

Abstract
The lysine (K)-rich mantle protein (KRMP) and shematrin protein families are unique to the organic matrices of pearl oyster shells. Similar to other proteins that are constituents of tough, extracellular structures, such as spider silk, shematrins and KRMPs, contain repetitive, low-complexity domains (RLCDs). Comprehensive analysis of available gene sequences in three species of pearl oyster using BLAST and hidden Markov models reveal that both gene families have large memberships in these species. The shematrin gene family expanded before the speciation of these oysters, leading to a minimum of eight orthology groups. By contrast, KRMPs expanded primarily after speciation leading to species-specific gene repertoires. Regardless of their evolutionary history, the rapid evolution of shematrins and KRMPs appears to be the result of the intrinsic instability of repetitive sequences encoding the RLCDs, and the gain, loss and shuffling of other motifs. This mode of molecular evolution is likely to contribute to structural characteristics and evolvability of the pearl oyster shell. Based on these observations, we infer that analogous RLCD proteins throughout the animal kingdom also have the capacity to rapidly evolve and as a result change their structural properties.
Keywords: Pinctada, shematrin, lysine (K)-rich mantle protein, oyster, mollusc, biomineralization
1. Introduction
The molluscan shell is an excellent example of the biofabrication of a highly complex and organized structure at nanoscale dimensions. The control of shell formation is provided, at least in part, by proteins that form an organic matrix within the shell. These proteins are secreted by epithelial cells lining a specialized organ, the mantle. It appears that the deposition of various shell layers is controlled by regionalized expression of genes within different zones of the mantle [1,2]. In both abalone (Haliotis) and pearl oyster (Pinctada) species, the outer prismatic shell layer is thought to be controlled by genes expressed in the mantle edge, whereas the inner nacreous (mother of pearl) layer is likely to be controlled by genes expressed more proximally, in the pallial zone [3–6]. Genes with zone-specific expression patterns have begun to be identified, but their functions are largely unknown [1,4,7–10].
The most highly expressed genes in the mantles of the three most commercially valuable pearl oyster species (Pinctada fucata, Pinctada maxima and Pinctada margaritifera) predominately belong to two families, the lysine (K)-rich mantle proteins (KRMPs) and the shematrins [5,11]. Both gene families encode secreted glycine-rich proteins that possess repetitive, low-complexity domains (RLCDs) and a basic C-terminal domain [12,13]. The repeats within shematrin genes are similar to those found in spider silks [12], and KRMP genes encode basic proteins (isoelectric points between 9.5 and 9.8) with conserved 5′ lysine-rich domains containing six characteristic lysine residues [13]. The incorporation of proteins from these gene families into the shell has been confirmed by proteomic techniques [7,12], and it is thought that proteins with these characteristics may be components of the silk-like gel observed within mollusc shells [14]. Although both KRMPs and shematrins originally were thought to be specific to the prismatic layer, the expression of members of both families in the mantle pallial and outer mantle fold indicates that these proteins may also have a role in the formation of the nacreous layer and periostracum [5,11,15].
RLCDs, particularly those that are glycine-rich, are commonly secreted by a wide range of organisms, including molluscs [16], insects [17] and plants [18,19]. Interestingly, these proteins are usually found in tough, extracellular structures, such as eggshells, cuticles or cell walls, suggesting that the RLCDs have a structural role. The exact function of these proteins is difficult to elucidate. For mollusc proteins, sequence similarity with other characterized proteins or in vitro crystallization studies have lead researchers to suggest that glycine-rich RLCDs may be cross-linked by quinone-tanning [13], form β-sheets [6], be involved in chitin-binding [20] or cause inhibition of CaCO3 precipitation [21]. Because the behaviour of these motifs in vivo is likely to be affected by multiple factors, such as interactions with other organic matrix components and differences in physiological conditions, more insight into the true functions of these proteins are likely to be obtained via reverse genetics. Knock-down of one KRMP gene in P. fucata by RNAi lead to the abnormal formation of prismatic tablets [22], however, the contributions of RLCDs and the mechanisms by which this phenotype was produced remain obscure.
The presence of RLCDs and high levels of expression of both KRMP and shematrin genes indicates that they are likely to have key roles in shell formation. Members of both families have been reported from P. fucata, P. maxima and P. margaritifera, however, the repetitive nature and rapid evolution of the genes makes alignment of the sequences and orthology assignments difficult [2,5]. The discovery of previously undescribed KRMP sequences in P. maxima [15] indicates that more family members may remain to be discovered. The recent availability of next-generation transcriptome data for several molluscs, including these three pearl oyster species, and the publication of the P. fucata draft genome [23] vastly increases the sequence data available, enabling a more thorough investigation into the gene complements of these animals. The phylogenetic relationships of the three species are also well understood; P. maxima and P. margaritifera are closely related, diverging from the P. fucata lineage approximately 14 Mya [24]. This knowledge, along with the sequence data, provides a powerful platform for analysing the evolution of key gene families involved in the shell formation process, and will lead to an understanding of the molecular mechanisms underlying the key morphological differences seen in the shells of these commercially important bivalves.
2. Material and methods
2.1. Sequence data
Publicly available transcriptome data from previous studies [7,11] were downloaded from DDBJ (P. fucata mantle edge, mantle pallial and pearl sac, http://trace.ddbj.nig.ac.jp/DRASearch/study?acc=DRP000399) and NCBI (P. margaritifera mantle, http://trace.ncbi.nlm.nih.gov/Traces/sra/?study=SRP002635). EST sequences from adult P. maxima mantle pallial have previously been reported [5], and were supplemented with 454 transcriptome data from juvenile whole mantle (F. Aguilera 2013, unpublished data). Mytilus galloprovincialis sequences were downloaded from MG-RAST (http://metagenomics.anl.gov/metagenomics.cgi?page=DownloadMetagenome&metagenome=4442949.3) [25], Crassostrea gigas from Sigenae (http://public-contigbrowser.sigenae.org:9090/Crassostrea_gigas/download) and Lottia gigantea from JGI (http://genome.jgi-psf.org/Lotgi1/Lotgi1.download.ftp.html). De novo assembly was performed using CLC Genomics Workbench v. 5.0.1 with default settings, followed by translation of all contigs and unmapped reads in all six frames to enable profile searching.
2.2. Initial identification of KRMP and shematrin sequences
Previously identified shematrin and KRMP sequences were downloaded from NCBI and manually aligned in Se-Al v. 2.0 [26]. These sequences were used as queries to identify similar sequences in the Pinctada spp. translated datasets by BLAST+ [27]. tBLASTn searches were supplemented by manual searching of sequences for common sequence motifs. All identified potential KRMP and shematrin homologues were added to a global KRMP or shematrin alignment. From this alignment, it was possible to distinguish groups of highly similar sequences, which likely represented allelic variants of a single gene. To confirm this, representative sequences from each group were used to query the P. fucata genome (http://marinegenomics.oist.jp/genomes/gallery?project_id=20) using a tBLASTn search against the pfu_1.00_genome database with an e-value cut-off of 50. Any identified genomic sequences with similarity to known shematrins or KRMPs were added to the global alignments. The likely intron/exon structure of these genes was determined by alignment to sequenced transcripts and/or by the program genscan [28].
2.3. Profile searching
The global KRMP and shematrin alignments were submitted to HMMER 3.0 (hmmer.org) for the generation of profile hidden Markov models (profile HMMs) for each gene family (see the electronic supplementary material, files S1–S4). Three profiles were generated for shematrin proteins, one based on an alignment of all sequences (shematrin-all), a second based on an alignment of shematrin-1 and shematrin-2 (shematrin1/2) type sequences, and a third based on an alignment of all shematrins except shematrin-1 and shematrin-2 (shematrin-other). A single profile was generated for the KRMPs. These profile HMMs were then used to query NCBI's non-redundant database (using the hmmsearch program at hmmer.org) to assess their effectiveness, before being used to search the P. maxima, P. margaritifera, P. fucata, M. galloprovincialis, C. gigas and L. gigantea translated datasets for KRMP or shematrin family members. Sequences identified by these profiles were aligned using ClustalX [29].
2.4. Phylogenetic analysis
The KRMP alignment was trimmed to include only the 5′ lysine-rich region and to remove any gaps. Two shematrin alignments were created, one containing the signal peptide and motif 2 from all shematrins excluding shematrins 4, 5 and 8, and a second containing the signal peptide and the basic domain from all shematrins. Incomplete sequences were removed from both alignments. Phylogenetic trees were constructed using the Phylip 3.66 package [30]. A neighbour-joining tree was produced using the JTT matrix with 1000 bootstraps, and a consensus tree was produced. Bayesian analysis was performed using MrBayes v. 3.2.1 [31], with two runs for 1 million generations (sampled every 100, first 250 trees discarded as burn-in) using the mixed amino acid substitution model and the gamma likelihood model for among-site variation. Trees were viewed and edited using FigTree [32]. All alignment files are available on request.
3. Results
3.1. Efficacy of identification of KRMP and shematrin sequences using profile hidden Markov models
Alignments of known and newly identified KRMP and shematrin sequences were used to generate profile HMMs representing each of these gene families. The effectiveness of these profile HMMs to identify family members was tested by applying them to NCBI's non-redundant database. The KRMP profile HMM produced 21 significant hits, all of which were previously identified KRMPs. Similarly, the shematrin1/2 profile HMM produced 18 significant hits, all of which were shematrins. Both the shematrin-all and shematrin-other profile HMMs produced false-positive hits, however, all of these except one had an e-value greater than ×10−10. All together, the profile HMMs were capable of identifying all known KRMP and shematrin sequences at an e-value of ×10−10 or lower, and are, therefore, likely to be useful for reliably identifying family members from datasets using this cut-off level. The profile HMMs were then used on transcription datasets from three species of pearl oyster (P. maxima, P. margaritifera and P. fucata).
To discover whether the shematrin and KRMP gene families are unique to pearl oysters, the KRMP and shematrin HMM profiles were used to screen 454 sequence data from mantle tissue of the mussel, M. galloprovincialis [33], Sanger-sequenced ESTs from the edible oyster C. gigas (including ESTs sequenced from mantle tissue) [34] and all gene models from the genome sequences of the limpet L. gigantea. No KRMP or shematrin sequences were discovered in any of these molluscs, indicating that these gene families are probably restricted to pearl oysters and possibly their closest relatives.
3.2. Shematrins
The P. fucata whole-genome assembly was queried via tBLASTn searches using previously identified shematrin sequences as queries. The expectation threshold was raised to 50 to allow the reporting of weak BLAST hits. In total, 13 genomic regions were identified that possessed open reading frames with shematrin-like characteristics (see the electronic supplementary material, table S1). Two of these appear to be alleles of the same locus (see the electronic supplementary material, table S2). All of the seven previously identified P. fucata shematrin genes [12] were represented. Two P. fucata shematrin-2 genes are reported on NCBI, each with slightly different sequences (accession nos BAE93434 and ABY54785). Both of these sequences are represented by genomic scaffolds, therefore, it is likely that they are independent genes. MSI31, a previously reported sequence identical to shematrin-2 at the N-terminus but with divergent ‘XSEEDY’ RLCDs in the C-terminus [6], is not represented in the genome and can be generated by a single nucleotide deletion at position 671, causing a frameshift.
Three genomic sequences do not correspond to any previously identified shematrin genes. One of these has a lower glycine content than the other shematrins, however, it possesses a signal peptide, a shematrin-like C-terminal basic motif (PKRKKY), and repetitive sequence structure, indicating that it belongs to this gene family (figure 1). This newly reported sequence has thus been named PfuShematrin8 (PfuShem8), in accordance with the naming scheme previously developed for this gene family in P. fucata [12]. The remaining two sequences have been named PfuShem9a and PfuShem9b owing to their high level of sequence similarity over their entire length. They also possess a signal peptide, a shematrin-like C-terminal basic motif (PKRKKY), and repetitive sequence structure, as well as a sequence motif shared between PfuShem1, PfuShem2a, PfuShem2b, PfuShem3 and PfuShem6 (figures 1 and 2).
Figure 1.

Alignment of Pinctada maxima, Pinctada margaritifera and Pinctada fucata shematrin sequences. The horizontal bar indicates the signal peptide. Dashes represent gaps in the alignment and blank lines represent missing sequence. An asterisk represents a stop codon. Orthology groups are indicated by thick vertical bars. (Online version in colour.)
Figure 2.
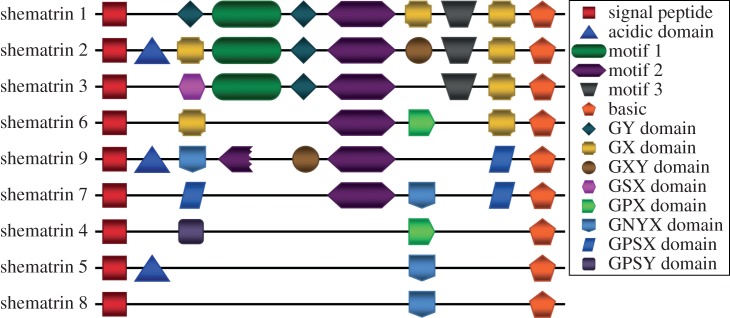
Schematic representation of sequence motifs in shematrin genes. (Online version in colour.)
PfuShem9a and PfuShem9b were the only two shematrin genes found on the same scaffold, where they are positioned in the same orientation and are separated by 1642 base pairs (bp). Although several of the genomic shematrin sequences are incomplete at either their 5′ or 3′ end, the genes are generally composed of two exons, with the intron located within the C-terminal basic domain. Two genes deviate from this stereotypical arrangement; PfuShem7 is encoded by a single exon, whereas PfuShem5 is encoded by four exons and does not have an intron in the basic domain (see the electronic supplementary material, figure S1).
Within the P. fucata transcriptome, the three shematrin profile HMMs identified 37 sequences from the mantle edge library, 827 from the mantle pallial library, and six from the pearl sac library. Upon alignment with the genomic sequences, transcripts representing all identified shematrin sequences except for PfuShem9a and PfuShem9b were found. No additional shematrin sequences were discovered.
The P. fucata shematrin sequences were used as queries to interrogate the P. margaritifera and P. maxima transcriptome datasets via tBLASTn. In P. margaritifera, four previously unreported shematrin sequences were identified, whereas five were identified in P. maxima. The three shematrin profile HMMs identified 2697 sequences from the P. margaritifera transcriptome, and 154 sequences from both the adult and juvenile P. maxima transcriptomes. All of these sequences represented either previously discovered shematrin genes, or those identified by BLAST searches as mentioned above. No additional shematrin sequences were identified by the shematrin profile HMMs.
For each species, an alignment of shematrin sequences was created from NCBI, BLAST searches and HMM searches. Sequences that were of poor quality (numerous ambiguous nucleotides in the nucleotide sequence) or possessed frameshift-inducing mutations were not included. For each sequence type, which here we infer represents a single gene, several variants were found. These variants are unlikely to be the result of sequencing error, as they usually consist of differing numbers of amino acid repeat units (greater than 6 nt), rather than small indels of a few nucleotides (see the electronic supplementary material, figure S2). We infer that these variants represent alleles. From each type, a representative sequence (usually the longest sequence) was selected and designated as a gene. If the difference between two similar sequences involved more than simple repeat variation (i.e. the generation of stretches of unique sequence), the two sequences were treated as separate genes. These sequences were then used to create an alignment of shematrin genes from all three species, presented in figure 1. A description of the relationships between the gene names in this figure and those of previously identified shematrin genes is provided in electronic supplementary material, table S3.
This more comprehensive understanding of the shematrin gene family allows the identification of sequence and motif similarities between family members that was previously obscured [5]. As well as the signal peptide and the basic C-terminal domain, several other motifs, including acidic domains and particular types of glycine-rich repeats, become apparent (see figure 2 and electronic supplementary material, figure S3). The levels of similarity between genes in the alignment and the particular motifs shared between genes from different species indicates that the shematrins fall into eight orthology groups (see black bars in figure 1), suggesting that the major diversification of the shematrin gene family occurred before the divergence of the three species. The P. fucata shematrin 9 sequences may represent a ninth orthology group or an early duplication within the P. fucata lineage, distinguishing between these alternatives will require identification of shematrin 9 sequences in P. maxima and/or P. margaritifera or identification of a reliable outgroup in order to determine an appropriate location in which to root phylogenetic trees. Only PmaxShem8 and P. maxima/margaritifera shematrin 9 genes have not been identified, however, this may simply be a consequence of the lack of whole-genome data for these species, and may not represent gene loss. Both P. fucata and P. maxima have had additional duplications of shematrin 2, as these are lineage-specific they have been named PfuShem2a/PfuShem2b and PmaxShem2α/PmaxShem2β to avoid false impressions of orthology. Pinctada maxima shematrin sequences have been submitted to NCBI (accession numbers KC494066-70, KC505164-7).
The only regions of the alignment that are conserved across all shematrin genes are the signal peptide and short basic domain, when concatenated these domains produce an alignment of 21 amino acids, which is not of sufficient length to build a reliable phylogenetic tree. Nonetheless, trees built with this alignment, and also with an alignment that includes the signal peptide and motif 2, but excluding shematrins 4, 5 and 8, support the orthologous groups outlined above (see the electronic supplementary material, figure S4).
3.3. KRMPs
The P. fucata whole-genome assembly was queried via tBLASTn searches using previously identified KRMP sequences as queries. As for the shematrin genes, the expectation threshold was raised to 50 to allow the reporting of weak BLAST hits. Twenty-two genomic regions were identified that possessed a clear open reading frame with KRMP-like characteristics (see the electronic supplementary material, table S4). Six of these appear to be alleles of the same locus (see the electronic supplementary material, table S5). All of the four previously identified P. fucata KRMP genes [13,35] were represented (two of these appear to represent variants of the same gene), whereas 13 unreported sequences were found. In several cases, multiple KRMP genes were found on the same scaffold (figure 3). All P. fucata KRMP sequences were encoded by a single exon.
Figure 3.
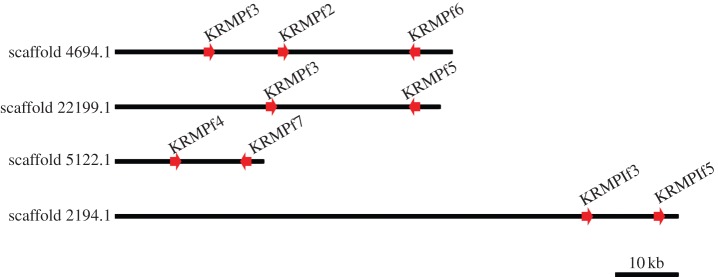
Genomic arrangement of clustered P. fucata KRMP genes. Direction of arrowheads indicate gene orientation. (Online version in colour.)
Within the P. fucata transcriptome, the KRMP profile HMM identified 22 sequences from the mantle edge library, 46 sequences from the mantle pallial library and six sequences from the pearl sac library. Upon alignment with the genomic sequences, transcripts representing most of the sequences identified from the genome were present. Sequences without transcript evidence include PfuKRMPf11, PfuKRMPf12, PfuKRMPlf2 and PfuKRMPlf3. No additional KRMP sequences were discovered in these transcriptomes.
The P. fucata KRMP sequences were used as queries to interrogate the P. margaritifera and P. maxima transcriptome datasets via tBLASTn. In P. margaritifera, four previously unreported KRMP sequences were identified, whereas five were identified in P. maxima. The KRMP profile HMM identified 1037 sequences from the P. margaritifera transcriptome, these sequences represented all previously reported KRMP sequences except for PmargKRMPr6 (KRMP11, ABP57449), and eight sequences that were not previously reported or discovered by BLAST. In P. maxima, the KRMP profile HMM identified 88 sequences from the juvenile and adult transcriptomes. The sole previously reported P. maxima KRMP sequence PmaxKRMPx3 (KRMP7, P86960) was identified as well as nine sequences that were not previously reported or discovered by BLAST.
For each species, each sequence type was represented by multiple transcripts with minor variations, such as the insertion or deletion of repeat elements. This was reminiscent of the situation for shematrin genes, therefore, the same rules were used to designate representative sequences for each sequence type, which likely correspond to individual genes. An alignment of these representative sequences from all three species was generated (figure 4). In contrast to the shematrin gene family, patterns of orthology were not evident from sequence alignment alone. Fortunately, all KRMP sequences possess a conserved lysine-rich domain with a stereotypical pattern of six cysteine residues. This conserved region was used to construct a neighbour-joining tree (figure 5; Bayesian analysis produced trees with similar topology, differing slightly at some terminal nodes, data not shown). This tree supports a division of the sequences into two major clades, the true KRMPs, containing most of the previously identified KRMP sequences, and the KRMP-like genes. The true KRMPs can be further divided into a P. fucata-specific radiation and a P. maxima/margaritifera-specific radiation. Some true KRMP genes fall outside these two groups and branch with low support at the base of the KRMP clade. This topology indicates a deep duplication of an ancestral KRMP gene prior to the divergence of P. fucata from P. maxima/P. margaritifera, giving rise to the KRMP and KRMP-like lineages. Additional, lineage-specific duplications have occurred subsequent to this divergence.
Figure 4.

Alignment of P. maxima, P. margaritifera and P. fucata KRMP sequences. The horizontal bar indicates the signal peptide. Dashes represent gaps and blank lines represent missing sequence. An asterisk represents a stop codon. Vertical bars and numbers refer to the groups described in figure 5. (Online version in colour.)
Figure 5.

Phylogenetic analysis of the relationship of KRMP genes produced from an alignment of the lysine-rich domain. Percentage of neighbour-joining bootstrap support is shown when over 50% (regular text); Bayesian posterior probabilities are shown when over 70% (bold text). The tree is unrooted owing to the lack of appropriate outgroup, branch lengths are to scale (scale = substitutions/site). (Online version in colour.)
The complex evolutionary history of the KRMP genes required the generation of a naming scheme that avoids providing false impressions of orthology. First, genes falling within the KRMP-like clade were designated KRMPl. A species-specific identifier was then added to the end of the sequence name (P. fucata: f, P. margaritifera: r, P. maxima: x), before each gene was assigned a unique number. Therefore, P. margaritifera possesses the gene PmargKRMPr6, and P. maxima possesses the gene PmaxKRMPx6. These two genes are not orthologues. A description of the relationships between the gene names proposed here and those of previously identified KRMP genes is provided in electronic supplementary material, table S6. P. maxima KRMP sequences have been submitted to NCBI (accession numbers KC494055-65).
3.4. Reliability of gene assignments
Although this study identifies many new and previously identified KRMP and shematrin genes, it is likely that more remain to be identified. The methods used here are conservative, and two sequences that are highly similar are classified as a single gene. It is likely that in many cases, these differences do represent true gene copies. As an example, this may be the case for PfuKRMPf3, which is found on two different scaffolds. On scaffold 4694.1, its immediate downstream neighbour is PfuKRMPf2, whereas on scaffold 22199.1 its neighbour is PfuKRMPf5 (figure 3). The surrounding genomic sequence of the gene is similar on both scaffolds; therefore, this may be the result of either the duplication of a genomic region or an assembly error. It is also possible that the methods used here did not identify divergent shematrin and KRMP genes, particularly in P. maxima and P. margaritifera for which no whole-genome information is available. HMMER is likely to be less effective in identifying short sequences as family members, such as those generated by next-generation sequencing technology. All KRMP and shematrin sequences analysed in this study can be found in electronic supplementary material, S1.
4. Discussion
4.1. KRMP and shematrin gene families have different evolutionary histories
The most striking similarity between shematrin and KRMP sequences is that of their composition—both gene families encode proteins that contain glycine-rich RLCDs. When the shematrin genes were first discovered, the glycine-rich repeats were likened to those found in the proteins that form spider silks and plant cell walls [12]. Although the similarities between these disparate proteins may seem to be coincidental, many other RLCD-containing proteins (and many with glycine-rich repeats, in particular) have been identified, most of which are involved in the formation of tough, extracellular structures. Although the evolutionary distance between the organisms possessing the structures makes it unlikely that these proteins are homologous, the similarities between them indicate that these RLCDs are functionally significant and have a high degree of evolvability.
The mantle transcriptomes of three closely related Pinctada species enables a more detailed analysis of the patterns of evolution of genes encoding RLCDs. Previous research has demonstrated that the parallel evolution of RLCDs is a key feature of molluscan shell evolution [5]. The secretomes of P. maxima and the gastropod Haliotis asinina were compared, and although shematrin and KRMP genes were not found in H. asinina, this gastropod's mantle transcriptome contains other, seemingly unrelated, RLCDs. The lack of similarity between the Pinctada and Haliotis transcriptomes, and even between shematrin and KRMP genes in different species of pearl oyster, supports the proposition that many proteins in the molluscan mantle secretome are rapidly evolving [4].
The shematrins and KRMPs share many similarities in addition to their sequence characteristics. Both gene families are of a similar size, are highly expressed in mantle transcriptomes, and, based on other molluscan genomes and transcriptomes, appear to be Pinctada-specific. Despite these similarities, reconstruction of the evolutionary histories of both gene families reveals differences in the timing of family divergence. Orthologues of members of the shematrin gene family are present in P. fucata, P. margaritifera and P. maxima, indicating that the vast majority of gene duplication and divergence events of this gene family occurred prior to the speciation of these pearl oysters (figure 6a). In contrast to this, orthology of KRMP genes is not evident. Although this may be due to rapid sequence divergence, the clustering of several of the genes into species-specific clades suggests that they have originated from more recent lineage- and species-specific duplications. In addition, there is little support for the position of some true KRMPs within the phylogenetic tree, indicating that they may have duplicated very soon after the origin of KRMP and KRMP-like clades (figure 5). It, therefore, appears that the current complement of KRMP genes has been generated by multiple duplications throughout the evolutionary history of pearl oyster species (figure 6b), in contrast to the shematrin gene family, which largely diversified before the separation of P. fucata and P. margaritifera/P. maxima lineages (figure 6a).
Figure 6.
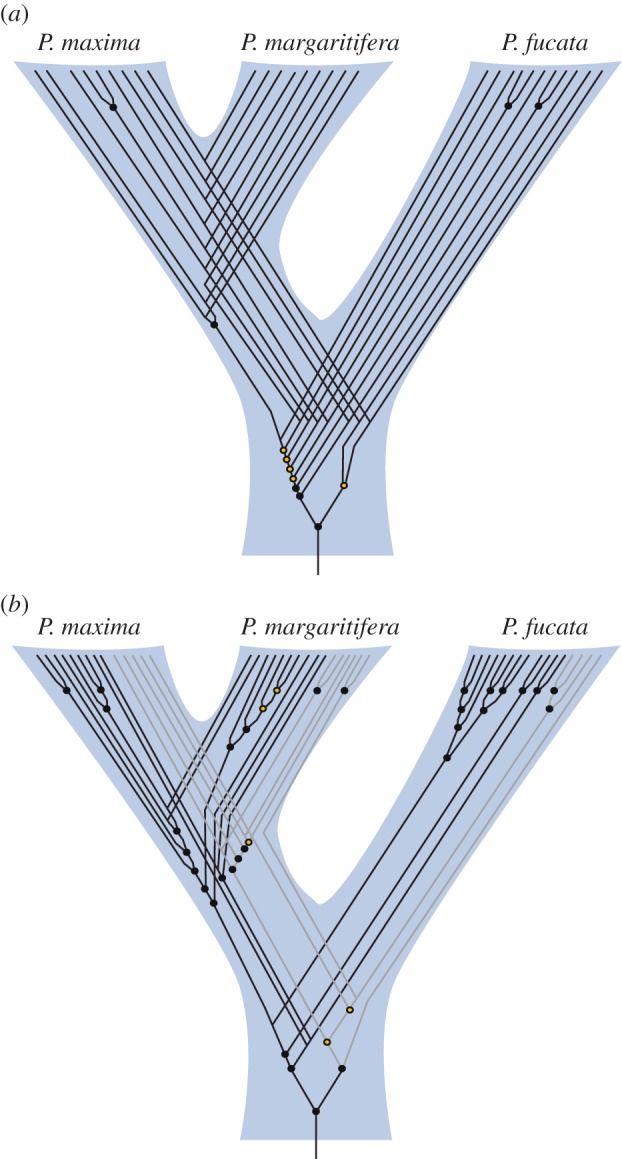
(a) Reconstruction of the evolutionary history of shematrin and (b) KRMP genes. Circles represent gene duplication events, lines represent individual genes. Closed circles indicate nodes with a neighbour-joining bootstrap value greater than 50% and/or Bayesian posterior probability greater than 70% in phylogenetic trees, open circles indicate lower support and that tree topology may differ from that shown (see figure 5 and electronic supplementary material, figure S4). For clarity, KRMP-like genes are represented by grey lines in B. The placement of the root (arbitrary owing to the lack of obvious outgroups) does not affect the duplication trends seen, i.e. primarily before the divergence of P. fucata from the P. maxima/margaritifera lineage for shematrins and primarily after this divergence for KRMPs. (Online version in colour.)
4.2. Repetitive low-complexity domains enable the rapid evolution of KRMPs and shematrins
This study reveals that the shematrin and KRMP gene families have undergone multiple duplications and extensive sequence divergence since the emergence of this clade of pearl oysters, supporting the proposition that these sequences are fast evolving. This diversification appears to be facilitated by the low-complexity, repetitive nature of the sequences, which would increase the likelihood of mispairing during replication [36]. Indeed, many of the sequence variants discovered by HMMER differed only by the insertion or deletion of a repeat element, and variation within repeat sequences of other shell matrix genes has been previously reported [37]. Rapid sequence divergence owing to the intrinsically unstable nature of repetitive coding sequences has also been reported for spider silks [38–40], and is, therefore, a key feature of proteins containing RLCDs.
In addition to the rapid expansion and evolution of shematrin and KRMP gene families, there appears to be little evidence of gene loss, at least in the shematrins for which the evolutionary reconstructions are the most reliable. All of the shematrin orthology groups (i.e. shematrins 1–8, and possibly shematrin 9) evolved before the diversification of the three Pinctada species and most have been maintained in all three species lineages for at least 14 million years. Furthermore, the majority of the shematrin genes have similar expression patterns [12], raising the question of why so many copies of these genes exist within pearl oyster genomes. Although the generation of these gene families may have occurred simply as a consequence of the innate evolvability of their repetitive sequence, there may also be selective advantages in increasing copy number, resulting in the retention of new gene copies. For example, there may be an advantage in expressing these genes in large amounts, and increasing the gene copy number could increase the number of transcripts produced. Transcriptome analyses of the mantles in all three Pinctada species support this contention, with shematrins and KRMPs being amongst the most highly represented transcripts in these mRNA pools [5,11]. There may also be undiscovered differences in the spatial or temporal expression of these genes. For example, PfuShem9a and PfuShem9b were not found within the mantle or pearl sac transcriptome data, which may reflect differing roles of these proteins.
Another possibility is that the genes generated from a duplication event have gained a novel function (neofunctionalization) or have partitioned the original functions of the ancestral gene (subfunctionalization) [41]. Each shematrin is characterized by a unique combination of motifs and RLCDs (figure 2), which may reflect different functions of the proteins. For example, the acidic domains found in shematrins 2, 5 and 9 may inhibit CaCO3 crystallization, as recombinant acidic peptides show inhibitory activity in vitro [20]. The presence of all shematrin genes in all three Pinctada spp. (with the possible exception of shematrins 8 and 9) is consistent with supposition that each shematrin, with its specific motifs and RLCD architecture, uniquely contributes to oyster shell formation.
This modular organization of rapidly evolving RLCDs and other motifs [2] enables the evolution of new architectures. For example, the differences between shematrins 1 and 2 are primarily owing to the presence of an acidic domain in shematrin 2 (which also has been lost in one of the products of a recent P. maxima shematrin 2 duplication; Pmaxshem2β, figure 1). Rearrangements of motifs can also be seen, for example, in the positions of the GX and GY domains of shematrins 1 and 2. Therefore, it appears that domain shuffling is an important process in the evolution of shematrin sequences. This shuffling is likely to occur via mispairing during replication rather than by exon shuffling, as the genes are encoded entirely (in the case of KRMPs) or almost entirely (in the majority of shematrins; electronic supplementary material, figure S1) within single exons.
Other differences between shematrins 1 and 2 involve a shift in the type of glycine-rich RLCD. Insights into how changes in RLCDs may occur can be provided by the P. maxima and P. margaritifera shematrin 4 sequences. The two genes are orthologues, and are significantly divergent in their C-terminal ends from PfuShem4. In P. margaritifera, part of this region is comprised of five repeats of the sequence ‘PSTGYAGYSYGY’, whereas in P. maxima, the repeat sequence is ‘P(T/S)AGYGGYSYGY’. This implies that, after the speciation event, sequence divergence has taken place which has subsequently been homogenized across the entire repeat region, presumably owing to gene conversion within the sequence [42].
From these observations, we propose that the ancestral shematrin gene minimally possessed a signal peptide and basic C-terminal sequence, as well as at least one glycine-rich RLCD. Subsequent duplications and divergences, including the loss and shuffling of various motifs and homogenization of repeat regions lead to the generation of the shematrin family, which consists of eight or nine orthology groups. The origin of the ancestral shematrin gene and the initial formation of the RLCDs is unknown and will require sequence information from additional Pterioidea species.
4.3. Evolution of shematrin and KRMP gene families and impacts on shell structure
Although the exact roles of shematrin and KRMP genes in shell formation are unknown, two lines of evidence suggest that they play key structural roles within the shell. First, knock-down of KRMP gene expression via RNAi causes defects in tablet morphology within the prismatic layer [22]. Second, the glycine-rich repeat regions within the genes are similar to those found in other structural proteins such as spider silks. Exactly how these glycine-rich regions contribute to the strength and/or elasticity of spider silks is unclear, and has traditionally been difficult to study owing to the size and repetitiveness of the protein [43]. Indeed, the shorter and less-repetitive KRMPs and shematrins may be useful tools for understanding the functionality of these domains in silk proteins.
These physical characteristics, in addition to the localization and high level of expression of the transcripts, suggest that shematrins and KRMPs are important structural components of molluscan shells. The fast-evolving nature of these genes is intriguing, as any critical shell component would be expected to be under stabilizing selective pressure. Although shematrins and KRMPs have not been detected in mantle transcriptomes of other molluscs, other RLCD proteins are present in these species, suggesting that parallel or convergent evolution is occurring [5]. The apparent structural requirement for these genes to have glycine-rich RCLDs also makes them prone to mispairing and thus highly evolvable. It is therefore likely that the diversification of these RLCD proteins has contributed to the diversity of structure and patterning observed within molluscan shells, and that similar evolutionary processes are operational in analogous RLCDs that confer physical properties on external structures fabricated by other organisms.
Acknowledgements
This study was supported by funding from the Australian Research Council to B.M.D.
References
- 1.Jolly C, Berland S, Milet C, Borzeix S, Lopez E, Doumenc D. 2004. Zonal localization of shell matrix proteins in mantle of Haliotis tuberculata (Mollusca, Gastropoda). Mar. Biotechnol. 6, 541–551 10.1007/s10126-004-3129-7 (doi:10.1007/s10126-004-3129-7) [DOI] [PubMed] [Google Scholar]
- 2.Marin F, Luquet G, Marie B, Medakovic D. 2008. Molluscan shell proteins: primary structure, origin, and evolution. Curr. Top. Dev. Biol. 80, 209–276 10.1016/S0070-2153(07)80006-8 (doi:10.1016/S0070-2153(07)80006-8) [DOI] [PubMed] [Google Scholar]
- 3.McDougall C, Green K, Jackson DJ, Degnan BM. 2011. Ultrastructure of the mantle of the gastropod Haliotis asinina and mechanisms of shell regionalization. Cells Tissues Organs 194, 103–107 10.1159/000324213 (doi:10.1159/000324213) [DOI] [PubMed] [Google Scholar]
- 4.Jackson DJ, McDougall C, Green K, Simpson F, Wörheide G, Degnan BM. 2006. A rapidly evolving secretome builds and patterns a sea shell. BMC Biol. 4, 40. 10.1186/1741-7007-4-40 (doi:10.1186/1741-7007-4-40) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jackson DJ, et al. 2010. Parallel evolution of nacre building gene sets in molluscs. Mol. Biol. Evol. 27, 591–608 10.1093/molbev/msp278 (doi:10.1093/molbev/msp278) [DOI] [PubMed] [Google Scholar]
- 6.Sudo S, Fujikawa T, Nagakura T, Ohkubo T, Sakaguchi K, Tanaka M, Nakashima K, Takahashi T. 1997. Structures of mollusc shell framework proteins. Nature 387, 563–564 10.1038/42391 (doi:10.1038/42391) [DOI] [PubMed] [Google Scholar]
- 7.Joubert C, Piquemal D, Marie B, Manchon L, Pierrat F, Zanella-Cléon I, Cochennec-Laureau N, Gueguen Y, Montagnani C. 2010. Transcriptome and proteome analysis of Pinctada margaritifera calcifying mantle and shell: focus on biomineralization. BMC Genomics 11, 613. 10.1186/1471-2164-11-613 (doi:10.1186/1471-2164-11-613) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takagi R, Miyashita T. 2010. Prismin: a new matrix protein family in the Japanese pearl oyster (Pinctada fucata) involved in prismatic layer formation. Zool. Sci. 27, 416–426 10.2108/zsj.27.416 (doi:10.2108/zsj.27.416) [DOI] [PubMed] [Google Scholar]
- 9.Kong Y, et al. 2009. Cloning and characterization of Prisilkin-39, a novel matrix protein serving a dual role in the prismatic layer formation from the oyster Pinctada fucata. J. Biol. Chem. 284, 10 841–10 854 10.1074/jbc.M808357200 (doi:10.1074/jbc.M808357200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takeuchi T, Endo K. 2006. Biphasic and dually coordinated expression of the genes encoding major shell matrix proteins in the pearl oyster Pinctada fucata. Mar. Biotechnol. 8, 52–61 10.1007/s10126-005-5037-x (doi:10.1007/s10126-005-5037-x) [DOI] [PubMed] [Google Scholar]
- 11.Kinoshita S, et al. 2011. Deep sequencing of ESTs from nacreous and prismatic layer producing tissues and a screen for novel shell formation-related genes in the pearl oyster. PLoS ONE 6, e21238. 10.1371/journal.pone.0021238.t004 (doi:10.1371/journal.pone.0021238.t004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yano M, Nagai K, Morimoto K, Miyamoto H. 2006. Shematrin: a family of glycine-rich structural proteins in the shell of the pearl oyster Pinctada fucata. Comp. Biochem. Physiol. B. Biochem. Mol. Biol. 144, 254–262 10.1016/j.cbpb.2006.03.004 (doi:10.1016/j.cbpb.2006.03.004) [DOI] [PubMed] [Google Scholar]
- 13.Zhang C, Xie L, Huang J, Liu X, Zhang R. 2006. A novel matrix protein family participating in the prismatic layer framework formation of pearl oyster, Pinctada fucata. Biochem. Biophys. Res. Commun. 344, 735–740 10.1016/j.bbrc.2006.03.179 (doi:10.1016/j.bbrc.2006.03.179) [DOI] [PubMed] [Google Scholar]
- 14.Addadi L, Joester D, Nudelman F, Weiner S. 2006. Mollusk shell formation: a source of new concepts for understanding biomineralization processes. Chemistry 12, 980–987 10.1002/chem.200500980 (doi:10.1002/chem.200500980) [DOI] [PubMed] [Google Scholar]
- 15.Gardner LD, Mills D, Wiegand A, Leavesley D, Elizur A. 2011. Spatial analysis of biomineralization associated gene expression from the mantle organ of the pearl oyster Pinctada maxima. BMC Genomics 12, 455. 10.1186/1471-2164-12-455 (doi:10.1186/1471-2164-12-455) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cao Q, Wang Y, Bayley H. 1997. Sequence of abductin, the molluscan ‘rubber’ protein. Curr. Biol. 7, R677–678 10.1016/S0960-9822(06)00353-8 (doi:10.1016/S0960-9822(06)00353-8) [DOI] [PubMed] [Google Scholar]
- 17.Robertson HM, Martos R, Sears CR, Todres EZ, Walden KK, Nardi JB. 1999. Diversity of odourant binding proteins revealed by an expressed sequence tag project on male Manduca sexta moth antennae. Insect Mol. Biol. 8, 501–518 10.1046/j.1365-2583.1999.00146.x (doi:10.1046/j.1365-2583.1999.00146.x) [DOI] [PubMed] [Google Scholar]
- 18.Fang RX, Pang Z, Gao DM, Mang KQ, Chua NH. 1991. cDNA sequence of a virus-inducible, glycine-rich protein gene from rice. Plant Mol. Biol. 17, 1255–1257 10.1007/BF00028742 (doi:10.1007/BF00028742) [DOI] [PubMed] [Google Scholar]
- 19.Keller B, Sauer N, Lamb CJ. 1988. Glycine-rich cell wall proteins in bean: gene structure and association of the protein with the vascular system. EMBO J. 7, 3625–3633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suzuki M, Nagasawa H. 2007. The structure–function relationship analysis of Prismalin-14 from the prismatic layer of the Japanese pearl oyster, Pinctada fucata. FEBS J. 274, 5158–5166 10.1111/j.1742-4658.2007.06036.x (doi:10.1111/j.1742-4658.2007.06036.x) [DOI] [PubMed] [Google Scholar]
- 21.Miyamoto H, Miyoshi F, Kohno J. 2005. The carbonic anhydrase domain protein nacrein is expressed in the epithelial cells of the mantle and acts as a negative regulator in calcification in the mollusc Pinctada fucata. Zool. Sci. 22, 311–315 10.2108/zsj.22.311 (doi:10.2108/zsj.22.311) [DOI] [PubMed] [Google Scholar]
- 22.Fang D, Xu G, Hu Y, Pan C, Xie L, Zhang R. 2011. Identification of genes directly involved in shell formation and their functions in pearl oyster, Pinctada fucata. PLoS ONE 6, e21860. 10.1371/journal.pone.0021860 (doi:10.1371/journal.pone.0021860) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takeuchi T, et al. 2012. Draft genome of the pearl oyster Pinctada fucata: a platform for understanding bivalve biology. DNA Res. 19, 117–130 10.1093/dnares/dss005 (doi:10.1093/dnares/dss005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cunha RL, Blanc F, Bonhomme F, Arnaud-Haond S. 2011. Evolutionary patterns in pearl oysters of the genus Pinctada (Bivalvia: Pteriidae). Mar. Biotechnol. 13, 181–192 10.1007/s10126-010-9278-y (doi:10.1007/s10126-010-9278-y) [DOI] [PubMed] [Google Scholar]
- 25.Meyer F, et al. 2008. The metagenomics RAST server—a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9, 386. 10.1186/1471-2105-9-386 (doi:10.1186/1471-2105-9-386) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rambaut A. 1996. Se-Al. v. 2.0a11. Edinburgh, UK: University of Edinburgh. See http://tree.bio.ed.ac.uk/software/seal/ [Google Scholar]
- 27.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10, 421. 10.1186/1471-2105-10-421 (doi:10.1186/1471-2105-10-421) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burge C, Karlin S. 1997. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94 10.1006/jmbi.1997.0951 (doi:10.1006/jmbi.1997.0951) [DOI] [PubMed] [Google Scholar]
- 29.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. 1997. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882 10.1093/nar/25.24.4876 (doi:10.1093/nar/25.24.4876) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Felsenstein J. 1995. PHYLIP (phylogeny inference package). Seattle WA: University of Washington; See http://evolution.genetics.washington.edu/phylip.html [Google Scholar]
- 31.Ronquist F, et al. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 10.1093/sysbio/sys029 (doi:10.1093/sysbio/sys029) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rambaut A. 2006. FigTree. 1.1.1. Edinburgh, UK: Edinburgh University; See http://tree.bio.ed.ac.uk/software/figtree/ [Google Scholar]
- 33.Craft JA, Gilbert JA, Temperton B, Dempsey KE, Ashelford K, Tiwari B, Hutchinson TH, Chipman JK. 2010. Pyrosequencing of Mytilus galloprovincialis cDNAs: tissue-specific expression patterns. PLoS ONE 5, e8875. 10.1371/journal.pone.0008875 (doi:10.1371/journal.pone.0008875) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fleury E, et al. 2009. Generation and analysis of a 29,745 unique expressed sequence tags from the Pacific oyster (Crassostrea gigas) assembled into a publicly accessible database: the GigasDatabase. BMC Genomics 10, 341. 10.1186/1471-2164-10-341 (doi:10.1186/1471-2164-10-341) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Masaoka T, Kobayashi T. 2009. Analysis of nucleotide variation and inheritance of lysine-rich matrix protein (KRMP) genes participating in shell formation of pearl oyster. DNA Polymorphism 17, 126–135 [Google Scholar]
- 36.Levinson G, Gutman GA. 1987. Slipped-strand mispairing: a major mechanism for DNA sequence evolution. Mol. Biol. Evol. 4, 203–221 [DOI] [PubMed] [Google Scholar]
- 37.Smith-Keune C, Jerry DR. 2009. High levels of intra-specific variation in the NG repeat region of the Pinctada maxima N66 organic matrix protein. Aquaculture Res. 40, 1054–1063 10.1111/j.1365-2109.2009.02199.x (doi:10.1111/j.1365-2109.2009.02199.x) [DOI] [Google Scholar]
- 38.Sezutsu H, Yukuhiro K. 2000. Dynamic rearrangement within the Antheraea pernyi silk fibroin gene is associated with four types of repetitive units. J. Mol. Evol. 51, 329–338 10.1007/s002390010095 (doi:10.1007/s002390010095) [DOI] [PubMed] [Google Scholar]
- 39.Manning RF, Gage LP. 1980. Internal structure of the silk fibroin gene of Bombyx mori II. Remarkable polymorphism of the organisation of crystalline and amorphous coding sequences. J. Biol. Chem. 255, 9451–9457 [PubMed] [Google Scholar]
- 40.Hayashi CY, Lewis RV. 2000. Molecular architecture and evolution of a modular spider silk protein gene. Science 287, 1477–1479 10.1126/science.287.5457.1477 (doi:10.1126/science.287.5457.1477) [DOI] [PubMed] [Google Scholar]
- 41.Force A, Lynch M, Pickett FB, Amores A, Yan YL, Postlethwait J. 1999. Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151, 1531–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Szostak JW, Orr-Weaver TL, Rothstein RJ, Stahl FW. 1983. The double-strand-break repair model for recombination. Cell 33, 25–35 10.1016/0092-8674(83)90331-8 (doi:10.1016/0092-8674(83)90331-8) [DOI] [PubMed] [Google Scholar]
- 43.Breslauer DN, Kaplan DL. 2012. Silks: properties and uses of natural and designed variants. Biopolymers 97, 319–321 10.1002/bip.22007 (doi:10.1002/bip.22007) [DOI] [PubMed] [Google Scholar]