

SUMMARY
Neuronal microtubules support intracellular transport, facilitate axon growth, and form a basis for neuronal morphology. While microtubules in non-neuronal cells are depolymerized by cold, Ca2+ or antimitotic drugs, neuronal microtubules are unusually stable. Such stability is important for normal axon growth and maintenance, while hyperstability may compromise neuronal function in aging and degeneration. Though mechanisms for stability were unclear, studies suggested that stable microtubules contain biochemically distinct tubulins that are more basic than conventional tubulins. Transglutaminase-catalyzed posttranslational incorporation of polyamines is one of the few modifications of intracellular proteins that add positive charges. Here we show that neuronal tubulin can be polyaminated by transglutaminase. Endogenous brain transglutaminase-catalyzed polyaminated tubulins have the biochemical characteristics of neuronal stable microtubules. Inhibiting polyamine synthesis or transglutaminase activity significantly decreases microtubule stability in vitro and in vivo. Together, this suggests that transglutaminase-catalyzed polyamination of tubulins stabilizes neuronal microtubules essential for unique neuronal structures and functions.
Keywords: Tubulin, Transglutaminase, Polyamine, Microtubule, Axon, Neuron, Stable Microtubules, Polyamination, Spermine, Spermidine, Putrescine
Neuronal microtubules (MTs) are biochemically and physiologically diverse. Multiple genes for α- and β-tubulins are expressed differentially during development and regeneration. Tubulins are also subject to posttranslational modifications, and contain a heterogeneous group of microtubule associated proteins (MAPs) (Luduena, 1998). Functional consequences of such diversity are thought to be generating MTs suited for unique demands of cells. Neurons are unusually polarized, with a single long axon and multiple branching dendrites. MTs in axons may be very long (>100s of microns in axons), and axonal MTs are maintained for weeks or months at considerable distances from sites of tubulin synthesis (>1 meter in some human nerves), imposing unusual constraints on neuronal MTs. Unlike MTs in non-neuronal cells which can be highly dynamic (Desai and Mitchison, 1997), axonal MTs are more stable, allowing them to act as a structural framework for the neuron, serve as tracks for organelle transport, maintain cell shape and connections, and define functional compartments (Brady, 1993). Moreover, MTs in axons are not continuous with a perikaryal microtubule organizing center or visible nucleating structure, (Yu and Baas, 1994). How can axonal MTs extend such distances and be stable for so long, yet retain the ability to be modified in response to physiological stimuli? A simple answer would be presence of a significant fraction of stable MTs. This stable MT fraction is not only important for cytoskeletal organization in early neuronal development (Kirkpatrick and Brady, 1994; Kirkpatrick et al., 2001) and axon maintenance through adulthood, but also structural rearrangement for axon regrowth and targeting during regeneration (Brady, 1993). More interestingly, increases in MT stability correlates with decreases in neuronal plasticity, both of which occur during aging and some neurodegenerative diseases. Therefore, learning about stable MT fragments, which are unique to neurons, is crucial to understand normal axonal development and neuronal differentiation; this may also aid in identifying novel therapeutic targets for neurodegeneration and regeneration.
The existence of a stable, biochemically distinct fraction of axonal tubulin was demonstrated some years ago (Brady et al., 1984; Sahenk and Brady, 1987). When preparing MTs from brain extracts, a substantial amount of tubulin remains in the pellet following low temperature depolymerization. This fraction is termed cold-insoluble, or cold-stable tubulin. A more extensive differential extraction using cold and Ca2+ extractions to produce labile, cold stable and cold/Ca2+ stable fractions was developed (fig 1A). The cold/Ca2+ fraction was enriched in axons. Using axonal transport to metabolically label MTs in rat optic nerve, the cold/Ca2+ stable tubulin fraction (P2) was examined by 2D-PAGE. A striking difference was found between tubulins in soluble and stable MTs: some tubulins in P2 exhibited a significant basic shift during isoelectric focusing (IEF) (Brady et al., 1984). This suggested that tubulins in stable MTs were biochemically distinct from those in cold-labile MTs. Specifically, cold-stable MTs contained tubulins significantly more basic than predicted from sequence or observed in cold-cycled MTs.
Figure 1. Effects of Difluoromethylornithine (DFMO) on microtubule stability in 3-month-old rat optic nerve.
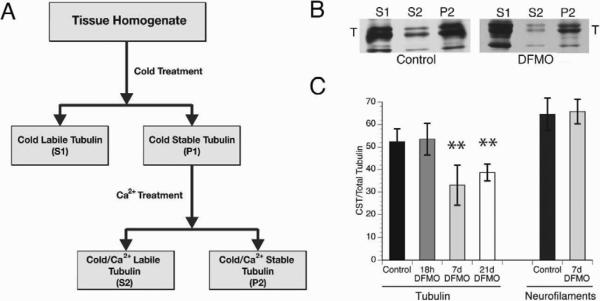
A) Scheme for fractionation of axonal tubulin in Soluble (S1), Cold Stable (S2) and Cold/Ca2+ Stable (P2, CST) tubulin. B) Fluorographs of tubulin fractions from optic nerve labeled by slow axonal transport (21 days after injection of 35S-methionine in the eye of control or DFMO treated rats (7D pretreatment). Tubulin and neurofilament triplet proteins are the major labeled protein species in the P2 fraction of control nerves. Tubulin consistently shifted from P2 to S1 with DFMO treatment, whereas neurofilaments remain in P2. C) Compared with control, DFMO treatments for both 7 and 21 days significantly decreased stable tubulin levels without affecting neurofilament fractionation (see suppl. table 1). Statistical analysis was by Student T-test. **denotes P<0.001. See also Table S1 and fig S1.
Stability of MTs has been related to differences in MAPs, specific tubulin isotypes and posttranslational modifications, but no factor was identified that is sufficient to make MTs stable to depolymerization by cold or elevated Ca2+. MAPs stabilize cycled MTs in vitro (Chapin and Bulinski, 1992), but the increase in stability is modest and MAPs partition with both stable and labile MTs (Brady et al., 1984). Similarly, detyrosination and acetylation of α-tubulin correlate with MT stability in many systems (Bulinski et al., 1988), but in vitro these modifications confer no measurable change in MT stability (Maruta et al., 1986; Webster et al., 1990) and are found in all cell types. Specific tubulin isotypes may contribute to MT stability (Falconer et al., 1994), but none partitions specifically with stable MTs and again differences in stability are modest. The native pI for highly conserved tubulin isoforms all fall within a narrow range (pI = 5.5–5.6 for mouse α-tubulins, pI = 4-8-4.9 for mouse β2–6 tubulins and pI = 5.6 for β1). MAPs do not associate with tubulin in IEF gels or change the charge on tubulins. Thus, no known tubulin isotype or modification can account for both the basic shift and exceptional stability of P2 tubulins, suggesting a novel posttranslational modification.
The unusual IEF behavior of tubulin in cold stable fractions suggests addition of positive charge to affected subunits, but most familiar modifications of cytoplasmic proteins are acidic or neutral, including phosphorylation, acetylation, detyrosination and glycosylation. One exception is covalent addition of a polyamine, such as putrescine (PUT), spermidine (SPD) and spermine (SPM), to a protein-bound glutamine residue by a transglutaminase (Mehta et al., 2006). Polyamines are abundant multivalent cations in many tissues, present at high levels in brain (Slotkin and Bartolome, 1986). Polyaminated proteins may exhibit unusual stability, increased insolubility and resistance to proteolysis (Esposito and Caputo, 2005). Ambron found that radioactive polyamines were covalently linked to various neuronal proteins in Aplysia, including a putative tubulin (Ambron and Kremzner, 1982). Polyamines and transglutaminase are abundant in brain, but their physiological roles in neurons are not well defined. However, increases in transglutaminase activity and polyamine levels correlate with neuronal differentiation and neurite outgrowth (Maccioni and Seeds, 1986; Slotkin and Bartolome, 1986).
The properties of polyamines and transglutaminase are consistent with a role for polyamination in stabilizing MTs. We tested the hypothesis that polyamination of axonal tubulins leads to generation of cold-stable MTs. When endogenous polyamine levels were lowered in rats with an irreversible inhibitor of polyamine synthesis, cold-stable tubulin levels significantly decreased. Both in vivo labeling of tubulin with radioactive PUT and in vitro transamidation with monodansylcadaverine (MDC, a fluorescent diamine) indicated that neuronal tubulin is a substrate for polyamination by transglutaminase. Polyamine modification sites were mapped by LC-MS-MS and were consistent with sequence specific incorporation of polyamines into neuronal tubulins by transglutaminase. MTs containing transglutaminase-catalyzed polyaminated tubulins were resistant to cold/Ca2+ depolymerization and had added positive charge, mimicking neuronal stable MTs, which are largely restricted to nervous tissues and highly enriched in axons in vivo. Further, a mouse model in which the major brain transglutaminase isoform 2 (TG2) was knocked out had deceased neuronal MT stability. Finally, the role of TG2 in stabilizing MTs was identified in mouse brains at different postnatal times as neurons mature and myelination of axons progresses. Transglutaminase-catalyzed polyamination of tubulin was essential for neurite growth and neuronal differentiation, as well as MT stability in culture. Together, these results indicated that transglutaminasecatalyzed polyamination of neuronal tubulins contributes to MT stability in axons and this posttranslational modification is important for neuronal development and maturation.
RESULTS
Inhibition of polyamine synthesis significantly decreases axonal MT stability
To determine whether polyamines are needed for cold/Ca2+ stable tubulin (CST), endogenous polyamine levels were lowered and CST evaluated in rat optic nerve. The rate-limiting enzyme in polyamine biosynthesis is ornithine decarboxylase (ODC) (Pegg and McCann, 1988). Difluoromethylornithine (DFMO) is a suicide inhibitor of ODC. Administered to rats as a 2% solution in drinking water, DFMO lowers total polyamine levels significantly in all tissues examined (Danzin et al., 1979). Rats treated with DFMO for 2–21d were used to determine effects of reduced neuronal polyamine on CST. After 18h, 7d and 21d of treatment, axonal MTs were labeled by injecting 35S-methionine into the vitreous of the eye and waiting 21d for axonal transport to deliver labeled tubulin to the optic nerve. Cold/Ca2+ fractionation of labeled optic nerve (fig 1A) showed a significant decrease in CST after DFMO treatment (fig 1B and C). Fluorographs of S1, S2 and P2 fractions from control and DFMO treated rats show a significant fraction of tubulin shifted from P2 to S1 fractions with DFMO treatment (fig. 1B). In control optic nerves, cold-insoluble tubulin was 52% of total radiolabeled axonal tubulin, but in 7d or 21d DFMO-treated nerves, this fraction was <40% (fig 1C, see also fig S1 and table S1) (p<0.001) suggesting that polyamines are required for generation of cold-insoluble tubulin in axons. To determine whether decreased polyamines generally reduced cytoskeleton stability in axons or was specific for MTs, neurofilament (NFM) fractionation was analyzed in parallel. There was no change in NFM fractionation after DFMO treatment (table S1).
In vivo labeling of tubulin with radiolabeled polyamines
Next, we determined if polyamines were covalently added to tubulin in vivo and if modified tubulin co-fractionated with cold-insoluble tubulin. 14C-PUT axonal transport labeling experiments were performed in rat optic nerves. Due to high levels of endogenous polyamines and low specific activity of 14C-PUT, endogenous polyamine levels were lowered by 18h DFMO pretreatment. When axonal proteins were fractionated 21d after 14C-PUT labeling, 70–80% of label was in P2 (not shown). Subsequent fractionation studies with higher specific activity 3H-PUT confirmed these results (fig 2A). In optic nerves labeled by axonal transport of 3H-PUT, the only proteins with significant incorporation of labeled polyamines had the MW of tubulin, although 35S-methionine labeled neurofilaments at the same time. Much lower levels of 3H-PUT were seen in S1, the cold labile MT fraction. This suggested that polyamination of tubulin can occur before formation of stable MTs. Polyaminated tubulin may help nucleate and stimulate polymerization of tubulins as well as stabilizing MTs after polymerization.
Figure 2. Covalent incorporation of radiolabeled PUT into rat optic nerve with tubulin as a putative substrate.

A) A protein of tubulin size was labeled with 3H-PUT (left) while 35S-Methionine labeled total proteins (right). 3H-PUT was incorporated into axonal tubulin mainly in cold/Ca2+ stable tubulin fractions (P2). 14C-PUT modified axonally transported P2 tubulin in a similar pattern (not shown). B) Upper panel showed gel filtration chromatography of 14C-labeled P2 proteins in rat optic nerve at 21-day ISI indicating a single broad peak of radioactivity. Lower panel showed that the peak of eluted radioactivity coincided with tubulin immunoreactivity, consistent with covalent incorporation of polyamines into tubulin in vivo.
14C-PUT labeled proteins in P2 were analyzed by gel filtration chromatography on a Toyopearl HW-55F (Supelco) column equilibrated in 6M guanidine-HCL in MES to determine if 14C-PUT co-eluted with tubulin. A single broad peak of 14C eluted 20–30 ml after the void volume coinciding with the peak of tubulin immunoreactivity. There was no peak of 14C corresponding to free polyamines (fig 2B), suggesting that polyamines in CST fractions were covalently incorporated into axonal tubulins.
Polyamination of neuronal tubulins and MTs in vitro
In vivo stabilization of neuronal MTs by polyamines could occur by covalent posttranslational modification of tubulins. To confirm modification of tubulins with polyamines and evaluate a potential role for transglutaminase, an enzyme known to catalyze polyamination, purified mouse brain tubulin/MTs were incubated with recombinant transglutaminase in vitro. In vitro transglutaminase activity was controlled by addition of Ca2+. Transglutaminase polyaminated both tubulin and polymerized MTs by a Ca2+-dependent reaction. Modification was visualized in two ways: First, monodansyl cadaverine (MDC), a fluorescent diamine and transglutaminase substrate (Lorand et al., 1971), was used as a polyamine analog, giving a fluorescent band at the apparent MW of tubulin in SDS-PAGE. This band was observed whether unassembled tubulin or assembled MTs were the substrate (fig 3A, see also fig S2). Second, modified tubulins were visualized by immunoblot when physiological polyamines (SPM and SPD) were used. Bands were identified as tubulin with the DM1A antibody against α-tubulin and polyamine modification identified with an anti-SPM/SPD antibody that recognizes both SPM and SPD (fig 3B). Both free tubulin and tubulin in MTs were modified by transglutaminase and SPM/SPD.
Figure 3. In vitro polyamination of neuronal tubulin and MTs and transglutaminase activity.

A) SDS-PAGE showed a fluorescent band at tubulin MW indicating covalent addition of Monodansylcadaverine (MDC) to purified mouse tubulin and MTs with transglutaminase (gpTG) activity. This modification required Ca2+ to activate transglutaminase and was also seen with pig brain tubulin and MTs. B) Spermine (SPM) and Spermidine (SPD) were also incorporated into neuronal tubulin. Immunoblotting for polyamine (red) and tubulin (green) showed polyaminated tubulins in yellow (merge), also see fig S2. Neuronal MTs polymerized in vitro showed comparable tubulin modifications (not shown). C) gpTG-catalyzed covalent addition of MDC was comparable for tubulin and taxol-stabilized MTs. Polyaminated proteins showed decreased solubility and were enriched in pellets. D) Coomassie blue stained gel (left) showed similar amounts of tubulin and taxol-stabilized MTs as substrates for polyamination in C. In the absence of polyamines, transglutaminase catalyzed cross-linking of tubulins (right), but cross-linked tubulins were primarily in supernatants and may not enter the stacking gel (not shown). E) Negative stain electron microscopy (EM) showed polymerized MTs with polyaminated tubulin. Several different magnification images show that many MTs are present in this fraction. F) Negative stain EM showed cross-linked tubulin aggregates by transglutaminase in the absence of polyamines.
Polyaminated tubulins/MTs showed reduced solubility as the bulk of modified tubulins were pelleted. When transglutaminase was incubated with tubulin without polyamines, we observed tubulin cross-linking, but cross-linked tubulin aggregates remained in supernatants rather than pelleting and much cross-linked tubulin failed to enter the gel (fig 3C and 3D). The presence of polyamines in reactions largely eliminated cross-linking. To determine whether modification of tubulin by transglutaminase favors disassembled or polymerized tubulin, similar amounts of free tubulins and taxol-stabilized MTs were incubated in vitro with transglutaminase and polyamines. There was no significant difference in incorporation of either MDC (fig 3C and D) or SPM/SPD (data not shown). Patterns of polyamine modification on unassembled tubulins (fig 3A~D), polymerized MTs (fig 3A) and taxol-stabilized polymerized MTs (fig 3C–D) were similar. Electron microscopy showed polyaminated tubulins polymerized in vitro at 37°C (fig 3E). In contrast, tubulin treated with transglutaminase without polyamines formed large aggregates with no evidence of MT formation (fig 3F). Since intracellular polyamine levels are high, the cross-linking is unlikely to occur. These data suggested that transglutaminase-catalyzed polyamination of tubulin occurs on both tubulin and MTs, and that polyaminated tubulins polymerize normally.
Identification of polyamination sites on neuronal tubulins
To verify covalent modification of tubulin and map modification sites, purified mouse brain tubulin/MTs modified in vitro by transglutaminase were analyzed by LC-MS-MS. Four different polyamines or analogues (PUT, SPD, SPM or DMC), in independent experiments, all modified tubulin in vitro similarly as identified by LC-MS-MS (fig 4). Similar results were observed with tubulin from P2 cold/Ca2+ stable MTs in vivo (Fig S3A–B). LC-MS-MS analysis of tubulin from S1 cold-labile MTs failed to detect significant amounts of polyamine-modified tubulins. Putative modification sites on both α and β–tubulins were mapped by MS1 spectra based on mass shift (fig 4A–B). Selective modification sites were not sensitive to specific polyamines used in vitro, but depended on the amino acid sequence. Putative modification sites were confirmed by MS/MS, based on specific ion shifts (fig 4B–E, see also fig S3C) and locations predicted relative to tubulin dimer structure (fig 4F and S3D). Conserved sites were identified in multiple tubulin isoforms. Targeted MS/MS analysis verified that the Q at position 13 of a conserved N-terminus β-tubulin tryptic peptide EIVHIQAGQCGNQIGAK (corresponding to the highly conserved Q15 in the β-tubulin sequence) was a primary modification site (fig 4A). Q15 is present in all mouse and human β-tubulin sequences (fig S3C). Based on a predicted tubulin dimer structure (Nogales et al., 1998), the Q15 residue is adjacent to the hydrolysable GTP in β–tubulin, allowing interaction between polyamine and GTP, where it might affect GTP binding and/or hydrolysis. Additional conserved sites were identified including sites on both α– and β–tubulins (data not shown). Modified residues on α–tubulins (Q31, Q128, Q133, Q256 and Q285) were of particular interest because they were on the surface between α–tubulin and β–tubulin in adjacent dimers. Sites in the interface between α and β-tubulin are consistent with a role of polyamine modification in MT stabilization.
Figure 4. Polyamination sites mapped on tubulins by Liquid Chromatography-Tandem Mass Spectrometry.

Trypsin-digested peptides from in vitro modified mouse brain tubulin and in vivo stable and labile MT fractions were subjected to LC-MS. Several putative modification sites from different tubulin isoforms were identified based on the mass shift. A representative modification site on a conserved glutamine residue (Q) in N-terminal β-tubulins is illustrated. A) A diagram showing sequence ions for peptide EIVHIQAGQCGNQIGAK (PEIVH). Based on accurate mass for sequence ions (see below), the peptide contained unmodified Qs at positions 6 and 9, leaving only Q at position 13 (in red, Q15 in the β-tubulin sequence) as a modification site with PUT in vitro. B) Targeted tandem MS spectrum on Ions derived from PUT modified PEIVH (PUT- PEIVH) with m/z 632.0043+ (peptide mass 1892.989, 1.06 ppm error) showed an accurate mass shift of 199.1321 from y4 to y5, suggesting an additional shift of 71.07 as a result of one PUT (mass: 88.15148) added to Q15 with a loss of one NH3 (mass: 17.0306) C, D) Chromatograms confirmed the presence of PUT- PEIVH in brain stable MT fraction (P8), suggesting the same modification occurs in vivo and that the modification is sequence specific. Detailed modification patterns on the precursor and its ion series were shown in D, peaks corresponding to SPM modified peptide (SPM- PEIVH) were observed in the same fractions. E) A summary of peak areas for polyaminated PEIVH from mouse brain MT fractions showed significantly higher levels of PUT- PEIVH and SPM- PEIVH in stable MT fractions (P) compared with labile MT fractions (S). F) Putative polyaminated sites on tubulins were shown with regard to the protein structure. Using coordinates from the Protein Structure database (MMDB ID: 8900; PDB ID: 1TUB) for predicted structures for taxotere-bound tubulin dimer (Nogales et al., 1998) the conserved Q15 residue is located adjacent to phosphates on the exchangeable GTP in β-tubulin. The structure was generated using Cn3D software (National Library of Medicine). This location suggests that positively charged polyamine might stabilize GTP and affect hydrolysis, consistent with a role in modulating microtubule dynamics. Circles are superimposed at Q15 to illustrate approximate dimensions of putrescine (red dashed circle) and spermine (grey circle). Note the potential for interacting with β-tubulin GTP (see also fig S3 and table S2–S4).
Biochemical characteristics of polyaminated brain tubulins catalyzed by endogenous transglutaminase
To determine whether neuronal transglutaminase and endogenous polyamines were sufficient to modify tubulins, we prepared a crude extract of endogenous transglutaminase from fresh 1mo mouse brains (fig S4). The transglutaminase fraction (S0) contains soluble brain tubulin and free polyamines. When endogenous transglutaminase was activated by reaction buffer, >70% soluble tubulin became cold/Ca2+ stable (aP2 in fig 5), but <20% of initially soluble tubulins were converted to cold/Ca2+ stable tubulin (ctrl in fig 5) in buffer lacking added Ca2+. Residual reactivity in control buffer may be due to endogenous Ca2+ activation of transglutaminase, or a higher sensitivity of DM1A for unmodified tubulins. Finally, in a mix of unmodified and polyaminated tubulins run on an iso-electric focusing (IEF), modified tubulins had more basic pIs than unmodified tubulins (fig 5E and S4), consistent with presence of added positive charge. Although the amount of modified tubulin was less than expected, it was sufficient to stabilize MTs, favoring the hypothesis that polyaminated tubulins nucleate tubulin dimer assembly and may occur as stable segments along MTs. Endogenous brain transglutaminase-catalyzed polyaminated tubulins share similar biochemical properties with CST in vivo, with regard to stability against cold/Ca2+, and presence of added positive charges. Thus, endogenous levels of polyamines and transglutaminase in brain are sufficient to modify and stabilize brain tubulin.
Figure 5. Biochemical similarities between in vitro polyaminated tubulins and in vivo neuronal cold stable tubulins.

Mouse brain soluble tubulins were polyaminated in vitro by endogenous mouse brain transglutaminase/polyamine and subjected to cold Ca2+ fractionation (fig S4). A) Coomassie blue stained gel; B) Immunoblots with DM1A (α-tubulin); and C) Tu27 (β-tubulin). Compared to controls without transglutaminase activation by addition of Ca2+, modified tubulins showed a remarkable increase in the cold/Ca2+ stable fraction (P2a). D) Quantitation of α-tubulin showed >70% soluble tubulins were converted to cold/Ca2+ stable tubulins (P fraction in aP2 group) after polyamination while most tubulin remained soluble with cold (ctrl S1) or Ca2+ (ctrl S2) in the control group. Statistical analysis was by Student T-test. **denotes P<0.001. E) 2D PAGE showed a shift towards a basic pI for polyaminated tubulins, consistent with added positive charge on in vivo cold/Ca2+ stable tubulin (see also fig S2).
Transglutaminase activity and TG2 protein distribution in vivo
Cold/Ca2+ stable MTs are a characteristic of nervous tissue with little or none detectable in nonneuronal tissues, except testes (fig 6A). Stable tubulin in testes may be associated with flagellar MTs and the role of polyaminated tubulin there remains to be determined.
Figure 6. Characterization of transglutaminase activity and TG2 protein levels in the nervous system.

A) Cold/Ca2+ stable tubulin was enriched in brain but was at or below levels of detection in non-neuronal tissues other than testes. The stable tubulin fraction in testes is presumably due to the stable microtubules in sperm flagella and may not be equivalent to the brain fraction. However, polyamines and TG2 are present in testes. B) Transglutaminase activity was present in various neural tissues including cerebral cortex (CC), brain stem (BS), spinal cord (SC), optic nerve (ON) and sciatic nerve (SN) from 5-week-old mice. C) Quantitative plots showed that transglutaminase activity was higher in axon-enriched compartments, like ON and SN, correlated with higher MT stability in axons. D) TG2 protein was also expressed differentially in various neural tissues. The same amount of total protein was loaded in each lane. Actin and GAPDH were loading controls. Actin was higher in axon-rich domains (ON and SN), while GAPDH was lower in axon-rich tissues as a fraction of total protein. E) Quantitative comparison shows TG2/actin ratio was lower in SN than ON but SN exhibited comparable levels of TG activity (C), thus other transglutaminase isoforms may contribute to axonal transglutaminase activity, particularly in the PNS. Statistical analysis was by Student T-test. **denotes P<0.001.
Transglutaminase activity in brain results from multiple gene products, including TG1, TG2, TG3 (Kim et al., 1999) and TG6 (Hadjivassiliou et al., 2008), but the primary cytoplasmic transglutaminase in brain is thought to be TG2 (Bailey and Johnson, 2004). To understand the role of TG2 in producing CST, we analyzed TG2 protein and enzymatic activity in brain grey matter enriched in perikarya/dendrites (cerebral cortex, brain stem and spinal cord) and in white matter enriched in axons (optic and sciatic nerves). CST levels were significantly higher (>50% of total tubulin) in adult optic nerves than in cerebrum, enriched in dendrites and perikarya. Axonal enrichment of CST suggested a spatial correlation between transglutaminase activity and CST levels. Transglutaminase activity was elevated in both optic and sciatic nerves (fig 6B and 6C), consistent with TG2 immunoreactivity (fig 6D and 6E). Sciatic nerve had less TG2 immunoreactivity than optic nerve (fig 6D and 6E) but equivalent enzyme activity (fig 6B and 6C), suggesting differential expression of transglutaminase isoforms in CNS and PNS. Quantification of TG2 protein in axonal tracts was normalized to actin, which is enriched in optic and sciatic nerve relative to cerebral cortex, brain stem and spinal cord, so relative TG2 levels in optic/sciatic nerves (fig 6D and 6E) are not directly comparable to other brain regions, but good spatial correlation existed between transglutaminase activity and CST distribution in nervous tissues.
inhibition of transglutaminase activity inhibited neurite growth
Since MT stability is essential for neuronal structure and function, transglutaminase-catalyzed polyamination of tubulin may affect neuronal morphology. To test this, SH-SY5Y neuroblastoma cells were differentiated by retinoic acid and BDNF in the presence of 10mM IR072 (fig S5), an irreversible transglutaminase inhibitor. Both transglutaminase activity and TG2 protein level were upregulated as cells differentiated and extended neurites (data not shown), correlating with increased MT stability (fig 7). Inhibition of transglutaminase had a significant effect on SH-SY5Y morphology (fig 7B) compared with cells treated with vehicle (fig 7A). IR072-treated cells remained viable but showed a phenotype more like undifferentiated SH-SY5Y cells, but no longer proliferated. Average neurite length was severely reduced from 76μm in control cells to 29μm with IR072 (fig 7C and fig 7D). Some control SH-SY5Y cells put out extremely long neurites (>150μm); but transglutaminase inhibition almost completely eliminated such long neurites. Cold/Ca2+ fractionations tested whether lack of transglutaminase activity reduced MT stability as well as neurite extension. IR072 decreased both cold stable and cold/Ca2+ stable tubulin levels, with more significant effects on cold/Ca2+ fractions (fig 7E). These suggested that transglutaminase is essential for early neurite development by generating stable tubulin/MTs and possibly by enhancing MT polymerization.
Figure 7. Effects of transglutaminase inhibition on neurite extension in SH-SY5Y cells.

To determine functions of polyamination and MT stability in neuronal development, we differentiated SH-SY5Y neuroblastoma in the presence of IR072 (fig S5), an irreversible transglutaminase inhibitor. A) Control SH-SY5Y cells were differentiated, fixed and stained with beta III tubulin antibody. Normal neurite extension and neuron-like phenotypes were observed. B) SH-SY5Y cells differentiated in the presence of the IR072 showed significant inhibition of neurite outgrowth and a phenotype more like undifferentiated SH-SY5Y cells. C) Length distribution of neurites for control SH-SY5Y cells showed an average of 76μm with some neurites >150μm. D) Length distribution of neurites for differentiated SH-SY5Y cells treated with IR072 showed a significant shift towards shorter neurites with an average length of 29μm with few neurites >90μm. The difference in mean neurite length for control and IR072 treated cells was statistically significant (P<0.00001). This suggests that transglutaminase activity is required for efficient elongation of neurites during differentiation of SH-SY5Y cells. E) Cold/Ca2+ fractionations were done on SH-SY5Y cells under 3 conditions (undifferentiated, differentiated, differentiated with IR072). Western blots with DM1A antibody (α-tubulin) showed that: 1) in undifferentiated cells with low transglutaminase activity and TG2 protein levels, <10% of total tubulin was cold stable, with no cold/Ca2+ stable tubulin. 2) In fully differentiated cells where both transglutaminase activity and TG2 proteins level peaked, cold stable tubulin level increased significantly to >40% of total tubulin and cold/Ca2+stable tubulin was >20% of total tubulin); 3) In differentiated cells treated with IR072 where transglutaminase activity was significantly reduced, cold/Ca2 stable tubulin was dramatically lowered <10%. This suggests a strong correlation between transglutaminase activity and microtubule stability, both of which may contribute to neurite development.
Altered CST formation in a TG2-KO mouse
Our data suggested a direct role for TG2 in CST formation in the CNS. To test this, we evaluated CST levels in brain and spinal cord of TG2-KO mice (Nanda et al., 2001), where no TG2 immunoreactivity was detectable (fig 8A, upper band). Total transglutaminase enzymatic activity was reduced to <30% of WT in both brain and spinal cord (fig 8B and 8C). Although transglutaminase activity and TG2 protein levels were comparable in brain for 5wk and 5mo WT mice, transglutaminase activity and TG2 protein levels decreased significantly in 5mo WT spinal cord relative to 5wk WT spinal cord. CST levels (black bar) and cold/Ca2+ stable tubulin levels (white bar) correlated with transglutaminase activity/TG2 protein levels in brain (fig 8D and 8E). CST levels were drastically reduced in 5wk TG2 KO mouse brain and spinal cord relative to age matched WT. The drop remained in 5mo TG2 KO brain, but CST and transglutaminase activity in 5mo TG2 KO spinal cord was comparable to age matched WTs where TG2 protein level is low. Compensation from other transglutaminase isoforms (mainly TG1 and TG3) in brain maintained some transglutaminase activity, and CST remains sufficient to maintain the fundamental structure and function of the CNS in this model. Future experiments knocking down other isoforms to reduce further total transglutaminase activity will be needed to see the phenotype due to complete elimination of transglutaminase and CST.
Figure 8. Neuronal MT stability in 5wk and 5mo TG2 KO mouse brains.

A) Immunoblots documented absence of TG2 immunoreactivity inTG2 KO mouse brain and spinal cord. In WT controls, brain TG2 was expressed at similar levels in both age groups but spinal cord TG2 levels dropped at 5mo relative to 5wk mice. B) transglutaminase activity was significantly reduced in TG2 KO mouse brains compared with age matched WT. Brain transglutaminase activities were similar between two ages paralleling TG2 protein levels. C) Spinal cord transglutaminase activity was much lower in TG2 KO mice than WT at 5wk, but transglutaminase activity was comparable in 5mo TG2 KO and WT spinal cord. D) Brain cold and cold/Ca2+ stable tubulin levels decreased drastically in TG2 KO mice for both age groups, consistent with changes in TG2 protein and transglutaminase activity. Residual transglutaminase activity due to other transglutaminase isoforms may contribute to cold/Ca2+ stable tubulin formation. E) Spinal cord cold stable tubulin and cold/Ca2+ stable tubulin levels decreased in 5wk TG2 KO mice as compared to age matched WT, but levels of stable tubulins in 5mo TG2 KO and WT groups were not significantly different, consistent with TG2 protein expression and transglutaminase activity. Other transglutaminase isoforms may make a greater contribution to MT stability in adult spinal cord. Statistical analysis was by Student T-test. **denotes P<0.001, *denotes P<0.005.
Upregulation of TG2 and CST during brain development
Increases in TG2 levels and activity between 5wk and 5mo (fig 8) suggested that TG2 and stable MTs play a role in neuronal maturation. Earlier studies indicated that microtubule stability increased with axonal maturation and myelination (Kirkpatrick and Brady, 1994); roughly ~2wk postnatal in mouse. To test whether stabilization of axonal MTs during this stage correlates with transglutaminase-catalyzed polyamination of neuronal tubulin, we examined transglutaminase activity and TG2 immunoreactivity in one hemisphere of mouse brains at 10d, 3wk and 3mo (fig 9A–D). Cold/Ca2+ fractionations were performed on the other hemisphere (fig 9E).
Figure 9. Characterization of transglutaminase activity, TG2 protein and microtubule stability in postnatal mouse brain development.
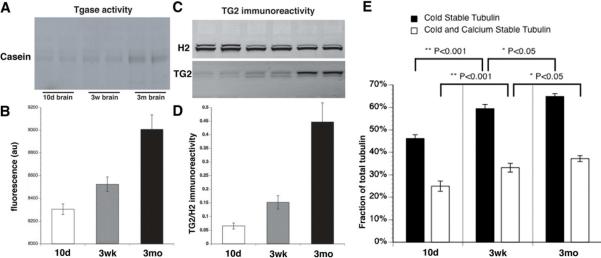
To evaluate changes in transglutaminase activity and TG2 protein as neurons develop and mature, mouse brain was analyzed at 10d (pre myelination); 3wk (active myelination) and 3mo (mature adult neurons). A) Fluorescent intensity of MDC incorporated into casein shows significantly increased transglutaminase activity during postnatal brain development. During this interval, neurite outgrowth plays a minimal role, but consolidation of synaptic connections and myelination progresses toward adult levels. Changes in transglutaminase activity during this time may be related to axonal maturation, correlated with myelination, B) Quantitative analysis show that transglutaminase activity increased 1.5 and 3 fold in 3wk and 3mo brains respectively, as compared with 10d mouse brain. C) Consistent with changes in transglutaminase activity, TG2 protein levels in western blots increased. Kinesin (H2) immunoreactivity was a loading control. D) Similar changes in TG2 protein and activity levels occurred during postnatal mouse brain development. E) Alteration of MT stability during postnatal mouse brain development. Cold/Ca2+ fractionations were performed on mouse brain from the 3 age groups and analyzed by immunoblot with DM1A antibody, which recognizes α-tubulin. Cold stable and cold/Ca2+ stable tubulin levels are 45% and 25% of total tubulin at 10d, respectively. At 3wk, cold stable tubulin increased to 58% of total tubulin, of which cold/Ca2+ stable tubulin was 32%. At 3mo, cold stable tubulin increased to 64%, while cold/Ca2+ tubulin increased to 36%. Differences are statistically significant (** P< 0.001 between 10-day and 3-week groups, * P<0.05 between 3-week and 3-month groups) n = 6.
Both transglutaminase activity (fig 9A–B) and TG2 immunoreactivity (fig 9C–D) were low in 10d brains, increased to a similar extent in 3wk brains, and continued to increase in 3mo brains. Correspondingly, cold stable and cold/Ca2+ stable tubulin levels rise concurrently (fig 9E), indicating a strong temporal and developmental correlation between transglutaminase activity, TG2 expression and MT stability in postnatal development of mouse brain. The result may be to stabilize microtubules in vivo during axon maturation and later stages of life, a process different from initiation and stabilization of early neurite development.
DISCUSSION
Regulation of MT polymer dynamics remains an important topic of study (Kueh and Mitchison, 2009). MTs are generally quite dynamic in non-neuronal cells, consistent with a need to rapidly reorganize MTs during division or migration (Desai and Mitchison, 1997). In contrast, neurons must balance two opposing properties of MTs: stability and dynamics. Stability is needed for axonal MTs to provide a structural framework and serve as tracks for axonal transport, while dynamics are needed for reorganization and repair during neurite growth and remodeling of synaptic connections (Brady, 1993). Most MTs remain intact for long periods of time in axons that may be >1meter in humans.
Consistent with increased stability of axonal MTs, a large fraction of neuronal tubulin pellets after extraction with cold, Ca2+ or antimitotic drugs: treatments that depolymerize most non-neuronal MTs. The morphological correlate of insoluble tubulin is stable segments of MTs (Sahenk and Brady, 1987), that are enriched in axons, continuous with labile MT polymer, and may serve as nucleation sites for adding tubulin dimers to MTs (Brady et al., 1984; Sahenk and Brady, 1987). Stable MTs provide a stable structural framework for neurons, while acting as axonal MT organizing centers to facilitate remodeling of MTs after stimulation or injury. However, the molecular basis for generation of this fraction was poorly understood and not explained by previously characterized tubulin modifications.
Stable axonal MTs can be isolated and characterized (Brady et al., 1984). Tubulin in these fractions is notable in two ways: 1) stable MTs were biochemically distinct from cold-labile MTs. 2) levels of axonally-transported cold insoluble tubulin correlated with axonal plasticity and maturation. Stable MTs are a higher fraction of axonal MTs than non-axonal MTs (dendrites and perikarya). Myelin deficient axons contain lower levels of CST (Kirkpatrick and Brady, 1994; Kirkpatrick et al., 2001), and young neurons contain lower levels than older ones (Brady and Black, 1986; Kirkpatrick and Brady, 1994; Kirkpatrick et al., 2001). These data suggested that cold-stable MTs play a role in neuronal maturation, making the biochemical basis of MT cold-stability a subject of much interest.
Analysis of purified stable MTs by 2D-PAGE revealed a striking difference between tubulins in labile and stable fractions: a shift in pI suggested that some tubulins in the stable fraction were more basic than primary sequence predicted, consistent with addition of positive charge. Most familiar posttranslational modifications of tubulins in brain were acidic (phosphorylation and glutamylation) or charge neutral (acetylation and detyrosination) (Janke and Kneussel, 2010). The positive charge implied a novel modification of tubulins.
A modification that adds positive charge to proteins is covalent addition of polyamines by transglutaminase. Consistent with this, a 56kDa protein in Aplysia neurons was polyaminated (Ambron and Kremzner, 1982). Given that polyaminated proteins may be less soluble, polyamination of tubulins might explain cold insolubility. The abundance of polyamines in post-mitotic neurons (Slotkin and Bartolome, 1986) and the presence of significant transglutaminase activity in brain (De Vivo et al., 2009) were consistent with this idea.
Polyamine levels are high in developing and adult nervous system (Shaw and Pateman, 1973), and affect neuron migration, axon outgrowth, and synapse formation (Slotkin and Bartolome, 1986). Polyamines are implicated in both normal brain function and neuropathology, but specific roles were poorly defined. The major physiological polyamines are PUT, SPM and SPD, derived from L-ornithine, a product of arginine degradation through activity of ODC, an abundant enzyme expressed in neuronal and nonneuronal cells (Pegg and McCann, 1988). SPD and SPM are produced by enzymatic addition of propylamine from S-adenosylmethionine to PUT and SPD respectively. High levels of polyamine in brain and rapid changes in ODC expression in response to various stimuli suggest that polyamine levels are well regulated.
Transglutaminases are a family of enzymes activated by Ca2+ that can catalyze cross-linking of peptides and proteins by formation of γ-glutamyl-ε-lysyl bonds (Griffin et al., 2002). However, in the presence of poly/di/monoamines, transglutaminases catalyze formation of γ-glutamyl amine bonds (Folk et al., 1980). There are 8 transglutaminase genes in human and mouse genomes, including secreted and intracellular forms (Esposito and Caputo, 2005). TG4 and Factor XIII are typically secreted and may serve extracellular functions, including a role in blood clotting. Modification of neuronal cytoskeletal elements requires an intracellular tissue-type transglutaminase. TG1, 2, 3, 5 and possibly 6 are likely to be intracellular. Intracellular transglutaminase functions are not well understood, but are assumed to modify or cross-link proteins.
TG2 is the primary intracellular tissue-type isoform in brain (Cooper et al., 2002). TG2 activity is found in mammalian CNS and PNS (Hand et al., 1993)]. Two features of brain transglutaminase are noteworthy: 1) brain transglutaminase activity increases during development and is linked to neuronal differentiation and neurite outgrowth; and 2) neuronal cytoskeletal elements are in vitro substrates of tissue-type transglutaminase from guinea pig liver (Miller and Anderton, 1986; Selkoe et al., 1982). Whether these proteins, particularly tubulin are indeed physiological substrates of brain transglutaminase and whether modified tubulin changes cytoskeletal properties remained to be addressed.
Eight independent lines of evidence support the idea that polyamination of neuronal tubulin by transglutaminase contributes to MT stability (see model in fig S6). First, lowering endogenous polyamine levels by inhibiting polyamine synthesis significantly decreases neuronal CST levels (fig 1 and table S1). The simplest interpretation is that decreasing polyamine levels by DFMO reduces polyamination of tubulin and cold/Ca2+ stable MT levels. Decreased polyamine levels may also regulate cold-insoluble tubulin indirectly by decreasing transglutaminase activity, consistent with studies of transglutaminase activity and polyamine levels in other systems (Melino et al., 1988). Regardless, both mechanisms suggest that polyamination of tubulin plays a role in stabilizing axonal MTs.
Second, radioactive polyamines incorporated into protein are delivered into axons with slow axonal transport of MTs (SCa). Radiolabeled polyamines fractionate with stable MTs through biochemical manipulations, migrate in SDS-PAGE with tubulin, and co-elute with tubulin-immunoreactive protein in gel filtration chromatography (fig 2).
Third, transglutaminase modifies purified brain tubulin, polymerized MTs and taxol-stabilized MTs in vitro by covalent addition of polyamines (fig 3). Both fluorescent analogues of polyamines (MDC) and physiological polyamines (SPM and SPD) can be linked to tubulin. MTs containing tubulins polyaminated by endogenous brain transglutaminase match endogenous CST in two key respects: They are resistant to cold/Ca2+ treatments that normally depolymerize MTs and exhibit increased positive charge (fig 5 and S2). Although transglutaminase can stabilize substrates through interor intra-molecular cross-linking (Esposito and Caputo, 2005) and intermolecular crosslinks can be generated in vitro, cross-linked tubulin is almost exclusively soluble (fig 3D) and does not polymerize (fig 3F), whereas polyaminated tubulin polymerizes into MTs that are similar to stable MTs in vivo. Little tubulin cross-linking is observed with physiological levels of polyamines. Polyamination of tubulin occurs on either free tubulins or preassembled MTs. Modification of soluble tubulin dimers may enhance polymerization by generating nucleating seeds and modification of assembled MTs may increase stability.
Fourth, both α and β tubulins have conserved polyamination sites. A prominent modification is the conserved Q15 of β-tubulin, positioned where it may affect GTP hydrolysis by tubulin. Mass spectrometry identified additional modification sites largely on interfaces where α/β tubulin dimers interact during polymerization. All identified locations are consistent with a role in facilitating and/or stabilizing MT formation.
Fifth, cold stable tubulin is a hallmark of nervous tissue; with little or none detectable in nonneuronal tissues (fig 6). In vivo transglutaminase activity and TG2 protein levels correlate with stable MT levels in nervous tissues. Axonal MTs (e.g. optic and sciatic nerve) are enriched in cold/Ca2+ insoluble tubulins and axon tracts exhibit a higher transglutaminase activity. Fractionation of different neuronal tissues reveals elevated transglutaminase activity in both optic and sciatic nerve (fig 6). The spatial correlation between neuronal tubulin stability and transglutaminase activity was consistent with a functional role for polyaminated tubulins in axons. Curiously, transglutaminase activity was comparable in both CNS and PNS axon-rich regions, but TG2 protein level was much higher in optic than in sciatic nerve. Although TG2 is the major tissue-type transglutaminase isoform in brain (Ruan and Johnson, 2007), other tissue-type transglutaminases may contribute to PNS transglutaminase activity.
Sixth, inhibiting transglutaminase activity reduces MT stability significantly in culture and inhibits neurite outgrowth in differentiating neuroblastoma cells (fig 7). Although transglutaminase activity and stable MT levels are relatively low in immature cells, they concurrently increase with differentiation, facilitating extension and stabilization of neurites. This suggests that polyamination of tubulins by transglutaminase has roles in neuronal differentiation and neurite development. The reorganization of cytoskeletal structures during this process may be modulated by tubulin polyamination, which may nucleate new microtubules and stabilize polymerized MTs.
Seventh, brain and spinal cord from TG2 KO mice have reduced MT stability, consistent with lower transglutaminase activity and absence of TG2 (fig 8). MT stability was significantly reduced where there was a drop in transglutaminase activity, e.g. in 5wk and 5mo TG2 KO brains and in 5wk old TG2 KO spinal cords. However, MT stability remains high in regions where transglutaminase activity was not significantly reduced. Thus, MT stability and transglutaminase activity were comparable in 5mo spinal cord for both TG2 KO and WT mice, although TG2 immunoreactivity was eliminated in TG2 KO. Maintenance of transglutaminase activity without detectable TG2 in TG2 KO mice suggests involvement of other transglutaminases in stabilizing MTs in spinal cord and to a lesser extent in brain. Thus, cold/Ca2+ insoluble tubulin levels were reduced, but not eliminated in TG2 KO mice. Identifying transglutaminase isoforms responsible for transglutaminase activity in TG2 KO brain and WT spinal cord requires further study. Regardless, correlations between transglutaminase activity and MT stability in WT and TG2 KO mouse models are consistent with our hypothesis that transglutaminase activity is a major contributor to stable MT formation in vivo.
Eighth, transglutaminase activity and TG2 protein levels correlate with MT stability during development and maturation in vivo. As axons mature and neuronal connections stabilize in response to various postnatal modifiers, such as myelination, cold/Ca2+ insoluble tubulin increase (Kirkpatrick and Brady, 1994; Kirkpatrick et al., 2001). Correspondingly, transglutaminase protein levels and enzymatic activity are elevated (fig 9). This suggests that developmental regulation of transglutaminase contributes to MT stabilization as the brain matures.
A role for transglutaminase activity and polyamines in stabilization of axonal MTs does not preclude other roles for transglutaminase and polyamines in the nervous system. Transglutaminases are proposed to play a role in neuronal development (Bailey and Johnson, 2004; Maccioni and Seeds, 1986; Mahoney et al., 2000; Tucholski et al., 2001) and signaling (Basso et al., 2012; Dai et al., 2008; Facchiano et al., 2010) as well as in neurodegenerative diseases (Bailey et al., 2005; Basso et al., 2012; De Vivo et al., 2009; Ruan and Johnson, 2007).
Transglutaminase activity and polyamine levels correlate with brain maturation, neuronal differentiation and formation of neurites (Bailey and Johnson, 2004), but underlying mechanisms were unclear. Here we propose that a key pathway for regulating neuronal development is modulation of MT stability by TG2-catalyzed polyamination of tubulins. Increases in both TG2 protein and transglutaminase activity increased stable MTs in postnatal brains (fig 9), concurrent with myelination and stabilization of neuronal circuitry. Molecular pathways responsible for effects of myelination on TG2 protein level and transglutaminase activity are under investigation. Transglutaminase activity may also contribute to changes in cellular morphology (Gentile et al., 1992). Depletion of polyamines results in disappearance of actin and MT bundles (Pohjanpelto et al., 1981). Inhibition of polyamine biosynthesis results in defects of neuronal morphogenesis, whereas exogenous polyamines stimulate adult neurogenesis (Malaterre et al., 2004). All these are consistent with our findings and suggest a common pathway based in part on alteration of MT dynamics and stability through polyamination of tubulin by TG2.
Stabilizing MTs by transglutaminase and polyamines has many positive aspects for normal cytoskeletal structure and function in developing brain, but may be a double-edge sword in the aging nervous system. Transglutaminase activity and polyamines levels increase in aging brain (Lesort et al., 2000; Slotkin and Bartolome, 1986) and are associated with neurodegenerative diseases including Huntington's (HD), Alzheimer's, and Parkinson's diseases (Jeitner et al., 2009; Lesort et al., 2000). Functional consequences for these increases may be complex. For example, exogenous polyamines and transglutaminase can be either neuroprotective or neurotoxic depending on dose and context. Similarly, changes in MT stability may be beneficial or detrimental depending on levels and circumstance. Stable MTs correlate positively with neuronal stability, but negatively with neuronal plasticity. The stable MTs fraction is modest but crucial in early development, facilitating axon growth and plasticity. As neurons mature, MT stability increases and neuronal plasticity decreases. As a consequence, neuronal connectivity may be stabilized, maintaining neuronal architecture, but continued declines in plasticity in aging may limit axonal recovery following injury or in neurodegenerative diseases by limiting MT dynamics. For example, Inhibiting transglutaminase in models of HD resulted in a modest improvement of lifespan and behavior in HD mouse models. Although transglutaminase may not be a direct component of molecular pathogenesis in HD, it may compromise the ability of neurons to respond to pathological changes by limiting sprouting and formation of new connections. Understanding changes in cytoskeletal dynamics and stability in development and neurodegeneration, including but not limited to regulating MT stability will greatly expand our knowledge of MT cytoskeleton in health and disease.
Materials and Methods
All chemicals used were ACS quality or better, from Sigma, Invitrogen, CalBiochem, or Polysciences. Animals used include: Sprague/Dawley rats (200–225 grams, Harlan), male C57BL/6 (Jackson Laboratories) and TG2 KO mice (Nanda et al., 2001). Axonal transport in rat optic nerve was labeled by intravitreal injection of 35S-methionine or polyamines (3H or 14C-PUT) as described previously (Brady and Lasek, 1982). An injection-sacrifice interval (ISI) of 21 days positioned the SCa wave containing stable and labile MTs in the optic nerve. Radioactive proteins were analyzed by SDS-PAGE and fluorography (Kirkpatrick et al., 2001). Our standard protocol for cold/Ca2+ fractionation of neuronal tubulins was used (Brady et al., 1984) (fig 1 and suppl. methods).
Following cold/Ca2+ fractionation, samples were separated on gradient gels as described and transferred to Immobilon-P membrane (Millipore). Primary antibodies include DM1A (1:20,000, Sigma) for α-tubulin, TGMO1 (1:4000) for TG2, H2 (1:50,000) (Pfister et al., 1989)) for kinesin heavy chain, Tu27 (1:10,000, provided by Dr. A. Frankfurter (Caceres et al., 1984)) for β-tubulin, A2066 (1:5000, sigma) for β-actin. pab0022 and pab0023 (1:1200, Covalab) for SPM/SPD (SPM/SPD).
For quantitative immunoblots, the secondary was rat anti-mouse IgG (1:1000, Jackson) detected with 125I-Protein A and measured by PhosphorImager (Molecular Dynamics) for quantitation with ImageQuant. For fluorescent quantitative immunoblots imaged by Typhoon 9410 (GE Healthcare), secondary antibodies were Cy5-anti-rat (1:400, Jackson) for TG MO1, Cy5-anti-rabbit (1:400, Invitrogen) for pab022 and pab023, and Cy3-anti-mouse (1:400, Jackson) for the rest.
Difluoromethylornithine (DFMO, Merrell Dow) was administered as a 2% solution in drinking water. DFMO was given to rats 16–18h prior to labeling of axonal transport with 35S-methionine, and continued for 48h, 7d, or all 21d of a 21d ISI. Control rats did not receive DFMO. To lower endogenous polyamine levels and facilitate protein labeling with labeled PUT (14C or 3H), DFMO was given 16–18h prior to labeling. Following a 21–60d ISI, rats were sacrificed and optic nerve-optic tract removed for cold/Ca2+ fractionation. P2 fractions were resuspended in 8M urea and run on a 60 ml Toyopearl HW-55F (Supelco) column equilibrated in 6M guanidine-HCL in 100 mM MES, pH 6.8. Total α-tubulin was detected with DM1A.
Tubulin was purified from mouse brains through two cycles of polymerization as described (Castoldi and Popov, 2003). For polymerized MTs, tubulin (50 μM) was incubated in BR buffer 1980 (BRB80) supplemented with 2 mM GTP at 37°C for 30min. For Taxol-stabilization, taxol was added stepwise equimolar to tubulin in warm BRB80 buffer supplemented with 1mM DTT and 1mM GTP and incubated at 37°C for 30min. Polymerized MTs were pelleted over a glycerol/BRB80 cushion (http://mitchison.med.harvard.edu/protocols.html).
In vitro polyamination assays used N,N'-dimethylcasein or tubulin/MTs as a transglutaminase substrate; with monodansylcadaverine (MDC) or polyamine mix (SPM/SPD); and guinea pig transglutaminase (similar to TG2 in brain) in reaction buffer (PH 7.5) ±10mM Ca2+ at 37°C for 60min. Reactions were stopped by 20mM cystamine and analyzed by SDS-PAGE. Fluorescence due to MDC incorporation into tubulin was detected by Gel Doc 2000 (Bio-Rad).
For endogenous transglutaminase activity assays, transglutaminase extract (150μg) was incubated with 0.2mg/ml N,N-dimethylcasein, 4mM MDC, 5mM DTT in Tris-HCl based reaction buffer (PH 7.5) with 10mM Ca2+ at 37°C for 60min. Transglutaminase extracts were prepared from mouse brain by homogenization in cold 50mM Tris-HCl, 1mM EDTA, 0.25M sucrose, 0.4mM DTT and protease inhibitor cocktail (Sigma). Homogenates were centrifuged at 16,000g at 4°C for 20min. Pellets were discarded and supernatant centrifuged at 100,000g at 4°C for 1h to produce the endogenous transglutaminase extract containing transglutaminase, soluble tubulins and polyamines. A tubulin pellet was obtained by centrifugation after stopping polyamination by cystamine and subjected to cold/Ca2+ fractionation.
Polyaminated and native tubulins were analyzed in 2D PAGE after solubilization in 8 M urea, 4% CHAPS, 65 mM DTE, 0.5% ampholytes, and bromophenol blue. Protein solution was cleared and absorbed onto a 7 cm immobilized pH gradient pH 3–10 IPG strip (IPG strip, GE Bioscience) and run for 60 kVhr at room temperature. Before SDS-PAGE, IPG strips were equilibrated and transferred to the top of 4–12% Nu-Page gels held in position with 0.5% agarose.
In vitro polyaminated tubulins were prepared fresh with MDC, PUT, SPM or SPD and digested overnight with trypsin, unmodified tubulin was a control. Samples were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS-MS, Thermo LTQ-FT Ultra using Agilent Zorbax SB300-C18 0.075 mm ID ×150 mm Capillary Analytical Column with 0.3 mm ID × 5 mm Zorbax SB300 C18 trapping 250 nL/min gradient, 5% ACN to 65% ACN. Nano ESI Positive Ion mode (Resolution 50,000 @m/z 400, Scan 400–1800 m/z) was performed. Top 3 +2 and greater charged ions for MS/MS from each MS scan were selected. Putative modified peptides were picked based on mass shift in MS1, and specific modification sites evaluated in MS2 spectra and confirmed through target runs. Searches using MASCOT (version 2.2.04, Matrix Sciences, Inc) and MassMatrix search engines against the SwissProtKB using mouse, carbamidomethylation and methionine oxidation as variable modifications in addition to PUT, SPM, SPD as variable modifications of Q. Searches were performed using 10ppm peptide tolerance and 0.6 Da fragment tolerance with decoy database searches to keep false discovery rates were below 5%.
Synthesis and purification of TG2 selective irreversible inhibitor, IR072 (fig S5), was as described previously (Chabot et al., 2010) using Fmoc chemistry for solid phase peptide synthesis. Details of synthesis and characterization of IR072 is given in supplemental methods.
For cell culture, SH-SY5Y cells (from ATCC) were grown in DMEM F12 medium (Gibco) supplemented with 10% FBS, PenStrep and 2mM glutamax. To produce a neuronal phenotype, cells were plated on poly-lysine coated wells or coverslips to 60% confluency. Differentiation was induced by 1) 10μM Retinoic acid in 2% FBS for 2d, followed by 10μM retinoic acid in serum free medium for 3d. 2) 50ng/ml BDNF in serum free medium for 5 days (Szebenyi et al., 2003). To test the role of transglutaminase in differentiation and neurite outgrowth, cells were treated with the TG2 inhibitor IR072 at 10μM in media during differentiation. Neurite outgrowth assays were a modification of previously described methods (Szebenyi et al., 2003), see supplemental methods. Statistical significance was determined by a paired sample t-test.
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of the Brady and Johnson Laboratories for their general support, Dr. Hua Xu for help with MassMatrix data analysis, Dr. Robert Graham for generating the TG2 KO mouse, and Gerardo Morfini, Gustavo Pigino, Hanwu Liu, Bin Wang and Bao-shiang Lee for advice and encouragement. The work was funded in part by grants from NINDS (NS23868, NS23320) to STB.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Ambron RT, Kremzner LT. Post-translational modification of neuronal proteins: Evidence for transglutaminase activity in R2, the giant cholinergic neuron of Aplysia. Proc Nat Acad Sci USA. 1982;79:3442–3446. doi: 10.1073/pnas.79.11.3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CD, Johnson GV. Developmental regulation of tissue transglutaminase in the mouse forebrain. J Neurochem. 2004;91:1369–1379. doi: 10.1111/j.1471-4159.2004.02825.x. [DOI] [PubMed] [Google Scholar]
- Bailey CD, Tucholski J, Johnson GV. Transglutaminases in neurodegenerative disorders. Prog Exp Tumor Res. 2005;38:139–157. doi: 10.1159/000084238. [DOI] [PubMed] [Google Scholar]
- Basso M, Berlin J, Xia L, Sleiman SF, Ko B, Haskew-Layton R, Kim E, Antonyak MA, Cerione RA, Iismaa SE, et al. Transglutaminase inhibition protects against oxidative stress-induced neuronal death downstream of pathological ERK activation. J Neurosci. 2012;32:6561–6569. doi: 10.1523/JNEUROSCI.3353-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady ST. Axonal Dynamics and Regeneration. In: Gorio A, editor. Neuroregeneration. Raven Press; New York City, NY: 1993. pp. 7–36. [Google Scholar]
- Brady ST, Black MM. Axonal transport of microtubule proteins: Cytotypic variation of tubulin and MAPs in neurons. Ann NY Acad Sci. 1986;466:199–217. doi: 10.1111/j.1749-6632.1986.tb38395.x. [DOI] [PubMed] [Google Scholar]
- Brady ST, Lasek RJ. Axonal Transport: A Cell Biological Method for studying proteins which associate with the cytoskeleton. Meth Cell Biol. 1982;25:365–398. doi: 10.1016/s0091-679x(08)61434-x. [DOI] [PubMed] [Google Scholar]
- Brady ST, Tytell M, Lasek RJ. Axonal tubulin and axonal microtubules: Biochemical evidence for cold stability. J Cell Biol. 1984;99:1716–1724. doi: 10.1083/jcb.99.5.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulinski JC, Richards JE, Piperno G. Posttranslational modifications of alpha tubulin: detyrosination and acetylation differentiate populations of interphase microtubules in cultured cells. J Cell Biol. 1988;106:1213–1220. doi: 10.1083/jcb.106.4.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caceres A, Binder LI, Payne MR, Bender P, Rebhun L, Steward O. Differential subcellular localization of tubulin and microtubule associated protein MAP2 in brain tissue as revealed by immunocytochemistry with monoclonal hybridoma antibodies. J Neurosci. 1984;4:394–410. doi: 10.1523/JNEUROSCI.04-02-00394.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castoldi M, Popov AV. Purification of brain tubulin through two cycles of polymerization-depolymerization in a high-molarity buffer. Protein Expr Purif. 2003;32:83–88. doi: 10.1016/S1046-5928(03)00218-3. [DOI] [PubMed] [Google Scholar]
- Chabot N, Moreau S, Mulani A, Moreau P, Keillor JW. Fluorescent probes of tissue transglutaminase reveal its association with arterial stiffening. Chem Biol. 2010;17:1143–1150. doi: 10.1016/j.chembiol.2010.06.019. [DOI] [PubMed] [Google Scholar]
- Chapin SJ, Bulinski JC. Microtubule stabilization by assembly-promoting microtubule-associated proteins: a repeat performance. Cell Motil Cytoskeleton. 1992;23:236–243. doi: 10.1002/cm.970230403. [DOI] [PubMed] [Google Scholar]
- Cooper AJ, Jeitner TM, Blass JP. The role of transglutaminases in neurodegenerative diseases: overview. Neurochem Int. 2002;40:1–5. doi: 10.1016/s0197-0186(01)00055-9. [DOI] [PubMed] [Google Scholar]
- Dai Y, Dudek NL, Patel TB, Muma NA. Transglutaminase-catalyzed transamidation: a novel mechanism for Rac1 activation by 5-hydroxytryptamine2A receptor stimulation. J Pharmacol Exp Ther. 2008;326:153–162. doi: 10.1124/jpet.107.135046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danzin C, Jung MJ, Grove J, Bey P. Effect of a-difluoromethylornithine, an enzyme-activated irreversible inhibitor of ornithine decarboxylase, on polyamine levels in rat tissues. Life Sci. 1979;24:519–524. doi: 10.1016/0024-3205(79)90173-5. [DOI] [PubMed] [Google Scholar]
- De Vivo G, Di Lorenzo R, Ricotta M, Gentile V. Role of the transglutaminase enzymes in the nervous system and their possible involvement in neurodegenerative diseases. Curr Med Chem. 2009;16:4767–4773. doi: 10.2174/092986709789909594. [DOI] [PubMed] [Google Scholar]
- Desai A, Mitchison TJ. Microtubule polymerization dynamics. Ann Rev Cell Dev Biol. 1997;13:83–117. doi: 10.1146/annurev.cellbio.13.1.83. [DOI] [PubMed] [Google Scholar]
- Esposito C, Caputo I. Mammalian transglutaminases. Identification of substrates as a key to physiological function and physiopathological relevance. The FEBS journal. 2005;272:615–631. doi: 10.1111/j.1742-4658.2004.04476.x. [DOI] [PubMed] [Google Scholar]
- Facchiano F, Deloye F, Doussau F, Innamorati G, Ashton AC, Dolly JO, Beninati S, Facchiano A, Luini A, Poulain B, Benfenati F. Transglutaminase participates in the blockade of neurotransmitter release by tetanus toxin: evidence for a novel biological function. Amino Acids. 2010;39:257–269. doi: 10.1007/s00726-009-0436-3. [DOI] [PubMed] [Google Scholar]
- Falconer MM, Vaillant A, Reuhl KR, Laferriere M, Brown DL. The molecular basis of microtubule stability in neurons. Neurotoxicology. 1994;15:109–122. [PubMed] [Google Scholar]
- Folk JE, Park MH, Chung SI, Schrode J, Lester EP, Cooper HL. Polyamines as physiological substrates for transglutaminases. J Biol Chem. 1980;255:3695–3700. [PubMed] [Google Scholar]
- Gentile V, Thomazy V, Piacentini M, Fesus L, Davies PJA. Expression of tissue transglutaminase in Balb-C 3T3 fibroblasts: Effects on cellular morphology and adhesion. J Cell Biol. 1992;119:463–474. doi: 10.1083/jcb.119.2.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin M, Casadio R, Bergamini CM. Transglutaminases: nature's biological glues. The Biochemical journal. 2002;368:377–396. doi: 10.1042/BJ20021234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hand D, Perry MJ, Haynes LW. Cellular transglutaminases in neural development. Int J Dev Neurosci. 1993;11:709–720. doi: 10.1016/0736-5748(93)90060-q. [DOI] [PubMed] [Google Scholar]
- Janke C, Kneussel M. Tubulin post-translational modifications: encoding functions on the neuronal microtubule cytoskeleton. Trends Neurosci. 2010;33:362–372. doi: 10.1016/j.tins.2010.05.001. [DOI] [PubMed] [Google Scholar]
- Jeitner TM, Pinto JT, Krasnikov BF, Horswill M, Cooper AJ. Transglutaminases and neurodegeneration. J Neurochem. 2009;109(Suppl 1):160–166. doi: 10.1111/j.1471-4159.2009.05843.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick LL, Brady ST. Modulation of the axonal microtubule cytoskeleton by myelinating Schwann cells. J Neurosci. 1994;14:7440–7450. doi: 10.1523/JNEUROSCI.14-12-07440.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick LL, Witt AS, Payne HR, Shine HD, Brady ST. Changes in microtubule stability and density in myelin-deficient shiverer mouse CNS axons. J Neurosci. 2001;21:2288–2297. doi: 10.1523/JNEUROSCI.21-07-02288.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kueh HY, Mitchison TJ. Structural plasticity in actin and tubulin polymer dynamics. Science. 2009;325:960–963. doi: 10.1126/science.1168823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesort M, Tucholski J, Miller ML, Johnson GV. Tissue transglutaminase: a possible role in neurodegenerative diseases. Prog Neurobiol. 2000;61:439–463. doi: 10.1016/s0301-0082(99)00052-0. [DOI] [PubMed] [Google Scholar]
- Lorand L, Lockridge OM, Campbell LK, Myhrman R, Bruner-Lorand J. Transamidating enzymes. II. A continuous fluorescent method suited for automating measurements of factor XIII in plasma. Analytical biochemistry. 1971;44:221–231. doi: 10.1016/0003-2697(71)90363-0. [DOI] [PubMed] [Google Scholar]
- Luduena R. Multiple forms of tubulin: different gene products and covalent modifications. Int Rev Cytol. 1998;178:207–275. doi: 10.1016/s0074-7696(08)62138-5. [DOI] [PubMed] [Google Scholar]
- Maccioni RB, Seeds NW. Transglutaminase and neuronal differentiation. Mol Cell Biochem. 1986;69:161–168. doi: 10.1007/BF00224763. [DOI] [PubMed] [Google Scholar]
- Mahoney SA, Wilkinson M, Smith S, Haynes LW. Stabilization of neurites in cerebellar granule cells by transglutaminase activity: identification of midkine and galectin-3 as substrates. Neuroscience. 2000;101:141–155. doi: 10.1016/s0306-4522(00)00324-9. [DOI] [PubMed] [Google Scholar]
- Malaterre J, Strambi C, Aouane A, Strambi A, Rougon G, Cayre M. A novel role for polyamines in adult neurogenesis in rodent brain. Eur J Neurosci. 2004;20:317–330. doi: 10.1111/j.1460-9568.2004.03498.x. [DOI] [PubMed] [Google Scholar]
- Maruta H, Greer K, Rosenbaum JL. The acetylation of alpha-tubulin and its relationship to the assembly and disassembly of microtubules. J Cell Biol. 1986;103:571–579. doi: 10.1083/jcb.103.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta K, Fok JY, Mangala LS. Tissue transglutaminase: from biological glue to cell survival cues. Front Biosci. 2006;11:173–185. doi: 10.2741/1789. [DOI] [PubMed] [Google Scholar]
- Melino G, Farrace MG, Ceru MP, Piacentini M. Correlation between transglutaminase activity and polyamine levels in human neuroblastoma cells. Effect of retinoic acid and alpha-difluoromethylornithine. Exp Cell Res. 1988;179:429–445. doi: 10.1016/0014-4827(88)90281-9. [DOI] [PubMed] [Google Scholar]
- Miller CCJ, Anderton BH. Transglutaminase and the neuronal cytoskeleton in Alzheimer's disease. J Neurochem. 1986;46:1912–1922. doi: 10.1111/j.1471-4159.1986.tb08513.x. [DOI] [PubMed] [Google Scholar]
- Nanda N, Iismaa SE, Owens WA, Husain A, Mackay F, Graham RM. Targeted inactivation of Gh/tissue transglutaminase II. J Biol Chem. 2001;276:20673–20678. doi: 10.1074/jbc.M010846200. [DOI] [PubMed] [Google Scholar]
- Nogales E, Wolf SG, Downing KH. Structure of the αβ tubulin dimer by electron crystallography. Nature. 1998;391:199–203. doi: 10.1038/34465. [DOI] [PubMed] [Google Scholar]
- Pegg AE, McCann PP. ISI Atlas of Science. ISI; Philadelphia, PA: 1988. Polyamine metabolism and function in mammalian cells and protozoans; pp. 11–18. [Google Scholar]
- Pfister KK, Wagner MC, Stenoien D, Bloom GS, Brady ST. Monoclonal antibodies to kinesin heavy and light chains stain vesicle-like structures, but not microtubules, in cultured cells. J Cell Biol. 1989;108:1453–1463. doi: 10.1083/jcb.108.4.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohjanpelto P, Virtanen I, Holtta E. Polyamine starvation causes disappearance of actin filaments and microtubules in polyamine-auxotrophic CHO cells. Nature. 1981;293:475–477. doi: 10.1038/293475a0. [DOI] [PubMed] [Google Scholar]
- Ruan Q, Johnson GV. Transglutaminase 2 in neurodegenerative disorders. Front Biosci. 2007;12:891–904. doi: 10.2741/2111. [DOI] [PubMed] [Google Scholar]
- Sahenk Z, Brady ST. Axonal Tubulin and Axonal Microtubules: Morphologic evidence for stable regions on axonal microtubules. Cell Mot Cytoskel. 1987;8:155–164. doi: 10.1002/cm.970080207. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Abraham C, Ihara Y. Brain transglutaminase: in vitro crosslinking of human neurofilament proteins into insoluble polymers. Proc Natl Acad Sci U S A. 1982;79:6070–6074. doi: 10.1073/pnas.79.19.6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw GG, Pateman AJ. The regional distribution of the polyamines spermidine and spermine in brain. J Neurochem. 1973;20:1225–1230. doi: 10.1111/j.1471-4159.1973.tb00091.x. [DOI] [PubMed] [Google Scholar]
- Slotkin TA, Bartolome J. Role of ornithine decarboxylase and the polyamines in nervous system development: a review. Brain Res Bull. 1986;17:307–320. doi: 10.1016/0361-9230(86)90236-4. [DOI] [PubMed] [Google Scholar]
- Szebenyi G, Morfini GA, Babcock A, Gould M, Selkoe K, Stenoien DL, Young M, Faber PW, MacDonald ME, McPhaul MJ, Brady ST. Neuropathogenic Forms of Huntingtin and Androgen Receptor Inhibit Fast Axonal Transport. Neuron. 2003;40:41–52. doi: 10.1016/s0896-6273(03)00569-5. [DOI] [PubMed] [Google Scholar]
- Tucholski J, Lesort M, Johnson GV. Tissue transglutaminase is essential for neurite outgrowth in human neuroblastoma SH-SY5Y cells. Neuroscience. 2001;102:481–491. doi: 10.1016/s0306-4522(00)00482-6. [DOI] [PubMed] [Google Scholar]
- Webster DR, Wehland J, Weber K, Borisy GG. Detyrosination of alpha tubulin does not stabilize microtubules in vivo. J Cell Biol. 1990;111:113–122. doi: 10.1083/jcb.111.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Baas PW. Changes in microtubule number and length during axon differentiation. J Neurosci. 1994;14:2818–2829. doi: 10.1523/JNEUROSCI.14-05-02818.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.