

Abstract
The glycosylation of microbial natural products often dramatically influences the biological and/or pharmacological activities of the parental metabolite. Over the past decade, crystal structures of several enzymes involved in the biosynthesis and attachment of novel sugars found appended to natural products have emerged. In many cases, these studies have paved the way to a better understanding of the corresponding enzyme mechanism of action and have served as a starting point for engineering variant enzymes to facilitate to production of differentially-glycosylated natural products. This review specifically summarizes the structural studies of bacterial enzymes involved in biosynthesis of novel sugar nucleotides.
1. Introduction
Glycosylation is a prevalent and critical reaction in cells, functioning in energy metabolism, maintenance of cell integrity, molecular recognition, pathogen virulence, molecular defense, signaling information storage and chemical defense 1–4. Many bacteria use glycosylated small molecules as chemical weapons to gain a selective advantage or as signaling molecules for intra- and interspecies communication 5. Scheme 1 presents just small set of representative glycosylated bacterial secondary metabolites. Given carbohydrates are capable of accessing a wide range of unique chemical space 6–9, the sugars attached to these metabolites can clearly compliment and expand inherent natural product chemical diversity. Such sugar attachments also dramatically influence numerous properties of the metabolite to which they are attached including pharmacological and pharmacokinetic properties such as solubility, distribution, metabolic stability and/or tissue, cellular and/or molecular specificity 2, 10, 11. Thus, the differential glycosylation of natural products has emerged as a viable strategy to produce bioactive compounds with improved activity 8, 12–16.
Scheme 1.

Representative glycosylated natural products of microbial origin. Appended sugars are highlighted in blue.
In nature, these sugar structures are generated through enzymatic modification of the functional groups of a common sugar nucleotide precursor via complex multi-enzyme pathways 13, 14. The inherent promiscuity of these enzymes, in conjunction with advances in bioengineering methodology, biochemical and structural studies, have enabled efforts to modify the glycosylation patterns of natural products through metabolic pathway engineering 17–22 and enzymatic glycodiversification 14, 23–29. Such studies are greatly augmented via a fundamental understanding of the intricate substrate-enzyme interactions, the substrate scope and the detailed catalytic mechanism of targeted enzyme-catalyzed transformation. The goal of this review is to summarize the structural biology studies of enzymes involved in the biosynthesis of key precursors of glycosylated microbial natural products – namely, novel sugar nucleotides. For an overview of the structural biology of the class of enzymes that catalyze the culminating glycosylation step in such pathways (glycosyltransferases, GTs), the reader is referred to recent reviews 24, 25, 30–35.
2. Fundamentals of bacterial sugar nucleotide biosynthesis
Nucleotide 5′-diphosphosugars (NDP-sugars) represent the most common form of sugar donor employed by glycosyltransferases of microbial biosynthetic pathways 36. Despite the enormous variety of glycosides found among microbial secondary metabolites, the biosynthesis of their corresponding NDP-sugar precursors are divergent and share a number of conceptual and strategic similarities. These pathways are initiated via the formation of nucleoside monophosphate (NMP) or nucleoside diphosphate (NDP) derivatives through the action of sugar-1-phosphate nucleotidyltransferases - enzymes which couple a sugar-1-phosphate (Sugar1P) with nucleoside triphosphate (NTP), via expulsion of phosphate (Pi) or pyrophosphate (PPi), to provide the desired NDP/NMP-sugar (Scheme 2). The corresponding Sugar1P originates from primary metabolic intermediates such as fructose-6-phosphate and glucose-6-phosphate. Glucose-6-phosphate is a biosynthetic precursor of many bacterial thymidine diphosphate (dTDP)-, cytidine diphosphate (CDP)-, and uridine diphosphate (UDP)-sugars. Fructose-6-phosphate is converted to mannose-6-phosphate by phosphomannoisomerase (PMI) in the biosynthesis of guanosine diphosphate (GDP)-sugars and to glucosamine-6-phosphate by glucosamine-6-phosphate synthase (GlmS) in the formation of UDP-glucosamine-based analogs. In all cases, the sugar-6-phosphates are converted to the corresponding Sugar1P by distinct but related phosphohexose mutases prior to nucleotidyltransfer 37 (Scheme 2). While structural information for many enzymes involved in sugar phosphate biosynthesis are known 38–42, they fall outside the scope of this review.
Scheme 2.
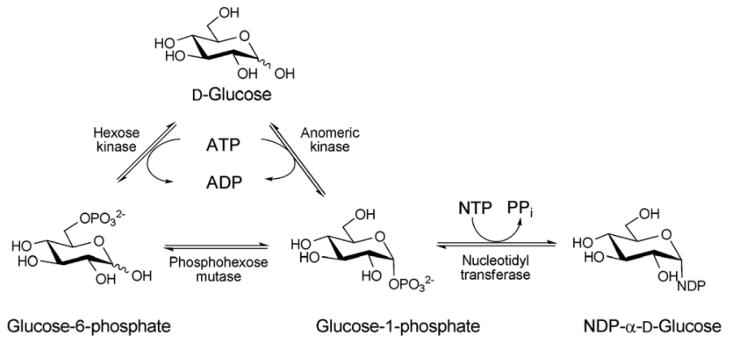
The biosynthesis of NDP-sugars from sugar-1-phosphate. The anomeric kinase reaction presented reflects reactions catalyzed by non-native engineered catalysts.
Among the NDP-sugars, dTDP-sugars are the predominant sugar donor form utilized in the biosynthesis of bacterial glycosylated natural products. Most dTDP-sugars from bacteria are 6-deoxyhexoses and many are also deoxygenated and/or functionalized at C2, C3, or C4 14. dTDP-sugars derive, almost exclusively, from glucose-1-phosphate (Glc1P), which is converted to a common intermediate (dTDP-4-keto-6-deoxy-D-glucose) in the biosynthesis of many diverse sugar nucleotides by the action of two ubiquitous enzymes – Glc1P thymidylyltransferase and dTDP-D-glucose 4,6-dehydratase. While genes encoding these common enzymes are sometimes found associated with secondary metabolite-encoding gene clusters, these core enzymes also participate in primary metabolism (particularly bacterial cell wall biosynthesis). The culminating C4-keto group of the common intermediate formed by this common two enzyme conversion is key to facilitating subsequent enzyme-catalyzed functionalization at C2, C3 and C4 (Scheme 3). Typical modifications proceeding from dTDP-4-keto-6-deoxy-D-glucose in bacterial secondary metabolism (and, in some cases also primary metabolism) include: 2-, 3- or 4-deoxygenation; 2-, 3-, 4- or 5-epimerization; 3- or 4-amination; and C-, N- or O-methylation 13, 14. In addition, amine oxidation, oxidation/decarboxylation, acetyltransfer, carbomoyltransfer and sulfur installation are among a range of downstream modifications known to further expand structural diversity 13, 43–47.
Scheme 3.
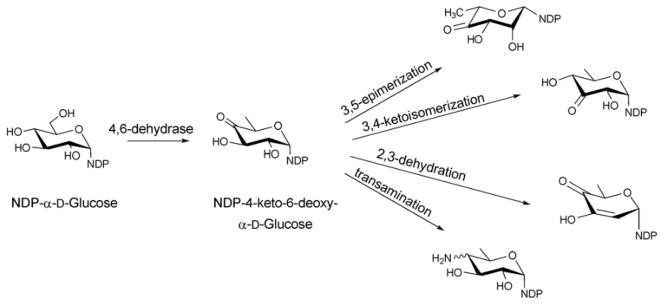
Reactions originating from the common intermediate, NDP-4-keto-6-deoxy-α-D-glucose, which is the product of the 4,6-dehydratase reaction of NDP-D-glucose.
Over the last two decades the structures of several sugar biosynthetic enzyme family members have emerged including examples for which structures of enzymes for an entire sugar nucleotide pathway have been elucidated (e.g., dTDP-L-rhamnose) 48–50. These structural studies have led to new mechanistic hypotheses 46, 51–53, offered a template to extend mechanistic studies via site-directed-mutagenesis 48, 53–58, and served as a foundation for structure-based engineering of new catalysts 35, 59–61. The structural studies highlighted within this review have been organized by reaction type with a primary focus upon the core enzymes involved in manipulating the sugar scaffold structure (specifically, sugar nucleotide formation and core sugar nucleotide modification reactions - deoxygenation, epimerization, oxidation/reduction and transamination). A section highlighting structural studies for enzymes that offer additional sugar nucleotide modification (methylation, acylation, N-oxidation, decarboxylation, formyl, enolpyruvyl transfer and pyranose/furanose intercoversion) has also been included. Table 1 lists the structures discussed as organized in the context of this review for quick reference. For additional details regarding the chemistry and mechanism of sugar nucleotide-modifying enzymes, the reader is referred to the many excellent recent reviews 13, 46–50, 55, 62–64.
3. Sugar nucleotide formation
3.1. Nucleotidyltransferases
Glycoside biosynthesis typically begins with the conjugation of α-D-glucose-1-phosphate (Glc1P) or α-D-mannose-1-phosphate (Man1P) to NMP (from NTP). This reaction, which, in most cases is dependent upon divalent metal, is catalyzed by a nucleotidyltransferase (also sometimes referred to as a sugar nucleotide pyrophosphorylase, EC 2.7.7.-) and proceeds with the concomitant loss of pyrophosphate. The three-dimensional structures of many examples of this class of enzyme are known, including thymidylyltransferases [dTDP-glucose pyrophosphorylases, RmlA/RfbA (EC 2.7.7.24)] from Salmonella enterica 59, Bacillus anthracis, Escherichia coli 65, Pseudomonas aeruginosa 54; uridylyltransferases [UDP-glucose pyrophosphorylase, UGPase, (EC 2.7.7.9)] from Escherichia coli 66, 67, 68, Helicobacter pylori 69,70; UDP-N-acetylglucosamine (UDP-GlcNAc) pyrophosphorylase GlmU (EC 2.7.7.23) from Escherichia coli 71, Streptococcus pneumoniae 72, Mycobacterium tuberculosis 73, 74, Haemophilus influenza 75; cytidylyltransferase [CDP-glucose pyrophosphorylase, CGPase, (EC 2.7.7.33)] from Salmonella typhi 76, 77; guanylyltransferase [GDP-mannose pyrophosphorylase, GMPase, (EC 2.7.7.22)] from Thermotoga maritima 78; adenylyltransferase [ADP-glucose pyrophosphorylase, AGPase, (EC 2.7.7.27)] from Agrobacterium tumefaciens 79.
Despite very little sequence homology, nucleotidyltransferases share a similar domain organization and common structural features. Though they do not seem to have any obligate multimeric preference (Figure 1A & 1B), most adopt a tetrameric structure 59, 66, 67, 69, 74, 78 wherein multimerization is generally mediated through C-terminal domain interactions (Figure 1A & 1B). Each monomer is composed of a conserved N-terminal catalytic domain and a C-terminal auxiliary domain (colored purple in Figure 2). The N-terminal catalytic domain is composed of an α/β/α sandwich reminiscent of the dinucleotide-binding Rossmann fold (Figure 1C). It consists of a twisted mixed β-sheet made up of seven β-strands arranged in the order 3214657 (Figure 1C). The central β-sheet is flanked by eight α-helices tightly packed against the β-sheet. Often there are additional secondary structure insertions, sometimes known as subdomains, between strands β5 and β6 and strands β2 and β3, depending upon the family (colored blue in Figure 2). For example, in the thymidylyltransferase RmlA 59, the insertion between β5 and β6 is a mixed two-stranded β-sheet flanked by α-helix (β5a and β5b), while cytidylyltransferase CGPase 76 has an extra mixed two stranded β-sheet (β2a and β2b) inserted between β2 and β3 (Figure 1F). The C-terminal auxiliary domains in this family sometimes form a left-handed β-helix motif (LβH), as in the CGPase, the GMPase, in GlmU (colored purple in Figure 2D, described in detail in section 5.2.1.) and in AGPases 72–74, 80, 81. adenylyltransferase AGPase (colored purple in Figure 2C) 79, 82, and guanylyltransferase GMPase (colored purple in Figure 2E) 78. The C-terminal auxiliary domains of RmlA, UGPase and CGPase (colored purple in Figures 2A 2B & 2F) are distinct and contain two or three α-helices as a main feature. In some enzymes this C-terminal domain presents a second enzymatic activity as exemplified by the bifunctional GMPase and GlmU, which exhibit phosphomannose isomerase (PMI) 78 and acetyltransferase activity (see section 5.2.1.) 74, respectively. The C-terminal domain can also regulate the nucleotidyltransferase activity by mediating enzyme oligomerization 59 and/or by binding allosteric regulators 82.
Figure 1.

Representative global structural features of nucleotidyltransferases A. Tetramer of dTDP-glucose pyrophosphorylase (RmlA) from Salmonella enterica where each monomer is represented by a distinct color (PDB 1IIN). B. Dimer of GDP-mannose pyrophosphorylase (GMPase) from Thermotoga maritime where each monomer is represented by a distinct color. (PDB 2X5Z). C. Monomer of CDP-glucose pyrophosphorylase (CGPase) from Salmonella typhi (PDB 1TZF) with secondary structures of core β-sheet and variable regions numbered. The core N-terminal Rossmann fold is colored in green and orange (β-strands), the variable regions are colored blue and red, CDP-Glucose is colored grey, the C-terminal domain is purple and the letters ‘C’ and ‘N’ represent the C- and N-terminus of the enzyme, respectively.
Figure 2.
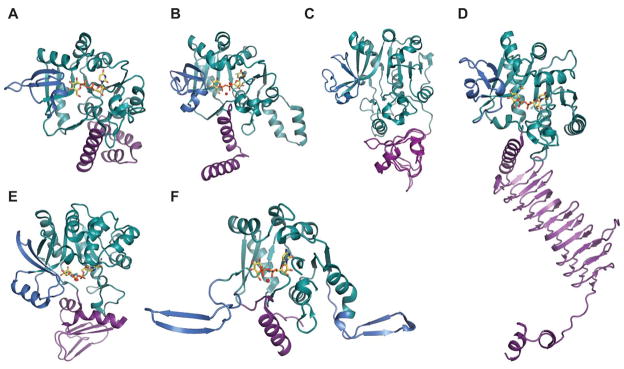
Representative monomeric features of nucleotidyltransferases. A. dTDP-glucose pyrophosphorylase RmlA from Salmonella enterica (PDB 1IIN). B. UDP-glucose pyrophosphorylase UGPase from Escherichia coli (PDB 2E3D). C. ADP-glucose pyrophosphorylase AGPase from Agrobacterium tumefaciens (PDB 3BRK). D. N-acetylglucosamine diphosphorylase uridylyltransferase GlmU from Mycobacterium tuberculosis. E. GDP-mannose pyrophosphorylase GMPase from Thermotoga maritime (PDB 2X5Z). F. CDP-glucose pyrophosphorylase CGPase from Salmonella typhi (PDB 1TZF). Rossmann fold is represented in green, variable regions colored blue, C-terminal domain in purple, ligands in yellow and Mg2+ as red sphere.
The general nucleotidyltransferase active site pocket is located in a deep cleft formed by the central β-sheet and flexible loops L1 and L3. Within this active site, the nucleotide base of the substrate is sandwiched between loop L1 after the strand β1, which contains the canonical signature motif G11-G12-X-G14-X-R16 (numbering in S.enterica RmlA), and loop L3 following β3. The O2, O3, and O4 hydroxyls of the sugar moiety interact with conserved residues within bacterial nucleotidyltransferases that eminate from the strand β6 and flexible loops L5, L5b, L6 (Figure 1C). The phosphates of the nucleotide are hydrogen-bonded by arginine and lysine side-chains (R16, K163 and R195 in S.enterica RmlA), and the ribose and pyranose are also anchored by several hydrogen-bonding interactions as shown in Figures 3A and 3B. Comparative analysis of the structures of apo, NTP and NDP-sugar complexes reveals substrate and product binding are associated with significant changes in both the conformation of loop regions lining the active site and in the overall orientation of the core domain relative to the subdomain involving a β-sheet (β5a and β5b) insertion between β5 and β6 (colored blue in Figure 2E) 78. For example, in the enzyme-product complex of GMPase, the phosphate backbone interacts with the side chains of two conserved aspartates (D109 and D260 in GMPase) via Mg2+ (Figure 3B) 78 whereas in the enzyme-NTP complex, the phosphate groups are pointed away from the sugar-binding site and interact with the main chain nitrogen atoms of the signature motif, GGXGXR. Upon Man1P binding, the subdomain containing β-sheet 5a and 5b (colored blue in Figure 2E) rotates by 10° toward Man1P, which results in the global closure of the sugar-binding site and restricts active site accessibility 78.
Figure 3.
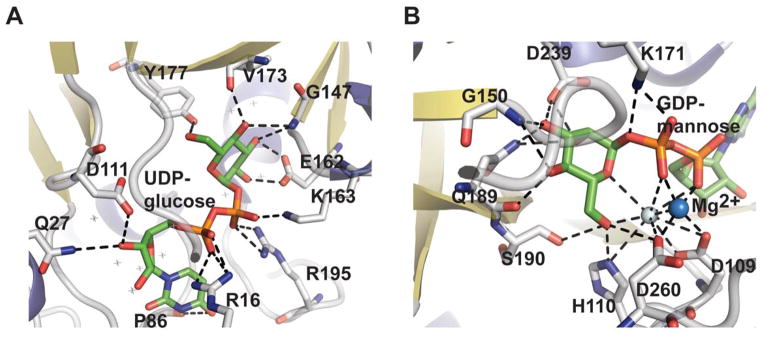
Nucleotidyltransferase active-site interactions. A. dTDP-glucose pyrophosphorylase RmlA from Salmonella enterica bound to UDP-glucose (PDB 1IIN). B. GDP-mannose pyrophosphorylase GMPase from Thermotoga maritime bound to GDP-mannose (PDB 2X5Z). NDP-sugar is colored green, Mg2+ is represented by a blue sphere, and water by a grey sphere.
Catalytic mechanisms for many nucleotidyltransferases have been thoroughly studied 54, 59, 65, 76–78. Typically, nucleotidyltransferases utilize an ordered sequential mechanism with NTP binding first. Isotopic-labeling is consistent with a SN2-type mechanism leading to stereochemical inversion of the NTP α-phosphate upon nucleophilic attack by the Sugar1P (Scheme 4) 83 ,59, 65, 76, 77. This general mechanism is also supported by RmlA structural studies in complex with reactants and products which revealed the Sugar1P nucleophile and departing pyrophosphate of the NTP to bind in a manner consistent with a SN2-type mechanism 59, 76. In this complex, the essential divalent metal is believed to play a role in transition-state stabilization/orientation and activation of the pyrophosphate leaving group 84, 65, 85.
Scheme 4.
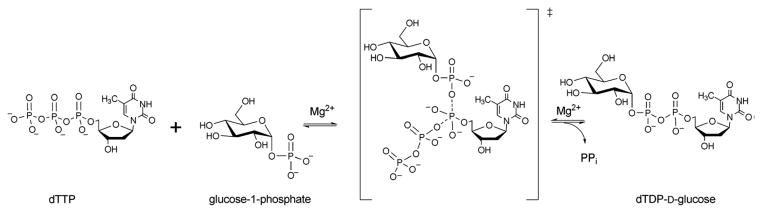
Reaction mechanism of glucose-1-phosphate thymidylytransferase.
4. Core sugar scaffold modification
4.1. Sugar dehydratases
Of the dehydratases (EC 4.2.1.-) involved in NDP-sugar core modification, the NDP-hexose-4,6-dehydratases are central to the biosynthesis of most deoxysugars. These enzymes catalyze a stepwise NAD(P)+-dependent C4-oxidation-C5/C6-elimination (dehydration)-C5/C6-ene reduction reaction sequence which enables the facile production of a NDP-4-keto-6-deoxysugar (most commonly of glucose origin). To date, NDP-hexose-4,6-dehydratases have been found to utilize the short-chain dehydrogenase/reductase (SDR) structural fold and include: dTDP-glucose-4,6-dehydratases (EC 4.2.1.46) RmlB from Salmonella enterica and Streptococcus suis and DesIV from Streptomyces venezuelae 55, 86, 87; CDP-glucose-4,6-dehydratase (EC 4.2.1.45) from Yersinia pseudotuberculosis and Salmonella typhi 88, 89; GDP-mannose-4,6-dehydratase (EC 4.2.1.47) from Escherichia coli, Pseudomonas aeruginosa and Aquifex aeolicus VF5 90–92; and UDP-GlcNAc-4,6 dehydratase (EC 4.2.1.115) FlaA1 from Helicobacter pylori 93. Dehydrases which subsequently act upon NDP-4-keto-6-deoxyhexoses to facilitate pyridoxamine 5′-phosphate (PMP)-mediated dehydration/deoxygenation adjacent to the C4 carbonyl adopt an aspartate aminotransferase (AAT) fold and include: the GDP-4-keto-6-deoxy-D-mannose-3-dehydratase ColD from Escherichia coli 94; and the CDP-6-deoxy-L-threo-D-glycero-4-hexulose 3-dehydrase E1 from Yersinia pseudotuberculosis 53.
4.1.1. SDR fold dehydratases
Dehydratases of SDR family do not exhibit an obligate multimeric preference and have been found as dimeric 55, 86, 87, 90, 94, 95, tetrameric 88, 91, and hexameric enzymes 93. The monomer-monomer association in these multimers occurs principally through hydrophobic interactions via a four-helix bundle (Figures 4A & 4B) and each monomer exhibits an α/β structure that can be divided into two domains. Of these, the larger N-terminal NAD(P)+-binding domain consists of a seven-stranded β sheet in the order 3214567 flanked by α helices, yielding a modified Rossmann fold (Figures 4C & 4D). The smaller C-terminal domain, responsible for binding the sugar substrate, contains four β strands and six α helices (Figures 4C & 4D). The interdomain cavity accommodates the active-site includes a characteristic GXGXXG motif and a conserved YXXXK which, in conjunction with a conserved S/T forms a catalytic triad. Based upon a comparison of ligand bound and apo enzyme (RmlB), substrate binding does not induce substantial conformation change 55.
Figure 4.

Representative global folds and monomeric structures of retaining and inverting dehydratases in the SDR structural family. Dimer (A) and monomer (C) of the retaining dehydratase RmlB bound to dTDP-Glucose and NAD from Salmonella enterica (PDB 1KEU). Dimer (B) and monomer (D) of the inverting dehydratase FlaA1 from Helicobacter pylori bound to UDP-GlcNAc and NADPH (PDB 2GN6). The core Rossmann fold is colored in blue/green, C-terminal domain highlighted in red, and purple signifies the helices involved in dimerization.
The active site of a typical SDR dehydratase is located within the cavity formed by the junction of the N- and C-terminus within each monomer. Cofactor binding is facilitated through a range of hydrophobic and hydrogen bonding interactions (Figure 5A) to present the nicotinamide ring in the syn conformation, consistent with pro-S hydride transfer from C4 of the dTDP-D-glucose to C4′ of the NAD(P)+. This cofactor-binding region contains the characteristic α/β/α motif wherein a conserved motif G8-X-G10-X-X-G13 (numbering in RmlB from S.enterica) contributes to interaction with nucleotide diphosphate group 55. In addition, one of the hydroxyl oxygens of nicotinamide ribosyl hydroxyls often hydrogen bond to lysine (K171 in RmlB from S.enterica) from the conserved motif, YXXXK. A conserved S/T (T133 in RmlB from S.enterica) (Figure 5B) has also been implicated in transition-state stabilization as a proton shuttle or possibly via involvement in a low barrier H-bond with the substrate 55. Sugar nucleotide binding is also mediated via H-bonding of phosphates of the nucleotide and arginine side-chains (Figures 5C & 5D) as well as conserved H-bonding interactions with the sugar moiety as shown in the Figures 5B 5C & 5D.
Figure 5.

SDR structural family dehydratase active-site interactions. The cofactor binding sites of Helicobacter pylori FlaA1 bound to UDP-GlcNAc and NADP (PDB 2GN6) and Salmonella enterica RmlB bound to NAD+ and dTDP-glucose (PDB 1KEU) are highlighted in panels (A) and (B), respectively. The corresponding sugar binding sites of FlaA1 and RmlB are highlighted in panels (C) and (D), respectively.
The mechanism of NDP-4-keto-6-deoxyhexose biosynthesis proceeds through three fundamental steps – C4 oxidation, C5/C6 dehydration and C5/C6 ene reduction (Scheme 5) 57, 96–99. C4 oxidation involves sugar C4-OH deprotonation, mediated by a conserved tyrosine (Y167 in S.enterica RmlB; Y141 in FlaA1) (Figures 5B & 5D), followed by C4 hydride transfer to nicotinamide 57, 96. During this step, a conserved lysine (K171 in S.enterica RmlB) and serine/threonine (T133 in S.enterica RmlB; T131 in FlaA1) (Figures 5B & 5D), stabilize the tyrosinate general base (Figures 5B & 5D). Formation of the C4 carbonyl enables a subsequent C5/C6 β-elimination and this dehydration reaction is mediated by a conserved general base (E135 in S.enterica RmlB; is K133 in FlaA1) and general acid (D134 in S.enterica RmlB; D132 in FlaA1) pair to afford the dTDP-4-keto-glucose-5,6-ene intermediate (Scheme 5) 97. The induced chair conformation reorganization, stabilized by hydrogen bonds between the C4 carbonyl oxygen and the side chains of Y167 and T133 (in S.enterica RmlB) (Scheme 5), enhance the electrophilicity and orientation of C6 for hydride transfer from NADH (with subsequent reprotonation at C5 from solvent). Thus, the net reaction affords an intramolecular transfer of hydride from C4 to C6 (ultimately, with inversion of absolute stereochemistry at C6) 55. While most NDP-4,6-dehydratases lead to retention of stereochemistry at C5, the UDP-GlcNAc-4,6-dehydratase FlaA1 (involved in the biosynthesis of pseudaminic acid) leads to inversion at C5 en route from UDP-GlcNAc to UDP-2-acetamido-2,6-dideoxy-β-L-arabino-hex-4-ulose. (Scheme 6). The structural equivalent of RmlB E135 in FlaA1 is K133 and has been proposed to facilitate water-mediated protonation on the opposite face molecule as an alternative to Y141 in RmlB (Scheme 6) 93.
Scheme 5.
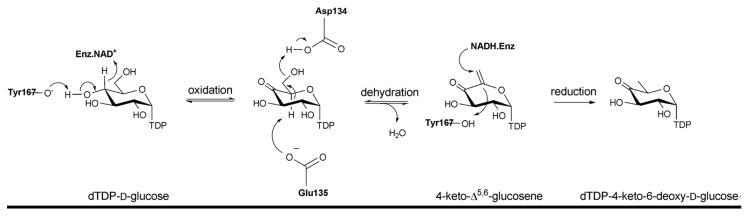
Reaction mechanism of dTDP-glucose-4,6-dehydratase (RmlB).
Scheme 6.
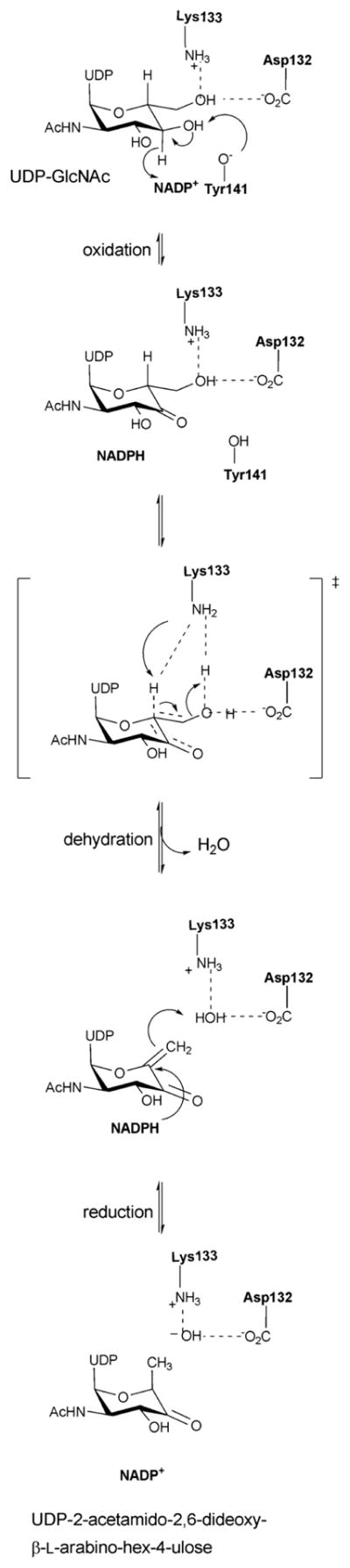
Reaction mechanism of an inverting 4,6-dehydratase (FlaA1).
4.1.2. AAT fold dehydratases
Dehydrases which subsequently act upon NDP-4-keto-6-deoxyhexoses to facilitate PMP-mediated dehydration or deoxygenation adjacent to the C4 carbonyl include ColD and E1 – both dimeric enzymes involved in the biosynthesis of 3,6-dideoxy sugars. While these catalysts adopt an aspartate aminotransferase (AAT) fold and, like typical aminotransferases, use PMP as a cofactor, the conserved transaminase lysine which typically forms an imine with PMP is replaced in these enzymes with a conserved histidine [H188 in ColD and H220 in E1] 53, 94. Although, they have same structural fold (Figure 6A), a key feature that distinguishes E1 from ColD is the presence of a [2Fe-2S] cluster critical to the final reductive deoxygenation step, the ligands for which are cysteine and a histidine (C251-X1-C253-X7-C261-X17-H278) 100. While a single mutation, H220K could convert E1 into a PLP/L-glutamate-dependent C4 aminotransferase, this E1 variant-catalyzed reaction was not catalytic due to an inability to regenerate PLP 53. Further mutagenesis revealed that four active site residues of E1 (D194H, Y217H, H220K, and F345H) and two active site residues of ColD (S187N, H188K) have successfully transformed each into a PLP/L-glutamate-dependent C4 aminotransferase 46, 53, 58.
Figure 6.

AATstructural family dehydrase global fold and active-site interactions. A. Overlay of ColD from Escherichia coli bound to hydrated PLP (red, PDB 2GMS) and E1 (H220K) from Yersinia pseudotuberculosis bound to PLP (blue, PDB 3BB8). The boxed flexible loop indicates the region that binds to the E1 [2Fe-2S] cluster. B. The active sites of ColD and E1 are represented in panels B and C, respectively. Letters ‘C’ and ‘N’ represent C and N-terminus, respectively.
Both ColD and E1 exhibit extensive dimer interface with each subunit folded into a large N-terminal cofactor binding domain and a small C-terminal domain. The N-terminal domain has a mixed β-sheet with seven or eight strands (strand order 17654238) surrounded by eight α-helices (Figure 6A). The smaller C-terminal domain consists of an antiparallel, three-stranded β-sheet surrounded by helices. Similar to sugar aminotransferases (SATs, section 4.4), both monomers contribute residues to the active site. The PLP-binding pocket is formed primarily by one subunit and a loop (consisting of residues 240–253 in ColD and 282–293 in E1) from the second subunit (residues colored in cyan in Figures 6B & 6C). While the aromatic stacking interactions (W88 in ColD and F120 in E1) help to orient the PLP, a network of H-bonding interactions with conserved serine, histidine and glutamate/aspartate from one subunit and an asparagine (S183/S215, H188/H220, E162/D194, and N248/N288 in ColD/E1) from a second subunit further anchor the cofactor in the active site (Figures 6B & 6C). Mutagenesis studies have confirmed that the [2Fe-2S] cluster in E1 is coordinated by three cysteine residues (C251, C253 and C261) 100 from a flexible loop (comprising residues 253–268; boxed in Figure 6A). The lack of electron density in this loop has been attributed to the putative “open” and “closed” conformational changes of the substrate binding pocket where the loop flexibility of E1 allows facilitates the formation of a reasonably tight binary complex with the reductase E3 to support the transfer of electrons from the reduced iron–sulfur center of E3 (via the iron–sulfur cluster of E1) to reduce the E1-bound substrate 46, 53, 101.
A catalytic mechanism for ColD has been proposed (Scheme 7A) in which a Schiff base, formed between the sugar C4 carbonyl and PMP, facilitates the H188 general acid/base-mediated loss of water to afford the Δ3,4-mannoseen intermediate. Intriguingly, upon hydrolysis of the cofactor in a manner reminiscent of standard transamination, the corresponding eneamine product is hydrolyzed by ColD to liberate ammonia and the corresponding NDP-3,6-dideoxy-4-keto-sugar 95. E1 shares many common mechanistic features with ColD (Scheme 7B) including the role of PMP and a conserved histidine (H220) in the early stage dehydration 46. However, distinct from ColD, E1 associates with a flavoprotein reductase (E3, a member of the ferredoxin-NADP+ reductase family) which provides reducing equivalents from NADH in a one electron fashion to reduce the corresponding Δ3,4-glucoseen intermediate in E1 (mediated via the E1 [2Fe-2S]) 46, 102–106. Upon reduction, the hydrolysis of the Schiff base gives rise to NDP-3,6-dideoxy-4-keto-sugar product and PMP to complete the cycle 46 (Scheme 7B).
Scheme 7.

A. Reaction mechanism of GDP-4,6-mannose-dehydratase ColD. B. Reaction mechanism of CDP-6-deoxy-L-threo-D-glycero-4-hexulose 3-dehydrase E1.
4.2. Sugar epimerases/isomerases
Sugar epimerases (EC 5.1.3.-) catalyze the stereochemical inversion of the configuration of an asymmetric carbon atom in a carbohydrate 48, 62, 63. Depending on the type of chemistry, NDP-sugar epimerases can be classified into three main groups 62: epimerases that epimerize at positions alpha to a carbonyl (typically the common C4 carbonyl) as exemplified by dTDP-6-deoxy-D-xylo-4-hexulose-3,5-epimerase (RmlC, EC 5.1.3.13) and GDP-4-keto-6-deoxy-mannose-epimerase/reductase (GMER, EC 1.1.1.271); those which epimerize via an oxidation/reduction at a single carbon as exemplified by CDP-tyvelose-2-epimerase (EC 5.1.3.10), UDP-galactose-4-epimerase (GALE, EC 5.1.3.2), and ADP-L-glycero-D-mannoheptose-6-epimerase (AGME, EC 5.1.3.20); and enzymes that epimerize at position adjacent to the sugar anomeric center via nucleotide elimination and re-addition as demonstrated by UDP-N-acetyl-D-glucosamine-2-epimerase (UDP-GlcNAc-2-epimerase). The structures for a range of sugar epimerases have been reported in the recent literature and cumulatively, this chemistry is accomplished by three main structural folds – a cupin fold 107, an extended SDR fold and a GT-B fold (most commonly associated with sugar-nucleotide-dependent glycosyltransferases 35.
4.2.1. Cupin superfamily
Epimerases/isomerases belonging to this structural fold invert one or two stereocenters alpha to a sugar carbonyl via simple keto-enol tautomerization 48. Structures for a number of members of this family have been reported including: dTDP-4-dehydrorhamnose 3,5-epimerase, (EC 5.1.3.13) RmlC (involved in dTDP-L-rhamnose pathway of Salmonella enterica, Streptococcus suis, Mycobacterium tuberculosis, and Pseudomonas aeruginosa) 62, 64, 108–112; dTDP-3-amino-4-keto-2,3,6-trideoxy-3-C-methyl-glucose-5-epimerase EvaD (involved in dTDP-epivancosamine of Amycolatopsis orientalis) 113, 114; dTDP-6-deoxy-D-xylo-4-hexulose 3-epimerase NovW (involved in the Streptomyces spheroids dTDP-noviose pathway) 115; and dTDP-4-keto-6-deoxy-D-glucose-3,4-ketoisomerase FdtA (involved in the Aneurinibacillus thermoaerophilus dTDP-3-acetamido-3,6-dideoxy-α-D-galactose pathway) 116. Of these FdtA, is somewhat unique as it utilizes the keto-enol tautomerization reaction to enable inversion of the initiating C4 carbonyl (in contrast to all other members which utilize a similar strategy to enable inversion adjacent to the initiating carbonyl).
‘Cupa’ in Latin means a small barrel and enzymes of this family are homodimers of ‘jelly-roll’ topology consistingmprised of a conserved beta-barrel fold (Figure 7A). The dimer interface is created by the antiparallel interaction of the β3 strand of one monomer with the β5 strand of the other monomer (Figure 7A). Each β barrel comprises 13 β strands and can be divided into three separate regions: the N-terminal, core, and C-terminal regions. The N terminus consists of an antiparallel β-sheet (β1–β3) and a two-turn α-helix. The core of the monomer consists of two twisted antiparallel β-sheets (β5–β13), which form a flattened barrel. One end of the barrel is open, and the entrance is lined with polar residues. The other side is obscured by β-strands that fold over the entrance. A number of hydrophobic residues in this part of the polypeptide chain seal the entrance to the barrel (Figures 9A & 9B). The C-terminal region consists of two or three small helices, depending on the enzyme (Figure 7). The active site conserved residues tyrosine, lysine and a histidine-glutamate/aspartate dyad (Y134, K74, H65-D171 in Pseudomonas aeruginosa RmlC) (Figure 9A) are critical to binding and catalysis.
Figure 7.

Global fold of epimerases belonging to cupin structural family. A. dTDP-4-dehydrorhamnose 3,5-epimerase RmlC from Pseudomonas aeruginosa bound to dTDP-4-keto-rhamnose (PDB 2IXK ). N-terminal domain colored purple, C-terminal domain colored blue, core-domain in yellow and green B. Amycolatopsis orientalis dTDP-3-amino-4-keto-2,3,6-trideoxy-3-C-methyl-glucose-5-epimerase EvaD from (PDB 1OFN). C. Aneurinibacillus thermoaerophilus dTDP-4-keto-6-deoxy-D-glucose-3,4-ketoisomerase FdtA bound to dTDP (PDB 2PA7). In panels B and C, red/green represents one monomer while blue/yellow represents another.
Figure 9.

Epimerase active site architecture. A. Interaction of Pseudomonas aeruginosa dTDP-4-dehydrorhamnose 3,5-epimerase RmlC bound to dTDP-4-keto-rhamnose (PDB 2IXK ). B. Nucleotide base interactions of the RmlC ligand-bound complex. C. Interaction of sugar within Escherichia coli UDP-galactose-4-epimerase bound to UDP-Glc and NAD (PDB 1XEL). D. Escherichia coli UDP-N-acetylglucosamine 2-epimerase bound to UDP (PDB 1F6D).
The general mechanism of cupin-based epimerases is initiated by proton abstraction from one face of the sugar ring by the catalytic dyad (H65 and D171 in Pseudomonas aeruginosa RmlC) (Figures 9A & 9B) to provide an enolate intermediate which is stabilized by K74. Reprotonation on the opposite face is facilitated by tyrosine (Y134 in Pseudomonas aeruginosa RmlC) or solvent (Scheme 8). RmlC, EvaD and NovW catalyze 3,5-, 5- and 3-epimerase activity, respectively 112, 113, 117. Based upon site directed mutagenesis of EvaD (M131F) the orientation the conserved Y133 was postulated to dictate regiospecificity 113. However, the subsequent structure of NovW (which catalyzes epimerization at C3) revealed an identical orientation for this conserved tyrosine 113, 117. The dual C3/C5 epimerization reaction catalyzed by RmlC proceeds in a step-wise fashion beginning with C5 inversion (Scheme 8) 112.
Scheme 8.
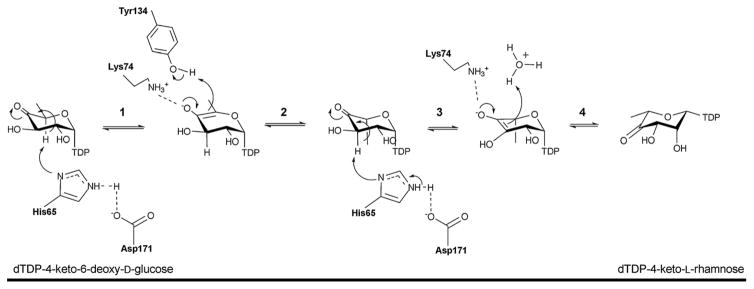
Reaction mechanism of the 3,5-epimerase RmlC. Step 1: C5 proton abstraction by the active site His-Asp diad; step 2: proton addition assisted by active site tyrosine and subsequent inversion; step 3: C3 proton abstraction by the active site His-Asp diad; step 4: proton addition and inversion.
4.2.2. SDR enzymes
Given the similar chemistries, it is perhaps not surprising that both dehydratases (discussed in section 4.1.1.) and epimerases can both adopt a SDR structural fold (Figure 8C). Epimerases within this structural family catalyze a NAD(P)+-dependent oxidation/reduction sequence that facilitates hydride removal from one face of the sugar (to afford a carbonyl) with re-introduction of the hydride from the opposing face of the sugar to ultimately afford stereochemical inversion at the target position (Scheme 9). Examples of this class include: GDP-4-keto-6-deoxy-D-mannose-3,5-epimerase/4-reductase (GMER) also known as GDP-L-fucose synthetase from Escherichia coli 118–120, UDP-galactose epimerase (GALE) from Escherichia coli 56, 121–124, ADP-L-glycero-D-mannoheptose-6-epimerase (AGME) from Escherichia coli and Helicobacter pylori 125–128 and CDP-tyvelose-2-epimerase from Salmonella typhi 129. As previously described for SDR folds, they do not have an obligate preference for the multimeric state (Figures 8A-C) and each monomer exhibits an α/β structure that can be divided into two domains. The larger N-terminal domain binds the nucleotide cofactor NAD(P)+ and consists of a seven-stranded β sheet in the order 3214567 flanked by α helices, yielding a modified Rossmann fold (Figure 8C). SDR epimerases typically display a T6-G7-X-X-G10-X-X-G13 (numbering in E. coli GALE) cofactor binding motif and a Y149-X-X-X-K153 (numbering in E. coli GALE) active site motif, with the tyrosine residue of this series serving as a critical general base (Figure 9C and Scheme 11). In addition, a nearby active site serine/threonine and/or an asparagine (S124 in E. coli GALE) is often important (Figure 9C). SDR epimerase substrate binding occurs in the C-terminal region, which is the main structural determinant of substrate specificity and, although substrate specificity is not well understood, the volume of the active site is a main contributor 130.
Figure 8.

Global fold of epimerases belonging to SDR structural family. A. Tetramer of CDP-tyvelose-2-epimerase from Salmonella enterica subsp. enterica serovar Typhi bound to CDP and NAD (PDB 1ORR). B. ADP-L-glycero-D-mannoheptose-6-epimerase from Escherichia coli (PDB 1EQ2). C. UDP-galactose epimerase from Escherichia coli bound to UDP-Glc and NAD (PDB 1XEL). For panels A and C, each monomer is represented by a distinct color. In panel B the Rossmann fold is highlighted in blue/green and the C-terminal domain in red/yellow.
Scheme 9.
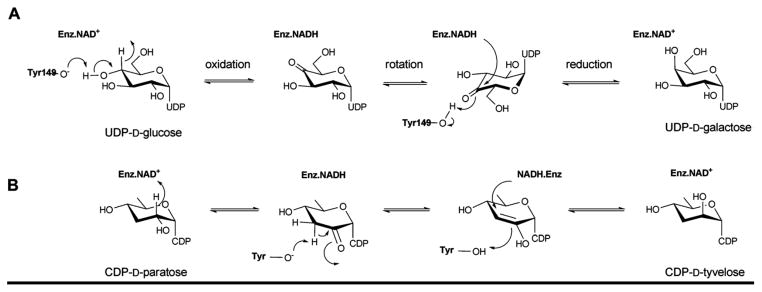
Reaction mechanism employed by UDP-galactose-4-epimerase, which catalyzes the interconversion of UDP-glucose into UDP-galactose (A), and CDP-tyvelose-2-epimerase, which interconverts CDP-tyvelose and CDP-paratose (B).
Scheme 11.
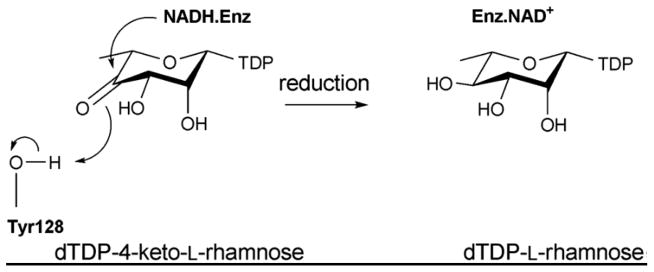
The mechanism of C4 reductase RmlD.
The catalytic mechanisms for UDP-galactose-4-epimerase and CDP-tyvelose-epimerase (Scheme 9) 41, 48, 56, 129 involve three fundamental steps: i) NAD(P)+-dependent oxidation of the C4 hydroxyl; ii) a rotation of the enzyme-bound keto-sugar intermediate by ~180° about the UDP-sugar bond; and iii) NAD(P)H-dependent reduction of the C4 carbonyl on the opposite face of the sugar. The conserved lysine (K153 in E. coli GALE) and serine/threonine (S124 in E. coli GALE) are believed to modulate the general acid/base function of tyrosine (Y149 in E. coli GALE) in this mechanism.
4.2.3. GT-B fold
Bacterial UDP-N-acetylglucosamine-2-epimerase catalyzes the reversible epimerization at C2 of UDP-N-acetylglucosamine (UDP-GlcNAc) to UDP-N-acetylmannosamine (UDP-ManNAc) 131, 132. This enzyme is a homodimer wherein dimerization is mediated by N-terminal helices α3, α4 and α5 (Figures 10A & 10B) with each monomer having two α/β/α domains that form a deep cleft at the domain interface (Figure 10B). The N and C-terminal domain both contain a Rossman fold with a seven-stranded and six stranded β-sheet, respectively (Figure 10B). Structures revealed both ‘open’ and ‘closed’ monomer conformations in dimer assembly and an observed 10° interdomain rotation, induced upon binding to substrate, activates one monomer for catalysis and may play a role in regulation 131.
Figure 10.

Global fold and monomeric structure of epimerases belonging to the GT-B structural family. Dimeric (A) and monomeric (B) structure of Escherichia coli UDP-N-acetylglucosamine 2-epimerase bound to UDP (PDB 1F6D). The labeled secondary structure elements (α3, α4, α5) signify the helices involved in dimerization.
The mechanism of UDP-N-acetylglucosamine-2-epimerase proceeds via the elimination of UDP from UDP-GlcNAc (Scheme 10). The anti elimination of UDP, which ultimately affords 2-acetamidoglucal and UDP, invokes a transient oxocarbenium species. The subsequent syn addition of UDP and C2 reprotonation provides UDP-ManNAc 133, 134. Based on the UDP-bound structure of UDP-GlcNAc epimerase, H213, which interacts with the NDP moiety in the active site, is assumed to act as a general acid (Figure 9D) while other conserved acidic residues (D95, E117 and E131) may be involved in stabilizing the oxocarbenium intermediate and/or as a general acid/base to promote C2 epimerization 62.
Scheme 10.
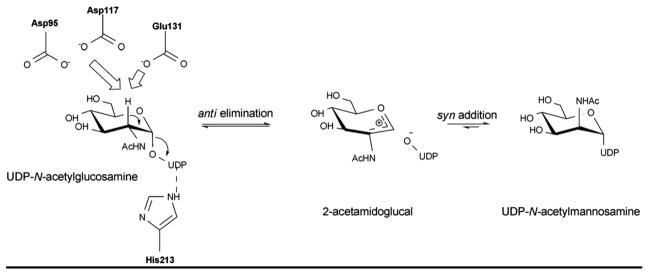
Reaction mechanism of UDP-GlcNAc-2-epimerase, the enzyme that converts UDP-GlcNAc to UDP-ManNAc.
4.3. Sugar ketoreductases
In the context of sugar nucleotide biosynthesis, ketoreductases (E.C. 1.1.1.-) are NAD(P)H-dependent enzymes which transfer hydride from NAD(P)H to a NDP-sugar carbonyl. NDP-sugar ketoreductases adopt two structural folds – the SDR fold (a fold discussed previously in sections 4.1.1. and 4.2.2.) and the glucose-fructose oxidoreductase (GFOR) fold. Representative examples include the Salmonella enterica serovar Typhimurium dTDP-6-deoxy-L-lyxo-4-hexulose C4-reductase (EC 1.1.1.133) RmlD [involved in dTDP-L-rhamnose biosynthesis 135] and the Actinomadura kijaniata dTDP-3,4-diketo-2,6-dideoxy-D-glucose C3-ketoreductase KijD10 [involved in dTDP-L-digitoxose biosynthesis 136], which adopt a SDR fold and GFOR fold, respectively.
4.3.1. RmlD
RmlD belongs to the previously discussed SDR structural family (sections 4.1.1. and 4.2.2.) 137 and contains the requisite Y128-X-X-X-K132 and glycine rich, G7-X-X-G10-X-X-G13 conserved motifs. RmlD is a homodimer and the dimerization is mediated by an intrafacial Mg2+ ion, also believed to be important for the proper orientation of cofactor and substrate binding domains. RmlD has several unusual features compared to other SDR enzymes: i) the dimer interface of RmlD is on the opposite face of Rossmann fold (involving helices α1, α6 and αC compared to enzymes such as RmlB (colored purple in Figure 11A); ii) dimerization is mediated mainly by hydrophilic interactions whereas hydrophobic interactions dominate the monomer/monomer interface in other SDR enzymes; iii) RmlD requires Mg2+ for full activity and does not discriminate between the cofactors NAD(H) and NADP(H), iv) large N-terminal cofactor binding domain lacks the structural elements β2-loop-α2, (colored orange in Figure 11B) resulting in a β-sheet with six β-strands in the Rossmann fold in the order 213456 (Figure 11A); and v) the small C-terminal substrate binding domain lacks C-terminal helices (colored in red Figure 11B) 135. The structural difference due to lack of extra structural elements found in RmlB (Figure 11B) is believed to contribute to the cofactor specificity distinction among RmlD and RmlB. Substrate binding by RmlD is accommodated by a solvent-exposed groove that extends from the cofactor binding site into the C-terminal domain to provide a deep cleft formed by two domains and the dimer interface. The hexa-hydrated Mg2+ is hydrogen bonded by three glutamic acid residues, Glu15, Glu190, and Glu292, from each monomer to mediate dimer interaction. Key residues involved in RmlD-catalyzed hydride transfer (Scheme 11) include the conserved Y128 (general acid) and K132 (which modulates the pKa of Y128) of the Y-X-X-X-K motif, as well as T104 (Figure 11C). The conserved serine/threonine has been implicated as part of a catalytic triad to enhance proton transfer via the formation of low-barrier hydrogen bonds 135.
Figure 11.

Comparison of sugar ketoreductase and sugar 4,6-dehydratase SDR folds. A. Salmonella enterica RmlD bound to dTDP-Rhamnose and NADPH (PDB 1KC3). B. Salmonella enterica RmlB bound to dTDP-Glucose and NAD+ (PDB 1KEU). Orange represents additional RmlB secondary structure (β2-L2-α2), red distinguishes an extra RmlB C-terminus helix and purple signifies secondary structures involved in dimerization C. The active site of RmlD.
4.3.2. KijD10
KijD10 belongs to the GFOR superfamily and crystallized as a tetramer or dimer of dimers 136. Each monomer comprises a N-terminal NADPH-binding Rossmann fold and a C-terminal mixed “open-faced” β-sheet-based substrate-binding domain. The N-terminal region contains a six-stranded parallel β-sheet flanked by two and three α-helices (one of which is contributed by the C-terminal domain), respectively (Figure 12A). The C-terminal domain, is dominated by a nine-stranded, mostly antiparallel β-sheet flanked on one side by three α-helices. The substrate binding site lies at the interface of the two domains and a characteristic of this fold is the presence cis-proline in the conserved E101-K102-P103 motif (Figure 12B) found within the active site cleft 138, 139. The enzyme adopts both “closed” and “open” states when bound to the same substrates (NADP+ and dTDP-benzene). In KijD10, the nicotinamide ring of the dinucleotide adopts a typical anti conformation, whereas the adenine ring is bound in a syn orientation (an unusual feature for this family of enzyme). Modeling dTDP-3-keto-6-deoxy-D-galactose within the active site implicated the Asp182 carboxylate to be important for substrate binding (Figure 12B). This aspartate belongs to the active site consensus sequence G177-G178-X-X-X-D182-X-X-X-(Y186/H) observed in related dehydrogenases and reductases. Based on site-directed mutagenesis studies, a mechanism for KijD10 been proposed whereby K102 (Figure 12B) participates as a general acid in protonation of the C3 oxygen. KijD10 is known to tolerate some sugar C2/C4 structural modification and is indiscriminate to C4 stereochemistry.
Figure 12.

Monomeric structure of an epimerase belonging to the GFOR structural family. A. Monomeric structure of Actinomadura kijaniata KijD10 from (PDB 3RC1) bound to NADP and dTDP-phenol. Helices within the N- and C- terminal domains are represented by distinct colors. B. KijD10 active site. Purple signifies residues within the conserved EKP motif, grey spheres highlight water molecules, and the side chain of Asp182 adopts two different configurations.
4.4. Sugar aminotransferases
Sugar aminotransferases (SATs) belong to the aspartate aminotransferase (AAT) type I family (EC 2.6.1.-) previously discussed in section 4.1.2. These enzymes catalyze the PLP-mediated transfer of an amino group from an amino acid donor (typically L-Glu, L-Gln, or L-Asp) to a NDP-ketosugar 46. Key signatures of these enzymes include a conserved lysine (K200, K193 and K183 in DesI, DesV and PseC, respectively), which binds PLP as an imine, and an invariant aspartate (D171, D164 and D154 DesI, DesV and PseC, respectively) which participates as a general acid to facilitate the transamination reaction 140, 141. SATs of this type are generally involved in the reversible formation of C3 or C4-amino-containing NDP-sugars and the overall reaction often favors the corresponding C3 or C4-keto NDP-sugar 46. Aminosugars deriving from the action of these enzymes are found in a wide variety of natural products and representatives include: macrolides (e.g., desosamine in erythromycin); anthracyclines (e.g, daunosamine in daunomycin); glycopeptides (e.g., vancosamine in vancomycin), polyenes (e.g., mycosamine in amphotericin B) to name just a few. During the last decade, crystal structures of several bacterial SATs have been solved: GDP-4-amino-6-deoxy-D-mannose transaminase (GDP-perosamine synthase) Per from Caulobacter crescentus 142; dTDP-4-amino-6-deoxy-D-glucose transaminase (EC 2.6.1.33) DesI from Streptomyces venezuelae 143; UDP-4-amino-L-arabinose transaminase (EC 2.6.1.87) ArnB from Salmonella typhimurium 144; UDP-2-acetamido-4-amino-6-deoxy-β-L-AltNAc transaminase PseC from Helicobacter pylori 145; UDP-4-amino-6-deoxy-D-GlcNAc transaminase PglE from Campylobacter jejuni 146; UDP-3-amino-2-acetamido-glucuronic acid transaminase WbpE from Pseudomonas aeruginosa 147; dTDP-3-amino-6-deoxy-D-glucose transaminase QdtB from Thermoanaerobacterium thermosaccharolyticum 148; and dTDP-3-amino-4,6-dideoxy-α-D-glucose transaminase (EC 2.6.1.89) DesV from Streptomyces venezuelae 149. In general, the enzymes share many common structural features45, 46.
These enzymes function as homodimers (Figure 13B) or higher-order oligomers (Figure 13A), with extensive dimer interfaces, featuring a large mixed β-sheet surrounded by α-helices (Figure 13C) involving two active sites per dimer. The active sites lie in the clefts formed by the dimer interface, and while each monomer contributes essential residues to both active sites, the active sites are generally independent. The overall architecture of the subunit comprises two α/β/α domains, a large N-terminal cofactor binding domain, and a small C-terminal domain. The large N-terminal domain features a central seven-stranded mixed β-sheet, with strand order 3245671, flanked on each side by α-helices. The smaller C-terminal domain consists of an antiparallel, three-stranded β-sheet surrounded by helices (Figure 13C). Additional protein-dependent secondary elements are generally inserted between the helices 7αb and 7αg of the C-terminal domain (Figure 13C). The N-terminus of the protein often contributes to the small domain as well (Figure 13C). In some enzymes these domains have been observed or predicted to move considerably upon substrate binding to create a “closed” conformation 45, 150. The inter-subunit interactions are extensive and involve several elements of secondary structure predominantly from the N-terminal domains of adjoined monomers.
Figure 13.

Representative global folds and monomeric structures for sugar aminotransferases belonging to the AAT family. A. Tetramer of Streptomyces venezuelae DesV, a 3-aminotransferase (PDB 2OGA). B. Dimer of Streptomyces venezuelae DesI, a 4-aminotransferase (PDB 2PO3). In panels A and B, each monomer is represented by a distinct color. C. Monomer of Helicobacter pylori PseC bound to UDP-GlcNAc and PMP (PDB 2FNU). D. Active site of PseC bound to UDP-GlcNAc and PMP.
The active sites of SATs are composed of residues from both halves of the dimer and contain several conserved residues. In the resting state, the PLP cofactor is covalently bound to the -amino group of a conserved lysine (K200, K193 and K183 in DesI, DesV and PseC, respectively) as an internal aldimine (Scheme 12)46. The major interaction of the NDP-sugar substrate in ligand-bound SATs is through a hydrogen-bonding network centered upon the nucleotide diphosphate with few apparent contacts to the pyranose 143, 145, 148. Consistent with this perceived relaxed sugar specificity, the C3-aminotransferases DesV and QdtB were demonstrated to accept C4 epimers as substrates 148, 151. Comparison of ligand-bound structures of the C4-aminotransferases PseC and DesI revealed their respective pyranose binding modes to differ by a 180° rotation 142, 143. This observed binding mode distinction is consistent with the axial versus equatorial amine installation displayed by PseC and DesI, respectively 148.
Scheme 12.

Reaction mechanism employed by aminotransferases. The first half of the reaction enables ‘amine loading’ (namely conversion of PLP to PMP) (A) while the second half of the reaction completes the cycle (B) to ultimately produce an amino sugar and regenerated PLP (bound as internal aldimine).
The reaction mechanism of SATs have been well characterized 140, 152–155 where the PLP cofactor serves as an electron sink throughout the reaction, and an invariant aspartate (D171, D164 and D154 DesI, DesV and PseC, respectively) helps to maintain this role by stabilizing the positively charged pyridinium ring of the cofactor 140. The catalysis is known to proceed via a ping pong mechanism in which the enzyme oscillates between PLP- and PMP-bound forms (Scheme 12A). The α-amino group of an amino donor (typically L-Glu, L-Gln, or L-Asp) attacks the C4′ atom of PLP internal aldimine (I), displacing lysine (K183 in PseC) to yield an external aldimine (II). SATs specifically orient the α-carbon/hydrogen bond perpendicular to the PLP π-system, thereby favoring this bond for cleavage. Proton extraction is mediated by the active site Lys (to provide quinoid intermediate III), which subsequently serves as general acid in C4′ protonation, yielding ketimine intermediate (IV). Subsequent attack by an activated water molecule leads to the formation of PMP and an oxoacid 153, 156, 157. At this stage, PMP is poised to react with a NDP-ketosugar and the process is reversed resulting in regeneration of the PLP-lysine internal aldimine and the transaminated product (Scheme 12B).
5. Additional sugar modification
5.1. Sugar O-/N-/C-methyltransferases
Sugar methyltransferases (SMTs) (EC 2.1.1.-) catalyze the transfer of a methyl group from S-adenosylmethionine (SAM) to a sugar-based nucleophile (typically N-, C- S- and O). In natural product biosynthesis, sugar C- and N-methylation typically occurs at the NDP-sugar stage (i.e., prior to glycosyltransferase-catalyzed transfer of the modified sugar to an aglycon) while sugar O-methylation commonly occurs post glycosylation (i.e., upon the final natural product glycoside) 14. The structures for a range of SMTs have emerged in recent years including: RebM [the rebeccamycin sugar 4-O-methyltransferase from Lechevalieria aerocolonigenes 158]; NovP [the novobiocin sugar 4-O-methyltransferase from Streptomyces spheroids 159]; MycE [the mycinamicin 2-O-methyltransferase from Micromonospora griseorubida 160]; TylM1 [the tylosin associated C3-N,N,-dimethyltransferase involved in the production of dTDP-D-mycaminose from Streptomyces fradiae 161]; DesVI [the pikromycin C3-N,N,-dimethyltransferase involved in the production of dTDP-D-desosamine from Streptomyces venezuelae 162]; and TcaB9 [the kijanimicin affiliated C3-methyltransferase involved in the biosynthesis of D-tetronitrose from Micromonospora chalcea 163].
Structurally, SMTs are classified as Class I MTs 164, 165 with some of them being monomeric (Figure 14C) 159, 161, 163, dimeric (Figure 14A and 14B) 158, 162 and even tetrameric 160. Despite their low sequence homology, all members of this family share a core Rossmann fold domain responsible for binding SAM comprising a seven stranded mixed β-sheet in the order 3214576 flanked by α-helices (Figure 14). Most SMTs contain auxiliary domains that are inserted throughout the core MT fold (Figure 14), the size and nature of which varies among the MTs and often contributes to substrate recognition. Examples include insertions at the N-terminus prior to the core domain, after strand β5, and between the strands β6 and β7. Sometimes, these auxiliary domains can form a ‘lid’ structure responsible for substrate binding and can also contribute to subunit-subunit interaction. The monomer/monomer interface among SMTs can vary. For example, the dimer interface in RebM (Figure 14A) is formed by reciprocal interactions between strands β6 from both monomers while in DesVI and TylM1 the dimerization is mediated by interaction of all four β-strands of the auxiliary domain (Figure 14B).
Figure 14.

Structure and active site architecture of sugar methyltransferases. A. Dimer of Lechevalieria aerocolonigenes RebM, a rebeccamycin sugar O-methyltransferase (PDB 3BUS). B. Dimer of Streptomyces venezuelae DesVI a N,N,-dimethyltransferase (PDB 3BXO). C. Micromonospora chalcea TcaB9, a C-methyltransferase (PDB 3NDJ) involved in the biosynthesis of D-tetronitrose. D. Active site of RebM. E. Active site of TcaB9 bound to dTDP-3-amino-2,3,6-trideoxy-4-keto-3-methyl-D-glucose. F. Overlay of the active sites of N-methyltransferases TylM1 (green, PDB 3PFG) and DesVI (yellow). Spheres represent water molecules.
Most SMT structures to date are cofactor-bound with SAH or SAM occupying the core Rossmann fold 164, 165. The SAM binding site is generally populated by hydrophobic residues. The amino acid portion of SAM interacts through the glycine-rich sequence E/D-X-G-X-G-X-G (D67-V-G69-C-G71-I-G73 in RebM). A loop following the strand β1, and an acidic loop after the strand β2, interacts with the ribose hydroxyls. The catalytic base histidine/aspartate (H140/H225 in RebM/MycE and D198 in NovP) responsible for deprotonation is located on a 310-helix (or sometimes just a loop) just after the β4-strand (Figures 14D & 14E). Substrate interaction is mediated by the secondary structure insertions after strand β5 and between the strands β6 and β7 - the size and type of which dictates the substrate specificity. Though structures bound to the substrate/product are available 160, 163, comparatively little is known about the structural factors which determine the regio-, stereo- and/or nucleophile-specificity of SMT-catalyzed reactions.
Typically, MTs catalyze SN2-like reactions, with inversion of methyl stereochemistry, involving oxygen-, nitrogen-, and carbon-based nucleophiles that require at least one proton transfer step prior to, in concert with, or after methyl group transfer. In O-SMTs, general acid/base catalysis contributes to rate acceleration wherein a catalytic base (H140/H278 in RebM/MycE and D198 in NovP) is involved in nucleophile deprotonation. With the exception of RebM, the SMTs discussed above require a divalent metal ion for catalysis with Mg+2 as the preferred metal. Molecular simulation and pH-rate studies suggest the divalent metal to function primarily to organize the substrate-binding site in SMTs, not as a general base 166, 167. The catalytic mechanism for C-MT (TcaB9) is thought to proceed via a similar mechanism wherein an active-site base, H225 (Figure 14E), abstracts the sugar C3 proton to initiate C3 C-methylation 168. Interestingly, this residue is strictly conserved among all C3 C-SMTs but less so in the corresponding N-SMTs 161, 163. Hence in N-SMTs TylM1 and DesVI, it is speculated that the proton on the C3 amino group is transferred to the water molecules lining the active site pocket and that the catalysis proceeds via approximation 161 (Figure 14F).
5.2. Sugar N-Acyltransferases
N-acyltransferases (EC 2.3.1.-) catalyze N-acylation using acetyl-CoA as an acyl donor. The structures of NDP-aminosugar acyltransferases that have emerged thus far fall into two major structural families - the GCN5-related N-acetyltransferase (GNAT) and left-handed β-helix motif (LβH) superfamilies. The current LβH family members include: a bifunctional UDP-N-acetylglucosamine pyrophosphorylase/glucosamine-1-phosphate N-acetyltransferase (EC 2.3.1.157) GlmU from Escherichia coli, Mycobacterium tuberculosis and Yersinia pestis 73, 74, 80, 84; QdtC (responsible for N-acetylation of dTDP-Quip3N in Thermoanaerobacterium thermosaccharolyticum) 169; PglD (responsible for N-acetylation of UDP-QuiNAc4N in Campylobacter jejuni) 170, 171; WlbB (EC 2.3.1.B6, an enzyme that catalyzes N-acetylation of UDP-GlcNAcNA in Bordetella petrii) 172; and the N-acyltransferase AntD (which catalyzes the acylation of the C4 amino group of dTDP-4-amino-4,6-dideoxyglucose using 3-hydroxy-3-methylbutyryl-CoA in Bacillus cereus en route to dTDP-D-Antrose 173. The sole GNAT example from NDP-sugar biosynthesis reported to date is the dTDP-fucosamine acetyltransferase WecD from Escherichia coli 174.
5.2.1. LβH fold
This LβH superfamily structural signature is dominated by a stunning parallel β-helix with repeating isoleucine-rich hexapeptide motifs and rare left-handed crossover connections 175 (Figure 15A). Members are typically trimeric wherein each monomer is dominated by a left-handed β-helix motif with a variable number of turns. This motif is characterized by imperfect, tandem repeated copies of the six-residue sequence, [L-I-V]-[G-A-E-D]-X-X-[S-T-A-V]-X which directs folding of a structural domain. Normally the LβH domain is accompanied by a second domain – e.g., a N-terminal pyrophosphorylase domain in GlmU 74, 80 (Figure 2D), a N-terminal α/β-domain for PglD (Figure 15D), a C-terminal α/β-domain (Figure 15C) for QdtC and a C-terminal β-sheet for WlbB (Figure 15B). The flat faces of the LβH are parallel β-sheets formed by stacks of short untwisted parallel β-strands. These strands participate in classical hydrogen bonding interactions with adjacent β-strands and the active site is located within a cleft between the LβH domains of two adjacent subunits (Figure 15B). Within the active site, acetyl-CoA is bound such that the pantetheine arm of the cofactor is arranged in an extended conformation and directed parallel to the 3-fold axis of the trimer (Figure 15A). The phosphoryl groups of the coenzyme project outward toward solvent, whereas the adenine ring, the pantothenate, and β-mercaptoethylamine units are buried within the trimer. The NDP-sugar is found deeper within the active where it adopts an extended conformation and abuts turns of the β-helix which orient the aminosugar for a direct SN2 attack upon the acetyl-CoA thioester. QtdC and WlbB bind their sugar nucleotides in a similar manner, primarily through interactions with the NDP and very minimal sugar contacts with the exception of a hydrogen bond between the sugar C3 amino group and a structurally conserved asparagine (N159 and N84 in QtdC and WlbB, respectively) 169, 172. Consistent with this, QdtC has been found to acetylate both dTDP-3-amino-3,6-dideoxy--D-glucose (dTDP-D-Quip3N) and dTDP-3-amino-3,6-dideoxy-α-D-galactose (dTDP-D-Fucp3N) 169. As expected, PglD binds its NDP-sugar substrate in a manner distinct from that observed for QtdC/WlbB to afford distinct regiospecificity.
Figure 15.

Representative global folds and monomeric structures for sugar N-acyltransferases belonging to the LβH family. A. Trimer of Campylobacter jejuni PglD (PDB 3BSW). B. Dimer interface of Bordetella petrii WlbB with subunit A and B substrates colored yellow and green, respectively (PDB 3MQH). C. Monomer of Thermoanaerobacterium thermosaccharolyticum QdtC bound to CoA and dTDP-3,6-dideoxy-3-amino galactose (PDB 3FSC). D. Overlay of monomer of PglD ligand-bound complexes - UDP-2-acetamido-4-amino-6-deoxy-glucose-bound (PDB 3BSS) and AcCoA-bound (PDB 3BSY). In panels C and D, the N-terminal domains are colored purple.
Ternary structures of the ‘pre’ and ‘post’ reaction states of WlbB (namely, with acetyl-CoA and CoA) have revealed the cofactor sulfur atom to move 2 Å upon departure of the acetyl group. A mechanism consistent with this structural information, mutagenesis studies and kinetics has been proposed (Scheme 13) 169, 172. In this proposed mechanism, the C3′ amino nitrogen of the substrate attacks the si face of the acetyl moiety of acetyl-CoA to produce a tetrahedral oxyanion intermediate which is stabilized and properly oriented by interaction with the structurally conserved N84 (N159 in QdtC and D94 in AntD). As the oxyanion intermediate collapses, the bond between the carbon and the sulfur of acetyl-CoA breaks allowing the sulfur to shift position and function as a general base by accepting a proton from the C3′ amino group. The proposed mechanism assumes that the amino group of the sugar binds in the active site in the unprotonated state and that the pKa of the CoA sulfur is sufficiently perturbed such that the sulfur can function as a general base 173. Though H125 has been suggested to play a role in PglD catalysis 170, the functional role of H125 remains open to debate.
Scheme 13.
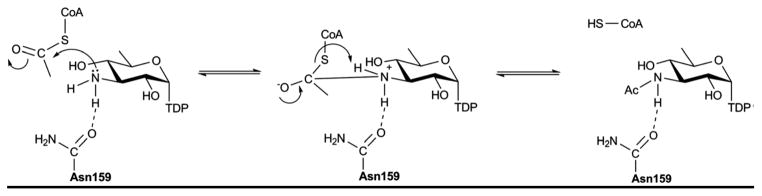
Proposed mechanism for the N-acetyltransferase QdtC.
5.2.2. GNAT fold
Members of the GNAT superfamily adopt an overall fold built around a central mixed β-sheet, which is typically composed of six β-strands 176, 177. WecD is a dimeric enzyme (Figure 16A) with each monomer adopting the GNAT N-acetyltransferase fold (Figure 16B) composed of two α/β domains. The N-terminal domain has a central, five-stranded mixed β-sheet structure and two α-helices. The C-terminal domain is comprises a seven-stranded mixed β-sheet and four α-helices. The strands β4 and β10 are long and extend across both domains and, although the two domains are distinct in the WecD structure, the β-strands of both domains extend toward each other to form a continuous, highly concave 10-stranded β-sheet with all α-helices, except α4 and α5, lining the outside of this sheet (Figure 16B). The dimer interface is created through interactions on the convex side of the β-sheet along strands β4 and β10 and also includes interactions from helices α3 and α7 (Figure 16A).
Figure 16.

Representative global fold and monomeric structure for a sugar N-acytransferase belonging to the GNAT family. Dimeric (A) and monomeric (B) structure of Escherichia coli WecD from bound to acetyl-CoA (PDB 2FT0).
The acetyl-CoA binding site is formed by residues from strands β8 and β9, the loop linking α4 and α5, as well as contributions from the loop β8-α6 and from helix α7. The loop between α4 and α5 is longer in WecD than in other GNAT proteins and affects the size and shape of the substrate-binding site 174. The NDP-sugar pyrophosphate forms hydrogen bonds to the backbone amide NH groups of G172 and G174 and to the side chain of R207. Interestingly, these two glycine residues, located in the loop region linking strand β8 and helix α6, are also part of the R/Q-X-X-G-X-G/A segment of the GNAT motif. Stacking interactions between the thymine base of nucleotides and aromatic side chains within WecD also contribute to substrate binding 174.
From the structure, WecD does not possess a residue that directly functions as a catalytic base. Various proposals have been put forward for how the substrate amino group is deprotonated in members of the GNAT family, including direct proton abstraction by an aspartate or glutamate residue via an activated water molecule or through a series of hydrogen-bonded water molecules that together form a “proton wire”. A structurally conserved active site tyrosine in many GNAT enzymes (Y208 in WecD) is positioned 3.0 Å from the sulfur atom of CoA. This residue has been suggested to stabilize and protonate the departing CoA thiolate anion and to assist in correctly orienting the acetyl group for transfer 174.
5.3. Oxidoreductases (N-oxidases)
Nitro, nitroso, and hydroxyamino sugars are found appended to a variety of natural products and generally derive from N-oxidase catalyzed oxidation of an appropriate NDP-aminosugar precursor 44. While hydroxylaminosugar formation en route to calicheamicin in Micromonospora echinospora is catalyzed by the P450 N-oxidase CalE10 44, 178, 179, N-oxidation in most other comparators are believed to be mediated by flavin containing monooxygenases (FMOs). FMO examples include: EvdC from Micromonospora carbonacea var. africana 180, 181 that mediates oxidation of dTDP-L-epi-vancosamine to the corresponding nitroso sugar 179, 182; RubN8 from Streptomyces achromogenes, involved in the biosynthesis of dTDP-D-rubranitrose and ultimately the antibiotic rubradirin 183; and KijD3 from Actinomadura kijaniata involved in the biosynthesis of dTDP-D-kijanose en route to the antibiotic kijanimycin 184. Recently, the structures of EvdC and KijD3 have been determined to place these N-oxidases into the class D FMOs, a family structurally related to the acyl-CoA dehydrogenases 182, 185. Studies to date suggest a stepwise process of successive oxidation [hydroxylaminosugar – nitrososugar – nitrosugar 44, 185] and it has been speculated that the formation of a nitroso/nitro product also depends upon the presence of an activating sugar carbonyl group adjacent to the targeted amine.
EvdC and KijD3 are tetrameric with each monomer adopting a three-domain fold (Figure 17A) comprising a N-terminal five α-helical bundle, an eight-stranded β-sheet, and a second five α-helical bundle at the C-terminus (Figure 17C) - a fold reminiscent of the acyl-CoA dehydrogenase superfamily. Interactions between adjacent C-terminal helical domains contribute the predominate contacts mediating tetramerization while the active site cleft is located at the junction of three domains. Residues contributing to the presumed active site emanate from all three domains but predominantly from the N-terminal helical domain and loops of the β-sheet domain. The active site is built along the length of helix Nα4, with contributions from Nα1- Nα3, Cα4, and the loops L1,L3 and L5 (Figure 17C) 182. The dTDP moiety is anchored to the protein via the side chains of E113, Q254, and R330 (Figure 17B). Based upon the structural differences observed between the KijD3-dTDP complex and apo-EvdC, it is speculated that the active site loops undergo some rearrangement upon substrate and/or cofactor binding 182.
Figure 17.

Structures of N-oxidases. A. Tetramer of Micromonospora carbonacea var. africana nitrososynthase EvdC (PDB 3MXL). B. dTDP binding site of Actinomadura kijaniata nitrosynthase KijD3 (PDB 3M9V). C. Monomer of KijD3. Green color distinguishes N-terminal α-helical domain, β-sheet domain is colored yellow, and purple highlights the C-terminal α-helical domain which is also the tetramerization interface.
In the proposed reaction mechanism of EvdC 179, 182, the four-electron flavin-mediated oxidation of an amine to a nitroso functional group involves a two-step process (A and B in Scheme 14) in which the amino sugar is first oxidized to the corresponding hydroxylamine via a classical flavin monooxygenase mechanism. In the second step, oxidation of the hydroxylamino sugar to nitroso sugar may be affected by an iterative oxidative process in which FAD would again be reduced by flavin reductase/NADPH prior to an additional round of oxidation.
Scheme 14.
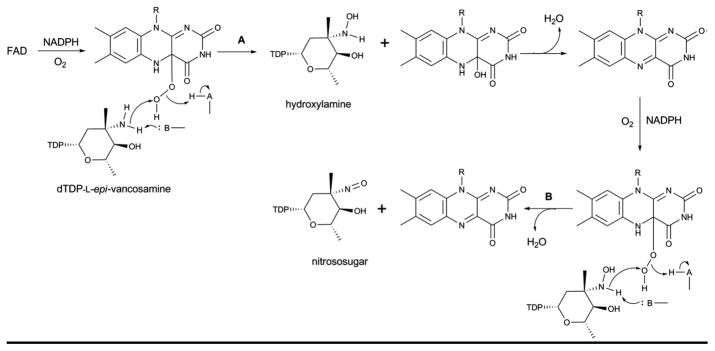
Proposed mechanism for FMO amine oxidase catalysis which provides for sequential oxidation (hydroxylamino – nitroso – nitro) of a target amino sugar.
5.4. ArnA (Dehydrogenase/Decarboxylase and Formyltransferase)
ArnA is one of the enzymes involved in the biosynthetic pathway of lipid A-4-amino-4-deoxy-L-arabinose (Ara4N), a main component of the outer membrane of gram-negative bacteria lipopolysaccharide (LPS). Modification of lipid A with Ara4N allows Gram-negative bacteria to resist the antimicrobial activity of cationic antimicrobial peptides and antibiotics such as polymyxin 186, 187. ArnA has two functionally independent domains - a dehydrogenase domain responsible for the NAD+-dependent oxidative decarboxylation of UDP-Glucuronic acid (UDP-GlcA) 188, and a transformylase domain that formylates UDP-4-amino-4-deoxy-L-arabinose (UDP-Ara4N) 189, 190. ArnA homologs - such CalS9 (calicheamicin biosynthesis, Micromonospora echinospora) 191, 192 and AtS9 (indolocarbazole AT2433 biosynthesis, Actinomadura melliaura) 193 - have been implicated in the biosynthesis of novel pentoses appended to natural products and recently have been confirmed to display similar dehydrogenase activity 194. Structures of full length Escherichia coli ArnA and that of each individual domain, both which are also independently functional, have been elucidated 188, 189, 195, 196.
ArnA is a hexamer or dimer of trimers when crystallized as the intact full two domain protein (Figure 18A). The hexamer adopts the shape of a three-blade propeller with the C-terminal decarboxylase domains forming the central core of the propeller and the N-terminal formyltransferase domains arranged on the periphery. The decarboxylase domains mediate most of the contacts between monomers within the hexamer and little interaction is observed between the dehydrogenase and formyltransferase domains. The intersubunit contacts are dominated by hydrogen bonding and ionic interactions.
Figure 18.

Global fold and dehydrogenase domain of E. coli ArnA. A. Hexameric structure of full-length ArnA bound to ATP and UDP-GlcA (PDB 1Z7E). B. Dehydrogenase (DH) domain of ArnA. C. Active site of DH domain of ArnA. The residues colored red and purple play a role in catalysis.
5.4.1. Dehydrogenase domain
The dehydrogenase domain of ArnA belongs to the SDR superfamily (previously described in sections 4.1.1, 4.2.2 & 4.3.1.). Like other enzymes belonging to this family that oxidize the C4-OH of sugar nucleotides (section 4.1.1.), it has a bilobal structure consisting of a large N-terminus subdomain and a small C-terminus subdomain (Figure 18B). The N-terminus subdomain has a Rossmann fold with a classical glycine-rich NAD+-binding motif G-X-(X)-G-X-X-G represented by amino acids G322-V-(N)-G325-F-I-G328 and the characteristic signature sequence Y463-X-X-X-K467 that, along with S433, form a catalytic triad. The active site is located in the cleft formed between two domains and a comparison of structures of the ArnA dehydrogenase domain in the presence and absence of ligand reveals a striking conformational change. Specifically, the loop that blocks the NAD+-binding site in the absence of UDP-GlcA moves ~17 Å to ultimately trap UDP-GlcA and allow NAD+ binding in the presence of the substrate 195.
The first step of the ArnA-catalyzed reaction is expected to mirror other SDR enzymes where the structurally conserved catalytic triad (T432-Y463 and K467, Figure 18C) facilitates UDP-GlcA C4 oxidization. The resulting 4-keto intermediate is believed to be unstable and the equilibrium for C4 oxidation favors the starting material. While decarboxylation is not spontaneous 189, irreversible decarboxylation helps drives the overall reaction (Scheme 15A). The identity and role of residues involved in the decarboxylation remains somewhat controversial with one study supporting conserved residues S433 and R619 (Figure 18C) to be important 188 and another noting conserved residues S433 and E434 (Figure 18C) as essential for oxidative decarboxylation of UDP-GlcUA (where E434 is proposed to function as a general base to deprotonate the sugar carboxylic acid) 189.
Scheme 15.

Proposed mechanism for the bifunctional activities of ArnA – dehydrogenase (A) and formyltransferase (B).
5.4.2. Formyltransferase domain
The structure of formyltransferase domain consists of two subdomains - a N-terminal subdomain with a Rossmann fold and a C-terminal subdomain resembling an oligonucleotide/oligosaccharide binding (OB) fold (Figure 19A) 196. The N-terminal subdomain, which contains the N-10-formyltetrahydrofolate binding site, is folded into a seven-stranded β-sheet (in the order 3214567 with strand-β6 oriented antiparallel) flanked by α-helices. The C-terminal subdomain has three large β-sheets (β8, β9 and β12) and two small β-sheets (β10 and β11) flanked by two α-helices. The enzyme has a conserved H104-X-S106-L107-L108-P109-X-X-X-G113 motif (reminiscent of N-10-formyltetrahydrofolate dehydrogenase FDH), where histidine, proline, and glycine are strictly conserved. This motif is located at the C-terminus of strand-β5 and in the loop following the strand-β5. A N102 H104 D140 triad has been identified as important for catalysis 196 wherein N102 is located on the strand-β5, His104 on the loop L5 (the loop after the strand β5) within the conserved motif at the beginning of the loop, and D140 is found on loop L6 (the loop after strand-β6). The side chain of the conserved H104 is stabilized by two hydrogen bonds (one by the side chain of S106 and another by a water molecule) that bring together all three catalytic residues (Figure 19B).
Figure 19.

The formyltransferase domain of E. coli ArnA. A. ArnA formyltransferase domain bound to UMP and N-5-formyltetrahydrofolate (FON) (PDB 2BLN). Helices in N- and C-terminal domains are colored differently. B. Active site of formyltransferase domain bound to UMP and N-5-Formyltetrahydrofolate. Grey spheres are water molecules.
A mechanism has been put forth which invokes all three residues of the catalytic triad in catalysis 196, 197. In the proposed mechanism (Scheme 15B), H104 and N102 activate the carbonyl carbon of the formyl group, which undergoes nucleophilic attack by the primary amine of UDP-Ara4N. The oxyanion of the putative tetrahedral intermediate is stabilized by H104 and N102. A water molecule, properly positioned in the active site by hydrogen bonding with the side chain of D140 (Figure 19B), works as a proton shuttle in mediating proton transfer from UDP-Ara4N to the nitrogen of the folate 198. The proton transfer is followed by decomposition of the tetrahedral intermediate and release of the products UDP-Ara4FN and tetrahydrofolate.
5.5. Enolpyruvyltransferase (MurA)
Enolpyruvyltransferase (MurA, also called MurZ, EC 2.5.1.7) catalyzes the transfer of an enolpyruvyl group from phosphoenolpyruvate (PEP) to UDP-N-acetylglucosamine (UDP-GlcNAc) to form UDP-N-acetylglucosamine enolpyruvate (UNAGEP). Both of these sugars form part of peptidoglycan essential for the integrity of the bacterial cell wall 199. One of the important naturally-occurring inhibitor of MurA is fosfomycin, which inhibits the enzyme by alkylating a catalytic cysteine residue 200. To date, structures of apo-MurA and ligand-bound form have been reported (Figure 20) 200–203.
Figure 20.

Structures of MurA where the active site flexible loop is colored red and domain linkers are colored magenta. A. Open, ligand-free form of E. cloacae MurA (PDB 1NAW). B. Half open conformation of H. influenzae MurA complexed with UDP-GlcNAc and fosfomycin (PDB 2RL2). C. Closed conformation of E. coli MurA complexed with UDP-GlcNAc and fosfomycin (PDB 1UAE). L1 and L2 are the two linker regions, loop La is an active site loop containing residues P111-P121. D. Active-site interactions within E. coli MurA complexed with UDP-GlcNAc and fosfomycin.
The structure of MurA enzyme is folded into two globular domains (N- and C-terminal domains) linked together by two linker regions [L1 and L22 consisting of residues S19-G20-A21 and L229-P230-D231 respectively (Figure 20)] with the interface cleft composing the active site. The two domains have similar secondary structure, consisting of an αβα motif arranged in such a way that the α-helices are surrounded by three mixed β-sheets exposed to the solvent. Several motifs containing conserved sequence L-X-X-L-G-A-Y-Z-Y (where Y= a polar residue and Z= a hydrophobic residue) have been found throughout the length of the enzyme 200 and a flexible ten amino acid-length loop, referred to as loop La (for active site), from the N-terminal domain (P112-P121 in E. coli and E. cloacae), moves toward the active site and closes the interdomain cleft like a lid upon ligand binding in the E. coli and E. cloacae enzymes 200, 202 (Figures 20A, 20B and 20C). This loop contains the conserved cysteine (Cys115 in MurA from E. cloacae and E. coli) critical for MurA activity and modified by fosfomycin 200, 203–206, a residue which is replaced by aspartate in fosfomycin-resistant strains 199, 204. In contrast, the active site loop conformational change has not been observed in H. influenzae MurA complex structures as both the ternary complex (with UDP-GlcNAc and fosfomycin) and binary complex (with UDP-GlcNAc) of H. influenzae MurA remain in a half open conformation [Figure 20B, 203].
UDP-GlcNAc interacts with the enzyme mainly through the NDP pyrophosphate via hydrogen-bonding with residues from both domains. Conserved interactions include participation of K22 in the formation of covalent adducts with PEP and fosfomycin 207, 208; C115 (Figure 20D) in the participation of catalysis and product release 205; D305 in the final deprotonation from the C3 atom of the tetrahedral intermediate 209; and D369 and L370 for specific interactions with fosfomycin (both residues mutated in resistant strains) 210.
In the proposed mechanism based upon biochemical and structural studies (Scheme 16), MurA binds UDP-GlcNAc and PEP sequentially. Substrate binding facilitates a large conformational change (open to closed form) wherein the closed form brings C115 into close proximity of PEP to enable the reaction. C115 has been proposed to function as a general acid/base catalyst in protonating/deprotonating C3 201, 204 and/or participate in product release 205. After the substrates bind to the active site, a proton is transferred to PEP resulting in the formation of an oxocarbenium ion, which is then attacked by the sugar C3 oxygen nucleophile. Subsequent elimination of phosphate from the tetrahedral intermediate leads to the final desired product 199.
Scheme 16.

Reaction (A) and proposed mechanism (B) catalyzed by MurA enzyme.
5.6. UDP-galactopyranose mutase
UDP-galactopyranose mutase (UGM, EC 5.4.99.9) is a flavoenzyme that catalyzes the reversible interconversion of UDP-galactopyranose (UDP-Galp) to UDP-galactofuranose (UDP-Galf). UDP-Galf is one of the main building blocks of the cell wall and extracellular matrix of many pathogenic bacteria, fungi, and protozoa and the absence of UGM in humans offers potential as a drug target 211, 212, 213. Unlike flavin dependent oxidoreductases, the redox state of the flavin in UGM is unchanged upon product formation. Although it is known that flavin must be in a reduced state, the precise role of the flavin in catalysis remains controversial. Studies determined that the reduced form of the flavin in UGM is anionic FADH (FADred) rather than neutral FADH2 214, 215. Recently structures of Escherichia coli 216, Klebsiella pneumoniae 217, 218, Mycobacterium tuberculosis217, and Deinococcus radiodurans 219, 220 UGMs have emerged.
Bacterial UGMs are homodimeric enzymes belonging to a mixed α/β class of proteins. Each monomer contains three domains. Domain 1 includes a Rossmann fold that binds flavin adenine dinucleotide (FAD), domain 2 is a bundle of α-helices that forms a major interface for the monomer-monomer interactions, and domain 3 features a twisted six-stranded β-sheet, situated between domain 1 and domain 2 which participates in substrate binding (Figure 21A). The FAD isoalloxazine binds in the crevice between domains 1 and 3 and the substrate binding site is located in a cleft adjacent to the isoalloxazine ring of FAD (Figure 21A). Superposition of monomers indicates domain 2 moves with respect to domain 1 using domain 3 as a molecular hinge to close on the substrate during the catalytic cycle 216. A mobile loop region is located at the entrance of substrate binding cleft, shown to close upon substrate binding. Structures of oxidized and reduced UGM in the presence of substrate show differential modes of substrate binding 218, 221. Comparison of the oxidized and reduced UGM substrate complexes reveals that flavin reduction results in a translocation of the mobile loop approximately 4 Å toward substrate (Figures 21B) and a shift in the relative orientation of the flavin and the UDP-Galp substrate positions the C1 of the galactose moiety directly adjacent to the nucleophilic N5 of the flavin. A comparison of UGM complexes with UDP-Galp and UDP-Glc implicates the C4-OH of galactose to engage in a hydrogen bond with the C4 carbonyl of the reduced flavin within this complex. It has been speculated that this H-bond may play an important role in enzyme’s ability to discriminate against UDP-Glc and may also provide a means to shuttle the proton from C4-OH to the nascent C5-OH after ring opening 218.
Figure 21.

A. Structure of UDP-galactopyranose mutase (UGM)-substrate complex from Klebsiella pneumonia (PDB 3INT). Domains 1 (FAD binding domain), 2 (substrate binding domain) and 3 (dimerization domain) are colored green/yellow, cyan and purple, respectively, the mobile loop is colored red, and substrate (UDP-Galp) and cofactor (FAD) are rendered as sticks and colored yellow. B. Two different views of the conformation of the mobile loop in the superposed structures of UGMs bound to UDP-Glc (green, 3GF4), UDP-Galp in oxidized state (magenta, PDB 3INR) and UDP-Galp in reduced state (yellow, PDB 3INT). C. The substrate binding site highlighting uridine and diphosphate interactions. D. The sugar and flavin binding region. Water molecules are illustrated as spheres.
The substrate and FAD are bound in the active site by a network of H-bonding and stacking interactions. Several conserved interactions have been demonstrated as essential for UGM activity 219, 222. These include stacking interactions of uridine with an aromatic residue (Y155 in K. pneumoniae) and stabilization of the negatively charged diphosphate backbone of the sugar nucleotide substrate by H-bonding interactions from the side-chains of the conserved arginines and tyrosines (R174, R280 Y185, Y314 and Y349 in K. pneumonia). Similarly, the sugar moiety of the substrate is stabilized through hydrogen bonding, some of which are mediated through water as shown in Figures 21C & D.
Studies indicate that the reaction mechanism of UGM proceeds via the formation of a covalent iminium intermediate (Scheme 17) 215, 218, 223, 224. This enables opening of the sugar ring and subsequent ring contraction to the furanose form. In the final reaction step, UDP serves as a nucleophile to displace the flavin, releasing product (Scheme 17). Three mechanistic hypotheses have been proposed to explain the formation of the iminium adduct: i) SN2 attack by N5 of FADred upon the anomeric carbon position of the substrate concerted with cleavage of the C1–OPβ bond (Scheme 17, path A); ii) a stepwise SN1-type substitution where elimination of UDP to produce an oxocarbenium intermediate precedes the nucleophilic attack by N5 of FAD (Scheme 17, path B); or iii) via covalent bond formation facilitated by single-electron transfer between a substrate and FADred radical pair (Scheme 17, path C) 215. Evidence for the participation of N5 in nucleophilic attack at C1 of the substrate to form the covalent adduct 218, 224 is provided by the close proximity of the anomeric carbon of the substrate and N5 of FADred in the ligand-bound UGM structure 218. Inability of 5-deaza-FAD to catalyze the reaction is also consistent with the proposed nucleophilic role of the flavin N5 215, 225. The intermediacy of the covalent iminium intermediate is supported by the ability to trap a covalent adduct during turnover via hydride reduction 218, 223. Indirect evidence for an oxocarbenium intermediate derives from a significant rate reduction observed with UDP-[2-F] Galf 226 and the inability of UGM to displace UDP from the linear substrate analog UDP-galactitol 227.
Scheme 17.

Proposed mechanism of UDP-galactopyranose mutase reaction.
6. Perspectives
As highlighted within this review, a wide range of structures have emerged and the new available structural information has contributed, in many cases, to better understanding of mechanism and/or specificity. Consistent with the divergent nature of sugar nucleotide biosynthetic pathways that derive from a series common core transformations, the structural scaffolds that support these core chemistries also utilize a relatively small set of common structural folds. As a result, dramatic shifts in chemical mechanism can be accomplished through very subtle residue substitutions or small secondary structure alterations – highlighting the potential of structure-based engineering and/or directed evolution of existing scaffolds to access new chemistries. Despite the considerable increase in available structures relevant to sugar nucleotide biosynthesis in recent years, there remains a deficiency of structural and/or biochemical understanding regarding the putative protein-protein interactions within these pathways. Such interactions have been invoked to explain how highly unstable sugar nucleotides may be ‘tunneled’ from one enzyme to the next to avoid degradation and also put forth as a mechanism of ‘co-locatization’ to avoid cross-talk among potential competing sugar nucleotide pathways within a cell. As the structural biology of individual catalysts within these pathways matures, the pursuit of such putative enzyme complexes may serve as one of the next frontiers in this exciting interdisciplinary field.
Table 1.
Representative structures, structural classes and enzyme-catalyzed reactions highlighted within this review.
| Organism | PDB (Variant) | Ligands | Ref | Enzyme & Reaction |
|---|---|---|---|---|
| Nucleotidyltransferases (EC 2.7.7.-) (Section 3.1 in this review) | ||||
| N-ter Rossmann fold and C-ter auxiliary domain | ||||
| Salmonella enterica | 1IIN 1IIM 1MP3(L89T) 1MP4(W224H) 1MP5(Y177F) 3PKP(Q83S) 3PKQ(Q83D) |
UDP-Glc TTP Apo Apo Apo dATP dGTP |
59, 61, 228, 229 | Glc1P thymidylyltransferase (RmlA ) (EC 2.7.7.24)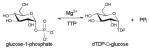
|
| Pseudomonas aeruginosa | 1FZW 1FXO 1G0R 1G1L 1G23 1G2V 1G3L |
Apo TMP Thymidine/Glc1P dTDP-Glc Glc1P TTP dTDP-Rha |
54 | |
| Escherichia coli | 1MC3 1H5T 1H5R 1H5S |
TTP dTDP-Glc/dTDP THM/Glc1P TMP |
84, 65 | |
| Bacillus anthracis | 3HL3 | Sucrose | ||
| Helicobacter pylori | 3JUJ 3JUK |
Apo UDP-Glc |
69 | Glc1P uridylyltransferase (UGPase) (EC 2.7.7.9) |
| Escherichia coli | 2E3D | Apo | 66, 67 | |
| Corynebacterium glutamicum | 2PA4 | UDP-Glc | 66, 67 | |
| Sphingomonas elodea | 2UX8 | Glc1P | 230 | |
| Streptococcus pneumoniae | 1HM8 1HM0 1HM9 |
AcCoA Apo UDP-GlcNAc/AcCoA |
231 | GlcNAc uridylyltransferase (GlmU) (EC 2.7.7.23)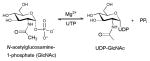
|
| Escherichia coli | 1FXJ 1FWY 1HV9 2OI5 2OI6 2OI7 |
Apo UDP-GlcNAC UDP-GlcNAC/CoA UDP-GlcNAc/AcCoA UDP-GlcNAC/CoA/GlcN1P UDP-GlcNAC/Desulfo-CoA/GlcNAc1P |
71, 80 | |
| Mycobacterium tuberculosis | 3D8V 3D98 3DK5 3DJ4 |
UDP-GlcNAC Apo Apo UDP-GlcNAC |
73, 74 | |
| Yersinia pestis Pestoides F | 3FWW | Apo | ||
| Salmonella enterica subsp. enterica serovar Typhi | 1WVC 1TZF |
CTP CDP-Glc |
76, 77 | Glc1P cytidylyltransferase (CGPase) (EC 2.7.7.33) |
| Agrobacterium tumefaciens | 3BRK | Apo | 79 | Glc1P adenylyltransferase (AGPase) (EC 2.7.7.27) |
| Thermotoga maritima | 2X5S 2X5Z 2X60 2X65 |
Apo GDP-Man GTP Man1P |
78 | Man1P guanylyltransferase (GMPase) ) (EC 2.7.7.22)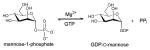
|
| Dehydratases (EC. 4.2.1.-) Section 4.1 in this review) | ||||
| SDR fold (Section 4.1.1. in the review) | ||||
| Salmonella enterica subsp. enterica serovar typhi | 1G1A 1KEU 1KEW |
NAD NAD/dTDP-Glc NAD/dTDP |
55, 86 | dTDP-glucose-4,6-dehydratases (RmlB) (EC 4.2.1.46) |
| Streptococcus suis | 1KET 1KER 1KEP 1OC2 |
NAD/dTDP NAD/dTDP-Glc NAD/dTDP-Xyl NAD/dTDP-Xyl |
55, 232 | |
| Streptomyces venezuelae | 1R66 1R6D |
NAD/dTDP NAD/dTDP-Glc |
87 | |
| Salmonella enterica subsp. enterica serovar typhi | 1WVG | CDP-Xyl | 89 | CDP-glucose-4,6-dehydratases (EC 4.2.1.45) |
| Yersinia pseudotuberculosis | 1RKX | NAD | 88 | |
| Pseudomonas aeruginosa | 1RPN | GDP/NADPH | 91 | GDP-mannose-4,6-dehydratases (GMD) (EC 4.2.1.47) |
| Escherichia coli | 1DB3 | Apo | 92 | |
| Aquifex aeolicus VF5 | 1Z95 2Z1M |
GDP/NADPH GDP/NADPH |
||
| Helicobacter pylori | 2GNA 2GN4 2GN6 2GN8 2GN9 |
NADP/UDP-Gal NADPH/UDP-GlcNAc NADP/UDP-GlcNAc NADP/UDP NADP/UDP-Glc |
93 | UDP-GlcNAc-4,6-dehydratases (FlaA1) (Inverting) (EC 4.2.1.115)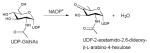
|
| AAT fold (Section 4.1.2. in the review) | ||||
| Escherichia coli | 2GMU 2GMS 2ROT(H188K) 3B8X(H188N) 3GR9(H188KS187N) |
PLP-glutamate ketimine Hydrated PLP PLP-glutamate diamine GDP-Perosamine PLP-glutamate ketimine |
52, 58, 94 | GDP-4-keto-6-deoxy-D-mannose 3-dehydratase (ColD) |
| Yersinia pseudotuberculosis | 3BCX 3BB8 (H220K) |
Apo PLP |
53 | CDP-6-deoxy-L-threo-d-glycero-4-hexulose 3-dehydrase (E1)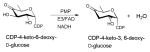
|
| Epimerases (EC 5.1.3.-) (Section 4.2 in this review) | ||||
| Cupin superfamily (Section 4..2.1. in the review) | ||||
| Salmonella enterica subsp. enterica serovar Typhi | 1DZR 1DZT |
Apo dTDP-phenol |
110 | dTDP-4-dehydrorhamnose 3,5-epimerase (RmlC) (EC 5.1.3.13)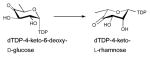
|
| Streptococcus suis | 1NXM 1NYW 1NZC 1IXL |
Apo dTDP-Glc dTDP-Xyl dTDP-Rha |
108, 109 | |
| Mycobacterium tuberculosis | 2IXC 1UPI 1PM7 |
dTDP-Rha Apo Apo |
111, 112 | |
| Pseudomonas aeruginosa | 2IXH 2IXI 2IXJ 2IXK |
dTDP-Rha dTDP-Xyl Apo dTDP-4-Keto Rha |
112 | |
| Bacillus anthracis str. Ames | 3RYK | dTDP | ||
| Amycolatopsis orientalis | 1WA4(M131F/L135A) 1OFN 1OI6 |
Apo Apo TMP |
113, 114 | dTDP-4-Keto-6-deoxy-D-glucose-5-epimerase (EvaD)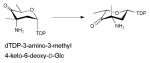
|
| Streptomyces caeruleus | 2C0Z | Apo | 115 | dTDP-6-deoxy-D-xylo-4-hexulose 3-epimerase (NovW)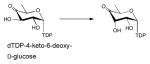
|
| Aneurinibacillus thermoaerophilus | 2PA7 2PAE(H49N) 2PAK(H51N) 2PAM (H49N/H51N) |
dTDP dTDP dTDP dTDP |
116 | dTDP-4-keto-6-deoxy-D-glucose-3,4-ketoisomerase (FdtA)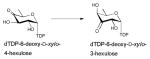
|
| SDR fold (Section 4.2.2. in the review) | ||||
| Escherichia coli | 1BWS 1BSV 1FXS 1GFS 1E6U 1E7Q(S107A) 1E7R(Y136E) 1E7S(K140R) |
NADPH NADPH NADP Apo NADP NADP NADP NADP |
118–120 | GDP-4-keto-6-deoxy-D-mannose-3,5-epimerase/4-reductase (GMER) (EC 1.1.1.271)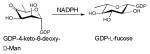
|
| Escherichia coli | 1A9Y(S124A/Y149F) 1A9Z(S124A/Y149F) 1KVQ(S124A) 1KVR 1KVS(S124T) 1KVT(S124V) |
NAD/UDP-Glc NAD/UDP-Glc NAD/UDP-Glc NAD/UDP NAD/UDP-Glc NAD/UDP-Glc |
56, 122, 123, 124 | UDP-glucose 4-epimerase (GALE) (EC 5.1.3.2 ) |
| Escherichia coli | 1EQ2 2X86(Y140F) 2X6T(Y140F) |
NADP/ADP-Glc NADP/ADP-Man NADP/ADP-Man |
126, 128, 233 | ADP-glyceromanno-heptose 6-epimerase (AGME) (EC 5.1.3.20)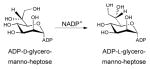
|
| Helicobacter pylori | 3SXP | NAD | 127 | |
| Salmonella enterica subsp. enterica serovar Typhi | 1ORR | NAD/CDP | 129 | CDP-paratose 2-epimerase (EC 5.1.3.10)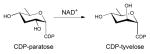
|
| GT-B fold (Section 4.2.3. in the review) | ||||
| Escherichia coli | 1F6D | UDP | 131, 234 | UDP-N-acetylglucosamine 2-epimerase (EC 5.1.3.14 )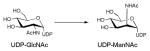
|
| Bacillus anthracis | 3BEO | UDP/UDP-GlcNAc | 132 | |
| Vibrio cholerae | 3DZC | Apo | ||
| Thermus Thermophilus HB8 | 1V4V | Apo | ||
| Ketoreductases (EC 1.1.1.-) (Section 4.3 in this review) | ||||
| SDR fold (Section 4..3.1. in the review) | ||||
| Salmonella enterica subsp. enterica serovar Typhi | 1KBZ 1KC0 1KC1 1KC3 1N2S |
Apo NADH NADPH NADPH/dTDP-L-Rha NAD |
135 | dTDP-4-dehydrorhamnose reductase (RmlD) (EC 1.1.1.133)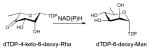
|
| Bacillus anthracis | 3SC6 | NADP | ||
| Glucose-Fructose OxidoReductase (GFOR) superfamily (Section 4..3.2. in the review) | ||||
| Actinomadura kijaniata | 3RBV 3RC1 3RC2 3RC7(Y186F) 3RC9(K102A) 3RCB(K102E) |
NADP NADP/dTDP-benzene NADP/dTDP-benzene NADP/dTDP-benzene NADP/dTDP-benzene NADP/dTDP-benzene |
136 | dTDP-C-3-ketoreductase (KijD10)
|
| Sugar aminotransferases (SAT) (EC 2.6.1.1.-) (Section 4.4 in this review) | ||||
| AAT fold (Section 4..4. in the review) | ||||
| Streptomyces venezuelae | 2PO3 | dTDP-4-amino-4,6-dideoxyglucose | 143 | dTDP-4-amino-6-deoxy-D-glucose transaminase (DesI) (EC2.6.1.33) |
| Streptomyces venezuelae | 2OGA 2OGE |
Ketamine with Glu internal aldimine |
149 | dTDP-3-amino-3,6-dideoxy-α-D-glucose transaminase (DesV) (EC 2.6.1.89)
|
| Thermoanaerobacterium thermosaccharolyticum | 3FRK | PLP:dTDP-3-aminoquinovose aldimine | 148 | dTDP-3-amino-6-deoxy-D-glucose transaminase (QdtB)
|
| Caulobacter crescentus | 3BN1 | internal aldimine | 142, 150 | GDP-4-amino-6-deoxy-D-mannose transaminase (Per)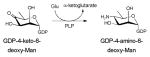
|
| CB15 | 3DR7 3DR4(K186A) |
GDP-3-deoxyperosamine | ||
| Helicobacter pylori | 2FN6 2FNI 2FNU |
Apo PLP PMP/UDP-GlcNAc |
145 | UDP-2-acetamido-4-amino-6-deoxy-β-L-AltNAc transaminase (PseC) |
| Salmonella enterica serovar Typhi | 1MDX 1MDO 1MDZ |
PLP/Oxaloglutaric acid PLP Cycloserine-PLP |
144 | UDP-4-amino-L-arabinose transaminase (ArnB) (EC 2.6.1.87) |
| Campylobacter jejuni | 1O69 | 2-amino-PLP | 146 | UDP-4-amino-6-deoxy-D-GlcNAc transaminase (PglE) |
| Pseudomonas aeruginosa | 3NYU 3NYS(K188A) 3NYT(K185A) |
Internal aldimine PLP PLP external aldimine adduct with UDP-3-amino-GlcNAcA |
147 | UDP-3-amino-2-acetamido-glucuronic acid transaminase (WbpE) |
| Sugar O-/N-/C-methyltransferases (SMTs) (EC 2.1.1.-) (Section 5.1 in this review) (Rossmann fold) | ||||
| Lechevalieria aerocolonigenes | 3BUS | SAH | 158 | Sugar 4′-O-MeT(RebM) |
| Streptomyces spheroids | 2WK1 | SAH | 159 | Novobiocin 4′-O-MeT (NovP)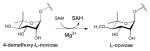
|
| Micromonospora griseorubida | 3SSO 3SSN 3SSM |
SAH SAH/Mycinamicin VI SAH |
160 | Mycinamicin 2-O-methyltransferase (MycE) |
| Streptomyces fradiae | 3PFH 3PFG 3PX3(H123A) 3PX2(H123N) |
SAH/dTDP-Quip3N SAM/dTDP-Phenol SAH/dTDP-Quip3N SAH/dTDP-Quip3N |
161 | C3-N,N,-dimethyltransferase (TylM1) |
| Streptomyces venezuelae | 3BXO | SAM/UDP-Phenol | 162 | Pikromycin C3-N,N,-dimethyltransferase (DesVI) |
| Micromonospora chalcea | 3NDJ 3NDI |
SAH/dTDP-3-amino-2,3,6-trideoxy-4-keto-3-methyl-D-glucose. SAH/dTMP |
163 | Kijanimicin affiliated C3-methyltransferase (tetronitrose) (TcaB9) |
| 5.2. Sugar N-Acetyltransferases | ||||
| left-handed-β-helix motif (LβH) superfamilies | ||||
| Escherichia coli | 2OI5 2OI6 2OI7 |
UDP-GlcNAc/AcCoA UDP-GlcNAc/CoA/GlcN-1-P UDP-GlcNAc/desulpho- CoA/GlcNAc-1-P |
80 | Glucosamine-1-phosphate N-acetyltransferase (GlmU) (EC 2.3.1.157; EC 2.7.7.23) |
| Mycobacterium tuberculosis | 2QKX 3D8V 3D98 3DJ4 3DK5 |
GlcNAc-1-P UDP-GlcNAc Apo UDP-GlcNAc Apo |
73, 74 | |
| Yersinia pestis | 3FWW | Apo | ||
| Thermoanaerobacterium thermosaccharolyticum | 3FSC 3FS8 3FSC |
CoA/dTDP-3,6-dideoxy-3-amino galactose AcCoA CoA/dTDP-3-amino-fucose |
169 | dTDP-Quip3N-3-N-acetyltransferase (QdtC) |
| Campylobacter jejuni | 3BSW 3BSY 3BSS 3VHE 3BFP 2NPO |
Apo AcCoA UDP-2-acetamido-4-amino-6-deoxy-glucose CoA Apo Apo |
170, 171 | UDP-QuiNAc4N-4-N acetyltransferase (PglD) |
| Bordetella petrii | 3MQH 3MQG |
CoA/UDP-3-amino-2-acetamido-2,3-dideoxy glucuronic acid AcCoA/UDP |
172 | UDP-GlcNAc3NA 3-N-acetyltransferase (WlbB) (EC 2.3.1.B6)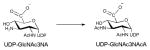
|
| Bacillus cereus | 3VBJ 3VBI 3VBK(S84A) 3VBL(S84C) 3VBN(D94A) 3VBP(D94N) |
dTDP/3-hydroxybutyryl-CoA dTDP-4-amino-4,6-dideoxyglucose/CoA dTDP-4-amino-4,6-dideoxyglucose/CoA dTDP-4-amino-4,6-dideoxyglucose, CoA dTDP, CoA dTDP, CoA |
173 | Acyltransferase (AntD)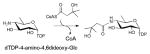
|
| GCN5-related N-acetyltransferase (GNAT) | ||||
| Escherichia coli | 2FT0 2FS5 |
AcCoA Apo |
174 | dTDP-fucosamine acetyltransferase (WecD) |
| 5.3. Oxidoreductases (N-oxidases) | ||||
| Micromonospora carbonacea var. africana | 3MXL | Apo | 182 | Nitrososynthase-EvdC or ORF36
|
| Actinomadura kijaniata | 3M9V | dTDP | 185 | Nitrosynthase-KijD3 |
| 5.4. Dehydrogenase/Decarboxylase and Formyltransferase (ArnA) (EC 1.1.1.305) | ||||
| Escherichia coli | 1U9J 2BLL 1YRW 1Z73(S433A) 1Z74(R619Y) 1Z75(R619M) 1Z7B(R619E) 1Z7E 2BLN |
Apo-DH domain Apo-DH domain Apo-FDH domain Apo-DH domain Apo-DH domain Apo-DH domain Apo-DH domain ATP, UDP-GlcA-Full-length UMP, N-5FDH- FDH domain |
188, 189, 195, 196 |
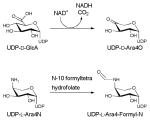
|
| 5.5. UDP-N-acetylglucosamine Enolpyruvyltransferase (MurA) (EC 2.5.1.7) | ||||
| Escherichia coli | 1UAE 1A2N(C115A) 3ISS(N67D) 2Z2C |
UDP-GlcNAc/fosfomycin Tetrahedral intermediate Enolpyruvyl-UNAM Cnicin |
201, 204, 206 |
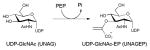
|
| Enterobacter cloacae | 1NAW 1DLG (C115S) 1EYN 1EJD 1EJC 1Q3G (D305A) 1RYW(C115S) 3LTH 3KQJ 3KR6 1YBG 3KQA |
Apo Apo ANS Apo Apo Tetrahedral intermediate UDP-GlcNAcEP UDP-GlcNAc, fosfomycin UDP-GlcNAc Fosfomycin Inhibitor T6361 Terreic acid |
202, 205, 209 | |
| Haemophilus influenzae | 2RL1 2RL2 |
UDP-GlcNAc UDP-GlcNAc, fosfomycin |
203 | |
| 5.6. UDP-galactopyranose mutase (UGM) (EC 5.4.99.9) | ||||
| Escherichia coli | 1I8T | FAD | 216 |
|
| Mycobacterium tuberculosis | 1V0J | FAD | 217 | |
| Klebsiella pneumoniae | 1WAM 2BI7 2BI8 3GF4 3INR 3INT 3KYB |
FADH− FAD FADH2 UDP-Glc/FAD UDP-Gal/FAD UDP-Gal/FADH2 FAD/FMN |
217, 218 | |
| Deinococcus radiodurans | 3MJ4 3HE3 3HDQ 3HDY |
FAD/UDP-Gal(phosphonate analog) FAD/UDP FAD/UDP-Gal FADH2/UDP-Gal |
219, 220 | |
Acknowledgments
The authors would like to thank all past and present members of the Thorson and Phillips laboratories for contributions relevant to this work. Support for this work was provided in part by NIH grants AI52218 (JST), CA84374 (JST), and PSI GM098248 (GNP and JST).
References
- 1.Kilcoyne M, Joshi L. Cardiovasc Hematol Agents, Med Chem. 2007;5:186–197. doi: 10.2174/187152507781058663. [DOI] [PubMed] [Google Scholar]
- 2.Kren V, Martinkova L. Curr Med Chem. 2001;8:1303–1328. doi: 10.2174/0929867013372193. [DOI] [PubMed] [Google Scholar]
- 3.Kren V, Rezanka T. FEMS Microbiol Rev. 2008;32:858–889. doi: 10.1111/j.1574-6976.2008.00124.x. [DOI] [PubMed] [Google Scholar]
- 4.Wrodnigg TM, Sprenger FK. Mini Rev Med Chem. 2004;4:437–459. doi: 10.2174/1389557043403918. [DOI] [PubMed] [Google Scholar]
- 5.Thorson JS, Hosted TJ, Jiang JQ, Biggins JB, Ahlert J. Curr Org Chem. 2001;5:139–167. [Google Scholar]
- 6.Le GT, Abbenante G, Becker B, Grathwohl M, Halliday J, Tometzki G, Zuegg J, Meutermans W. Drug Discov Today. 2003;8:701–709. doi: 10.1016/s1359-6446(03)02751-x. [DOI] [PubMed] [Google Scholar]
- 7.Nicotra F, Cipolla L, La Ferla B, Airoldi C, Zona C, Orsato A, Shaikh N, Russo L. J Biotechnol. 2009;144:234–241. doi: 10.1016/j.jbiotec.2009.05.014. [DOI] [PubMed] [Google Scholar]
- 8.Cipolla L, Araujo AC, Bini D, Gabrielli L, Russo L, Shaikh N. Expert Opin Drug Discov. 2010;5:721–737. doi: 10.1517/17460441.2010.497811. [DOI] [PubMed] [Google Scholar]
- 9.Cipolla L, La Ferla B, Airoldi C, Zona C, Orsato A, Shaikh N, Russo L, Nicotra F. Future Med Chem. 2010;2:587–599. doi: 10.4155/fmc.10.8. [DOI] [PubMed] [Google Scholar]
- 10.Butler MS. J Nat Prod. 2004;67:2141–2153. doi: 10.1021/np040106y. [DOI] [PubMed] [Google Scholar]
- 11.Weymouth-Wilson AC. Nat Prod Rep. 1997;14:99–110. doi: 10.1039/np9971400099. [DOI] [PubMed] [Google Scholar]
- 12.Gantt RW, Peltier-Pain P, Thorson JS. Nat Prod Rep. 2011;28:1811–1853. doi: 10.1039/c1np00045d. [DOI] [PubMed] [Google Scholar]
- 13.Thibodeaux CJ, Melancon CE, Liu HW. Nature. 2007;446:1008–1016. doi: 10.1038/nature05814. [DOI] [PubMed] [Google Scholar]
- 14.Thibodeaux CJ, Melancon CE, 3rd, Liu HW. Angew Chem Int Ed Engl. 2008;47:9814–9859. doi: 10.1002/anie.200801204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oh TJ, Mo SJ, Yoon YJ, Sohng JK. J Microbiol Biotechnol. 2007;17:1909–1921. [PubMed] [Google Scholar]
- 16.Blanchard S, Thorson JS. Curr Opin Chem Biol. 2006;10:263–271. doi: 10.1016/j.cbpa.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 17.Volchegursky Y, Hu Z, Katz L, McDaniel R. Mol Microbiol. 2000;37:752–762. doi: 10.1046/j.1365-2958.2000.02059.x. [DOI] [PubMed] [Google Scholar]
- 18.Hutchinson CR. Curr Opin Microbiol. 1998;1:319–329. doi: 10.1016/s1369-5274(98)80036-2. [DOI] [PubMed] [Google Scholar]
- 19.Butler AR, Bate N, Kiehl DE, Kirst HA, Cundliffe E. Nat Biotechnol. 2002;20:713–716. doi: 10.1038/nbt0702-713. [DOI] [PubMed] [Google Scholar]
- 20.Hutchinson E, Murphy B, Dunne T, Breen C, Rawlings B, Caffrey P. Chem Biol. 2010;17:174–182. doi: 10.1016/j.chembiol.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 21.Pageni BB, Oh TJ, Liou K, Yoon YJ, Sohng JK. J Microbiol Biotechnol. 2008;18:88–94. [PubMed] [Google Scholar]
- 22.Salas JA, Mendez C. Trends Microbiol. 2007;15:219–232. doi: 10.1016/j.tim.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 23.Yang J, Hoffmeister D, Liu L, Fu X, Thorson JS. Bioorg Med Chem. 2004;12:1577–1584. doi: 10.1016/j.bmc.2003.12.046. [DOI] [PubMed] [Google Scholar]
- 24.Monica MP. Curr Opin Chem Biol. 2011;15:226–233. doi: 10.1016/j.cbpa.2010.11.022. [DOI] [PubMed] [Google Scholar]
- 25.Erb A, Weiss H, Harle J, Bechthold A. Phytochem. 2009;70:1812–1821. doi: 10.1016/j.phytochem.2009.05.019. [DOI] [PubMed] [Google Scholar]
- 26.Williams GJ, Gantt RW, Thorson JS. Curr Opin Chem Biol. 2008;12:556–564. doi: 10.1016/j.cbpa.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luzhetskyy A, Mendez C, Salas JA, Bechthold A. Curr Top Med Chem. 2008;8:680–709. doi: 10.2174/156802608784221514. [DOI] [PubMed] [Google Scholar]
- 28.Williams GJ, Zhang C, Thorson JS. Nat Chem Biol. 2007;3:657–662. doi: 10.1038/nchembio.2007.28. [DOI] [PubMed] [Google Scholar]
- 29.Fu X, Albermann C, Zhang C, Thorson JS. Org Lett. 2005;7:1513–1515. doi: 10.1021/ol0501626. [DOI] [PubMed] [Google Scholar]
- 30.Bowles D, Isayenkova J, Lim EK, Poppenberger B. Curr Opin Plant Biol. 2005;8:254–263. doi: 10.1016/j.pbi.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 31.Breton C, Šnajdrová L, Jeanneau C, Koča J, Imberty A. Glycobiology. 2006;16:29R–37R. doi: 10.1093/glycob/cwj016. [DOI] [PubMed] [Google Scholar]
- 32.Xiaoqiang W. FEBS Lett. 2009;583:3303–3309. doi: 10.1016/j.febslet.2009.09.042. [DOI] [PubMed] [Google Scholar]
- 33.Pesnot T, Jorgensen R, Palcic MM, Wagner GK. Nat Chem Biol. 2010;6:321–323. doi: 10.1038/nchembio.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wagner GK, Pesnot T. Chembiochem. 2010;11:1939–1949. doi: 10.1002/cbic.201000201. [DOI] [PubMed] [Google Scholar]
- 35.Chang A, Singh S, Phillips GN, Jr, Thorson JS. Curr Opin Biotechnol. 2011;22:800–808. doi: 10.1016/j.copbio.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu HW, Thorson JS. Annu Rev Microbiol. 1994;48:223–256. doi: 10.1146/annurev.mi.48.100194.001255. [DOI] [PubMed] [Google Scholar]
- 37.Shackelford GS, Regni CA, Beamer LJ. Protein Sci. 2004;13:2130–2138. doi: 10.1110/ps.04801104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mouilleron S, Badet-Denisot MA, Badet B, Golinelli-Pimpaneau B. Arch Biochem Biophys. 2011;505:1–12. doi: 10.1016/j.abb.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 39.Sellick CA, Campbell RN, Reece RJ. Int Rev Cell Mol Biol. 2008;269:111–150. doi: 10.1016/S1937-6448(08)01003-4. [DOI] [PubMed] [Google Scholar]
- 40.Holden HM, Thoden JB, Timson DJ, Reece RJ. Cell Mol Life Sci. 2004;61:2471–2484. doi: 10.1007/s00018-004-4160-6. [DOI] [PubMed] [Google Scholar]
- 41.Thoden JB, Holden HM. J Biol Chem. 2005;280:21900–21907. doi: 10.1074/jbc.M502411200. [DOI] [PubMed] [Google Scholar]
- 42.Thoden JB, Timson DJ, Reece RJ, Holden HM. J Biol Chem. 2005;280:9662–9670. doi: 10.1074/jbc.M412916200. [DOI] [PubMed] [Google Scholar]
- 43.Tanner ME. Curr Org Chem. 2001;5:169–192. [Google Scholar]
- 44.Timmons SC, Thorson JS. Curr Opin Chem Biol. 2008;12:297–305. doi: 10.1016/j.cbpa.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holden HM, Cook PD, Thoden JB. Curr Opin Struct Biol. 2010;20:543–550. doi: 10.1016/j.sbi.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Romo AJ, Liu HW. Biochim Biophys Acta. 2011;1814:1534–1547. doi: 10.1016/j.bbapap.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.He XM, Liu HW. Annu Rev Biochem. 2002;71:701–754. doi: 10.1146/annurev.biochem.71.110601.135339. [DOI] [PubMed] [Google Scholar]
- 48.Field RA, Naismith JH. Biochemistry. 2003;42:7637–7647. doi: 10.1021/bi0345079. [DOI] [PubMed] [Google Scholar]
- 49.Naismith JH. Chem Soc Rev. 2006;35:763–770. doi: 10.1039/b602313d. [DOI] [PubMed] [Google Scholar]
- 50.Naismith JH. Biochem Soc Trans. 2004;32:647–654. doi: 10.1042/BST0320647. [DOI] [PubMed] [Google Scholar]
- 51.Beyer N, Alam J, Hallis TM, Guo Z, Liu HW. J Am Chem Soc. 2003;125:5584–5585. doi: 10.1021/ja030088r. [DOI] [PubMed] [Google Scholar]
- 52.Cook PD, Thoden JB, Holden HM. Protein Sci. 2006;15:2093–2106. doi: 10.1110/ps.062328306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smith P, Szu PH, Bui C, Liu HW, Tsai SC. Biochemistry. 2008;47:6329–6341. doi: 10.1021/bi702449p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blankenfeldt W, Asuncion M, Lam JS, Naismith JH. EMBO J. 2000;19:6652–6663. doi: 10.1093/emboj/19.24.6652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Allard ST, Beis K, Giraud MF, Hegeman AD, Gross JW, Wilmouth RC, Whitfield C, Graninger M, Messner P, Allen AG, Maskell DJ, Naismith JH. Structure. 2002;10:81–92. doi: 10.1016/s0969-2126(01)00694-3. [DOI] [PubMed] [Google Scholar]
- 56.Liu Y, Thoden JB, Kim J, Berger E, Gulick AM, Ruzicka FJ, Holden HM, Frey PA. Biochemistry. 1997;36:10675–10684. doi: 10.1021/bi970430a. [DOI] [PubMed] [Google Scholar]
- 57.Gerratana B, Cleland WW, Frey PA. Biochemistry. 2001;40:9187–9195. doi: 10.1021/bi0108249. [DOI] [PubMed] [Google Scholar]
- 58.Cook PD, Kubiak RL, Toomey DP, Holden HM. Biochemistry. 2009;48:5246–5253. doi: 10.1021/bi9005545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barton WA, Lesniak J, Biggins JB, Jeffrey PD, Jiang J, Rajashankar KR, Thorson JS, Nikolov DB. Nat Struct Biol. 2001;8:545–551. doi: 10.1038/88618. [DOI] [PubMed] [Google Scholar]
- 60.Hoffmeister D, Thorson JS. Chembiochem. 2004;5:989–992. doi: 10.1002/cbic.200400003. [DOI] [PubMed] [Google Scholar]
- 61.Moretti R, Thorson JS. J Biol Chem. 2007;282:16942–16947. doi: 10.1074/jbc.M701951200. [DOI] [PubMed] [Google Scholar]
- 62.Allard ST, Giraud MF, Naismith JH. Cell Mol Life Sci. 2001;58:1650–1665. doi: 10.1007/PL00000803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Samuel J, Tanner ME. Nat Prod Rep. 2002;19:261–277. doi: 10.1039/b100492l. [DOI] [PubMed] [Google Scholar]
- 64.Dong C, Beis K, Giraud MF, Blanchard S, Allard ST, Major LL, Kerr ID, Whitfield C, Naismith JH. Biochem Soc Trans. 2003;31:532–536. doi: 10.1042/bst0310532. [DOI] [PubMed] [Google Scholar]
- 65.Zuccotti S, Zanardi D, Rosano C, Sturla L, Tonetti M, Bolognesi M. J Mol Biol. 2001;313:831–843. doi: 10.1006/jmbi.2001.5073. [DOI] [PubMed] [Google Scholar]
- 66.Thoden JB, Holden HM. Protein Sci. 2007;16:1379–1388. doi: 10.1110/ps.072864707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thoden JB, Holden HM. Protein Sci. 2007;16:432–440. doi: 10.1110/ps.062626007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thoden JB, Ruzicka FJ, Frey I, Rayment PA, Holden HM. Biochemistry. 1997;36:1212–1222. doi: 10.1021/bi9626517. [DOI] [PubMed] [Google Scholar]
- 69.Kim H, Choi J, Kim T, Lokanath N, Ha S, Suh S, Hwang HY, Kim K. Mol Cells. 2010;29:397–405. doi: 10.1007/s10059-010-0047-6. [DOI] [PubMed] [Google Scholar]
- 70.Kim H, Wu CA, Kim DY, Han YH, Ha S, Suh S, Kim K. Acta Crystallogr D Biol Crystallogr. 2004;60:1447–1449. doi: 10.1107/S0907444904012727. [DOI] [PubMed] [Google Scholar]
- 71.Brown K, Pompeo F, Dixon S, Mengin-Lecreulx D, Cambillau C, Bourne Y. EMBO J. 1999;18:4096–4107. doi: 10.1093/emboj/18.15.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kostrewa D, D’Arcy A, Takacs B, Kamber M. J Mol Biol. 2001;305:279–289. doi: 10.1006/jmbi.2000.4296. [DOI] [PubMed] [Google Scholar]
- 73.Verma SK, Jaiswal M, Kumar N, Parikh A, Nandicoori VK, Prakash B. Acta Crystallogr F. 2009;65:435–439. doi: 10.1107/S1744309109010252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang Z, Bulloch EM, Bunker RD, Baker EN, Squire CJ. Acta Crystallogr D Biol Crystallogr. 2009;65:275–283. doi: 10.1107/S0907444909001036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mochalkin I, Lightle S, Zhu Y, Ohren JF, Spessard C, Chirgadze NY, Banotai C, Melnick M, McDowell L. Protein Sci. 2007;16:2657–2666. doi: 10.1110/ps.073135107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Koropatkin NM, Holden HM. J Biol Chem. 2004;279:44023–44029. doi: 10.1074/jbc.M407755200. [DOI] [PubMed] [Google Scholar]
- 77.Koropatkin NM, Cleland WW, Holden HM. J Biol Chem. 2005;280:10774–10780. doi: 10.1074/jbc.M414111200. [DOI] [PubMed] [Google Scholar]
- 78.Pelissier MC, Lesley SA, Kuhn P, Bourne Y. J Biol Chem. 2010;285:27468–27476. doi: 10.1074/jbc.M109.095182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cupp-Vickery JR, Igarashi RY, Perez M, Poland M, Meyer CR. Biochemistry. 2008;47:4439–4451. doi: 10.1021/bi701933q. [DOI] [PubMed] [Google Scholar]
- 80.Olsen LR, Vetting MW, Roderick SL. Protein Sci. 2007;16:1230–1235. doi: 10.1110/ps.072779707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McCoy JG, Bitto E, Bingman CA, Wesenberg GE, Bannen RM, Kondrashov DA, Phillips GN., Jr J Mol Biol. 2007;366:830–841. doi: 10.1016/j.jmb.2006.11.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jin X, Ballicora MA, Preiss J, Geiger JH. EMBO J. 2005;24:694–704. doi: 10.1038/sj.emboj.7600551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Giraud MF, Naismith JH. Curr Opin Struct Biol. 2000;10:687–696. doi: 10.1016/s0959-440x(00)00145-7. [DOI] [PubMed] [Google Scholar]
- 84.Sivaraman J, Sauve V, Matte A, Cygler M. J Biol Chem. 2002;277:44214–44219. doi: 10.1074/jbc.M206932200. [DOI] [PubMed] [Google Scholar]
- 85.Mizanur RM, Zea CJ, Pohl NL. J Am Chem Soc. 2004;126:15993–15998. doi: 10.1021/ja046070d. [DOI] [PubMed] [Google Scholar]
- 86.Allard ST, Giraud MF, Whitfield C, Graninger M, Messner P, Naismith JH. J Mol Biol. 2001;307:283–295. doi: 10.1006/jmbi.2000.4470. [DOI] [PubMed] [Google Scholar]
- 87.Allard ST, Cleland WW, Holden HM. J Biol Chem. 2004;279:2211–2220. doi: 10.1074/jbc.M310134200. [DOI] [PubMed] [Google Scholar]
- 88.Vogan EM, Bellamacina C, He X, Liu HW, Ringe D, Petsko GA. Biochemistry. 2004;43:3057–3067. doi: 10.1021/bi035547f. [DOI] [PubMed] [Google Scholar]
- 89.Koropatkin NM, Holden HM. Acta Crystallogr D Biol Crystallogr. 2005;61:365–373. doi: 10.1107/S0907444904033876. [DOI] [PubMed] [Google Scholar]
- 90.Fruscione F, Sturla L, Duncan G, Van Etten JL, Valbuzzi P, De Flora A, Di Zanni E, Tonetti M. J Biol Chem. 2008;283:184–193. doi: 10.1074/jbc.M706614200. [DOI] [PubMed] [Google Scholar]
- 91.Webb NA, Mulichak AM, Lam JS, Rocchetta HL, Garavito RM. Protein Sci. 2004;13:529–539. doi: 10.1110/ps.03393904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Somoza JR, Menon S, Schmidt H, Joseph-McCarthy D, Dessen A, Stahl ML, Somers WS, Sullivan FX. Structure. 2000;8:123–135. doi: 10.1016/s0969-2126(00)00088-5. [DOI] [PubMed] [Google Scholar]
- 93.Ishiyama N, Creuzenet C, Miller WL, Demendi M, Anderson EM, Harauz G, Lam JS, Berghuis AM. J Biol Chem. 2006;281:24489–24495. doi: 10.1074/jbc.M602393200. [DOI] [PubMed] [Google Scholar]
- 94.Cook PD, Holden HM. Biochemistry. 2007;46:14215–14224. doi: 10.1021/bi701686s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cook PD, Holden HM. J Biol Chem. 2008;283:4295–4303. doi: 10.1074/jbc.M708893200. [DOI] [PubMed] [Google Scholar]
- 96.Gross JW, Hegeman AD, Gerratana B, Frey PA. Biochemistry. 2001;40:12497–12504. doi: 10.1021/bi011138c. [DOI] [PubMed] [Google Scholar]
- 97.Gross JW, Hegeman AD, Vestling MM, Frey PA. Biochemistry. 2000;39:13633–13640. doi: 10.1021/bi001963d. [DOI] [PubMed] [Google Scholar]
- 98.Melo A, Elliott WH, Glaser L. J Biol Chem. 1968;243:1467. [PubMed] [Google Scholar]
- 99.Gabriel O, Lindquis Lc. J Biol Chem. 1968;243:1479–1484. [PubMed] [Google Scholar]
- 100.Agnihotri G, Liu YN, Paschal BM, Liu HW. Biochemistry. 2004;43:14265–14274. doi: 10.1021/bi048841w. [DOI] [PubMed] [Google Scholar]
- 101.Johnson DA, Gassner GT, Bandarian V, Ruzicka FJ, Ballou DP, Reed GH, Liu HW. Biochemistry. 1996;35:15846–15856. doi: 10.1021/bi961370w. [DOI] [PubMed] [Google Scholar]
- 102.Miller VP, Thorson JS, Ploux O, Lo SF, Liu HW. Biochemistry. 1993;32:11934–11942. doi: 10.1021/bi00095a025. [DOI] [PubMed] [Google Scholar]
- 103.Lo SF, Miller VP, Lei Y, Thorson JS, Liu HW, Schottel JL. J Bacteriol. 1994;176:460–468. doi: 10.1128/jb.176.2.460-468.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Thorson JS, Liu HW. J Am Chem Soc. 1993;115:12177–12178. [Google Scholar]
- 105.Thorson JS, Liu HW. J Am Chem Soc. 1993;115:7539–7540. [Google Scholar]
- 106.Thorson JS, Lo SF, Liu HW. J Am Chem Soc. 1993;115:5827–5828. [Google Scholar]
- 107.Agarwal G, Rajavel M, Gopal B, Srinivasan N. Plos One. 2009;4:e5736. doi: 10.1371/journal.pone.0005736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dunwell JM, Culham A, Carter CE, Sosa-Aguirre CR, Goodenough PW. Trends Biochem Sci. 2001;26:740–746. doi: 10.1016/s0968-0004(01)01981-8. [DOI] [PubMed] [Google Scholar]
- 109.Dong C, Major L, Allen A, Blankenfeldt W, Maskell D, Naismith J. Structure. 2003;11:715–723. doi: 10.1016/s0969-2126(03)00098-4. [DOI] [PubMed] [Google Scholar]
- 110.Giraud MF, Leonard GA, Field RA, Berlind C, Naismith JH. Nat Struct Biol. 2000;7:398–402. doi: 10.1038/75178. [DOI] [PubMed] [Google Scholar]
- 111.Kantardjieff KA, Kim CY, Naranjo C, Waldo GS, Lekin T, Segelke BW, Zemla A, Park MS, Terwilliger TC, Rupp B. Acta Crystallogr D Biol Crystallogr. 2004;60:895–902. doi: 10.1107/S0907444904005323. [DOI] [PubMed] [Google Scholar]
- 112.Dong C, Major LL, Srikannathasan V, Errey JC, Giraud MF, Lam JS, Graninger M, Messner P, McNeil MR, Field RA, Whitfield C, Naismith JH. J Mol Biol. 2007;365:146–159. doi: 10.1016/j.jmb.2006.09.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Merkel AB, Major LL, Errey JC, Burkart MD, Field RA, Walsh CT, Naismith JN. J Biol Chem. 2004;279:32684–32691. doi: 10.1074/jbc.M404091200. [DOI] [PubMed] [Google Scholar]
- 114.Merkel AB, Temple GK, Burkart MD, Losey HC, Beis K, Walsh CT, Naismith JH. Acta Crystallogr D Biol Crystallogr. 2002;58:1226–1228. doi: 10.1107/s0907444902007382. [DOI] [PubMed] [Google Scholar]
- 115.Jakimowicz P, Tello M, Meyers CLF, Walsh CT, Buttner MJ, Field RA, Lawson DM. Proteins. 2006;63:261–265. doi: 10.1002/prot.20818. [DOI] [PubMed] [Google Scholar]
- 116.Davis ML, Thoden JB, Holden HM. J Biol Chem. 2007;282:19227–19236. doi: 10.1074/jbc.M702529200. [DOI] [PubMed] [Google Scholar]
- 117.Tello M, Jakimowicz P, Errey JC, Freel Meyers CL, Walsh CT, Buttner MJ, Lawson DM, Field RA. Chem Commun. 2006:1079–1081. doi: 10.1039/b515763c. [DOI] [PubMed] [Google Scholar]
- 118.Rosano C, Bisso A, Izzo G, Tonetti M, Sturla L, De Flora A, Bolognesi M. J Mol Biol. 2000;303:77–91. doi: 10.1006/jmbi.2000.4106. [DOI] [PubMed] [Google Scholar]
- 119.Rizzi M, Tonetti M, Vigevani P, Sturla L, Bisso A, Flora AD, Bordo D, Bolognesi M. Structure. 1998;6:1453–1465. doi: 10.1016/s0969-2126(98)00144-0. [DOI] [PubMed] [Google Scholar]
- 120.Somers WS, Stahl ML, Sullivan FX. Structure. 1998;6:1601–1612. doi: 10.1016/s0969-2126(98)00157-9. [DOI] [PubMed] [Google Scholar]
- 121.Bauer AJ, Rayment I, Frey PA, Holden HM. Proteins. 1992;12:372–381. doi: 10.1002/prot.340120409. [DOI] [PubMed] [Google Scholar]
- 122.Thoden JB, Holden HM. Biochemistry. 1998;37:11469–11477. doi: 10.1021/bi9808969. [DOI] [PubMed] [Google Scholar]
- 123.Holden HM, Rayment I, Thoden JB. J Biol Chem. 2003;278:43885–43888. doi: 10.1074/jbc.R300025200. [DOI] [PubMed] [Google Scholar]
- 124.Thoden JB, Heheman HD, Wesenberg G, Chapeau MC, Frey PA, Holden HM. Biochemistry. 1997;199736:6294–6304. doi: 10.1021/bi970025j. [DOI] [PubMed] [Google Scholar]
- 125.Read JA, Ahmed RA, Morrison JP, Coleman WG, Tanner ME. J Am Chem Soc. 2004;126:8878–8879. doi: 10.1021/ja0485659. [DOI] [PubMed] [Google Scholar]
- 126.Deacon AM, Ni YS, Coleman WG, Jr, Ealick SE. Structure. 2000;8:453–462. doi: 10.1016/s0969-2126(00)00128-3. [DOI] [PubMed] [Google Scholar]
- 127.Shaik MM, Zanotti G, Cendron L. Biochim Biophys Acta. 2011;1814:1641–1647. doi: 10.1016/j.bbapap.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 128.Kowatz T, Morrison JP, Tanner ME, Naismith JH. Protein Sci. 2010;19:1337–1343. doi: 10.1002/pro.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Koropatkin NM, Liu HW, Holden HM. J Biol Chem. 2003;278:20874–20881. doi: 10.1074/jbc.M301948200. [DOI] [PubMed] [Google Scholar]
- 130.Bhatt VS, Guo CY, Guan W, Zhao G, Yi W, Liu ZJ, Wang PG. Protein Sci. 2011;20:856–866. doi: 10.1002/pro.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Campbell RE, Mosimann SC, Tanner ME, Strynadka NCJ. Biochemistry. 2000;39:14993–15001. doi: 10.1021/bi001627x. [DOI] [PubMed] [Google Scholar]
- 132.Velloso LM, Bhaskaran SS, Schuch R, Fischetti VA, Stebbins CE. EMBO Rep. 2008;9:199–205. doi: 10.1038/sj.embor.7401154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Glaser L. Biochim Biophys Acta. 1960;41:534–536. doi: 10.1016/0006-3002(60)90056-1. [DOI] [PubMed] [Google Scholar]
- 134.Sommar KM, Ellis DB. Biochim Biophys Acta. 1972;268:590–595. doi: 10.1016/0005-2744(72)90356-7. [DOI] [PubMed] [Google Scholar]
- 135.Blankenfeldt W, Kerr ID, Giraud MF, McMiken HJ, Leonard G, Whitfield C, Messner P, Graninger M, Naismith JH. Structure. 2002;10:773–786. doi: 10.1016/s0969-2126(02)00770-0. [DOI] [PubMed] [Google Scholar]
- 136.Kubiak RL, Holden HM. Biochemistry. 2011;50:5905–5917. doi: 10.1021/bi200514b. [DOI] [PubMed] [Google Scholar]
- 137.Jornvall H, Persson B, Krook M, Atrian S, Gonzalez-Duarte R, Jeffery J, Ghosh D. Biochemistry. 1995;34:6003–6013. doi: 10.1021/bi00018a001. [DOI] [PubMed] [Google Scholar]
- 138.Kingston RL, Scopes RK, Baker EN. Structure. 1996;4:1413–1428. doi: 10.1016/s0969-2126(96)00149-9. [DOI] [PubMed] [Google Scholar]
- 139.Thoden JB, Holden HM. Biochemistry. 2011;50:1483–1491. doi: 10.1021/bi101871f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Eliot AC, Kirsch JF. Annu Rev Biochem. 2004;73:383–415. doi: 10.1146/annurev.biochem.73.011303.074021. [DOI] [PubMed] [Google Scholar]
- 141.Paiardini A, Bossa F, Pascarella S. Protein Sci. 2004;13:2992–3005. doi: 10.1110/ps.04938104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cook PD, Holden HM. Biochemistry. 2008;47:2833–2840. doi: 10.1021/bi702430d. [DOI] [PubMed] [Google Scholar]
- 143.Burgie ES, Holden HM. Biochemistry. 2007;46:8999–9006. doi: 10.1021/bi700751d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Noland BW, Newman JM, Hendle J, Badger J, Christopher JA, Tresser J, Buchanan MD, Wright TA, Rutter ME, Sanderson WE, Muller-Dieckmann HJ, Gajiwala KS, Buchanan SG. Structure. 2002;10:1569–1580. doi: 10.1016/s0969-2126(02)00879-1. [DOI] [PubMed] [Google Scholar]
- 145.Schoenhofen IC, Lunin VV, Julien JP, Li Y, Ajamian E, Matte A, Cygler M, Brisson JR, Aubry A, Logan SM, Bhatia S, Wakarchuk WW, Young NM. J Biol Chem. 2006;281:8907–8916. doi: 10.1074/jbc.M512987200. [DOI] [PubMed] [Google Scholar]
- 146.Badger J, Sauder JM, Adams JM, Antonysamy S, Bain K, Bergseid MG, Buchanan SG, Buchanan MD, Batiyenko Y, Christopher JA, Emtage S, Eroshkina A, Feil I, Furlong EB, Gajiwala KS, Gao X, He D, Hendle J, Huber A, Hoda K, Kearins P, Kissinger C, Laubert B, Lewis HA, Lin J, Loomis K, Lorimer D, Louie G, Maletic M, Marsh CD, Miller I, Molinari J, Muller-Dieckmann HJ, Newman JM, Noland BW, Pagarigan B, Park F, Peat TS, Post KW, Radojicic S, Ramos A, Romero R, Rutter ME, Sanderson WE, Schwinn KD, Tresser J, Winhoven J, Wright TA, Wu L, Xu J, Harris TJR. Proteins: Struct, Funct, Bioinf. 2005;60:787–796. doi: 10.1002/prot.20541. [DOI] [PubMed] [Google Scholar]
- 147.Larkin A, Olivier NB, Imperiali B. Biochemistry. 2010;49:7227–7237. doi: 10.1021/bi100805b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Thoden JB, Scha ffer C, Messner P, Holden HM. Biochemistry. 2009;48:1553–1561. doi: 10.1021/bi8022015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Burgie ES, Thoden JB, Holden HM. Protein Sci. 2007;16:887–896. doi: 10.1110/ps.062711007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Cook PD, Carney AE, Holden HM. Biochemistry. 2008;47:10685–10693. doi: 10.1021/bi801309q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Schoenhofen IC, McNally DJ, Vinogradov E, Whitfield D, Young NM, Dick S, Wakarchuk WW, Brisson JR, Logan SM. J Biol Chem. 2006;281:723–732. doi: 10.1074/jbc.M511021200. [DOI] [PubMed] [Google Scholar]
- 152.Jansonius JN. Curr Opin Struct Biol. 1998;8:759–769. doi: 10.1016/s0959-440x(98)80096-1. [DOI] [PubMed] [Google Scholar]
- 153.Soda K, Yoshimura T, Esaki N. Chem Rec. 2001;1:373–384. doi: 10.1002/tcr.1021. [DOI] [PubMed] [Google Scholar]
- 154.Percudani R, Peracchi A. EMBO Rep. 2003;4:850–854. doi: 10.1038/sj.embor.embor914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Michael DT. Arch Biochem Biophys. 2005;433:279–287. doi: 10.1016/j.abb.2004.09.037. [DOI] [PubMed] [Google Scholar]
- 156.Johan NJ. Curr Opin Struct Biol. 1998;8:759–769. doi: 10.1016/s0959-440x(98)80096-1. [DOI] [PubMed] [Google Scholar]
- 157.Hayashi H, Kagamiyama H. Biochemistry. 1995;34:9413–9423. doi: 10.1021/bi00029a017. [DOI] [PubMed] [Google Scholar]
- 158.Singh S, Mccoy JG, Zhang C, Bingman CA, Phillips GN, Thorson JS. J Biol Chem. 2008;283:22628–22636. doi: 10.1074/jbc.M800503200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Garcia IG, Stevenson CEM, Uson I, Meyers CLF, Walsh CT, Lawson DM. J Mol Biol. 2010;395:390–407. doi: 10.1016/j.jmb.2009.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Akey DL, Li S, Konwerski JR, Confer LA, Bernard SM, Anzai Y, Kato F, Sherman DH, Smith JL. J Mol Biol. 2011;413:438–450. doi: 10.1016/j.jmb.2011.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Carney AE, Holden HM. Biochemistry. 2011;50:780–787. doi: 10.1021/bi101733y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Burgie ES, Holden HM. Biochemistry. 2008;47:3982–3988. doi: 10.1021/bi800063j. [DOI] [PubMed] [Google Scholar]
- 163.Bruender NA, Thoden JB, Kaur M, Avey MK, Holden HM. Biochemistry. 2010;49:5891–5898. doi: 10.1021/bi100782b. [DOI] [PubMed] [Google Scholar]
- 164.Martin JL, McMillan FM. Curr Opin Struct Biol. 2002;12:783–793. doi: 10.1016/s0959-440x(02)00391-3. [DOI] [PubMed] [Google Scholar]
- 165.Schubert HL, Blumenthal RM, Cheng X. Trends Biochem Sci. 2003;28:329–335. doi: 10.1016/S0968-0004(03)00090-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Thakker DR, Boehlert C, Kirk KL, Antkowiak R, Creveling CR. J Biol Chem. 1986;261:178–184. [PubMed] [Google Scholar]
- 167.Zheng YJ, Bruice TC. J Am Chem Soc. 1997;119:8137–8145. [Google Scholar]
- 168.Chen H, Zhao Z, Hallis TM, Guo Z, Liu H-w. Angew Chem Int Ed Engl. 2001;40:607–610. [PubMed] [Google Scholar]
- 169.Thoden JB, Cook PD, Schaffer C, Messner P, Holden HM. Biochemistry. 2009;48:2699–2709. doi: 10.1021/bi802313n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Olivier NB, Imperiali B. J Biol Chem. 2008;283:27937–27946. doi: 10.1074/jbc.M801207200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Rangarajan ES, Ruane KM, Sulea T, Watson DC, Proteau A, Leclerc S, Cygler M, Matte A, Young NM. Biochemistry. 2008;47:1827–1836. doi: 10.1021/bi702032r. [DOI] [PubMed] [Google Scholar]
- 172.Thoden JB, Holden HM. Biochemistry. 2010;49:4644–4653. doi: 10.1021/bi1005738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kubiak RL, Holden HM. Biochemistry. 2012;51:867–878. doi: 10.1021/bi201650c. [DOI] [PubMed] [Google Scholar]
- 174.Hung M-N, Rangarajan E, Munger C, Nadeau G, Sulea T, Matte A. J Bacteriol. 2006;188:5606–5617. doi: 10.1128/JB.00306-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Raetz CR, Roderick SL. Science. 1995;270:997–1000. doi: 10.1126/science.270.5238.997. [DOI] [PubMed] [Google Scholar]
- 176.Dyda F, Klein DC, Hickman AB. Annu Rev Biophys Biomol Struct. 2000;29:81–103. doi: 10.1146/annurev.biophys.29.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Vetting MW, SdCLP, Yu M, Hegde SS, Magnet S, Roderick SL, Blanchard JS. Arch Biochem Biophys. 2005;433:212–226. doi: 10.1016/j.abb.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 178.Johnson HD, Thorson JS. J Am Chem Soc. 2008;130:17662–17663. doi: 10.1021/ja807557a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Hu Y, Al-Mestarihi A, Grimes CL, Kahne D, Bachmann BO. J Am Chem Soc. 2008;130:15756–15757. doi: 10.1021/ja8051415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Cooper R, Horan AC, Gentile F, Gullo V, Loebenberg D, Marquez J, Patel M, Puar MS, Truumees I. J Antibiot. 1988;41:13–19. doi: 10.7164/antibiotics.41.13. [DOI] [PubMed] [Google Scholar]
- 181.Ganguly AK, Girijavallabhan VM, Miller GH, Sarre OZ. J Antibiot. 1982;35:561–570. doi: 10.7164/antibiotics.35.561. [DOI] [PubMed] [Google Scholar]
- 182.Vey JL, Al-Mestarihi A, Hu Y, Funk MA, Bachmann BO, Iverson TM. Biochemistry. 2010;49:9306–9317. doi: 10.1021/bi101336u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kim CG, Lamichhane J, Song KI, Nguyen VD, Kim DH, Jeong TS, Kang SH, Kim KW, Maharjan J, Hong YS, Kang JS, Yoo JC, Lee JJ, Oh TJ, Liou K, Sohng JK. Arch Microbiol. 2008;189:463–473. doi: 10.1007/s00203-007-0337-3. [DOI] [PubMed] [Google Scholar]
- 184.Zhang H, White-Phillip JA, Melancon CE, 3rd, Kwon HJ, Yu WL, Liu HW. J Am Chem Soc. 2007;129:14670–14683. doi: 10.1021/ja0744854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Bruender NA, Thoden JB, Holden HM. Biochemistry. 2010;49:3517–3524. doi: 10.1021/bi100318v. [DOI] [PubMed] [Google Scholar]
- 186.Gunn JS, Ryan SS, Van Velkinburgh JC, Ernst RK, Miller SI. Infect Immun. 2000;68:6139–6146. doi: 10.1128/iai.68.11.6139-6146.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Gunn JS. J Endotoxin Res. 2001;7:57–62. [PubMed] [Google Scholar]
- 188.Gatzeva-Topalova PZ, May AP, Sousa MC. Biochemistry. 2004;43:13370–13379. doi: 10.1021/bi048551f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Williams GJ, Breazeale SD, Raetz CR, Naismith JH. J Biol Chem. 2005;280:23000–23008. doi: 10.1074/jbc.M501534200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Breazeale SD, Ribeiro AA, McClerren AL, Raetz CR. J Biol Chem. 2005;280:14154–14167. doi: 10.1074/jbc.M414265200. [DOI] [PubMed] [Google Scholar]
- 191.Bililign T, Shepard EM, Ahlert J, Thorson JS. Chembiochem. 2002;3:1143–1146. doi: 10.1002/1439-7633(20021104)3:11<1143::AID-CBIC1143>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 192.Ahlert J, Shepard E, Lomovskaya N, Zazopoulos E, Staffa A, Bachmann BO, Huang K, Fonstein L, Czisny A, Whitwam RE, Farnet CM, Thorson JS. Science. 2002;297:1173–1176. doi: 10.1126/science.1072105. [DOI] [PubMed] [Google Scholar]
- 193.Gao Q, Zhang C, Blanchard S, Thorson JS. Chem Biol. 2006;13:733–743. doi: 10.1016/j.chembiol.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 194.Simkhada D, Oh T-J, Pageni B, Lee H, Liou K, Sohng J. Appl Microbiol Biotechnol. 2009;83:885–895. doi: 10.1007/s00253-009-1941-8. [DOI] [PubMed] [Google Scholar]
- 195.Gatzeva-Topalova PZ, May AP, Sousa MC. Structure. 2005;13:929–942. doi: 10.1016/j.str.2005.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Gatzeva-Topalova PZ, May AP, Sousa NC. Biochemistry. 2005;44:5328–5338. doi: 10.1021/bi047384g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Warren MS, Marolewski AE, Benkovic SJ. Biochemistry. 1996;35:8855–8862. doi: 10.1021/bi9528715. [DOI] [PubMed] [Google Scholar]
- 198.Klein C, Chen P, Arevalo JH, Stura EA, Marolewski A, Warren MS, Benkovic SJ, Wilson IA. J Mol Biol. 1995;249:153–175. doi: 10.1006/jmbi.1995.0286. [DOI] [PubMed] [Google Scholar]
- 199.Gautam A, Rishi P, Tewari R. Appl Microbiol Biotechnol. 2011;92:211–225. doi: 10.1007/s00253-011-3512-z. [DOI] [PubMed] [Google Scholar]
- 200.Skarzynski T, Mistry A, Wonacott A, Hutchinson SE, Kelly VA, Duncan K. Structure. 1996;4:1465–1474. doi: 10.1016/s0969-2126(96)00153-0. [DOI] [PubMed] [Google Scholar]
- 201.Skarzynski T, Kim DH, Lees WJ, Walsh CT, Duncan K. Biochemistry. 1998;37:2572–2577. doi: 10.1021/bi9722608. [DOI] [PubMed] [Google Scholar]
- 202.Schönbrunn E, Eschenburg S, Krekel F, Luger K, Amrhein N. Biochemistry. 2000;39:2164–2173. doi: 10.1021/bi991091j. [DOI] [PubMed] [Google Scholar]
- 203.Yoon H-J, Lee SJ, Mikami B, Park H-J, Yoo J, Suh SW. Proteins: Struct, Funct, Bioinf. 2008;71:1032–1037. doi: 10.1002/prot.21959. [DOI] [PubMed] [Google Scholar]
- 204.Kim DH, Lees WJ, Kempsell KE, Lane WS, Duncan K, Walsh CT. Biochemistry. 1996;35:4923–4928. doi: 10.1021/bi952937w. [DOI] [PubMed] [Google Scholar]
- 205.Eschenburg S, Priestman M, Schonbrunn E. J Biol Chem. 2005;280:3757–3763. doi: 10.1074/jbc.M411325200. [DOI] [PubMed] [Google Scholar]
- 206.Jackson SG, Zhang F, Chindemi P, Junop MS, Berti PJ. Biochemistry. 2009;48:11715–11723. doi: 10.1021/bi901524q. [DOI] [PubMed] [Google Scholar]
- 207.Samland AK, Amrhein N, Macheroux P. Biochemistry. 1999;38:13162–13169. doi: 10.1021/bi991041e. [DOI] [PubMed] [Google Scholar]
- 208.Samland AK, Etezady-Esfarjani T, Amrhein N, Macheroux P. Biochemistry. 2001;40:1550–1559. doi: 10.1021/bi001490a. [DOI] [PubMed] [Google Scholar]
- 209.Eschenburg S, Kabsch W, Healy ML, Schönbrunn E. J Biol Chem. 2003;278:49215–49222. doi: 10.1074/jbc.M309741200. [DOI] [PubMed] [Google Scholar]
- 210.Takahata S, Ida T, Hiraishi T, Sakakibara S, Maebashi K, Terada S, Muratani T, Matsumoto T, Nakahama C, Tomono K. Int J Antimicrob Agents. 2010;35:333–337. doi: 10.1016/j.ijantimicag.2009.11.011. [DOI] [PubMed] [Google Scholar]
- 211.Richards MR, Lowary TL. Chembiochem. 2009;10:1920–1938. doi: 10.1002/cbic.200900208. [DOI] [PubMed] [Google Scholar]
- 212.Peltier P, Euzen R, Daniellou R, Nugier-Chauvin C, Ferrières V. Carbohydr Res. 2008;343:1897–1923. doi: 10.1016/j.carres.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 213.Pan F, Jackson M, Ma Y, McNeil M. J Bacteriol. 2001;183:3991–3998. doi: 10.1128/JB.183.13.3991-3998.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Zhang Q, Liu H-w. J Am Chem Soc. 2000;122:9065–9070. [Google Scholar]
- 215.Sun HG, Ruszczycky MW, Chang W-c, Thibodeaux CJ, Liu H-w. J Biol Chem. 2012;287:4602–4608. doi: 10.1074/jbc.M111.312538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Sanders DAR, Staines AG, McMahon SA, McNeil MR, Whitfield C, Naismith JH. Nat Struct Mol Biol. 2001;8:858–863. doi: 10.1038/nsb1001-858. [DOI] [PubMed] [Google Scholar]
- 217.Beis K, Srikannathasan V, Liu H, Fullerton SWB, Bamford VA, Sanders DAR, Whitfield C, McNeil MR, Naismith JH. J Mol Biol. 2005;348:971–982. doi: 10.1016/j.jmb.2005.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Gruber TD, Westler WM, Kiessling LL, Forest KT. Biochemistry. 2009;48:9171–9173. doi: 10.1021/bi901437v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Partha SK, Sadeghi-Khomami A, Slowski K, Kotake T, Thomas NR, Jakeman DL, Sanders DAR. J Mol Biol. 2010;403:578–590. doi: 10.1016/j.jmb.2010.08.053. [DOI] [PubMed] [Google Scholar]
- 220.Partha SK, van Straaten KE, Sanders DAR. J Mol Biol. 2009;394:864–877. doi: 10.1016/j.jmb.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 221.Gruber TD, Borrok MJ, Westler WM, Forest KT, Kiessling LL. J Mol Biol. 2009;391:327–340. doi: 10.1016/j.jmb.2009.05.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Chad JM, Sarathy KP, Gruber TD, Addala E, Kiessling LL, Sanders DAR. Biochemistry. 2007;46:6723–6732. doi: 10.1021/bi7002795. [DOI] [PubMed] [Google Scholar]
- 223.Soltero-Higgin M, Carlson EE, Gruber TD, Kiessling LL. Nat Struct Mol Biol. 2004;11:539–543. doi: 10.1038/nsmb772. [DOI] [PubMed] [Google Scholar]
- 224.Yuan Y, Bleile DW, Wen X, Sanders DAR, Itoh K, Liu H-w, Pinto BM. J Am Chem Soc. 2008;130:3157–3168. doi: 10.1021/ja7104152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Huang Z, Zhang Q, Liu H. Bioorg Chem. 2003;31:494–502. doi: 10.1016/j.bioorg.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 226.Zhang Q, Liu H. J Am Chem Soc. 2001;123:6756–6766. doi: 10.1021/ja010473l. [DOI] [PubMed] [Google Scholar]
- 227.Itoh K, Huang Z, Liu HW. Org Lett. 2007;9:879–882. doi: 10.1021/ol0631408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Barton WA, Biggins JB, Jiang J, Thorson JS, Nikolov DB. Proc Natl Acad Sci U SA. 2002;99:13397–13402. doi: 10.1073/pnas.192468299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Moretti R, Chang A, Peltier-Pain P, Bingman CA, Phillips GN, Jr, Thorson JS. J Biol Chem. 2011;286:13235–13243. doi: 10.1074/jbc.M110.206433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Aragão D, Fialho AM, Marques AR, Mitchell EP, Sá-Correia I, Frazão C. J Bacteriol. 2007;189:4520–4528. doi: 10.1128/JB.00277-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Sulzenbacher G, Gal L, Peneff C, Fassy F, Bourne Y. J Biol Chem. 2001;276:11844–11851. doi: 10.1074/jbc.M011225200. [DOI] [PubMed] [Google Scholar]
- 232.Beis K, Allard ST, Hegeman AD, Murshudov G, Philp D, Naismith JH. J Am Chem Soc. 2003;125:11872–11878. doi: 10.1021/ja035796r. [DOI] [PubMed] [Google Scholar]
- 233.Morrison JP, Read JA, Coleman WG, Jr, Tanner ME. Biochemistry. 2005;44:5907–5915. doi: 10.1021/bi050106c. [DOI] [PubMed] [Google Scholar]
- 234.Samuel J, Tanner ME. Biochimi Biophys Acta. 2004;1700:85–91. doi: 10.1016/j.bbapap.2004.03.017. [DOI] [PubMed] [Google Scholar]