

Abstract
The NS5B RNA-dependent RNA polymerase is an attractive target for the development of novel and selective inhibitors of hepatitis C virus replication. In order to identify novel structural hits as anti-HCV agents, we performed structure-based virtual screening of our in-house library followed by rational drug design, organic synthesis and biological testing. These studies led to the identification of pyrazolobenzothiazine scaffold as a suitable template for obtaining novel anti-HCV agents targeting the NS5B polymerase. The best compound of this series was the meta-fluoro-N-1-phenyl pyrazolobenzothiazine derivative 4a, which exhibited an EC50= 3.6 µM, EC90= 25.6 µM and CC50 > 180 µM in the Huh 9–13 replicon system, thus providing a good starting point for further hit evolution.
Keywords: Hepatitis C virus, RNA-dependent RNA polymerase, structure-based drug discovery, virtual screening, NS5B inhibitors, pyrazolobenzothiazines
Introduction
Hepatitis C virus (HCV) is a small, enveloped RNA virus that belongs to the Flaviviridae family. Worldwide, over 170 million people have become chronically infected with HCV;1 the overall prevalence is ~3%, with rates up to 20%, for example in Egypt.2 The virus may remain undetected for >15 years, a period during which transmission can occur. Due to intensive donor screening, transfusion-associated cases have become very rare, and most of the new infections can be attributed to high risk drug-related behavior (60%) and unsafe injection practices. Following a usually mild and subclinical acute infection, a chronic stage develops in 70–85% of the cases.2 Chronic HCV infection is one of the leading causes of liver cirrhosis and hepatocellular carcinoma (primary liver cancer), and the first indication for liver transplantation in industrialized countries.3 The standard treatment of HCV infection used to be a regimen of pegylated interferon alpha (pegIFN-α) in combination with ribavirin (RBV). During treatment, a high number of patients suffer from severe adverse effects and the sustained viral response rate unfortunately is rather low for individuals infected with genotype 1, the most prevalent genotype in North America, Europe and Asia. Recently, the first two direct-acting antiviral agents (DAAs) targeting the NS3/4A protease, telaprevir and boceprevir, have been approved by the Food and Drug Administration and European Medicines Agency for the treatment of patients infected with HCV genotype 1.4 Any of these two drugs, combined with pegINF-α and RBV in a triple combination therapy, induce a significantly improved sustained virological response rate both in treatment-naive patients as well as those that have a treatment history.5 However, despite improved response rates, combination treatment including either boceprevir or telaprevir shows long-term toxic effects, is associated with high costs, results in an increased pill burden on the patient, suffers from many drug interactions, and has a low barrier to resistance if used as monotherapy.5 Therefore, there is still a need for alternative treatment options, which are preferably pegINF-α and RBV-free and which target multiple steps of the virus replication cycle or different sites on a viral protein. In principle, every step of the HCV lifecycle, from receptor binding and endocytosis, to fusion, uncoating, translation, polyprotein processing, RNA replication, virion assembly, maturation, transport and release, can be a target for the development of new and selective anti-HCV drugs.6
Since the elucidation of the molecular genetics of the virus, the non-structural HCV proteins NS3/4A protease and NS5B polymerase have drawn most attention as drug targets. In contrast to the protease, drugs targeting the NS5B polymerase have yet to reach the market. NS5B is a RNA-dependent-RNA polymerase which catalyzes the synthesis of progeny viral RNA strands, and because of its apparent sequence and structural difference with human DNA and RNA polymerases, poses an attractive target for the development of selective inhibitors.7, 8 To date, a variety of nucleoside and non-nucleoside HCV polymerase inhibitors have been reported.9, 10 The non-nucleoside inhibitors (NNIs) can be divided into five groups based on the distinct allosteric binding site on the HCV polymerase they target, namely thumb site I (TSI), thumb site II, (TSII), palm site I (PSI), palm site II (PSII), and palm site III (PSIII).11 Because of the number of drugable pockets on NS5B and the lack of cross-resistance between inhibitors that target these different binding sites, multiple NS5B-targeting drugs may likely become part of future combination therapy. For many of these pockets, protein-ligand crystal structures, together with biochemical evidence, are described in literature, thus offering a solid base to set up exploratory projects aimed at identifying novel anti-NS5B chemotypes.
In continuation of our efforts towards identification of new chemical entities that inhibit HCV replication,12, 13 in this study we have focused our attention on the allosteric PSI, located at the junction of the thumb and palm domains and in close proximity to the active site. Several chemotypes that bind at this site have been reported in literature;10, 11 among them, hydroxyquinoline-benzothiadiazine (BTDZ) and hydroxyquinoline-benzothiazine (BTZ) classes represented by derivatives 1 and 2, respectively (Figure 1), are the most investigated PSI-NNIs with promising candidates in clinical development.14
Figure 1.

Chemical structures of 1 and 2 as representative of BTDZ and BTZ PSI-NNIs, respectively.
Given the scenario of PSI-NNIs as highly effective antiviral agents, we herein describe the rational design, synthesis, anti-NS5B activity and inhibitory effect on HCV replication of novel pyrazolobenzothiazine-based compounds in a quest to discover new selective anti-HCV agents.
Results and Discussion
Structure-based identification of the pyrazolobenzothiazine scaffold
The first goal of this study was to investigate in-depth the binding requirements for potent NS5B inhibitory activity at PSI. This aspect was important to gain useful information regarding the identification of new potential PSI-NNIs. Towards this, we focused our attention on the BTDZ and BTZ classes (e.g., compounds 1 and 2) (Figure 1) which, beside being potent inhibitors in both NS5B polymerase and cellular replicon system assays, are well characterized from the structure-activity relationships (SAR) standpoint.14 Several studies have reported detailed structural analysis of the ligand/NS5B contacts for different inhibitors of the BTDZ and BTZ series.14 All these inhibitors share similar contacts with the NS5B residues, although the more potent compounds establish some additional hydrogen bond interactions. Of note, all of the available X-ray structures reveal some highly conserved water molecules which mediate the protein-inhibitor hydrogen-bond interactions.
Using as reference the co-crystal structure of NS5B with compound 2,15 we have summarized the key binding requirements as follows (Figure 2): i) a hydrophobic group (HYD-1) occupying a pocket mainly defined by residues Met414, Gly410, Gln446, Asn411 and Tyr415; ii) a N-alkyl/aryl lipophilic tail (HYD-2) filling a deep pocket shaped by Tyr448, Cys366, Pro197, Tyr415, Leu384, Arg200 and Ser368; iii) a hydrogen bond acceptor group (HBA-1) involved in a direct interaction with the backbone N-H of Tyr448 and in a water-mediated interaction with Gly449; iv) one of the sulfone oxygens of the BTZ/BTDZ moiety (HBA-2) establishing a water-mediated interaction with Ser556 or Ser288; v) the phenyl portion of the benzothiadiazine ring system (ETF) making an edge-to-face aromatic interaction with Phe193; and vi) the methylsulfonamide group (HBD-HBA) making strong interactions with Asn291 and Asp318. Moreover, one of the sulfonamide oxygen atoms of the methylsulfonamide moiety forms a third H-bond network with residue Ser288 via the structural water molecule mentioned in point iv.
Figure 2.

Schematic representation of the interactions between NS5B polymerase and compound 2 (PDB ID 3G86) as representative of BTZ/BTDZ inhibitors. NS5B residues lying within a distance of 4 Å from the bound ligand are shown and color coded as follows: red-acidic; green-hydrophobic; purple-basic; cyan-polar; white-Gly. Waters are displayed as gray circles, while interactions between ligand atoms and protein residues are marked with lines: solid pink, H-bonds to the protein backbone; dotted pink, H-bonds to protein side chains; green, π-π stacking interactions. The six chemical features of PSI-NNIs are highlighted as well: HBA, hydrogen bond acceptor; HBD, hydrogen bond donor; HYD, hydrophobic regions; ETF, group involved in edge-to-face interaction.
Taking into account the above observations, structure-based drug discovery (SBDD) approaches were performed to discover novel anti-NS5B chemotypes from an in-house library of 2692 molecules synthesized and/or published by our research group. The compound collection was prepared for docking using the LigPrep process16 and submitted to a structure-based virtual screening workflow against the crystal structure of NS5B in complex with inhibitor 2 (PDB ID 3G86) by means of the Glide software.17 All water molecules found in the crystal structure were removed, with the exception of the conserved water molecules involved in direct interactions with the ligand (Figure 2). In order to validate the docking performance, test calculations using 2 were carried out, extracting the ligand from the corresponding NS5B complex and then docking it back into the allosteric pocket of the enzyme crystal structure. The best docking pose of 2 (XP GScore = −9.4 kcal/mol) agreed well with its experimental binding conformation, with a root-mean square (rms) deviation value of 0.3 Å.
The top-ranked compounds were visually inspected searching for virtual hits endowed with high docking score and also able to create the key interactions with the PSI residues. Unfortunately, none of the in-house compounds possessed both these characteristics. However, our attention was attracted by the pyrazolobenzothiazine-3-carboxymethyl ester derivative 3a18 (rank position #94, XP GScore = −5.8 kcal/mol) (Figure 3), a compound already reported by our research group as an intermediate in the preparation of anti-inflammatory agents.19
Figure 3.

A) Simplified 2D representation of the predicted interactions between compound 3a and NS5B amino acids (color legend as in Figure 2). B) Docking pose of compound 3a (green) together with the experimental position of BTZ derivative 2 (violet). Water molecules are displayed in cyan, whereas hydrogen bonds are represented as yellow dashed lines. Both pictures show protein residues within 4 Å of the docked compound.
Interestingly, compound 3a was predicted to bind in a fashion somewhat similar to that of known PSI-NNIs (Figure 3), establishing some of the already mentioned key ligand-NS5B interactions (Figure 2). The sulfonyl function of the molecule, corresponding to the HBA-1 feature, was engaged in a direct hydrogen bonding interaction with Tyr448; the same moiety also established a water-mediated interaction with Gly449. The previously described hydrophobic features HYD-1 and HYD-2, were represented here by the phenyl ring of the pyrazolobenzothiazine nucleus and the N-phenyl tail, respectively. Of note, the pyrazole ring showed an additional π−π interaction with the side chain of Tyr448.
Computer-driven design of pyrazolobenzothiazine derivatives
The interaction between compound 3a and NS5B polymerase was evaluated using surface plasmon resonance (SPR), while its effect on the catalytic activity of the enzyme was determined in a functional assay and its effect on transcription was evaluated in a cell-based HCV subgenomic replicon system. The analysis of the interaction with NS5B (Con-1 strain, genotype 1b) indicated that the affinity was very low with a dissociation equilibrium constant KD > 300 µM. In the functional anti-NS5B (CΔ21, genotype 1b) assay, 50 µM of the compound resulted in 11% inhibition. A preliminary screening analysis for its ability to inhibit HCV replication (genotype 1b), using a HCV subgenomic replicon containing human hepatoma cells (Huh 5–2), showed that compound 3a exhibited ~40% inhibition of HCV RNA replication at non-toxic concentration. Albeit the modest biological results obtained for 3a did not allow to classify it as hit, the interesting information raised from the proposed binding conformation suggested that this chemotype was worthy of further optimization. Therefore, taking into account the key chemical features of PSI-NNIs (Figure 2), we planned suitable chemical modifications of derivative 3a with the objective of obtaining potent NS5B small-molecule inhibitors.
Since it was not straight forward to synthesize molecules possessing the six desired features at the same time, we decided to design a series of pyrazolobenzothiazine-based compounds (4a-j, 5a and 6a), (Scheme 1 and Table 1) which could potentially contain all the binding requirements except HBA-2. In particular, superimposition of the docked conformation of compound 3a on the BTZ inhibitor 2-NS5B co-crystal structure (Figure 3) suggested replacement of methyl with para-methanesuphonamidephenyl moiety from the C-3 carboxylic group. Thus, derivative 4a was designed where the ester linkage was replaced by the more chemically stable amide bridge.
Scheme 1a.

aReagents and conditions: (i) ArNHNH2 HCl, MeOH, reflux; (ii) 10% acq NaOH, MeOH, reflux; (iii) a) SOCl2, reflux; b) ArNH2 or ArOH, Et3N, dry DMF, 40 °C; (iv) H2, Raney-Ni, DMF, rt, atm pressure.
Table 1.
Biological results obtained for derivatives 3a, 4a-j, 5a, and 6a.
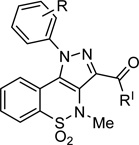 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| cpds | R | R' | SPR on NS5B |
NS5B functional assay |
Replicon assay on Huh 5-2 cells |
LLE (KD)g | LLE (IC50)h | |||
| KD (µM)a |
IC50 (µM)b |
EC50 (µM)c |
EC90 (µM)d |
CC50 (µM)e |
SIf | |||||
| 3a | 3-F | OMe | >300 | NDi, j | 172 ± 10 | NDi | >323 | 2 | - | - |
| 4a | 3-F | 75±12 | 21.0±2.8 | 7.5 ± 2.5 | 42 ± 2 | >370 | >49 | 1.85 | 2.40 | |
| 4b | 2-F | 162±63 | 14.2±0.6 | 6.4 ± 0.2 | NDi | >231 | >36 | 1.61 | 2.66 | |
| 4c | 4-F | NDi | 3.9±0.3 | 23 ± 13 | NDi | >231 | >10 | - | 3.11 | |
| 4d | H | 53±6 | 19.8±3.6 | 14 ± 1 | NDi | >239 | >17 | 2.18 | 2.61 | |
| 4e | 3-NO2 | 14±6 | NDi, j | >220 | >220 | >220 | ≥1 | 3.41 | - | |
| 4f | 2-NO2 | 35±3 | 20±0.4 | 17.8 ± 0.1 | NDi | >220 | >12.4 | 2.84 | 3.09 | |
| 4g | 4-NO2 | NDi | 7.7±1.3 | 7.9 ± 1.0 | NDi | 55 ± 8 | 7.0 | - | 3.80 | |
| 4h | 3-NH2 | 163±8 | NDi, j | 9.2 ± 0.9 | NDi | 20 ± 6 | 2.2 | 2.52 | - | |
| 4i | 2-NH2 | 106±42 | 39.9±1.7 | 26 ± 2 | NDi | >232 | >9 | 2.66 | 3.08 | |
| 4j | 4-NH2 | 165±16 | 18.2±2.1 | 12 ± 1 | NDi | 25 ± 5 | 2 | 2.53 | 3.48 | |
| 5a | 3-F | 161±103 | NDi, j | 19 ± 2 | NDi | >270 | >14 | 0.99 | - | |
| 6a | 3-F | 63±8 | NDi, j | 88 ± 10 | NDi | >185 | >2 | 1.87 | - | |
KD= Dissociation equilibrium constant. At this concentration 50% of all binding site are occupied. Values as averages and standard deviation (SD) from duplicate or triplicate runs.
IC50 = concentration of compound that inhibits 50% enzyme activity in vitro. The reported values represent the means ± SD of data derived from two independent experiments performed in duplicate.
EC50 = the effective concentration required to inhibit virus induced cytopathic effect by 50%. The reported values represent the means ± SD of data derived at least from three independent experiments.
EC90 = the effective concentration required to inhibit virus induced cytopathic effect by 90%. The reported value represents the means ± SD of data derived at least from three independent experiments.
CC50 = is the concentration required to reduce the bioreduction of MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxy-phenyl)-2-(4-sulfophenyl)-2H-tetrazolium) into formazan by 50%. The reported value represents the means ± SD of data derived at least from three independent experiments.
SI = selectivity index (ratio of CC50 to EC50).
LLE(KD) = pKD – predicted logP.
LLE(IC50) = pIC50 – predicted logP.
ND = not determined.
Compounds that did not reach the 50% inhibition and show a maximum % inhibition @ 50 µM: 3a 10.6%, 4e 33.7%, 4h 45.8%, 6a 21.4%, 5a 31.5%.
Starting from compound 4a, the deep hydrophobic cavity occupied by the chemical feature HYD-2 was explored by moving the fluorine atom from meta to ortho (4b) and para (4c) positions, or by designing the unsubstitued N-1 phenyl derivative 4d. The fluorine atom was also replaced with strong electron withdrawing (i.e. NO2, 4e-g) or electron donating (i.e. NH2, 4h-j) substituents. Finally, the methanesuphonamide group was substituted by an amino group obtaining compound 5a. This change was aimed at assessing the impact of the mesyl moiety in this series of compounds. Finally, the direct optimized version of compound 3a, where the methyl fragment of the C-3 carboxylic group was substituted by the para-methanesuphonamidephenyl moiety, was also planned (6a).
Before starting with the chemical synthesis of the new pyrazolobenzothiazine-based compounds, we sought to validate our hypothesis by performing automated docking studies of the designed derivatives using the computational protocol described above. The docking results predicted that 4a-j, 5a and 6a may potentially interact with the NS5B residues of PSI in a fashion similar to known NNIs, as discussed below. For instance, the proposed binding mode of pyrazolobenzothiazine derivative 4a (XP GScore = −7.4 kcal/mol) is shown in Figure 4, where a schematic representation of the predicted NS5B-compound 4a interactions is also illustrated.
Figure 4.

A) Schematic representation of the predicted interactions between compounds 4a and the NS5B residues (color legend as in Figure 2). B) Docked conformation of compound 4a (green) together with the X-ray conformation of BTZ derivative 2 (violet). Water molecules are displayed in cyan, while hydrogen bonds are represented as yellow dashed lines. Both figures illustrated NS5B residues lying within a distance of 4 Å from the docked compound.
Beside the ligand-NS5B interactions already highlighted for the parent compound 3a (Figure 3), derivative 4a was able to establish the additional desired π−π (EFT) and three-point (HBD-HBA) interactions through its para-methanesuphonamide phenyl group, in agreement with the SBDD. Further, since NS5B harbors multiple allosteric sites, we next investigated the binding mode of the rationally designed pyrazolobenzothiazine derivatives at the other pockets. For this purpose, molecular docking studies at PSII (PDB ID 3FQL),20 PSIII (PDB ID 3LKH),21 TSI (PDB ID 2BRK)12b, 22 and TSII (PDB ID 3FRZ)12b, 23 were performed. At first, the ability of the docking setup to reproduce the co-crystallized pose of each inhibitor within the respective NS5B site was validated. The pyrazolobenzothiazine derivatives were then docked into the allosteric pockets, and the visual inspection of the results revealed their inability to properly occupy those binding sites.
Together, this data suggested that the newly designed compounds 4a-j, 5a and 6a may bind at PSI, thus providing a rationale to synthesize and test these molecules as potential inhibitors of NS5B polymerase and HCV replication.
Synthesis of pyrazolobenzothiazine derivatives
The synthetic route employed for the preparation of targets compounds 4a-j, 5a and 6a is depicted in Scheme 1. Starting from key intermediate 7,19 the pyrazolobenzothiazine-3-carboxymethyl esters 3a,19 8b-c, 8d,19 8e-g were prepared by condensation with the appropriate commercially available phenylhydrazines hydrochloride, in MeOH at reflux. According to our previous works19, 24 this reaction led to the formation of the sole N-1 phenylpyrazolobenzothiazine regioisomers. In fact, in all 1H NMR spectra of derivatives 3a, and 8b-g the presence of H-9 proton shielded by the anisotropic effect of N-1 phenyl ring is a proof consistency of the assigned structure. In order to unambiguously demonstrate the reaction regioselectivity, a bidimensional NOESY spectrum was performed for derivative 8e, as an exhaustive example. The results indicated two diagnostic interactions: the first one was observed between the H-9 proton of pyrazolobenzothiazine nucleus and the H-2' proton of the N-1 phenyl ring whereas the second one occurred between the same H-9 proton and the H-4' proton (Figure 5). No interactions were found between the N-1 phenyl ring and the C-3 methyl ester.
Figure 5.

NOESY experiments for 8e showing two main interactions: H-9→H-2' and H-9→H-4'.
The esters intermediates 3a,19 and 8b-g were hydrolyzed under basic conditions at reflux to give the pyrazolobenzothiazine-3-carboxylates 9a-c, 9d,19 and 9e-g. They were then converted in the corresponding carbonyl chloride, under reflux of SOCl2, and immediately reacted with N-(4-aminophenyl)methanesulfonamide,25 using Et3N as scavenger, to afford the target amide derivatives 4a-g. On the other hand, nitro derivatives 4e-g were reduced to the corresponding amino targets 4h-j employing H2 flux and Raney-Ni as catalyst.
Using the same procedures, the carbonyl chloride of 9a was reacted with N-(4-hydroxyphenyl)methanesulfonamide26 to give the target ester derivative 6a; intermediate 9a was also converted into amino derivative 5a by reaction with 4-nitroaniline followed by catalytic reduction with H2/Raney-Ni.
Biochemical and biological studies
The synthesized compounds 4a-j, 5a and 6a were evaluated by a combination of three biochemical and biological methods to determine the binding affinity for NS5B polymerase, the ability to inhibit the enzyme and the anti-HCV activity. The SPR biosensor analysis is a direct binding method that provides information on the interaction between the compounds and the enzyme; the affinity is an important SAR characteristic, but does not provide the functional information that is obtained from the enzyme activity-based inhibition or replicon assays. The latter is the most complex assay of the three, providing a measure not only of effects on polymerase activity but other features related to viral replication. Ideally, a good compound shows a low KD, IC50 and EC50 values. In recent years, ligand efficiency indices have been shown to be useful tools in reviewing the potency data, where potency is either a measure of binding (KD) or biological activity (Ki, IC50 or EC50). Ligand efficiency (LE)27 refers to the potency of a compound averaged by its non-hydrogen atom count and is used to try to avoid unnecessary increases in molecular weight as a lever to increase potency. Although LE is probably the most widely used and intuitive metric, it does not take into account lipophilicity. Considering the crucial role of lipophilicity in drug discovery, the concept of ligand-lipophilic efficiency (LLE or LipE) has recently become a popular measure linking potency and lipophilicity in an attempt to estimate druglikeness.28 For a given compound, LLE is defined as pPotency – logP or logD, where pPotency is –log(Potency), logP is the octanol/water partition coefficient and logD is the distribution coefficient. LogP value of a substance is most appropriate for neutral (un-ionized) compounds, while logD (the pH-dependent lipophilicity descriptor) should be used when working with ionizable compounds. Since our pyrazolobenzothiazines did not contain a functionality likely to be ionized at the pH of the used assays, logP value was chosen as the relevant parameter. In this study, LLE values were calculated to support the SAR understanding and to evaluate the druglikeness of the compounds by employing logP calculated using the program QikProp (see also Supporting Information Table 1S).29
These data are reported in Table 1.
SPR interaction analysis
The interaction between the synthesized compounds and HCV-NS5B genotype 1b was assayed using a SPR-based biosensor as reported in Experimental Section.
No compounds were found to affect the immobilized enzyme irreversibly or linger in the instrument flow system to potentially interfere with subsequent measurements. An appropriate level of structural integrity and functionality of the detection surface was maintained throughout the experiments, as determined by injections of a positive control substance (filibuvir, PF00868554).23
The apparent affinities were determined for all compounds except two, i.e. 4c and 4g. The results from derivative 4c suggested that this compound might interact with the enzyme, but possible solubility issue prevented a proper determination of the affinity. On the other hand, there was no detectable interaction between compound 4g and the target.
Starting from pyrazolobenzothiazine 3a, the introduction of the para-methanesuphonamidephenyl moiety at the C-3 position permitted an increased affinity to be observed for almost all the synthesized compounds (KD values ranged from 14 to 165 µM and corresponding LLE parameters from 0.99 to 3.41). In particular, the analogues bearing an amide or ester linkage (compounds 4a and 6a, respectively) exhibited increased NS5B affinity when compared to the parent compound 3a. Deletion of the methansulfonyl fragment negatively affected the binding affinity, as highlighted by comparing both KD and LLE values of derivative 4a (KD = 75 µM, LLE = 1.85) and 5a (KD = 161 µM and LLE = 0.99). Both these observations were in agreement with the rationale of our design. NS5B affinity was also influenced by the position of the substituent on the N-1 phenyl ring. Indeed, SPR data combined with lipophilic efficiency index analysis revealed that in the fluoro and nitro subsets, the meta substitution (4a and 4e) was preferred over the ortho substitution (4b and 4f). In particular, meta-nitro N-1-phenyl derivative 4e (KD = 14 µM and LLE = 3.41) displayed both the best affinity and LLE value within this pyrazolobenzothiazine series.
NS5B polymerase assay
The anti-NS5B RdRp activity of all the synthesized compounds was evaluated by the standard primer-dependent elongation reaction employing poly rA/U12 template-primer and recombinant HCV NS5BCΔ21 according to previously described procedures.30 Compound 131 was included as a reference NS5B inhibitor and yielded an IC50 value of 0.085 µM 0.006 µM, consistent with previously reported data.32 All compounds were first screened at 50 µM concentration to identify NS5B inhibitor candidates exhibiting ≥50% inhibition of NS5B RdRp activity at this concentration. This investigation led to the identification of eight compounds (4a-4d, 4f, 4g, 4i and 4j) satisfying this criterion, while four compounds (4e, 4h, 5a and 6a) exhibited, at the same concentration, ~21–46% inhibition of NS5B RdRp activity. The eight selected compounds were further screened for their NS5B inhibition potency and yielded IC50 values ranging from 3.9 µM to 40 µM, with corresponding LLE values ranging from 2.4 to 3.8 (Table 1). The enzymatic data, in agreement with the SPR analysis, thus highlighted that the rational introduction of the para-methanesuphonamidephenyl moiety at the C-3 position was a valid strategy to increase the inhibitory activity of the starting compound 3a; which did not reach 50% inhibition but only showed 11% inhibition at 50 µM. Furthermore, the enzyme inhibition results, similarly to that observed by the SPR data analysis, clearly showed how the deletion of the methansulfonyl resulted detrimental in this series of pyrazolonbenzothiazines (compare 4a with an IC50 value of 21 µM versus 5a with 31.5% inhibition at 50 µM).
Looking at both the NS5B inhibition data and LLE values, further considerations can be proposed. The position of the substituent on the N-1 phenyl ring influenced the anti-NS5B activity. In particular, in this series the substitution at the para position (4c, 4g and 4j) was preferred over the ortho (4b, 4f and 4i) and meta (4a, 4e and 4h) positions. For instance, para-fluoro N-1-phenyl pyrazolobenzothiazine 4c displayed the most potent activity in this series with an IC50 of 3.9 µM and LLE of 3.11, while its ortho (4b) and meta (4a) analogues showed IC50 of 14.2 µM and 21 µM, and LLE of 2.66 and 2.40, respectively. These data clearly suggest that, although our compounds exhibited lower anti-NS5B potency compared to the representative BTDZ 1, the pyrazolonbenzothiazine nucleus merits further investigation representing a new chemical scaffold unrelated to any known class of NS5B NNIs.
Anti-HCV activity
The selective antiviral activity of the target compounds (4a-j, 5a, and 6a) was evaluated in the HCV genotype 1b subgenomic replicon system (Huh 5-2). From the dose-response curves, the concentration of the compound that inhibits virus replication by 50% (i.e., EC50) as well as the concentration of compound that reduces host cell metabolism by 50% (i.e., CC50) was derived. These values in turn allowed us to calculate the selectivity index (i.e., SI = CC50/EC50), a measure for the therapeutic potential of the compound in the assay system (Table 1).
A preliminary analysis of the data obtained from the replicon assay apparently showed that, with the exception of derivatives 4g, 4h, and 4j, the pyrazolobenzothiazines herein reported did not possess any significant cytotoxicity. Furthermore, the nontoxic compounds were able to inhibit HCV replication in a micromolar range.
However, even though an EC50 may be obtained from the dose-response curve, following interpretation of the dose-response curve, compounds were only considered as selective inhibitors in the replicon assay when significant inhibition (>70%) of virus RNA replication was observed at concentrations that did not exert an adverse effect on host cell metabolism. The antiviral response observed for the compounds that did not match these criteria most likely had to be attributed to a pleiotropic or aspecific effect of the compound on the host cell metabolism. In addition, the cell and monolayer morphology of the selected compounds was checked microscopically, as the microtiter plate readout data did not necessarily allow to identify minor changes that can be detected visually. Even though the dose-response curves (data not shown) of several compounds (derivatives 4a-c and 5a) matched the hit selection criteria, following microscopic inspection, it was apparent that the compounds induced changes at the level of cell and monolayer morphology. Even though these observations pointed towards an aspecific or pleiotropic effect being responsible for the observed anti-HCV activity, it however cannot be excluded from this assay that the compounds in addition did not inhibit HCV replication as well.
Taking into account the above general considerations, some preliminary SAR can be proposed. A good activity coupled with no apparent cytotoxic effects was maintained within the N-1-fluorophenyl subset, with compound 4a being the most interesting derivative (EC50 = 7.5 µM) since notably it was also able to reach 90% inhibition of HCV replication at non-toxic concentration (EC90 = 42 µM). To better evaluate compound 4a as true anti-HCV agent, it was validated at the level of viral RNA replication in the Huh 9–13 replicon system by means of real-time quantitative RT-PCR. In this assay, derivative 4a showed a selective antiviral effect (EC50 = 3.6 µM; EC90 = 25.6 µM) without any cytostatic effect (CC50 > 180 µM, SI > 50). A dose-response curve matching the hit selection criteria was obtained, suggesting selective anti-HCV activity. However, based on the microscopic observations, no final conclusion can be drawn.
Highlights from the LLE analysis
The introduction of the concept of LE has been useful as it provides a measure of the binding energy of the ligand per atom. This has been extended to also account for lipophilic contributions, in the LLE metric. The magnitude of LLE is thought to be correlated with druggability and it can be used to select and prioritize compounds with high affinity per atom and low lipophilicity. The new pyrazolobenzothiazine derivatives were therefore evaluated using this metric. The LLE calculated from KD and IC50 values was useful for ranking and identified compounds 4e (3.4) and 4g (3.80) as the ones with the highest LLE, respectively (see also Supporting Information Figure S1 and S2). These results can be compared to the calculated mean LLE values for a large and diverse data set representing different stages of drug discovery recently published by Tarcsay et al.33 Although LLE greater than 5 is usually considered optimal for a promising drug candidate, LLE values of 2.5 were found for both fragment and HTS hits, while fragment and HTS leads showed higher values of 3.9 and 3.6, respectively. The study further reported LLE values greater than 5 for compounds that entered phase II trials and marketed drugs. In our work, several synthesized derivatives had LLE values greater than 2.5 and low logP values (see Supporting Information Figure S1 and S2), thereby suggesting that the pyrazolobenzothiazine chemotype is noteworthy of further investigation.
Another surprising finding was that, although 4a was the anti-HCV hit compound validated by the strict criteria of the replicon system, curiously this pyrazolobenzothiazine did not rank among the derivatives which exhibited the highest LLE values based on calculations using either KD or IC50. In contrast, both compound 4e and 4g that showed the highest LLE values were not detected as genuine anti-HCV agents in the replicon system. This may be an effect of the higher complexity of the replicon assay compared to the SPR and enzyme inhibition assays. It is influenced by the pharmacokinetic properties of a compound (e.g. the ability to enter the cell) and its capability to interact also with a multitude of other cell components. Even though the concept of LLE was originally introduced specifically using pIC50 and pKi presumably to avoid the complication due to the use of cell system, it was later expanded to include pEC50.34 LLE for 4a using its EC50 data determined by RT-PCR assay was 3.21, thus ranging between the LLE values for hit compounds (mean value of 2.5) and lead compounds (mean values of 3.6–3.9).
Additional pyrazolobenzothiazine analogues will have to be explored to identify the features that are associated with high potency in the replicon system and that give higher LLE.
Conclusions
Although a wide variety of compounds have been reported as NS5B inhibitors, drugs active against this enzyme have not yet been approved by the FDA. Thus, there is still a compelling need to identify new anti-HCV chemotypes acting on NS5B polymerase. In this context, structure-based virtual screening of our in-house library led to the identification of the pyrazolobenzothiazine 3a as a suitable starting point for the rational design of new potential NS5B inhibitors. A simple synthetic route for novel pyrazolobenzothiazines bearing a para-methanesuphonamidephenyl moiety at C-3 position has been developed, leading to the preparation of twelve derivatives. Biochemical and biological testing confirmed that our strategy was successful in the search for a new anti-HCV chemotype most likely targeting the NS5B polymerase. In summary, the results showed that 4a represented the best hit within this first series of pyrazolobenzothiazines, having relatively good affinity and inhibitory effects in both enzymatic and replicon assays. Further investigations are warranted and are currently in progress to develop pyrazolobenzothiazine derivatives with improved activity and to better elucidate their antiviral mechanism of action.
Experimental section
Molecular Modeling
The crystal structure of NS5B RdRP complexed with inhibitor 2 (PDB ID 3G86) was retrieved from the RCSB Protein Data Bank and used for our modeling studies on PSI. Water molecules, except those involved in mediating hydrogen bonds between the ligands and the protein were deleted. Schrodinger Protein Preparation Wizard,35 a workflow designed to ensure chemical correctness and to optimize a protein structure for further analysis, was then used to obtain a satisfactory starting structure. In particular, hydrogen atoms were added, and bond orders and charges were assigned; the orientation of hydroxyl groups on Ser, Thr and Tyr, the side chains of Asn and Gln residues, and the protonation state of His residues were optimized. Steric clashes were relieved by performing a small number of minimization steps, not intended to minimize the system completely. In our study, the minimization (OPLS 2500 force field) was stopped when the RMSD of the non-hydrogen atoms reached 0.30 Å.
Docking studies were performed using the software Glide version 5.6.17 The prepared protein structure was used to generate the receptor grid, which was centered on the crystallographic positions of PSI-NNI 2.
Prior to docking experiments, the in-house library and the designed compounds (4a-j, 5a and 6a) were built using the Schrodinger Maestro interface36 and then submitted to the LigPrep utility,16 which rapidly produces low energy 3D structures taking into account ionization states, tautomers, sterochemistries, and ring conformations at the desired pH. For our study, a pH range of 6–8 was set.
The PSI-NNI 2 was instead extracted from the complex with NS5B, and its crystallographic coordinates were transformed in order to avoid biasing the docking toward the known NS5B- bound conformation. After modeling the ligand enolic moiety as anion due to its ionization at physiological pH, the compound was submitted to Polak–Ribiere conjugate gradient minimization [0.0005 kJ/(Å mol) convergence].
All compounds were flexibly docked in a stepwise manner with Glide SP (Standard Precision) and XP (Extra Precision) as scoring functions. Docking experiments were performed using a 0.80 factor to scale the VdW radii of the nonpolar ligand atoms with partial atomic charge less than 0.15 (absolute value). Poses were discarded as duplicates when rms deviation in the ligand-all atoms was less than 1.5 Å and maximum atomic displacement was less than 2.0 Å. Three bound conformations per ligand were retained from SP docking, and these were refined, rescored and minimized using GlideXP. The best XP scoring bound conformation for each ligand was retained. Docking experiments at PSII (PDB ID 3FQL),20 PSIII (PDB ID 3LKH),12b, 21 TSI (PDB ID 2BRK)12b, 22 and TSII (PDB ID 3FRZ)23 were performed using the same protocol described for PSI.
Figure 2, 3A and 4A were generated using the Ligand Interaction Diagram tool of Maestro GUI,36 whereas Figure 3B and 4B were prepared using the software PyMOL.37
LogP values of the pyrazolobenzothiazines were computed by using Schrodinger QikPro program29 (descriptor QPlogPo/w), and the LLE calculations were performed by using Excel spreadsheet.
Chemistry
All starting materials were commercially available, unless otherwise indicated. Reagents and solvents were purchased from common commercial suppliers and were used as such. Organic solutions were dried over anhydrous Na2SO4 and concentrated with a rotary evaporator at low pressure. All reactions were routinely checked by thin-layer chromatography (TLC) on silica gel 60F254 (Merck) and visualized by using UV or iodine. Column chromatography separations were carried out on Merck silica gel 60 (mesh 70–230), flash chromatography on Merck silica gel 60 (mesh 230–400). Melting points were determined in capillary tubes (Buchi Electrotermal model 9100) and are uncorrected. Yields were of purified products and were not optimized. 1H NMR spectra were recorded at 200 or 400 MHz (Bruker Avance DRX-200 or 400, respectively) while 13C NMR spectra were recorded at 100 MHz (Bruker Avance DRX-400) as well as 2D 1H NMR NOESY run in phase sensitive mode. Chemical shifts are given in ppm (δ) relative to TMS. Spectra were acquired at 298 K. Data processing was performed with standard Bruker software XwinNMR and the spectral data are consistent with the assigned structures. Elemental analyses were performed on a Fisons elemental analyzer, model EA1108CHN, and data for C, H, and N are within 0.4% of the theoretical values.
General procedure for condensation of key intermediate 7 with phenylhydrazines hydrochloride. Method A
To a refluxing solution of key intermediate 719 (1 mmol) in MeOH (20 mL), the appropriate hydrazine hydrochloride (1.2 mmol), dissolved in hot MeOH (2 mL), was added at once. The reaction was maintained at reflux for 3–15 h, then concentrated under reduced pressure to half volume and cooled to room temperature The precipitated formed was filtered under vacuum to give the pyrazolobenzothiazine-3-carboxymethyl esters 8b-g, pure by TLC (CHCl3/MeOH 98:2).
Methyl 1-(2-fluorophenyl)-4-methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylate 5,5-dioxide (8b)
Following the general procedure method A and using the 2-(fluorophenyl)hydrazine hydrochloride, compound 8b was obtained in 71% yield (reaction time 5 h) as yellowish crystalline solid: mp 174–175 °C. 1H NMR (200 MHz, acetone-d6): δ 3.30 (s, 3H, NCH3), 4.00 (s, 3H, OCH3), 7.10 (d, J = 7.8 Hz, 1H, H-9), 7.50–7.90 (m, 6H, H-7, H-8, H-3', H-4', H-5' and H-6'), 8.05 (dd, J = 1.5 and 7.6 Hz, 1H, H-6).
Methyl 1-(4-fluorophenyl)-4-methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylate 5,5-dioxide (8c)
Following the general procedure method A and using the 4-(fluorophenyl)hydrazine hydrochloride, compound 8c was obtained in 86% yield (reaction time 10 h) as white crystalline solid: mp 215–216 °C. 1H NMR (200 MHz, CDCl3): δ 3.35 (s, 3H, NCH3), 4.05 (s, 3H, OCH3), 7.00 (dd, J = 0.7 and 7.8 Hz, 1H, H-9), 7.25–7.35 (m, 2H, H-3' and H-5'), 7.45–7.70 (m, 4H, H-7, H-8, H-2' and H-6'), 8.10 (dd, J = 1.4 and 7.8 Hz, 1H, H-6).
Methyl 4-methyl-1-(3-nitrophenyl)-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylate 5,5-dioxide (8e)
Following the general procedure method A and using the 3-(nitrophenyl)hydrazine hydrochloride, compound 8e was obtained in 90% yield (reaction time 12 h) as yellow woolly solid: mp 283–284 °C. 1H NMR (200 MHz, DMSO-d6): δ 3.20 (s, 3H, NCH3), 4.00 (s, 3H, OCH3), 7.15 (d, J = 7.6 Hz, 1H, H-9), 7.65 (dt, J = 1.5 and 7.9 Hz, 1H, H-8), 7.80 (dt, J = 1.5 and 7.9 Hz, 1H, H-7), 7.90 (t, J = 8.1 Hz, 1H, H-5'), 8.00–8.10 (m, 2H, H4' and H-6), 8.45 (t, J = 1.9 Hz, 1H, H-2') 8.55 (m, 1H, H-6').
Methyl 4-methyl-1-(2-nitrophenyl)-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylate 5,5-dioxide (8f)
Following the general procedure method A and using the 2-(nitrophenyl)hydrazine hydrochloride (reaction time 15 h); after flash column chromatography purification (CHCl3/MeOH 99:1), compound 8f was obtained in 15% yield as yellow woolly solid: mp 277–278 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.10 (s, 1H, NCH3), 3.75 (s, 1H, OCH3), 6.90 (d, J = 7.8 Hz, 1H, H-9), 7.60 (dt, J = 1.3 and 7.8 Hz, 1H, H-8), 7.70 (dt, J = 1.0 and 7.8 Hz, 1H, H-7), 7.80–7.85 (m, 1H, H-4'), 7.90–8.00 (m, 3H, H-6, H-5' and H-6'), 8.30–8.35 (m, 1H, H-3').
Methyl 4-methyl-1-(4-nitrophenyl)-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylate 5,5-dioxide (8g)
Following the general procedure method A and using the 4-(nitrophenyl)hydrazine hydrochloride, compound 8g was obtained in 40% yield (reaction time 6 h) as yellow solid: mp 275–276 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.10 (s, 3H, NCH3), 3.90 (s, 3H, OCH3), 7.10 (d, J = 7.8 Hz, 1H, H-9), 7.65 (dt, J = 1.3 and 7.8 Hz, 1H, H-8), 7.70 (dt, J = 1.1 and 7.8 Hz, 1H, H-7), 7.80–7.85 (m, 2H, H-2' and H-6'), 8.00 (dd, J = 1.1 and 7.8 Hz, 1H, H-6), 8.40–8.45 (m, 2H, H-3' and H-5').
General procedure of basic hydrolysis. Method B
A stirred mixture of appropriate pyrazolobenzothiazine methyl esters 3a,19 8b-c, 8d,19 and 8e-g (1 mmol) in acq. 10% NaOH (6 mL) and MeOH (6 mL) was refluxed for 1 h and then concentrated to one-third of the volume under reduced pressure. The mixture was poured into ice/water and acidified with 2N HCl (pH = 3). The precipitated formed was then filtered under vacuum, washed with Et2O and dried to give the pyrazolobenzothiazine-3-carboxylic acids 9a-g used as it in the next reaction step.
1-(3-Fluorophenyl)-4-methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylic acid 5,5-dioxide (9a)
Following the general procedure method B, compound 9a was obtained from 3a,19 in 88% yield as white solid: mp 243–245 °C. 1H NMR (200 MHz, acetone-d6): δ 3.30 (s, 3H, NCH3), 7.20 (dd, J = 1.2 and 7.7 Hz, 1H, H-9), 7.45–7.60 (m, 3H, H-7, H-8 and H-6'), 7.65–7.85 (m, 3H, H-2', H-4' and H-5'), 8.05 (dd, J = 1.4 and 7.7 Hz, 1H, H-6).
1-(2-Fluorophenyl)-4-methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylic acid 5,5-dioxide (9b)
Following the general procedure method B, compound 9b was obtained from 8b, in 89% yield as white solid: mp 224–225 °C. 1H NMR (200 MHz, acetone-d6): δ 3.30 (s, 3H, NCH3), 7.10 (d, J = 7.3 Hz, 1H, H-9), 7.45–7.95 (m, 6H, H-7, H-8, H-3', H-4', H-5' and H-6'), 8.05 (dd, J = 1.4 and 7.7 Hz, 1H, H-6).
1-(4-Fluorophenyl)-4-methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylic acid 5,5-dioxide (9c)
Following the general procedure method B, compound 9c was obtained from 8c, in 88% yield as white solid: mp 248–250 °C. 1H NMR (200 MHz, acetone-d6): δ 3.30 (s, 3H, NCH3), 7.20 (dd, J = 1.0 and 7.5 Hz, 1H, H-9), 7.45–7.55 (m, 2H, H-7 and H-8), 7.60–7.80 (m, 4H, H-2', H-3', H-5' and H-6'), 8.05 (dd, J = 1.4 and 7.6 Hz, 1H, H-6).
4-Methyl-1-(3-nitrophenyl)-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylic acid 5,5-dioxide (9e)
Following the general procedure method B, compound 9e was obtained from 8e, in 99% yield as yellow solid: mp 184–185 °C. 1H NMR (200 MHz, DMSO-d6): δ 3.05 (s, 3H, NCH3), 7.10 (d, J = 7.8 Hz, 1H, H-9), 7.65 (dt, J = 1.2 and 7.8 Hz, 1H, H-8), 7.75 (dt, J = 1.2 and 7.8 Hz, 1H, H-7), 7.90 (t, J = 8.1 Hz, 1H, H-5'), 8.00–8.10 (m, 2H, H-6 and H-4'), 8.40 (t, J = 2.0 Hz, 1H, H-2'), 8.50 (m, 1H, H-6'), 13.80 (bs, 1H, CO2H). NMR NOESY spectra showed two relevant NOE cross-peaks: H-9→H-2'; H-9→H-4'.
4-Methyl-1-(2-nitrophenyl)-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylic acid 5,5-dioxide (9f)
Following the general procedure method B, compound 9f was obtained from 8f, in 60% yield as yellow solid: mp 220–221 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.05 (s, 3H, NCH3), 6.85 (d, J = 7.5 Hz, 1H, H-9), 7.60 (t, J = 7.5 Hz, 1H, H-8), 7.70 (t, J = 7.5 Hz, 1H, H-7), 7.80 (dd, J = 2.0 and 9.0 Hz, 1H, H-6'), 7.85–8.00 (m, 3H, H-6, H-4' and H-5'), 8.30 (dd, J = 2.1 and 7.5 Hz, 1H, H-3').
4-Methyl-1-(4-nitrophenyl)-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylic acid 5,5-dioxide (9g)
Following the general procedure method B, compound 9g was obtained from 8g, in 98% yield as yellow solid: mp 226–228°C. 1H NMR (200 MHz, DMSO-d6): δ 3.05 (s, 3H, NCH3), 7.15 (dd, J = 1.0 and 6.7 Hz, 1H, H-9), 7.65–7.80 (m, 2H, H-7 and H-8), 7.85–7.95 (m, 2H, H-2' and H-6'), 8.05 (dd, J = 1.4 and 6.9 Hz, 1H, H-6), 8.45–8.55 (m, 2H, H-3' and H-5').
General procedure for amidation reaction with N-(4-aminophenyl)methanesulfonamide. Method C
A mixture of intermediates 9a-g (1 mmol) in an excess of SOCl2 was refluxed under stirring for 1 h. The solvent was distilled under reduced pressure and the residues was washed three time with dry benzene. The corresponding pyrazolobenzothiazine-3-carbonyl chloride was immediately dissolved in dry DMF (2 mL) and added dropwise, under nitrogen flux, to a solution of N-(4-aminophenyl)methanesulfonamide25 (2 mmol) and Et3N (2 mmol) in dry DMF (7 mL). The mixture was heated at 40 °C for 24 h. The solvent was then concentrated to half volume and the mixture was poured into ice-water, acidified with 2N HCl (pH = 3) and the precipitated formed was filtered under vacuum. The solid obtained was crystallized by EtOH/DMF (2:1) to give the target pyrazolobenzothiazine-3-carboxyamides 4a-g.
1-(3-Fluorophenyl)-4-methyl-N-{4-[(methylsulfonyl)amino]phenyl}-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxamide 5,5-dioxide (4a)
Following the general procedure method C, compound 4a was obtained from 9a, in 70% yield as white solid: mp 285–287 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.00 (s, 3H, SO2CH3), 3.15 (s, 3H, NCH3), 7.05 (d, J = 7.6 Hz, 1H, H-9), 7.20–7.25 (m, 2H, H-3" and H-5"), 7.40–7.50 (m, 1H, H-7), 7.55 (dt, J = 1.7 and 8.5 Hz, 1H, H-8), 7.65–7.80 (m, 6H, H-2', H-4', H-5', H-6', H-2" and H-6"), 8.00 (dd, J = 1.1 and 6.6 Hz, 1H, H-6), 9.60 (s, 1H, SO2NH), 10.40 (s, 1H, CONH). 13C NMR (100 MHz, DMSO-d6), δ 39.15, 39.36, 113.89 (d, JC-F = 20 Hz, C-2'), 117.50 (d, JC-F = 20 Hz, C-4'), 121.28, 122.13, 122.45 (d, JC-F = 2 Hz, C-6'), 123.35, 124.90, 125.16, 126.77, 130. 62, 130.74, 130.86, 132.29 (d, JC-F = 10 Hz, C-5'), 133.47, 134.70, 135.03, 139.71, 140.52 (d, JC-F = 10 Hz, C-1'), 158.72, 162.73 (d, JC-F = 245 Hz, C-3'). Anal. Calcd (%) for C24H20FN5O5S2: C 53.13, H 3.72, N 12.93. Found C, 53.30; H, 3.60; N 12.88.
1-(2-Fluorophenyl)-4-methyl-N-{4-[(methylsulfonyl)amino]phenyl}-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxamide 5,5-dioxide (4b)
Following the general procedure method C, compound 4b was obtained from 9b, in 40% yield as white solid: mp 264–266 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.00 (s, 3H, SO2CH3), 3.30 (s, 3H, NCH3), 6.95 (d, J = 7.7 Hz, 1H, H-9), 7.20–7.25 (m, 2H, H-3" and H-5"), 7.55–7.60 (m, 2H, H-7 and H-8), 7.65–7.80 (m, 5H, H-4', H-5', H-6', H-2" and H-6"), 7.95 (t, J = 7.8 Hz 1H, H-3'), 8.00 (d, J = 7.5 Hz, 1H, H-6), 9.60 (s, 1H, SO2NH), 10.40 (s, 1H, CONH). 13C NMR (100 MHz, DMSO-d6): δ 39.26, 39.38, 117.62 (d, JC-F = 37 Hz, C-3'), 121.15, 122.08, 123.24, 123.30, 125.22, 126.03, 126.59 (bs, C-6'), 127.10 (d, JC-F = 20 Hz, C-1'), 129.61 (bs, C-5'), 130.74, 130.89, 131.97, 133.29 (d, JC-F = 10 Hz, C-4'), 133.71, 134.60, 134.95, 140.31, 156.34 (d, JC-F = 490 Hz, C-2'), 158.55. Anal. Calcd (%) for C24H20FN5O5S2: C, 53.13; H, 3.72; N, 12.93. Found C, 52.97; H, 3.80; N, 13.01.
1-(4-Fluorophenyl)-4-methyl-N-{4-[(methylsulfonyl)amino]phenyl}-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxamide 5,5-dioxide (4c)
Following the general procedure method C, compound 4c was obtained from 9c, in 40% yield as white solid: mp 298–300 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.00 (s, 3H, SO2CH3), 3.30 (s, 3H, NCH3), 7.00 (d, J = 7.7 Hz, 1H, H-9), 7.20–7.25 (m, 2H, H-3" and H-5"), 7.45–7.55 (m, 2H, H-7 and H-8), 7.65–7.80 (m, 6H, H-2', H-3', H-5', H-6', H-2" and H-6"), 8.00 (d, J = 7.5 Hz, 1H, H-6), 9.60 (s, 1H, SO2NH), 10.40 (s, 1H, CONH). 13C NMR (100 MHz, DMSO-d6): δ 39.23, 39.34, 117.50 (d, JC-F = 20 Hz, C-3' and C-5'), 121.28, 122.12, 123.45, 124.76, 125.19, 126.51, 128.82 (d, JC-F = 10 Hz, C-2' and C-6'), 130.64, 130.76, 130.90, 133.49, 134.65, 135.09, 135.72 (d, JC-F = 3 Hz, C-1'), 139.55, 158.80, 162.89 (d, JC-F = 250 Hz, C-4'). Anal. Calcd (%) for C24H20FN5O5S2: C, 53.13; H, 3.72; N, 12.93. Found C, 53.02; H, 3.86; N, 12.90.
N-{4-[(Methylsulfonyl)amino]phenyl}-1-phenyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxamide 5,5-dioxide (4d)
Following the general procedure method C, compound 4d was obtained from 9d,19 in 70% yield as pale pink solid: mp 280–283 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.00 (s, 3H, SO2CH3), 3.30 (s, 3H, NCH3), 6.95 (d, J = 7.8 Hz, 1H, H-9), 7.20–7.25 (m, 2H, H-3" and H-5"), 7.65–7.80 (m, 9H, H-7, H-8, H-2', H-3', H-4', H-5', H-2" and H-6"), 8.00 (d, J = 7.70 Hz, 1H, H-6), 9.60 (s, 1H, SO2NH), 10.40 (s, 1H, CONH). 13C NMR (100 MHz, DMSO-d6): δ 39.26, 39.38, 121.32, 122.15, 123.61, 124.75, 125.20, 126.36, 126.62, 130.57, 130.61, 130.89, 130.92, 133.39, 134.67, 135.14, 139.35, 139.54, 158.89. Anal. Calcd (%) for C24H21N5O5S2: C, 55.05; H, 4.04; N, 13.38. Found C, 54.97; H, 3.89; N, 13.61.
N-{4-[(Methylsulfonyl)amino]phenyl}-1-(3-nitrophenyl)-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxamide 5,5-dioxide (4e)
Following the general procedure method C, compound 4e was obtained from 9e, in 50% yield as white solid: mp 233–235 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.00 (s, 3H, SO2CH3), 3.30 (s, 3H, NCH3), 7.05 (d, J = 8.0 Hz, 1H, H-9), 7.20–7.25 (m, 2H, H-3" and H-5"), 7.65 (dt, J = 1.2 and 8.0 Hz, 1H, H-8), 7.75–7.80 (m, 3H, H-7, H-2" and H-6"), 7.90 (t, J = 8.1 Hz, 1H, H-5'), 8.00–8.10 (m, 2H, H-6 and H-6'), 8.50 (m, 1H, H-4'), 8.60 (t, J = 2.1 Hz, 1H, H-2'), 9.60 (s, 1H, NHSO2), 10.50 (s, 1H, NHCO). 13C NMR (100 MHz, DMSO-d6): δ 39.05, 39.37, 121.05, 121.28, 122.18, 123.17, 125.01, 125.15, 125.24, 127.11, 130.73, 130.86, 130.90, 131.98, 132.26, 133.53, 134.76, 134.99, 139.86, 140.16, 148.96, 158.63. Anal. Calcd (%) for C24H20N6O7S2: C, 50.70; H, 3.55; N, 14.78. Found C, 51.05; H, 3.52; N, 14.46.
N-{4-[(methylsulfonyl)amino]phenyl}-1-(2-nitrophenyl)-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxamide 5,5-dioxide (4f)
Following the general procedure method C, compound 4f was obtained from 9f, in 80% yield as white solid: mp 245–247 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.00 (s, 3H, SO2CH3), 3.25 (s, 3H, NCH3), 6.95 (d, J = 7.8 Hz, 1H, H-9), 7.15–7.20 (m, 2H, H-3" and H-5"), 7.65 (t, J = 7.8 Hz, 1H, H-8), 7.70–7.80 (m, 3H, H-7, H-2" and H-6"), 7.90 (d, J = 7.7 Hz, 1H, H-6'), 7.95–8.05 (m, 3H, H-6, H-4' and H-5'), 8.40 (d, J = 7.6 Hz, 1H, H-3'), 9.55 (s, 1H, NHSO2), 10.55 (s, 1H, NHCO). 13C NMR (100 MHz, DMSO-d6): δ 39.34, 39.35, 121.20, 122.24, 122.84, 124.14, 125.35, 126.39, 126.94, 130.56, 131.04, 131.11, 132.09, 132.17, 132.90, 133.80, 134.73, 134.91, 135.92, 140.50, 145.66, 158.44. Anal. Calcd (%) for C24H20N6O7S2: C, 50.7; H, 3.55; N, 14.78. Found C, 50.40; H, 3.50; N, 15.13.
N-{4-[(methylsulfonyl)amino]phenyl}-1-(4-nitrophenyl)-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxamide 5,5-dioxide (4g)
Following the general procedure method C, compound 4g was obtained from 9g, in 60% yield as white solid: mp 321–323 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.00 (s, 3H, SO2CH3), 3.25 (s, 3H, NCH3), 7.15–7.20 (m, 3H, H-6, H-3" and H-5"), 7.70 (t, J = 7.6 Hz, 1H, H-8), 7.75–7.80 (m, 3H, H-7, H-2" and H-6"), 7.95–8.00 (m, 2H, H-2' and H-6'), 8.10 (d, J = 7.6 Hz, 1H, H-6), 8.45–8.50 (m, 2H, H-3' and H-5'), 9.75 (s, 1H, SO2NH), 10.60 (s, 1H, CONH). 13C NMR (100 MHz, DMSO-d6): δ 39.22, 39.31, 121.24, 122.12, 123.19, 125.17, 125.51, 125.93, 126.85, 127.46, 130.68, 130.75, 130.93, 133.61, 134.77, 134.96, 140.58, 143.82, 147.97, 158.58. Anal. Calcd (%) for C24H20N6O7S2: C, 50.70; H, 3.55; N, 14.78. Found C, 50.60; H, 3.50; N 14.93.
General procedure for reduction of nitro group. Method D
A stirred solution of nitro derivatives 4e-g (1 mmol) in DMF (60 mL) was hydrogenated over a catalytic amount of Raney nickel at room temperature and atmospheric pressure for 5 h. The mixture was then filtered over Celite, the filtrate was evaporated to dryness and the residue was crystallized by EtOH/DMF (2:1) to give the target aminoderivatives 4h-j.
1-(3-Aminophenyl)-4-methyl-N-{4-[(methylsulfonyl)amino]phenyl}-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxamide 5,5-dioxide (4h)
Following the general procedure method D, compound 4h was obtained from 4e, in 50% yield as whitish solid: mp 266–267 °C. 1H NMR (400 MHz, CDCl3): δ 3.00 (s, 3H, SO2CH3), 3.30 (s, 3H, NCH3), 5.80 (s, 2H, NH2), 6.80–6.85 (m, 2H, H-2' and H-4'), 6.85 (d, J = 7.8 Hz, 1H, H-9), 7.10 (d, J = 8.1 Hz, 1H, H-6'), 7.20–7.30 (m, 2H, H-3" and H-5"), 7.30 (t, J = 8.1 Hz, 1H, H-5'), 7.45 (dt, J = 1.1 and 7.8 Hz, 1H, H-8), 7.55 (dt, J = 0.8 and 7.8 Hz, 1H, H-7), 7.65–7.70 (m, 2H, H-2" and H-6"), 8.00 (dd, J = 1.1 and 7.8 Hz, 1H, H-6), 8.75 (s, 1H, SO2NH). 13C NMR (100 MHz, DMSO-d6): δ 39.25, 39.33, 110.67, 112.61, 115.49, 121.28, 122.12, 123.77, 124.89, 125.01, 126.31, 130.24, 130.46, 130.70, 130.80, 133.35, 134.58, 135.15, 138.96, 140.08, 150.73, 158.95. Anal. Calcd for C24H22N6O5S2: C, 53.52; H, 4.12; N, 15.60. Found C, 53.39; H, 4.06; N 15.81.
1-(2-Aminophenyl)-4-methyl-N-{4-[(methylsulfonyl)amino]phenyl}-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxamide 5,5-dioxide (4i)
Following the general procedure method D, compound 4i was obtained from 4f, in 50% yield as whitish solid: mp 260–262 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.00 (s, 3H, SO2CH3), 3.25 (s, 3H, NCH3), 5.50 (s, 2H, NH2), 6.75 (t, J = 8.0 Hz, 1H, H-5'), 6.90–7.15 (m, 3H, H-3', H-6' and H-9), 7.20–7.25 (m, 2H, H-3'' and H-5''), 7.30 (t, J = 8.0 Hz, 1H, H-4'), 7.60–7.70 (m, 2H, H-7 and H-8), 7.75–7.80 (m, 2H, H-2'' and H-6''), 8.00 (d, J = 7.0 Hz, 1H, H-6), 9.55 (bs, 1H, SO2NH), 10.50 (s, 1H, CONH). 13C NMR (100 MHz, DMSO-d6): δ 39.14, 39.23, 116.67, 116.79, 121.30, 122.13, 123.77, 123.86, 124.04, 124.87, 125.91, 128.55, 130.36, 130.87, 131.01, 131.83, 133.46, 134.62, 135.13, 139.42, 145.61, 158.99. Anal. Calcd for C24H22N6O5S2: C, 53.52; H, 4.12; N ,15.60. Found C, 53.80; H, 4.09; N, 15.35.
1-(4-Aminophenyl)-4-methyl-N-{4-[(methylsulfonyl)amino]phenyl}-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxamide 5,5-dioxide (4j)
Following the general procedure method D, compound 4j was obtained from 4g, in 80% yield as whitish solid: mp 198–201 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.00 (s, 3H, SO2CH3), 3.30 (s, 3H, NCH3), 5.75 (s, 2H, NH2), 6.65–6.75 (m, 2H, H-3' and H-5'), 7.05–7.10 (m, 1H, H-9), 7.15–7.25 (m, 4H, H-2', H-6', H-3" and H-5"), 7.65–7.70 (m, 2H, H-7 and H-8), 7.75–7.80 (m, 2H, H-2" and H-6") 7.95–8.00 (m, 1H, H-6), 9.65 (s, 1H, SO2NH), 10.40 (s, 1H, CONH). 13C NMR (100 MHz, DMSO-d6): δ 39.25, 39.51, 114.31, 121.31, 122.09, 124.00, 124.49, 125.04, 125.88, 127.33, 127.43, 130.28, 130.45, 130.83, 133.26, 134.52, 135.22, 138.57, 150.81, 159.03. Anal. Calcd for C24H22N6O5S2: C, 53.52; H, 4.12; N ,15.60. Found C, 53.85; H, 4.14; N, 15.27.
4-[(Methylsulfonyl)amino]phenyl 1-(3-fluorophenyl)-4-methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxylate 5,5-dioxide (6a)
Following the general procedure method C and replacing the N-(4-aminophenyl)methanesulfonamide25 with the N-(4-hydroxyphenyl)methanesulfonamide,26 compound 6a was obtained from 9a in 65% yield as white solid: mp 252–254 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.00 (s, 3H, SO2CH3), 3.30 (s, 3H, NCH3), 7.00 (d, J = 7.7 Hz, 1H, H-9), 7.25–7.30 (m, 4H, H-2", H-3", H-5" and H-6"), 7.40 (dt, J = 1.0 and 8.0 Hz, 1H, H-8), 7.50 (dt, J = 1.9 and 7.7 Hz, 1H, H-7), 7.55–7.75 (m, 4H, H-2', H-4', H-5', and H-6'), 8.00 (dd, J = 1.0 and 7.7 Hz, 1H, H-6), 9.80 (s, 1H, SO2NH). 13C NMR (100 MHz, DMSO-d6): δ 39.07, 39.36, 113.78 (d, JC-F = 25 Hz, C-2'), 117.97 (d, JC-F = 20 Hz, C-4'), 121.45, 122.50 (bs, C-6'), 123.11, 123.15, 124.91, 125.20, 128.35, 130.80, 130.85, 130.97, 132.52 (d, JC-F = 10 Hz, C-5'), 133.65, 135.69, 136.90, 140.40 (d, JC-F = 10 Hz, C-1'), 146.46, 159.04, 162.74 (d, JC-F = 245 Hz, C-3'). Anal. Calcd (%) for C24H19FN4O6S2: C, 53.13; H, 3.53; N, 10.33. Found C, 53.02; H, 3.50; N, 10.47.
1-(3-Fluorophenyl)-4-methyl-N-(4-nitrophenyl)-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxamide 5,5-dioxide (10a)
Following the general procedure method C and replacing the N-(4-aminophenyl)methanesulfonamide25 with the 4-nitroaniline, compound 10a was obtained from 9a, in 35% yield as yellowish solid: mp 265–266 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.20 (s, 3H, NCH3), 7.05 (d, J = 7.6 Hz, 1H, H-9), 7.45–7.55 (m, 2H, H-7 and H-8), 7.65–7.75 (m, 4H, H-2', H-4', H-5' and H-6'), 8.00 (dd, J = 1.4 and 7.6 Hz, 1H, H-6), 8.05–8.10 (m, 2H, H-2" and H-6"), 8.20–8.25 (m, 2H, H-3" and H-5"), 11.05 (s, 1H, NH).
N-(4-aminophenyl)-1-(3-fluorophenyl)-4-methyl-1,4-dihydropyrazolo[4,3-c][1,2]benzothiazine-3-carboxamide 5,5-dioxide (5a)
Following the general procedure method D, compound 5a was obtained from 10a, in 70% yield as white solid: mp 235–236 °C. 1H NMR (400 MHz, DMSO-d6): δ 3.15 (s, 3H, NCH3), 5.00 (s, 2H, NH2), 6.50–6.55 (m, 2H, H-3" and H-5"), 7.10 (d, J = 7.4 Hz, 1H, H-9), 7.40–7.50 (m, 3H, H-8, H-2" and H-6"), 7.60 (t, J = 7.7 Hz, 1H, H-7), 7.65–7.75 (m, 4H, H-2', H-4', H-5' and H-6'), 8.00 (d, J = 7.2 Hz, 1H, H-6), 10.10 (s, 1H, NH). 13C NMR (100 MHz, DMSO-d6): δ 39.12, 113.76 (d, JC-F = 20 Hz, C-2'), 114.07, 117.40 (d, JC-F = 20 Hz, C-4'), 122.34 (bs, C-6'), 122.73, 123.44, 124.90, 125.14, 126.56, 127.52, 130.38, 130.65, 130.82, 132.20 (d, JC-F = 10 Hz, C-5'), 133.43, 140.21, 140.58 (d, JC-F = 10 Hz, C-1'), 146.00, 158.09, 162.74 (d, JC-F = 245 Hz, C-3'). Anal. Calcd for C24H18FN5O3S: C, 59.60; H, 3.91; N, 15.11. Found C, 59.87; H, 4.01; N, 14.74.
Biochemical and biological assays
SPR Interaction analysis
The analysis of the interaction between the synthesized compounds and the ectodomain of HCV NS5B from a Con 1-strain isolate, subtype 1b.38 The C-terminal 21 amino acid transmembrane region was exchanged for a hexa histidine tag coding sequence. Experiments were performed using SPR biosensor analysis with a S51 instrument from Biacore (GE Healthcare).
The enzyme was immobilized by standard amine coupling to a CM5 biosensor chip. The compounds were typically injected over the protein surface for 60 s at 30 µL/min using in concentration series between 6 and 200 µM in running buffer (20 mM Tris pH 7.4 at room temperature, 130 mM NaCl, 0.05 % (v/v) Tween20) to a final DMSO concentration of 5 % (v/v), using twofold dilutions. The structural integrity of the immobilized protein was assessed with filibuvir (PF-00868554)23 as a positive control substance. Raw data was processed according to standard procedures, including reference and blank subtraction along with solvent correction to eliminate any artifacts such as non-specific binding and discrepancies in buffer composition. Thereafter, each concentration series was subjected to global non-linear regression in the Biacore Evaluation software (GE Healthcare). The 1:1 Langmuir binding model (Eq. 1) was used to determine the dissociation equilibrium constant (KD) and estimate the maximal signal (Rmax) from signal versus compound concentrations plots:
| (Eq. 1) |
c = compound concentration
NS5B polymerase assay
Recombinant NS5B 1b bearing an N-terminal histidine-tag and 21-amino acid truncation at its C-terminus was purified from the plasmid pThNS5BCΔ21 (NIH1b strain) expressed in Escherichia coli DH5α employing Ni-NTA chromatography.30 The compounds were dissolved in dimethylsulfoxide (DMSO) as 10 mM stocks and serially diluted in DMSO immediately prior to the assay. Inhibition of HCV NS5B by the compounds was evaluated by the standard primer dependent elongation assay as previously described.13a Preliminary screening of the was first investigated at 50 µM concentration to identify NS5B inhibitors. Reactions were carried out in a buffer containing 20 mM Tris-HCl (pH 7.0), 100 mM NaCl, 100 mM sodium glutamate, 0.5 mM DTT, 0.01% BSA, 0.01% Tween-20, 5% glycerol, 20 U/mL RNase Out, 0.25 µM poly rA/U12, 20 µM UTP, 1–2 µCi [α-32P]UTP, 100 ng of NS5BCΔ21 and 1.0 mM MnCl2 in a total volume of 25 µL. The assay mix was incubated at 30°C for 1 h and stopped by quenching with chilled 5% (v/v) trichloroacetic acid (TCA) supplemented with 0.5 mM pyrophosphate. RNA products were precipitated on GF-B filters and quantified by liquid scintillation counting. NS5B activity in the presence of DMSO control was set at 100% and that in the presence of the compounds was determined relative to this control. Compounds exhibiting ≥50% inhibition at 50 µM concentration were then investigated for their IC50 values. Dose-response curves of 8–12 concentrations of the compounds in duplicate in two independent experiments were plotted using nonlinear regression analysis and IC50 values were determined using Graphpad Prism software.
Cells and viruses
The Huh 5–2 and Huh 9–13 HCV subgenomic replicon-containing cells were provided by Prof R Bartenschlager (University of Heidelberg, Heidelberg, Germany).
Antiviral assays
Huh 5.2 cells, containing the hepatitis C virus genotype 1b I389luc-ubi-neo/NS3-3’/5.1 replicon39 were sub-cultured in DMEM supplemented with 10% FCS, 1% non-essential amino acids, 1% penicillin/streptomycin and 2% Geneticin at a ratio of 1:3 to 1:4, and grown for 3–4 days in 75 cm2 tissue culture flasks. One day before addition of the compound, cells were harvested and seeded in assay medium (DMEM, 10% FCS, 1% non-essential amino acids, 1% penicillin/streptomycin) at a density of 6 500 cells/well (100 µL/well) in 96-well tissue culture microtiter plates for evaluation of anti-metabolic effect and CulturPlate (Perkin Elmer) for evaluation of the antiviral effect. The microtiter plates were incubated overnight (37 °C, 5% CO2, 95–99% relative humidity), yielding a non-confluent cell monolayer.
The evaluation of the anti-metabolic as well as antiviral effect of each compound was performed in parallel. Four-step, 1-to-5 compound dilution series were prepared for the first screen, to collect data for a more detailed dose-response curve, an eight-step, 1-to-2 dilution series was used. Following assay setup, the microtiter plates were incubated for 72 hours (37 °C, 5% CO2, 95–99% relative humidity). For the evaluation of anti-metabolic effects, the assay medium was aspirated, replaced with 75 µL of a 5% MTS solution in phenol red-free medium and incubated for 1.5 hours (37 °C, 5% CO2, 95–99% relative humidity). Absorbance was measured at a wavelength of 498 nm (Safire2, Tecan), and optical densities (OD values) were converted to percentage of untreated controls. For the evaluation of antiviral effects, assay medium was aspirated and the cell monolayers were washed with PBS. The wash buffer was aspirated, and 25 µL of Glo Lysis Buffer (Promega) was added allowing for cell lysis to proceed for 5 min at room temperature. Subsequently, 50 µL of Luciferase Assay System (Promega) was added, and the luciferase luminescence signal was quantified immediately (1000 ms integration time/well, Safire2, Tecan). Relative luminescence units were converted into percentage of untreated controls.
The EC50 and EC90 (values calculated from the dose-response curve) represent the concentrations at which 50% and 90% inhibition, respectively, of viral replication is achieved. The CC50 (value calculated from the dose-response curve) represents the concentration at which the metabolic activity of the cells is reduced by 50 % as compared to untreated cells.
A concentration of compound is considered to elicit a genuine antiviral effect in the HCV replicon system when the anti-replicon effect is well above the 70% threshold at concentrations where no significant anti-metabolic activity is observed.
Compounds that reproducibly matched the above-outlined selection criteria were evaluated for selective antiviral activity in the Huh 9–13 replicon system. A similar assay setup was used as described above; the antiviral and anti-metabolic effect of the compounds was evaluated in parallel. The anti-metabolic activity of the compounds was quantified as outlined above. For the evaluation of the antiviral effect, assay medium was aspirated and the plates with dry monolayer were stored at −80 °C awaiting extraction. Following thawing of the plates at room temperature, the cell monolayer was lysed with 100 µL of cell-to-cDNA lysis buffer (Invitrogen). Lysis of the cells was allowed to proceed for 10 min at room temperature after which all liquid was transferred to a PCR plate (Axygen). The PCR plate was incubated for 15min at 75 °C (T3, Biometra). The lysate was diluted 1:2 with RNase/DNase-free water, after which 5 µL was transferred to a real-time PCR plate (Applied Biosystems). Replicon RNA content was quantified using a real-time quantitative one-step RT-PCR method (RT-qPCR). Per sample, 20 µL master mix was added containing 12.5 µL 2x RT-qPCR mix (Low Rox One-Step RT-qPCR master mix, Abgene), 0.125 µL of a 60 µM forward primer solution (5’-CCA GAT CAT CCT GAT CGA CCA G-3’, final [ ] of 300 nM), 0.125 µL of a 60 µM reversed primer solution (5’-CCG GCT ACC TGC CCA TTC-3’, final [ ] of 300 nM), 0.3 µL of a 5 µM probe solution (5’-ACA TCG CAT CGA GCG AGC ACG TAC-3’, final [ ] of 60 nM) and 6.825 µL of DNase/RNase-free water (ACROS). The samples were analyzed using a SDS7500F (Applied Biosystems, standard thermocycling profile: 30 min at 48 °C, 10 min at 95 °C, 40 cycles of 15 sec at 95 °C and 1 min at 60 °C). Replicon RNA quantities were converted to percentage of untreated controls, allowing to calculate EC50 and EC90 values. Similar as for the Huh 5–2 assay, a compound is only considered to be a selective inhibitor of HCV replication when clear inhibition of virus replication is observed at concentrations that do not elicit a significant anti-metabolic effect on the host cells.
Supplementary Material
Acknowledgements
This work was supported by the Swedish Research Council (VR) to U.H.D and the National Institute of Health Research Grant CA153147 to N.K.-B. We thank Roberto Bianconi, Stijn Delmotte, Mieke Flament and Tom Bellon for excellent technical assistance.
Abbreviation used
- BTDZ
hydroxyquinoline-benzothiadiazine
- BTZ
hydroxyquinoline-benzothiazine
- DAAs
directacting antiviral agents
- DMEM
Dulbecco's modified Eagle medium
- ETF
edge-to-face
- FCS
fetal calf serum
- HYD
hydrophobic regions
- LLE
ligand-lipophilic efficiency
- MTS
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxy-phenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- NNIs
nonnucleoside inhibitors
- pegIFN-α
pegylated interferon alpha
- RdRp
RNA-dependent RNA polymerase
- PSI
palm site I
- PSII
palm site II
- PSIII
palm site III
- RBV
ribavirin
- SPR
surface plasmon resonance
- TSI
thumb site I
- TSII
thumb site II
Footnotes
ASSOCIATED CONTENT
Supporting Information
Values of pKD, pIC50 and calculated logP for compounds 4a-j, 5a, and 6a. Plots of calculated logP versus pIC50 and pKD. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
All authors have given approval to the final version of the manuscript.
References and notes
- 1.Sy T, Jamal MM. Epidemiology of Hepatitis C Virus (HCV) Infection. Int. J. Med Sci. 2006;3:41–46. doi: 10.7150/ijms.3.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.WHO. [accessed September 30, 2012];Factsheet No 164, July 2011. http://www.who.int/mediacentre/factsheets/fs164/en/
- 3.Seeff LB. Natural History of Chronic Hepatitis C. Hepatology. 2002;36:S35–S46. doi: 10.1053/jhep.2002.36806. [DOI] [PubMed] [Google Scholar]
- 4.Scheridan C. New Merck and Vertex Drugs Raise Standard of Care in Hepatitis C. Nat. Biotechnol. 2011;29:553–554. doi: 10.1038/nbt0711-553. [DOI] [PubMed] [Google Scholar]
- 5.Ferenci P, Reddy KR. Impact of HCV Protease-Inhibitor-Based Triple Therapy for Chronic HCV Genotype 1 Infection. Antivir. Ther. 2011;16:1187–1201. doi: 10.3851/IMP1934. [DOI] [PubMed] [Google Scholar]
- 6.Pawlotsky JM, Chevaliez S, McHutchison JG. The Hepatitis C Virus Life Cycle as a Target for New Antiviral Therapies. Gastroenterology. 2007;132:1979–1998. doi: 10.1053/j.gastro.2007.03.116. [DOI] [PubMed] [Google Scholar]
- 7.Behrens SE, Tomei L, De Francesco R. Identification and Properties of the RNA-Dependent RNA Polymerase of Hepatitis C Virus. EMBO J. 1996;15:12–22. [PMC free article] [PubMed] [Google Scholar]
- 8.Wu JZ, Hong Z. Targeting NS5B RNA-Dependent RNA Polymerase for Anti-HCV Chemotherapy. Curr. Drug Targets Infect. Disord. 2003;3:207–219. doi: 10.2174/1568005033481114. [DOI] [PubMed] [Google Scholar]
- 9.Sofia MJ, Chang W, Furman PA, Mosley RT, Ross BS. Nucleoside, Nucleotide, and Non-Nucleoside Inhibitors of Hepatitis C Virus NS5B RNA-Dependent RNA-Polymerase. J. Med. Chem. 2012;55:2481–2531. doi: 10.1021/jm201384j. [DOI] [PubMed] [Google Scholar]
- 10.Mayhoub AS. Hepatitis C RNA-Dependent RNA Polymerase Inhibitors: a Review of Structure-Activity and Resistance Relationships; Different Scaffolds and Mutations. Bioorg. Med. Chem. 2012;20:3150–3161. doi: 10.1016/j.bmc.2012.03.049. [DOI] [PubMed] [Google Scholar]
- 11.Barreca ML, Iraci N, Manfroni G, Cecchetti V. Allosteric Inhibition of the Hepatitis C Virus NS5B Polymerase: in Silico Strategies for Drug Discovery and Development. Future Med. Chem. 2011;3:1027–1055. doi: 10.4155/fmc.11.53. [DOI] [PubMed] [Google Scholar]
- 12.a) Manfroni G, Paeshuyse J, Massari S, Zanoli S, Gatto B, Maga G, Tabarrini O, Cecchetti V, Fravolini A, Neyts J. Inhibition of Subgenomic Hepatitis C Virus RNA Replication by Acridone Derivatives: Identification of an NS3 Helicase Inhibitor. J. Med . Chem. 2009;52:3354–3365. doi: 10.1021/jm801608u. [DOI] [PubMed] [Google Scholar]; b) Manfroni G, Meschini F, Barreca ML, Leyssen P, Samuele A, Iraci N, Sabatini S, Massari S, Maga G, Neyts J, Cecchetti V. Pyridobenzothiazole Derivatives as New Chemotype Targeting the HCV NS5B Polymerase. Bioorg. Med. Chem. 2012;20:866–876. doi: 10.1016/j.bmc.2011.11.061. [DOI] [PubMed] [Google Scholar]
- 13.a) Nichols DB, Fournet G, Gurukumar KR, Basu A, Lee JC, Sakamoto N, Kozielskie F, Musmuca I, Joseph B, Ragno R, Kaushik-Basu N. Inhibition of Hepatitis C Virus NS5B Polymerase by S-Trityl-L-Cysteine Derivatives. Eur. J. Med. Chem. 2012;49:191–199. doi: 10.1016/j.ejmech.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Musmuca I, Caroli A, Mai A, Kaushik-Basu N, Arora P, Ragno R. Combining 3-D Quantitative Structure-activity Relationship with Ligand Based and Structure-based Alignment Procedures for in Silico Screening of New Hepatitis C virus NS5B Polymerase Inhibitors. J. Chem. Inf. Model. 2010;50:662–676. doi: 10.1021/ci9004749. [DOI] [PubMed] [Google Scholar]
- 14.Das D, Hong J, Chen SH, Wang G, Beigelman L, Seiwert SD, Buckman BO. Recent Advances in Drug Discovery of Benzothiadiazine and Related Analogs as HCV NS5B Polymerase Inhibitors. Bioorg. Med. Chem. 2011;19:4690–4703. doi: 10.1016/j.bmc.2011.06.079. [DOI] [PubMed] [Google Scholar]
- 15.de Vicente J, Hendricks RT, Smith DB, Fell JB, Fischer J, Spencer SR, Stengel PJ, Mohr P, Robinson JE, Blake JF, Hilgenkamp RK, Yee C, Adjabeng G, Elworthy TR, Tracy J, Chin E, Li J, Wang B, Bamberg JT, Stephenson R, Oshiro C, Harris SF, Ghate M, Leveque V, Najera I, Le Pogam S, Rajyaguru S, Ao-Ieong G, Alexandrova L, Larrabee S, Brandl M, Briggs A, Sukhtankar S, Farrell R, Xu B. Non-Nucleoside Inhibitors of HCV Polymerase NS5B. Part 2: Synthesis and Structure-Activity Relationships of Benzothiazine-Substituted Quinolinediones. Bioorg. Med. Chem. Lett. 2009;19:3642–3646. doi: 10.1016/j.bmcl.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 16.LigPrep, version 2.5. New York, NY: Schröodinger, LLC; 2011. [Google Scholar]
- 17.a) Glide, version 5.6. New York, NY: Schröodinger, LLC; 2010. [Google Scholar]; b) Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shaw DE, Shelley M, Perry JK, Francis P, Shenkin PS. Glide: A New Approach for Rapid, Accurate Docking and Scoring, 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]; c) Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]; d) Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 18.The synthesis of compound 3a has been reported previously by us (see ref. 19). Compound 3a was re-synthesized and its identity was verified by 1H and 13C NMR; the purity was determined by combustion analysis and was found to be >95%. Compound 3a: 1H NMR (400 MHz, CDCl3): δ 3.27 (s, 3H, NCH3), 4.00 (s, 3H, OCH3), 7.05 (d, J = 8.0 Hz, 1H, H-9), 7.20–7.30 (m, 1H, H-2'), 7.35–7.40 (m, 2H, H-4' and H-5'), 7.45–7.55 (m, 2H, H-8 and H-6'), 7.65 (t, J = 8.0 Hz, 1H, H-7), 8.05 (dd, J = 0.9 and 7.9 Hz, 1H, H-6). 13C NMR (100 MHz, CDCl3): δ38.81, 52.63, 113.31 (d, JC-F = 20 Hz, C-2'), 117.01 (d, JC-F = 20 Hz, C-4'), 121.24 (d, JC-F = 3.4 Hz, C-6'), 123.418, 124.36, 125.15, 128.27, 129.72, 130.44, 131.17 (d, JC-F = 8.9 Hz, C-5'), 131.34, 132.21, 136.72, 140.32 (d, JC-F = 10 Hz, C-1'), 160.81, 162.88 (d, JC-F = 249 Hz, C-3'). Anal. Calcd for C18H14FN3O4S: C, 55.81; H, 3.64; N, 10.85. Found C, 55.92; H, 3.76; N, 10.65.
- 19.Cecchetti V, Fravolini A, Schiaffella F, De Regis M, Orzalesi G, Volpato I. Synthesis and Pharmacological Activity of Pyrazolo-1,2-benzothiazine- and Isoxazolo-1,2-benzothiazinecarboxamides. Il Farmaco. 1983;38:35–44. [PubMed] [Google Scholar]
- 20.Hang JQ, Yang Y, Harris SF, Leveque V, Whittington HJ, Rajyaguru S, Ao-Ieong G, McCown MF, Wong A, Giannetti AM, Le Pogam S, Talamás F, Cammack N, Nájera I, Klumpp K. Slow binding inhibition and mechanism of resistance of non-nucleoside polymerase inhibitors of hepatitis C virus. J. Biol. Chem. 2009;284:15517–15529. doi: 10.1074/jbc.M808889200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng CC, Shipps GW, Jr, Yang Z, Kawahata N, Lesburg CA, Duca JS, Bandouveres J, Bracken JD, Jiang CK, Agrawal S, Ferrari E, Huang HC. Inhibitors of Hepatitis C Virus Polymerase: Synthesis and Characterization of Novel 2-oxy-6-Fluoro-N-((S)-1-hydroxy-3-phenylpropan-2-yl)-benzamides. Bioorg. Med. Chem. Letters. 2010;20:2119–2124. doi: 10.1016/j.bmcl.2010.02.054. [DOI] [PubMed] [Google Scholar]
- 22.Di Marco S, Volpari C, Tomei L, Altamura S, Harper S, Narjes F, Koch U, Rowley M, De Francesco R, Migliaccio G, Carfi A. Interdomain Communication in Hepatitis C Virus Polymerase Abolished by Small Molecule Inhibitors Bound to a Novel Allosteric Site. J. Biol. Chem. 2005;280:29765–29770. doi: 10.1074/jbc.M505423200. [DOI] [PubMed] [Google Scholar]
- 23.Li H, Tatlock J, Linton A, Gonzalez J, Jewell T, Patel L, Ludlum S, Drowns M, Rahavendran SV, Skor H, Hunter R, Shi ST, Herlihy KJ, Parge H, Hickey M, Yu X, Chau F, Nonomiya J, Lewis C. Discovery of (R)-6-cyclopentyl-6-(2-(2,6-diethylpyridin-4-yl)ethyl)-3-((5,7-dimethyl-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)methyl)-4-hydroxy-5,6-dihydropyran-2-one (PF-00868554). as a Potent and Orally Available Hepatitis C Virus Polymerase Inhibitor. J. Med. Chem. 2009;52:1255–1258. doi: 10.1021/jm8014537. [DOI] [PubMed] [Google Scholar]
- 24.Sabatini S, Gosetto F, Serritella S, Manfroni G, Tabarrini O, Iraci N, Brincat JP, Carosati E, Villarini M, Kaatz GW, Cecchetti V. Pyrazolo[4,3-c][1,2]benzothiazines 5,5-dioxide: a Promising New Class of Staphylococcus Aureus NorA Efflux Pump Inhibitors. J. Med. Chem. 2012;55:3568–3572. doi: 10.1021/jm201446h. [DOI] [PubMed] [Google Scholar]
- 25.Waslei J, Rosenthal GJ, Sun X, Strong S, Qiu J. Novel Pyrrole Inhibitors of S-Nitrosoglutathione Reductase as Therapeutic Agents. PTC WO 2010/019903 A1. 2010 [Google Scholar]
- 26.Wang Y, Guziec FS. A Convenient Reductive Deamination (Hidrodeamination) of Aromatic Amines. J. Org. Chem. 2001;66:8293–8296. doi: 10.1021/jo010681w. [DOI] [PubMed] [Google Scholar]
- 27.a) Hopkins AL, Groom CR, Alex A. Ligand Efficiency: a Useful Metric for Lead Selection. Drug Discov. Today. 2004;9:430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]; b) Reynolds CH, Tounge BA, Bembenek SD. Ligand Binding Efficiency: Trends, Physical basis, and Implications. J. Med. Chem. 2008;51:2432–2438. doi: 10.1021/jm701255b. [DOI] [PubMed] [Google Scholar]
- 28.Leeson PD, Springthorpe B. The Influence of Drug-Like Concepts on Decision-Making in Medicinal Chemistry. Nat. Rev. Drug Discov. 2007;6:881–890. doi: 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- 29.QikPro, version 3.5. New York, NY: Schröodinger, LLC; 2012. [Google Scholar]
- 30.a) Kaushik-Basu N, Bopda-Waffo A, Talele TT, Basu A, Costa PR, da Silva AJ, Sarafianos SG, Noel F. Identification and Characterization of Coumestans as Novel HCV NS5B Polymerase Inhibitors. Nucleic Acids Res. 2008;36:1482–1496. doi: 10.1093/nar/gkm1178. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chen Y, Bopda-Waffo A, Basu A, Krishnan R, Silberstein E, Taylor DR, Talele TT, Arora P, Kaushik-Basu N. Characterization of Aurintricarboxylic Acid as a Potent Hepatitis C Virus Replicase Inhibitor. Antivir. Chem. Chemother. 2009;20:19–36. doi: 10.3851/IMP1286. [DOI] [PubMed] [Google Scholar]
- 31.Compound 1 was synthesized by us following the experimental procedure reported in ref. 32. Its identity was verified by 1H NMR; the purity was determined by combustion analysis and was found to be >95%. Compound 1: 1H NMR (400 MHz, CDCl3): δ 1.00 (d, J = 6.5 Hz, 6H, CH3), 1.55–1.65 (m, 2H, CH2), 1.85 (spt, J = 6.5 Hz, 1H CH), 4.30–4.40 (m, 2H, NCH2), 7.30–7.50 (m, 4H, Ar-H), 7.60–7.80 (m, 2H, Ar-H), 8.00 (dd, J = 0.8 and 7.9 Hz, 1H, Ar-H), 8.30 (dd, J = 1.6 and 8.1 Hz, 1H, Ar-H), 14.50 (bs, 1H, NH), 15.10 (s, 1H, OH). Anal. Calcd for C21H21N3O4S·0.3 EtOAc: C, 60.85; H, 5.41; N, 9.53. Found C, 61.25; H, 5.14; N, 9.15.
- 32.Tedesco R, Shaw AN, Bambal R, Chai D, Concha NO, Darcy MG, Dhanak D, Fitch DM, Gates A, Gerhardt WG, Halegoua DL, Han C, Hofmann GA, Johnston VK, Kaura AC, Liu N, Keenan RM, Lin-Goerke J, Sarisky RT, Wiggall KJ, Zimmerman MN, Duffy KJ. 3-(1,1-Dioxo-2H-(1,2,4)-benzothiadiazin-3-yl)-4-hydroxy- 2(1H)-quinolinones, Potent Inhibitors of Hepatitis C Virus RNA-dependent RNA Polymerase. J. Med. Chem. 2006;49:971–983. doi: 10.1021/jm050855s. [DOI] [PubMed] [Google Scholar]
- 33.Tarcsay A, Nyiri K, Keseru GM. Impact of Lipophilic Efficiency on Compound Quality. J. Med. Chem. 2012;55:1252–1260. doi: 10.1021/jm201388p. [DOI] [PubMed] [Google Scholar]
- 34.a) Christiansen E, Due-Hansen ME, Urban C, Grundmann M, Schroder R, Hudson BD, Milligan G, Cawthorne MA, Kostenis E, Kassack MU, Ulven T. Free Fatty Acid Receptor 1 (FFA1/GPR40). Agonists: Mesylpropoxy Appendage Lowers Lipophilicity and Iimproves ADME Properties. J. Med. Chem. 2012;55:6624–6628. doi: 10.1021/jm3002026. [DOI] [PubMed] [Google Scholar]; b) Cid JM, Tresadern G, Vega JA, de Lucas AI, Matesanz E, Iturrino L, Linares ML, Garcia A, Andres JI, Macdonald GJ, Oehlrich D, Lavreysen H, Megens A, Ahnaou A, Drinkenburg W, Mackie C, Pype S, Gallacher D, Trabanco AA. Discovery of 3-Cyclopropylmethyl-7-(4-phenylpiperidin-1-yl)-8-trifluoromethyl[1,2,4]triazolo[4,3-a]pyridine (JNJ-42153605): a Positive Allosteric Modulator of the Metabotropic Glutamate 2 Receptor. J. Med. Chem. 2012;55:8770–8789. doi: 10.1021/jm3010724. [DOI] [PubMed] [Google Scholar]
- 35.Schrödinger Suite. 2011. [Google Scholar]; Epik version 2.2. New York, NY: Schröodinger, LLC; 2011. [Google Scholar]; Impact version 5.7. New York, NY: Schröodinger, LLC; 2011. [Google Scholar]; Prime version 2.3. New York, NY: Schröodinger, LLC; 2011. [Google Scholar]
- 36.Maestro, version 9.2. New York, NY: Schröodinger, LLC; 2011. [Google Scholar]
- 37.The PyMOL Molecular Graphics System, Version 1.5.0.4. Schröodinger, LLC; [Google Scholar]
- 38.Winquist J, Abdurakhmanov E, Baraznenok V, Henderson I, Vrang L, Danielson UH. Resolution of the Interaction Mechanisms and Characteristics of Non-nucleoside Inhibitors of Nepatitis C Virus Polymerase. Antivir. Res. doi: 10.1016/j.antiviral.2012.12.027. [DOI] [PubMed] [Google Scholar]
- 39.Vrolijk JM, Kaul A, Hansen BE, Lohmann V, Haagmans BL, Schalm SW, Bartenschlager R. A Replicon-Based Bioassay for the Measurement of Interferons in Patients with Chronic Hepatitis C. J. Virol. Methods. 2003;110:201–209. doi: 10.1016/s0166-0934(03)00134-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.