of the AD phenotype. A strong activity of PFKFB3, the master regulator of glycolysis, will decrease this risk. The model thus predicts an inverse relation between cancer and AD. This prediction is consistent with the cross-sectional and case–control studies of individuals who are afflicted with the disease.
In table 1, we contrast cancer and AD in terms of the various hallmarks that characterize the diseases.
Table 1.
Hallmarks of the diseases.
| disease | cellular | demographic | bioenergetic |
|---|---|---|---|
| cancer | insensitivity to growth signals, sustained angiogenesis | limitless replicative potential | upregulation of glycolysis (Warburg effect) |
| AD | loss of dendritic branches, synaptic loss | apoptotic cell death | upregulation of OxPhos activity (inverse Warburg effect) |
6. Cancer and Alzheimer's: therapeutic strategies
Cancer is characterized by the Warburg effect: upregulation of glycolysis. The malignancy of the disease is determined by the intensity of selection for cells that use predominantly glycolytic activity. AD is characterized by the inverse Warburg effect: the upregulation of OxPhos activity. The severity of the disease is determined by the intensity of selection for neurons with enhanced OxPhos activity.
The selective mechanisms we have described suggest certain therapeutic strategies to impede the transition towards malignant states of cancer, and severe states of AD. These strategies will depend on metabolic interventions that will decrease the intensity of selection that favours higher glycolytic activity in the case of cancer cells, and increased OxPhos activity in the case of neurons.
6.1. Cancer cells
The overexpression of PFKFB3 will enhance glycolytic activity, in view of the master regulatory function of this enzyme. Because the resource condition in cancer cells is described by large variation in abundance, we conclude that upregulation of PFKFB3 will increase the intensity of selection in favour of cancer cells.
This suggests that PFKFB3 is an important therapeutic target for cancer. The downregulation of PFKFB3 in cancer cells is predicted to reduce the activity of glycolytic enzymes and therefore will reduce the intensity of selection for the glycolytic profile. Accordingly, inhibiting the activity of PFKFB3 will reduce the transition towards malignancy.
6.2. Alzheimer's disease
The changes in expression of PFKFB3 in the astrocytes are unknown. However, if astrocyte levels of this enzyme decrease with age, then this will result in decreased lactate production. Lactate, the metabolic source for the type (2) neurons, will now become a limited resource, and consequently the intensity of selection on the type (2) neurons will increase. Accordingly, downregulation of PFKFB3 will result in intense selection for neurons with the degenerate phenotype.
These observations suggest that upregulation of PFKFB3 in astrocytes will upregulate glycolysis, thereby reversing the age-related decline in astrocyte lactate production. The resulting increased lactate will reduce the degree to which lactate is limited in availability as an energy substrate for neurons, thereby reducing the competition between dysfunctional neurons for this substrate. Thus, upregulation of PFKFB3, specifically in astrocytes, constitutes a potential therapeutic strategy for AD.
Upregulation of PFKFB3 in neurons to upregulate glycolysis has also been studied. Initial experimental support for the possibility that low PFKFB3 in neurons might be relevant to neurodegeneration comes from observations that the spatial distribution of beta-amyloid deposition corresponds to that of aerobic glycolysis [39], and that the subset of neurons that survive in AD and are resistant to beta-amyloid toxicity are those that exhibit increased glycolysis [37]. However, upregulation of glycolysis by increasing PFKFB3 in neurons in addition to increasing glycolysis, also markedly increases oxidation of glucose through the pentose phosphate pathway, resulting in oxidative stress and apoptotic cell death [40]. In contrast to this toxicity associated with upregulation of PFKFB3 in neurons, we hypothesize that upregulation of this enzyme specifically in astrocytes may prove to be protective. Whether or not upregulation of PFKFB3 in astrocytes might carry an increased risk of malignancy remains a concern that requires additional investigation.
7. Conclusion
The analysis in this paper contends that both cancer and AD are metabolic diseases—a predictable consequence of the process of ageing and the effect of metabolic instability.
Cancer is characterized by a metabolic shift—an upregulation of glycolysis (the Warburg effect). AD is characterized by an inverse shift—an upregulation of OxPhos (the inverse Warburg effect).
The proliferation of cancer cells and the transition towards AD implicate a common mechanism—the entropic selection principle. This principle entails that the outcome of selection between cells using OxPhos and glycolysis will be predicted by the metabolic rate, which is positively correlated with evolutionary entropy, and contingent on the resource abundance and resource diversity: cells with upregulated OxPhos will have a selective advantage when resources are constant and limited, whereas cells with upregulated glycolysis will be favoured when resources undergo large variation in abundance.
The entropic selection principle, when integrated with the metabolic processes underlying cancer and AD, predicts an inverse relationship between the incidence of the two diseases: individuals with cancer are less likely to develop AD in the future, and individuals with AD are less vulnerable to cancer. These predictions are consistent with recent epidemiological studies.
The elucidation of a common dynamic process underlying both sporadic forms of the disorders suggests a new class of therapeutic strategies for regulating the propagation of these diseases.
Acknowledgements
The authors thank Robert Cumming, Jack Jhamandas and Luc Pellerin for critical comments.
References
- 1.Roe CM, Behrens MI, Xiong C, Miller JP, Morris JC. 2005. Alzheimer disease and cancer. Neurology 64, 895–898 10.1212/01.WNL.0000152889.94785.51 (doi:10.1212/01.WNL.0000152889.94785.51) [DOI] [PubMed] [Google Scholar]
- 2.Warburg O. 1956. Origin of cancer cells. Oncologia 9, 75–83 10.1159/000223920 (doi:10.1159/000223920) [DOI] [PubMed] [Google Scholar]
- 3.Vander Heiden MG, Cantley LC, Thompson CB. 2009. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 10.1126/science.1160809 (doi:10.1126/science.1160809) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seyfried TN, Shelton LM. 2010. Cancer as a metabolic disease. Nutr. Metab. 7, 7. 10.1186/1743-7075-7-7 (doi:10.1186/1743-7075-7-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanahan D, Weinberg RA. 2000. The hallmarks of cancer. Cell 100, 57–70 10.1016/S0092-8674(00)81683-9 (doi:10.1016/S0092-8674(00)81683-9) [DOI] [PubMed] [Google Scholar]
- 6.Demetrius LA, Simon DK. 2012. An inverse-Warburg effect and the origin of Alzheimer's disease. Biogerontology 13, 583–594 10.1007/s10522-012-9403-6 (doi:10.1007/s10522-012-9403-6) [DOI] [PubMed] [Google Scholar]
- 7.Lin MT, Beal MF. 2006. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795 10.1038/nature05292 (doi:10.1038/nature05292) [DOI] [PubMed] [Google Scholar]
- 8.Moreira PI, Carvalho C, Zhu X, Smith MA, Perry G. 2010. Mitochondrial dysfunction is a trigger of Alzheimer's disease pathophysiology. Biochim. Biophys. Acta 1802, 2–10 10.1016/j.bbadis.2009.10.006 (doi:10.1016/j.bbadis.2009.10.006) [DOI] [PubMed] [Google Scholar]
- 9.Roe CM, et al. 2010. Cancer linked to Alzheimer disease but not vascular dementia. Neurology 74, 106–112 10.1212/WNL.0b013e3181c91873 (doi:10.1212/WNL.0b013e3181c91873) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Behrens MI, Lendon C, Roe CM. 2009. A common biological mechanism in cancer and Alzheimer's disease? Curr. Alzheimer Res. 6, 196–204 10.2174/156720509788486608 (doi:10.2174/156720509788486608) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Driver JA, et al. 2012. Inverse association between cancer and Alzheimer's disease: results from the Framingham Heart Study. Br. Med. J. 344, e1442. 10.1136/bmj.e1442 (doi:10.1136/bmj.e1442) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Plun-Favreau H, Lewis PA, Hardy J, Miguel Martins L, Wood NW. 2010. Cancer and neurodegeneration: between the devil and the deep blue sea. PLoS Genet. 6, e1001257. 10.1371/journal.pgen.1001257 (doi:10.1371/journal.pgen.1001257) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Demetrius L. 2004. Caloric restriction, metabolic rate, and entropy. J. Gerontol. 59, B902–B915 10.1093/gerona/59.9.B902 (doi:10.1093/gerona/59.9.B902) [DOI] [PubMed] [Google Scholar]
- 14.Demetrius LA, Coy JF, Tuszynski J. 2010. Quantum metabolism, the Warburg effect and the proliferation of cancer. Theor. Biol. Med. Model. 7, 2. 10.1186/1742-4682-7-2 (doi:10.1186/1742-4682-7-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hayflick L. 2007. Biological aging is no longer an unsolved problem. Ann. NY Acad. Sci. 1100, 1–13 10.1196/annals.1395.001 (doi:10.1196/annals.1395.001) [DOI] [PubMed] [Google Scholar]
- 16.Harold FM. 1986. A study of bioenergetics. London, UK: WH Freeman and Company [Google Scholar]
- 17.Lehninger AL. 1965. Bioenergetics: the molecular basis of biological energy transformations. Biology Teaching Monograph Series New York, NY: W. A. Benjamin [Google Scholar]
- 18.Calder WA. 1983. Size, function, and life history III. Cambridge, MA: Harvard University Press [Google Scholar]
- 19.Peters RH. 1983. The ecological implications of body size. Cambridge, UK: Cambridge University Press [Google Scholar]
- 20.Demetrius L. 2003. Quantum statistics and allometric scaling of organisms. Physica A 322, 477–490 10.1016/S0378-4371(03)00013-X (doi:10.1016/S0378-4371(03)00013-X) [DOI] [Google Scholar]
- 21.Demetrius L, Tuszynski JA. 2010. Quantum metabolism explains the allometric scaling of metabolic rates. J. R. Soc. Interface 7, 507–514 10.1098/rsif.2009.0310 (doi:10.1098/rsif.2009.0310) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Demetrius L. 1997. Directionality principles in thermodynamics and evolution. Proc. Natl Acad. Sci. USA 94, 3491–3498 10.1073/pnas.94.8.3491 (doi:10.1073/pnas.94.8.3491) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Demetrius L, Legendre S, Harremöes P. 2009. Evolutionary entropy: a predictor of body size, metabolic rate and maximal life span. Bull. Math. Biol. 71, 800–818 10.1007/s11538-008-9382-6 (doi:10.1007/s11538-008-9382-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pellerin L. 2008. Brain energetics (thought needs food). Curr. Opin. Clin. Nutr. Metab. Care 11, 701–705 10.1097/MCO.0b013e328312c368 (doi:10.1097/MCO.0b013e328312c368) [DOI] [PubMed] [Google Scholar]
- 25.Fuller S, Steele M, Münch G. 2010. Activated astroglia during chronic inflammation in Alzheimer's disease: do they neglect their neurosupportive roles? Mutat. Res. 690, 40–49 10.1016/j.mrfmmm.2009.08.016 (doi:10.1016/j.mrfmmm.2009.08.016) [DOI] [PubMed] [Google Scholar]
- 26.Pellerin L, Bouzier-Sore A-K, Aubert A, Serres S, Merle M, Costalat R, Magistretti PJ. 2007. Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia 55, 1251–1262 10.1002/glia.20528 (doi:10.1002/glia.20528) [DOI] [PubMed] [Google Scholar]
- 27.Bolaños J, Almeida A, Moncada S. 2010. Glycolysis: a bioenergetic or a survival pathway? Trends Biochem. Sci. 35, 145–149 10.1016/j.tibs.2009.10.006 (doi:10.1016/j.tibs.2009.10.006) [DOI] [PubMed] [Google Scholar]
- 28.Zhu X, Perry G, Moreira PI, Aliev G, Cash AD, Hirai K, Smith MA. 2006. Mitochondrial abnormalities and oxidative imbalance in Alzheimer's disease. J. Alzheimer’s Dis. 9, 147–153 [DOI] [PubMed] [Google Scholar]
- 29.Swerdlow RH. 2012. Mitochondria and cell bioenergetics: increasingly recognized components and a possible etiologic cause of Alzheimer's disease. Antioxidants Redox Signal. 16, 1434–1455 10.1089/ars.2011.4149 (doi:10.1089/ars.2011.4149) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hardy JA, Higgins GA. 1992. Alzheimer's disease: the amyloid cascade hypothesis. Science 256, 184–185 10.1126/science.1566067 (doi:10.1126/science.1566067) [DOI] [PubMed] [Google Scholar]
- 31.Selkoe DJ. 1991. The molecular pathology of Alzheimer's disease. Neuron 6, 487–498 10.1016/0896-6273(91)90052-2 (doi:10.1016/0896-6273(91)90052-2) [DOI] [PubMed] [Google Scholar]
- 32.Jhamandas JH, MacTavish D. 2004. Antagonist of the amylin receptor blocks β-amyloid toxicity in rat cholinergic basal forebrain neurons. J. Neurosci. 24, 5579–5584 10.1523/jneurosci.1051-04.2004 (doi:10.1523/jneurosci.1051-04.2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jhamandas JH, Li Z, Westaway D, Yang J, Jassar S, MacTavish D. 2011. Actions of β-amyloid protein on human neurons are expressed through the amylin receptor. Am. J. Pathol. 178, 140–149 10.1016/j.ajpath.2010.11.022 (doi:10.1016/j.ajpath.2010.11.022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prins ND, Visser PJ, Scheltens P. 2010. Can novel therapeutics halt the amyloid cascade? Alzheimer's Res. Ther. 2, 3. 10.1186/alzrt26 (doi:10.1186/alzrt26) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Imbimbo BP, Giardina GA. 2011. γ-Secretase inhibitors and modulators for the treatment of Alzheimer's disease: disappointments and hopes. Curr. Top. Med. Chem. 11, 1555–1570 10.2174/156802611795860942 (doi:10.2174/156802611795860942) [DOI] [PubMed] [Google Scholar]
- 36.Nunomura A, et al. 2001. Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 60, 759–767 [DOI] [PubMed] [Google Scholar]
- 37.Newington JT, Pitts A, Chien A, Arseneault R, Schubert D, Cumming RC. 2011. Amyloid beta resistance in nerve cell lines is mediated by the Warburg effect. PLoS ONE 6, e19191. 10.1371/journal.pone.0019191 (doi:10.1371/journal.pone.0019191) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mattson MP, Magnus T. 2006. Aging and neuronal vulnerability to oxidative stress in the brain. Front. Aging Neurosci. 2, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vlassenko AG, Vaishnavi SN, Couture L, Sacco D, Shannon BJ, Mach RH, Morris JC, Raichle ME, Mintun MA. 2010. Spatial correlation between brain aerobic glycolysis and amyloid-β (Aβ) deposition. Proc. Natl Acad. Sci. USA 107, 17 763–17 767 10.1073/pnas.1010461107 (doi:10.1073/pnas.1010461107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Herrero-Mendez A, Almeida A, Fernández E, Maestre C, Moncada S, Bolaños JP. 2009. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat. Cell Biol. 11, 747–753 10.1038/ncb1881 (doi:10.1038/ncb1881) [DOI] [PubMed] [Google Scholar]