

Abstract
It has repeatedly been shown that statins decrease morbidity and mortality in patients with atherosclerosis, thus supporting their use for the primary and secondary prevention of ischemic heart disease. Different pathological pathways that are triggered in the setting of acute coronary syndrome (ACS), such as endothelial dysfunction, activation of inflammatory and coagulation cascades, and thrombus formation, are known to be inhibited by statins, thereby justifying the use of these agents in patients with ACS. Several recent prospective controlled clinical trials have demonstrated the safety and, in some cases, the efficacy of statins when administered early after ACS. An increasing number of publications have reported, however, that statins may confer a beneficial effect not only in early secondary prevention, but also in the direct treatment of ACS (ie, when statins are administered as first-line treatment in clinically unstable patients). This therapeutic option is supported by the following: numerous experimental studies demonstrating a protective effect of statins under conditions of acute ischemia; analysis of different registries and trials, which has demonstrated a more favourable prognosis for statin-treated patients at the time of acute myocardial ischemia; and small clinical trials reporting a lower periprocedural infarction rate during coronary intervention or lower levels of several prognostic biomarkers, in addition to a lower incidence of cardiovascular events associated with statin therapy. Nevertheless, confirmation of this hypothesis in large prospective controlled clinical trials will be necessary before the implementation of statins as first-line therapy in unstable patients with ACS, irrespective of blood cholesterol levels.
Keywords: Acute coronary syndrome, Myocardial infarction, Statin
3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors (statins) lower low-density lipoprotein (LDL) cholesterol levels by decreasing endogenous cholesterol synthesis. Statins are used worldwide in patients with hypercholesterolemia and coronary artery disease (CAD). There is strong evidence supporting their efficacy and use for both primary and secondary prevention, and in reducing mortality and nonfatal cardiovascular event rates. This effect can, however, be only partially explained by lipid-lowering mechanisms. During the past decade, substantial effort has been devoted to the investigation of the nonlipid-mediated action of statins, ie, their ‘pleiotropic effects’. It has been shown that inhibition of HMG-CoA reductase not only decreases cholesterol synthesis, but also the production of several isoprenoid metabolites, resulting in the inhibition of small GTP-binding proteins (eg, Ras, Rac, RhoA) responsible for various nonlipid-mediated activities (1) (Figure 1). Statins, therefore, also exert anti-inflammatory, antithrombotic and antioxidant effects, increase nitric oxide (NO) production and improve endothelial dysfunction (2).
Figure 1).
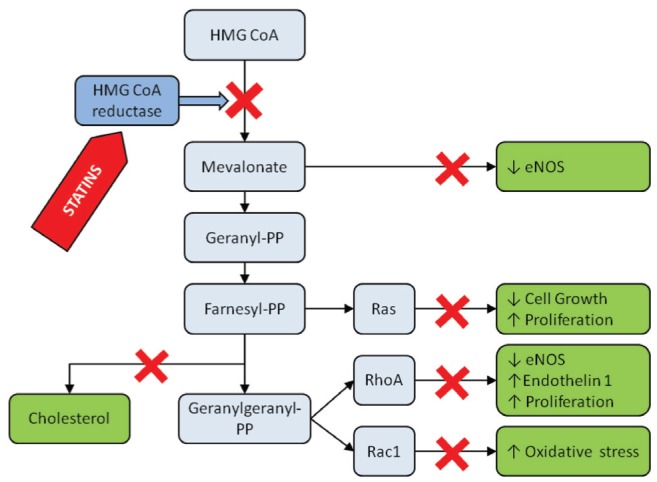
Schematic illustration of the mevalonate pathway and the effects of the inhibition of HMG CoA inhibition by statins. ↓ Decrease; ↑ Increase; HMG CoA 3-hydroxy-3-methylglutaryl coenzyme A; NOS Nitric oxide synthase; PP Pyrophosphate
For many years, coronary atherosclerosis has been known to be associated with lipid metabolism disorders. However, it has been shown that ‘classical’ risk factors, such as hypercholesterolemia, hypertension, diabetes and cigarette smoking, cannot fully explain the development of coronary stenosis in all patients suffering from CAD. Intensive research on the pathogenesis of coronary plaque development and rupture led to the hypothesis of possible involvement of other mechanisms in this process. Inflammation has been described as a particularly important factor in the development of CAD. The increased level of some inflammatory markers, such as C-reactive protein (CRP), has been shown to be one of the strongest risk factors for cardiovascular events. The activation of inflammatory pathways also likely plays a key role in coronary plaque destabilization and rupture, with subsequent thrombosis development clinically manifested as acute coronary syndrome (ACS). Statins have been shown to modulate several mechanisms involved in the pathogenesis of ACS including inflammation, oxidative stress, endothelial dysfunction and thrombosis (1–3) (Figure 2). The heightened interest in the use of statins for the treatment of ACS is, therefore, completely justified.
Figure 2).
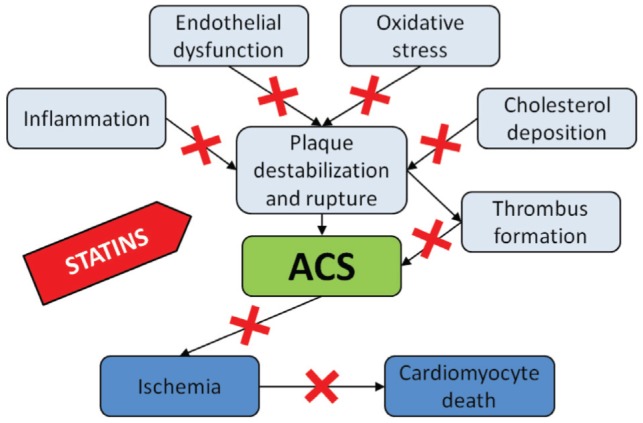
‘Pleiotropic’ effects of statins. ACS Acute coronary syndrome
VULNERABLE PLAQUE
During recent years, marked progress has been made in understanding the pathogenesis of ACS. Several pathways have been found to participate in the development of coronary instability, with the activation of inflammatory mechanisms likely playing an important role in plaque destabilization. Statins have been shown to simultaneously influence multiple factors that participate in this inflammatory process. A brief overview of the current knowledge of the pathogenesis of vulnerable plaque development will provide a fundamental theoretical background, which is essential for a better understanding of the possible therapeutic targets for statins.
Based on pathological and angiographic studies, plaque fissuring or rupture was determined to be a trigger for coronary thrombosis, which is the cause of ACS in the majority of cases. Atherosclerotic plaque that is susceptible to disruption is identifiable by certain histological features: it usually contains a large lipid pool; an accumulation of macrophages; and a thin fibrous cap (4–7). The mediators contributing to plaque vulnerability may be divided into extrinsic and intrinsic factors (8,9). Extrinsic factors actually trigger plaque disruption and include circumferential stress, hemodynamic shear stress, vasospasm or a prothrombotic milieu (9–12). Intrinsic factors are responsible for susceptibility to plaque rupture or directly cause it. Intrinsic factors include the size and composition of the lipid core, neovascularization, endothelial erosion/fissure, low cap thickness, inflammation, increased levels of matrix degradation enzymes, a decreased number of smooth muscle cells (SMCs) and low collagen content, outward remodelling and nodular calcification (9).
Several cell types are intimately involved in the pathogenesis of vulnerable plaque development.
Macrophages
Lesional macrophages overexpress various enzymes, signalling molecules, proinflammatory cytokines and other active substances. Proteinases produced by macrophages include members of the matrix metalloproteinase (MMP) family, represented by collagenases MMP-1, MMP-8 and MMP-13. Collagenases are believed to be responsible for fibrous cap degradation (4,13), which is associated with the loss of tensile stress tolerance (14). Other enzymes expressed by macrophages are myeloperoxidases, which play a major role in the release of reactive oxygen species, resulting in oxidative stress (OS). OS may activate other cell types such as endothelial cells (ECs) and different MMPs (MMP-2, MMP-9 and MMP-14) (15–17). Macrophages in atheromas also secrete several pro-inflammatory cytokines: interleukin (IL)-1 beta, IL-2, tumor necrosis factor-alpha and interferon gamma; and growth factors: vascular endothelial growth factor, macrophage colony-stimulating factor, which lead to activation and/or proliferation of other cell types such as SMC or ECs (18). Moreover, lesional macrophages also express tissue factor (TF), a potent initiator of the coagulation cascade, and plasminogen activator inhibitor 1, a fibrinolysis inhibitor (19–20). These agents may accelerate thrombus formation after fibrous cap disruption (4). Recently, lipoprotein-associated phospholipase A2 (Lp-PLA2) was introduced, not only as a risk factor markedly associated with higher incidence of cardiovascular events, but also as a potentially important pathogenic factor participating in the progression of atherosclerosis (21). Lp-PLA2 is an enzyme produced by macrophages, foam cells, mast cells and T-lymphocytes in atherosclerotic plaques, and by liver cells (22). Lp-PLA2 is responsible for the hydrolysis of oxidized phospholipids, which leads to the production of highly proinflammatory products (eg, lysophosphatidylcholine and oxidized nonesterified fatty acids) (22). In the blood, Lp-PLA2 is predominantly associated with low-density lipoprotein (LDL) (22). This enzyme has been found in both stable and vulnerable atherosclerotic plaques; a significantly higher concentration of Lp-PLA2 in vulnerable and ruptured plaques has been reported. In these unstable plaques, Lp-PLA2 is localized not only in the necrotic core, but also in the subendothelial space of the regions within the thin fibrous cap or at the site of plaque rupture (23,24). Secretory phospholipase A2 (sPLA2) is another enzyme participating in the development of atherosclerotic lesions. Both Lp-PLA2 and sPLA2 cleave the oxidized fatty acid side chain of oxidized phospholipids; in contrast to Lp-PLA2, sPLA2 is a relatively small molecule and is expressed in hepatocytes, macrophages, platelets and vascular smooth muscle cells (25–27). Similarly to Lp-PLA2, however, sPLA2 has been detected in plaque areas with massive lipid accumulation, leukocyte infiltration, cellular necrosis and calcifications (25). Elevated levels of sPLA2 mass and/or activity are associated with increased risk of cardiovascular events (25–27).
Endothelium
Another major contributor to coronary plaque vulnerability are EC. Expression of different surface molecules (eg, vascular cell adhesion molecule 1 [VCAM-1], selectin E and selectin P) and production of proinflammatory substances (eg, monocyte chemoattractant protein 1) by EC lead to leukocyte recruitment and the formation of macrophage-rich atheromas (28–31). In atheromas, the expression of endothelial NO synthase (eNOS), which produces NO from L-arginine, is also decreased (32).
SMCs
Proliferation of SMCs, similar to the production of different components of the extracellular matrix (ECM) by SMCs, is considered to be important for plaque stability (33). Advanced atherosclerotic lesions are, however, characterized by a paucity of SMC proliferation and decreased collagen content (10,13,33).
T lymphocytes
Different types of T lymphocytes have been detected in atherosclerotic plaques. Interactions between various phenotypes of CD4+ T lymphocytes (Th cells) likely play an important role in unstable coronary plaque development. While the Th1 response, characterized by the production of interferon-gamma and IL-12, decreases SMC proliferation and ECM synthesis, the Th2 response (production of IL-4, IL-5 and IL-10) inhibits the development of atherosclerosis, possibly by inhibiting apoptosis and downregulating the Th1 response (9,34,35).
PLEIOTROPIC EFFECTS OF STATINS
As mentioned above, statins are highly effective for the treatment of dyslipidemias. Statins significantly reduce total cholesterol (TC), LDL-cholesterol and triglyceride levels, and increase high-density lipoprotein (HDL)-cholesterol levels after several weeks of therapy (36,37). However, statins also exert a variety of nonlipid effects known as ‘pleiotropic effects’. During the past several years, it has repeatedly been observed that statins influence several mechanisms that are intimately involved in the development of vulnerable plaque (Figure 2). Discovery of these pleiotropic effects, therefore, strongly supports the concept of using statins for the treatment of ACS.
Anti-inflammatory effects of statins
As described above, inflammation is probably intimately involved in the pathogenesis of atherosclerosis and in the development of coronary plaque vulnerability. Increased levels of a variety of circulating markers of inflammation (CRP, serum amyloid A, heat-shock protein 65, IL-6 and circulating adhesion molecules ICAM-1 and VCAM-1) have been reported to be related to the severity of atherosclerosis and to the poorer prognosis of patients with ischemic heart disease. CRP levels, one of the most important risk factors for cardiovascular events, have repeatedly been shown to be reduced by different statins in human trials – a reduction that was independent of their lipid-lowering effect (38–46). The best evidence of a direct effect of statins on CRP production comes, however, from experimental studies showing reduction in CRP levels by statins even in cultured human hepatocytes (47). Furthermore, statins suppressed the secretion of some cytokines, such as IL 1, IL-6, IL-8, tumour necrosis factor-alpha and monocyte chemoattractant protein-1 (48–55), as well as the production of MMPs (50,56–58). Moreover, reductions in the levels of circulating adhesion molecules ICAM-1, P-selectin, and E-selectin after statin therapy has been described (59–61). It has been shown that long-term, intensive statin therapy may also decrease Lp-PLA2 levels by more than 20% (62); this is, however, largely due to the fact that most Lp-PLA2 is bound to apoB-containing particles, the concentration of which is reduced by statins.
Antioxidant effects of statins
Reactive oxygen species are directly involved in the degradation of NO and enhance endothelial dysfunction; moreover, oxidation of LDL contributes to foam cell formation by increasing the accumulation of cholesterol in macrophages and stimulating thrombosis and inflammation (63). It has been demonstrated that fluvastatin exerts a superoxide or hydroxyl radical scavenging activity, and reduces susceptibility of LDL to oxidation (64–67), while cerivastatin has superoxide scavenging activity and preserves active NO (68–69). Another member of the statin group, atorvastatin, has been shown to decrease free radical-induced lipid peroxidation in plasma and increase total antioxidant status (70). Furthermore, it has been shown that statins inhibit reactive oxygen species production via inhibition of NADPH oxidase activation (71).
The effect of statins on endothelial dysfunction
Endothelial dysfunction can be described as an imbalance between the compensatory mechanisms responsible for vasoconstriction and vasodilation. Endothelium-derived vasorelaxation is mediated by NO and, in contrast, endothelin-1 (ET-1) and angiotensin are potent vasoconstrictive agents (63).
Effects of statins on NO:
NO plays a protective role in CAD via its vasodilating effect, modification of the inflammatory response, decrease of SMC proliferation, and leukocyte and platelet activation (63,72,73). Furthermore, NO reduces the endothelial expression of adhesion molecules, monocyte adhesion to the endothelium, and decreases production of IL-6 and IL-8 (63,74). Increases in NO production, mediated by upregulation of eNOS, have been observed after administration of statins in different experimental models, even in cell cultures in the absence of any lipid medium (68,69,75–77). A possible explanation for this effect is the reduced level of cholesterol-synthesis metabolites, such as mevalonate, geranyl-pyrophosphate, farnesyl-pyrophosphate or geranylgeranyl-pyrophosphate, with subsequent inhibition of Ras, Rho and Rac proteins, leading to suppressed NADPH oxidase activation, reduced eNOS messenger RNA (mRNA) degradation and resulting in higher eNOS level and activity (63,77–81) (Figure 1).
Effects of statins on ET-1:
ET-1 is synthesized by ECs and has an action opposite to NO, ie, it stimulates vasoconstriction and vascular cell proliferation, and acts as a platelet activator (63,82). In experimental models, the activation of ET-1 expression promotes the development of atherosclerosis (83). Administration of statins induces downregulation of pre-pro ET-1 mRNA levels (84); however, it remains unclear whether this is a direct effect of statins on ET-1 synthesis or whether it is mediated by statin-induced increase of NO production that can exert a similar effect on ET-1 (63,84).
In clinical settings, the effect of statins on endothelial dysfunction has been assessed mostly by the measurement of flow-mediated dilation (FMD), a parameter that is impaired in patients with atherosclerosis (85,86). Administration of statins in patients with hypercholesterolemia and/or CAD has been reported to significantly improve FMD independent of any cholesterol-lowering effect (85–89). Clear evidence of the direct, lipid-independent effect of statins on endothelial dysfunction was shown by Landmesser et al (90) in their study comparing the effects of simvastatin and ezetimib: despite a similar reduction in circulating LDL by both agents, simvastatin significantly improved FMD and the number of functionally active endothelial progenitor cells.
Antithrombotic effects of statins
It has been repeatedly demonstrated in cultured monocytes/macrophages, SMCs and endothelial cells that statins can reduce the level and activity of TF, which initiates the coagulation cascade by activating factors IX and X (57,91–95). This effect of statins was also confirmed in vivo in animal models and in human studies (57,95–99). On the other hand, statins have been shown to decrease total levels of the TF pathway inhibitor (TFPI), a potent anticoagulant agent (100–103); however, statins do not influence the level of free TFPI (100,102,103). Because the anticoagulant activity of TFPI is related only to the free fraction of TFPI (103,104), this molecule appears to play a relatively marginal role in the global anticoagulant activity of statins (104).
Based on studies evaluating the effect of statins on prothrombin fragment (F1+2), fibrinopeptide A and thrombin-antithrombin III complexes, statins have been found to significantly decrease thrombin formation (98,105–110). It has been shown that this effect results not only in the inhibition of platelet-dependent thrombin formation but also in decreased expression of TF (92,104).
Clinical trials have reported contradictory data on the effect of statins on fibrinogen levels, one of the important risk factors for cardiovascular events (111–115). This discrepancy could be at least partially explained by the different methods used in these studies (104). Factor VII, factor VIII and von Willebrand factor have also been identified as predictors of increased risk for CAD (116). Although data in the literature regarding the effect of statins on these factors are inconsistent, the majority of studies have demonstrated favourable results (104).
Statins, however, influence not only coagulation but also fibrinolytic activity. In vitro studies have repeatedly demonstrated that administration of statins results in an increase in the level, activity and synthesis of tissue plasminogen activator and, simultaneously, in a decrease in the level, activity and synthesis of plasminogen activator inhibitor-1 (76,115,117–123). Although there is general agreement among the in vitro studies concerning the fibrinolysis-stimulating role of statins, clinical trials have produced less convincing results (104). These clinical studies are, however, inconsistent with respect to inclusion criteria, type of statin and dosage used, duration of treatment and methodological approaches (104).
STATINS IN EARLY SECONDARY PREVENTION
There is overwhelming evidence generated from large prospective trials, such as the Scandinavian Simvastatin Survival Study (4S)(124), the Cholesterol and Recurrent Events (CARE) trial (125) or the Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) trial (126), supporting the beneficial role of statins in the therapy of stable forms of CAD. The discovery of the nonlipid effects of statins in recent years has, however, been immediately reflected in the efforts to also use statins in less stable patients. Different observational studies have been published demonstrating the favourable effects of statin therapy when started soon after the onset of ACS. Furthermore, several randomized prospective trials have been performed to evaluate the effect of statins when administered early after the onset of ACS.
Observational studies
A large, prospective cohort study using data from the Swedish Register of Cardiac Intensive Care (RIKS-HIA) demonstrated that early initiation of statin therapy in patients with acute myocardial infarction (AMI) was associated with a reduced one-year mortality rate (risk ratio [RR] 0.75; P=0.001) (127). Post hoc analysis of two randomized trials – Global Use of Strategies to Open Occluded Coronary Arteries (GUSTO IIb) and Platelet Glycoprotein IIb/IIIa in Unstable Angina: Receptor Suppression Using Integrilin Therapy (PURSUIT) – showed that patients with ACS discharged on lipid-lowering drugs demonstrated a survival benefit at six months (RR 0.48; P<0.001) (128). In the Orofiban in Patients with Unstable Coronary Syndromes/Thrombolysis in Myocardial Infarction (OPUS/TIMI 16) study (129), mortality at one month was reduced in patients treated with lipid-lowering drugs (RR 0.30; P<0.001). The analysis of the Sibrafiban Versus Aspirin to Yield Maximum Protection from Ischemic Heart Events Post-Acute Coronary Syndromes (SYMPHONY) trial demonstrated lower mortality in statin-treated patients at three months (RR 0.58; P<0.05) (130). Subgroup analysis of the Platelet Receptor Inhibition in Ischemic Syndrome Management (PRISM) (131) study demonstrated a more favourable prognosis for statin-treated patients compared with patients without statin therapy. Surprisingly, the worst prognosis in this analysis was described in patients in whom statin therapy was withdrawn after hospital admission for ACS. Early use of statins in patients with AMI was associated with a reduced in-hospital mortality rate in the Maximized Individual Therapies in Acute Myocardial Ischemia (MITRA) registry (132). It has been shown that statin therapy also improved prognosis in patients undergoing percutaneous coronary intervention (133). It should, however, be emphasized that the risk reduction in most of the above mentioned analyses was far greater than anything observed in large randomized controlled trials; it is, therefore, very likely that there were unaccounted confounders influencing the results.
Randomized trials
Recently, several randomized trials evaluating statin therapy started early after ACS onset have been published. A brief summary of their design and results for the primary end points are presented in Table 1.
TABLE 1.
Large randomized trials evaluating early statin therapy after the onset of acute coronary syndrome
| Trial (reference) | Drugs studied | Primary end point | Results |
|---|---|---|---|
| MIRACL (134) | Atorvastatin/placebo | MACE | RR 0.84; P=0.048 |
| PACT (135) | Pravastatin/placebo | MACE | P not significant |
| FLORIDA (138) | Fluvastatin/placebo | Ischemia | P not significant |
| A to Z (141) | Simvastatin/placebo | MACE | P not significant |
| PROVE-IT (142) | Atorvastatin/pravastatin | MACE | RR 0.84; P=0.005 |
FLORIDA Fluvastatin on Risk Diminishing After Acute Myocardial Infarction; MACE Major adverse cardiac events; MIRACL Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering; PACT Pravastatin in Acute Coronary Treatment; PROVE-IT Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22
The Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering (MIRACL) trial (134) randomly assigned 3086 patients with unstable angina or non-Q-wave AMI to atorvastatin 80 mg or placebo within 24 h to 96 h after hospital admission; treatment continued for 16 weeks. The investigators reported a 16% relative reduction in the combined end points of death, recurrent nonfatal AMI, resuscitated cardiac arrest and recurrent ischemia requiring repeated hospitalization (14.8% versus 17.4%; RR 0.84; P=0.048). This benefit was largely driven by a reduction in the endpoint of recurrent ischemia requiring hospitalization (6.2% versus 8.4%; RR 0.74; P=0.02). Limitations of this study from the current point of view were the exclusion criterion of coronary intervention undergone or planned; only conservatively treated patients were included in this study.
The Pravastatin in Acute Coronary Treatment (PACT) trial (135) evaluated the effect of pravastatin administration within 24 h of the onset of symptoms in patients with ACS. The recruitment of 10,000 patients was planned; however, the study was stopped early. A total of 3408 patients were randomly assigned to pravastatin 20 mg, 40 mg or placebo. After 30 days of follow-up, no significant difference in the occurrence of major adverse cardiac events (MACE) was observed.
In the Prevention of Ischemic Events by Early Treatment of Cerivastatin Study (PRINCESS) (136), patients were randomly assigned within 48 h of admission for ACS to cerivastatin or placebo. This trial was prematurely stopped after cerivastatin was withdrawn from the worldwide market. Data obtained from a total of 3600 patients who were followed-up for 4.5 months were analyzed; no significant difference in the occurrence of MACE was found between the groups.
The Lescol Intervention Prevention Study (LIPS) (137) enrolled 1677 patients who were scheduled for a first percutaneous coronary intervention (PCI) procedure for stable angina, unstable angina or silent ischemia. Patients were randomly assigned to fluvastatin 40 mg twice daily or placebo at an average of 2.7 days after the procedure. Statin therapy resulted in a reduction of the occurrence of MACE at a median follow-up period of 3.9 years (21.4% versus 26.7%; RR 0.78; P=0.01). It should, however, be mentioned that only approximately 49% of patients in both groups were categorized as ‘unstable angina’ and the number of patients with confirmed ACS was not published.
The Fluvastatin on Risk Diminishing After Acute Myocardial Infarction (FLORIDA) trial (138) randomly assigned 540 patients with AMI to fluvastatin or placebo within 14 days of the index MI. The primary end point was reduction in cardiac ischemia, which was determined by ambulatory Holter monitoring. No differences in the occurrence of ischemia were observed and, similarly, there were no differences in mortality rates or the rate of MACE after one year.
The Pravastatin Turkish Trial (PTT) (139) was a small study that randomly assigned 150 patients with AMI to pravastatin 40 mg or no lipid-lowering drug within 6 h of hospital admission. All patients were treated with intravenous fibrinolytic therapy. At six months, no difference was found in mortality; the pravastatin-treated group, however, experienced fewer recurrent ischemic events compared with the placebo group.
The Lipid-Coronary Artery Disease (L-CAD) trial (140) was primarily designed to examine the effect of pravastatin on angiographic regression of atherosclerosis. In the L-CAD study, 126 patients who had undergone PCI for ACS were randomly assigned to pravastatin 20 mg to 40 mg (with or without cholestyramine and/or nicotinic acid) or to usual care determined by a family physician, and patients were enrolled by hospital discharge. After two years of follow-up, fewer patients in the pravastatin group experienced MACE compared with the usual care group (OR 0.28; P=0.005); it should, however, be mentioned that the event reduction in this small open-label study is far greater than in large, placebo-controlled trials, again indicating possible unaccounted confounders that influenced the results.
Phase Z of the A to Z trial (141) compared an early intensive strategy with a delayed conservative strategy of simvastatin therapy in patients with ACS. Within five days of the onset of symptoms, a total of 4497 patients were randomly assigned to simvastatin 40 mg daily for one month followed by 80 mg daily thereafter, or to placebo for four months followed by simvastatin 20 mg daily. The rates of MACE were not significantly different between the early intensive and the delayed conservative group, either at four months or at two years.
The Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 (PROVE IT-TIMI 22) trial (142) randomly assigned 4162 patients within 10 days of hospital admission for ACS to pravastatin 40 mg daily (standard therapy) or atorvastatin 80 mg daily (intensive therapy). After a median follow-up period of two years, the rate of MACE was significantly lower in the intensive therapy group compared with the standard therapy group (22.4% versus 26.3%; RR 0.84; P=0.005).
Recently, several meta-analyses of randomized clinical trials evaluating statin therapy started early after ACS onset have been published. They have clearly shown that administration of statins does not influence the rates of nonfatal myocardial infarction, the number of cerebrovascular events or mortality rate within the first four months. Thereafter, however, the beneficial effect of statin therapy on cardiovascular events became detectable, reaching statistical significance at six months and persisting for two years of follow-up (HR 0.84 [95% CI 0.76 to 0.94]) (143–146).
It can be concluded that these prospective trials have shown safety and, in some cases, also benefit of the early initiation of statin therapy after ACS. It is important to emphasize that atorvastatin 80 mg is the only agent/dose proven in randomized controlled trials to have benefit after ACS, either against placebo (MIRACL) or against another statin regimen (PROVE-IT). Despite this evidence, now available for more than 10 years, it is surprising that only about one-half of patients with ACS are treated with an intensive statin regimen, according to a recent analysis (147). Thus, there remains a gap between evidence and clinical practice in the use of statins after ACS .
STATINS IN ACS THERAPY
It is necessary to stress that all of the above-mentioned trials were designed for early (in some cases ‘very early’) secondary prevention, but not for initial, emergency department therapy of ACS. In all these studies, patients were randomly assigned at least several hours and, in most cases, even several days after hospital admission. During such a long period of time, the majority of patients were already clinically stable (clinical stabilization was also an inclusion criterion in some of the trials). In the context of the above-described pathogenesis of vulnerable plaque development and mechanisms of the pleiotropic effects of statins, it is obvious that there are relevant reasons to test administration of these drugs not only for secondary prevention, but also directly for the treatment of ACS when administered to patients who are particularly unstable.
As discussed above, the pleiotropic effects of statins may affect different pathogenic pathways participating in the development of vulnerable plaque and in the pathogenesis of ACS. Expanding this suggestion, statins may significantly contribute to plaque stabilization, reduction of thrombus formation and acceleration of fibrinolysis. It may, therefore, be fruitful to investigate possible additional favourable effects of immediate, first-line statin therapy in the emergency department or catheterization laboratory at the time of coronary instability, rather than initiation after the acute event or at hospital discharge (148).
Animal studies
It has repeatedly been demonstrated in animal models that treatment with statins berfore the onset of myocardial ischemia reduces ischemia-reperfusion injury. In 1999, Lefer et al (149) demonstrated the cardio-protective effect of simvastatin in a model of isolated perfused rat heart, when simvastatin was administered before the induction of ischemia and before any effects on circulating lipoproteins. Simvastatin inhibited leukocyte-endothelium interactions and improved contractile parameters. Ueda et al (150) reported that pravastatin treatment decreased infarct size after experimentally induced ischemia and ischemic preconditioning in hypercholesterolemic rabbits. In another study, Lefer et al (151) demonstrated reduction in infarct size after simvastatin treatment in a diabetic mouse model. It has also been suggested that this protective effect may be at least partly mediated by the stimulation of eNOS and the subsequent increased production of NO (151,152).
While these experimental studies demonstrate the impact of prophylactic therapy, they do not address the question of whether patients with ACS may benefit from the initiation of statin therapy after the onset of ischemia or before reperfusion. Bauersachs et al (153) failed to demonstrate a reduction in infarct size when the statin was administered 24 h after the onset of myocardial ischemia, a time frame similar to the protocol described in of most of the above-mentioned randomized clinical trials. A study by Hayashidani et al (154) was among the first to demonstrate a significant benefit of statin therapy started immediately after the onset of ischemia. The authors described lower mortality rates in mice subjected to coronary artery ligation and fluvastatin therapy initiated immediately following the procedure. Furthermore, fluvastatin in this study attenuated left ventricular remodelling, decreased the incidence of heart failure and reduced the activity of MMPs. Bell and Yellon (155) demonstrated the beneficial effect of atorvastatin administered at the time of reperfusion: in the experimental model of isolated perfused mouse heart, atorvastatin reduced ischemia-reperfusion injury (infarct size) when added to the perfusion solution. The results of this in vitro study were confirmed by Wolfrum et al (156) in an in vivo study of experimental infarction in rats. The investigators administered simvastatin intravenously 3 min before restoration of flow after a temporary coronary artery occlusion; simvastatin treatment decreased infarct size by 42%. The two most recent studies have also shown another important aspect of the immediate effect of statin therapy: they demonstrated an acute increase in eNOS activity, which is probably not related to the up-regulation of eNOS, by stabilizing eNOS mRNA. To stabilize eNOS mRNA in a cell-culture system (75), several hours of statin treatment is required. The acute stimulation of eNOS activity is, however, at least partially mediated by the PI 3-kinase/Akt pathway, which is capable of phosphorylating eNOS within minutes (155,156). Zheng and Hu (157) demonstrated the protective effect of simvastatin when it was added to reperfusion solution in an isolated perfused rat heart model. In the same model, our group observed that acute administration of simvastatin during reperfusion protects myocardial contractile parameters from ischemia-reperfusion injury (158). In contrast, long-term pretreatment with simastatin in this study was associated with only a marginal effect.
Inflammatory markers
A recent prospective pilot trial evaluated the effect of statins as a part of first-line therapy in patients with ACS. Forty-four consecutive patients with ACS without ST segment elevation were randomly assigned at admission to cerivastatin 0.3 mg or no statin therapy (159). Twenty-four hours after hospital admission (and after the initiation of statin therapy), a significant reduction in the level of inflammatory markers CRP and IL-6 was observed in the cerivastatin-treated group compared with the untreated group. These results were confirmed by other authors that observed reduction of inflammatory markers by statins administered in first-line therapy of ACS. Luo et al (160) compared the effect of simvastatin 20 mg and placebo on the levels of CRP and IL-6 in 50 patients with ACS. After three weeks of therapy, they found significantly lower levels of both biomarkers in the simvastatin group. Macin et al (161) randomly assigned 90 ACS patients to atorvastatin 40 mg and placebo. They described significantly lower CRP levels in the atorvastatin group at discharge and at 30 days.
Lipid parameters
For many years, ACS was recognized as a strong stress factor associated with marked spontaneous changes in lipid parameters, and it has been speculated that the effect of statins on cholesterol levels can be detected only after several weeks of therapy. Recently, however, it has been shown that the current management of ACS results in only clinically insignificant spontaneous changes in lipoprotein levels during the course of ACS (162). In general, 90% of the eventual LDL reduction with statins is seen 14 days after initiation of treatment; however, smaller effects may be observed much sooner. Our group has studied the immediate effect of statins on lipid parameters in 64 ACS patients randomly assigned at admission to fluvastatin 80 mg or standard therapy without statin during the initial days of therapy. We observed significant reduction of TC, LDL-cholesterol and HDL-cholesterol levels by 14.5%, 17.2% and 10%, respectively, only 24 h after initiation of statin therapy (163). We observed similar alterations in lipid levels in the analysis of a group of 114 ACS patients who were treated with atorvastatin 80 mg (164). The acute reduction in HDL levels in the above studies is in contrast to the increases in HDL levels observed with long-term statin therapy; this may not be the result of statin therapy but just the acute phase response. The acute effect of statins on lipid levels was also investigated in several other studies with nonacute patients. Michelena et al (165) observed a reduction in TC and LDL levels after three days of therapy with simvastatin 80 mg in stable patients with high cardiovascular risk. Similarly, Marchesi et al (87) described decreases in TC and LDL levels after one week of atorvastatin therapy in hypercholesterolemic females. On the other hand, Tsunekawa et al (88) did not detect any difference after three-day cerivastatin therapy in diabetic patients; however, relatively low daily doses were used (0.15 mg).
These results indicate that statin therapy initiated early in ACS patients can influence lipid parameters as quickly as inflammatory markers. It is possible that rapid reduction of cholesterol levels may help to promote early coronary plaque stabilization by statins.
Clinical outcomes
Based on the experimental and surrogate clinical findings mentioned above, there are strong reasons to believe that administration of statins as first-line therapy for ACS may also improve clinical outcomes. Current evidence supporting the clinical efficacy of statins administered directly for the treatment of ACS, however, remains limited.
The first multicentre, randomized, double-blind, placebo-controlled study that focused on the effect of statin therapy initiated as first-line therapy of ACS was the Fluvastatin in the therapy of Acute Coronary Syndrome (FACS) trial (166). One-hundred fifty-six ACS patients were randomly assigned at the time of hospital admission to fluvastatin 80 mg or placebo. Study medication was administered for 30 days, after which time patients in both groups were encouraged to continue with open-label statin therapy. The primary end points were CRP, IL-6 and pregnancy-associated plasma protein A (PAPP-A) levels, which remained the same in both groups. In contrast, fluvastatin therapy was associated with a significant reduction in the cardiovascular event rate at one year (combined secondary end point) (11.5% versus 24.4%; OR 0.40 [95% CI 0.17 to 0.95]; P=0.038). Despite the small study population and other limitations, results of the FACS trial indicate possible benefits from initiation of statin therapy as early as possible in ACS patients. A favourable effect of atorvastatin 80 mg administered as first-line treatment of ACS in patients with ST elevation was recently reported by Kim et al (167). In their STATIN STEMI trial, 171 patients were randomly assigned to atorvastatin 80 mg or atorvastatin 10 mg before primary PCI. The authors found improved coronary flow, faster ST segment resolution and a trend toward a lower cardiovascular event rate in the intensive atorvastatin group.
CONCLUSION
Statins were introduced to clinical practice as lipid-lowering drugs for the treatment of high blood cholesterol levels. They were shown to be highly effective in hypercholesterolemic patients for primary and secondary prevention of CAD. Their efficacy in secondary prevention was demonstrated in large prospective morbidity and mortality clinical trials involving patients with stable CAD; these studies enrolled patients at least several months after the onset of ACS. Later, it was observed that statins exert a favourable effect, not only in hypercholesterolemia, but also in patients with normal or low cholesterol levels and, therefore, nonlipid-mediated effects of statins have been suggested. The discovery of the pleiotropic effects of statins opened the possibility for new indications for statin treatment. Several randomized trials have demonstrated the safety and, in some cases, the efficacy of statin therapy if initiated early after the onset of ACS. Extension of knowledge regarding the pleiotropic effects of statins, together with the increasing understanding of the pathogenesis of ACS have, however, shifted the initiation of statin therapy closer to the onset of symptoms. Recent experimental studies, similar to the first clinical trials, have generated promising results that have supported the concept of the cardioprotective effect of statin administration as first-line therapy for ACS. Confirmation of this approach by a large randomized trial is needed, however, to determine whether statins should be administered alongside acetylsalicylic acid in the earliest phase of treatment for ACS.
Acknowledgments
This work was supported by the grant from the Czech Ministry of Health, Nr. NT12153. The author thanks Prof Gregory G Schwartz for his invaluable help with manuscript preparation.
REFERENCES
- 1.Zhou Q, Liao JK. Pleiotropic effects of statins – basic research and clinical perspectives. Circ J. 2010;74:818–26. doi: 10.1253/circj.cj-10-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blum A, Shamburek R. The pleiotropic effects of statins on endothelial function, vascular inflammation, immunomodulation and thrombogenesis. Atherosclerosis. 2009;203:325–30. doi: 10.1016/j.atherosclerosis.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 3.Lardizabal JA, Deedwania PC. The anti-ischemic and anti-anginal properties of statins. Curr Atheroscler Rep. 2011;13:43–50. doi: 10.1007/s11883-010-0147-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aikawa M, Libby P. The vulnerable atherosclerotic plaque: Pathogenesis and therapeutic approach. Cardiovasc Pathol. 2004;13:125–38. doi: 10.1016/S1054-8807(04)00004-3. [DOI] [PubMed] [Google Scholar]
- 5.Davies MJ. Acute coronary thrombosis – the role of plaque disruption and its initiation and prevention. Eur Heart J. 1995;(16 Suppl):L:3–7. doi: 10.1093/eurheartj/16.suppl_l.3. [DOI] [PubMed] [Google Scholar]
- 6.Davies MJ. Detecting vulnerable coronary plaques. Lancet. 1996;347:1422–3. doi: 10.1016/s0140-6736(96)91677-3. [DOI] [PubMed] [Google Scholar]
- 7.Ambrose JA, Hjemdahl-Monsen CE, Borrico S, Gorlin R, Fuster V. Angiographic demonstration of a common link between unstable angina pectoris and non-Q-wave acute myocardial infarction. Am J Cardiol. 1988;61:244–7. doi: 10.1016/0002-9149(88)90924-1. [DOI] [PubMed] [Google Scholar]
- 8.Pasterkamp G, Schoneveld AH, Hijnen DJ, et al. Atherosclerotic arterial remodeling and the localization of macrophages and matrix metalloproteases 1, 2 and 9 in the human coronary artery. Atherosclerosis. 2000;150:245–53. doi: 10.1016/s0021-9150(99)00371-8. [DOI] [PubMed] [Google Scholar]
- 9.Dickson BC, Gotlieb AI. Towards understanding acute destabilization of vulnerable atherosclerotic plaques. Cardiovasc Pathol. 2003;12:237–48. doi: 10.1016/s1054-8807(03)00072-3. [DOI] [PubMed] [Google Scholar]
- 10.Falk E, Shah PK, Fuster V. Coronary plaque disruption. Circulation. 1995;92:657–71. doi: 10.1161/01.cir.92.3.657. [DOI] [PubMed] [Google Scholar]
- 11.Rabbani R, Topol EJ. Strategies to achieve coronary arterial plaque stabilization. Cardiovasc Res. 1999;41:402–17. doi: 10.1016/s0008-6363(98)00279-x. [DOI] [PubMed] [Google Scholar]
- 12.Bank AJ, Versluis A, Dodge SM, Douglas WH. Atherosclerotic plaque rupture: A fatigue process? Med Hypotheses. 2000;55:480–4. doi: 10.1054/mehy.2000.1096. [DOI] [PubMed] [Google Scholar]
- 13.Shah PK, Falk E, Badimon JJ, et al. Human monocyte-derived macrophages induce collagen breakdown in fibrous caps of atherosclerotic plaques. Potential role of matrix-degrading metalloproteinases and implications for plaque rupture. Circulation. 1995;92:1565–9. [PubMed] [Google Scholar]
- 14.Loree HM, Kamm RD, Stringfellow RG, Lee RT. Effects of fibrous cap thickness on peak circumferential stress in model atherosclerotic vessels. Circ Res. 1992;71:850–8. doi: 10.1161/01.res.71.4.850. [DOI] [PubMed] [Google Scholar]
- 15.Heinecke JW. Oxidized amino acids: Culprits in human atherosclerosis and indicators of oxidative stress. Free Radic Biol Med. 2002;32:1090–101. doi: 10.1016/s0891-5849(02)00792-x. [DOI] [PubMed] [Google Scholar]
- 16.Sugiyama S, Okada Y, Sukhova GK, Virmani R, Heinecke JW, Libby P. Macrophage myeloperoxidase regulation by granulocyte macrophage colony-stimulating factor in human atherosclerosis and implications in acute coronary syndromes. Am J Pathol. 2001;158:879–91. doi: 10.1016/S0002-9440(10)64036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rajagopalan S, Meng XP, Ramasamy S, Harrison DG, Galis ZS. Reactive oxygen species produced by macrophage-derived foam cells regulate the activity of vascular matrix metalloproteinases in vitro. Implications for atherosclerotic plaque stability. J Clin Invest. 1996;98:2572–9. doi: 10.1172/JCI119076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Libby P. Atherosclerosis: The new view. Sci Am. 2002;286:46–55. doi: 10.1038/scientificamerican0502-46. [DOI] [PubMed] [Google Scholar]
- 19.Wilcox JN, Smith KM, Schwartz SM, Gordon D. Localization of tissue factor in the normal vessel wall and in the atherosclerotic plaque. Proc Natl Acad Sci U S A. 1989;86:2839–43. doi: 10.1073/pnas.86.8.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lupu F, Bergonzelli GE, Heim DA, et al. Localization and production of plasminogen activator inhibitor-1 in human healthy and atherosclerotic arteries. Arterioscler Thromb. 1993;13:1090–100. doi: 10.1161/01.atv.13.7.1090. [DOI] [PubMed] [Google Scholar]
- 21.Colley KJ, Wolfert RL, Cobble ME. Lipoprotein associated phospholipase A(2): Role in atherosclerosis and utility as a biomarker for cardiovascular risk. EPMA J. 20112:27–38. doi: 10.1007/s13167-011-0063-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Epps KC, Wilensky RL. Lp-PLA – a novel risk factor for high-risk coronary and carotid artery disease. J Intern Med. 2011;269:94–106. doi: 10.1111/j.1365-2796.2010.02297.x. [DOI] [PubMed] [Google Scholar]
- 23.Zalewski A, Macphee C. Role of lipoprotein-associated phospholipase A2 in atherosclerosis: Biology, epidemiology, and possible therapeutic target. Arterioscler Thromb Vasc Biol. 2005;25:923–31. doi: 10.1161/01.ATV.0000160551.21962.a7. [DOI] [PubMed] [Google Scholar]
- 24.Kolodgie FD, Burke AP, Skorija KS, et al. Lipoprotein-associated phospholipase A2 protein expression in the natural progression of human coronary atherosclerosis. Arterioscler Thromb Vasc Biol. 2006;26:2523–9. doi: 10.1161/01.ATV.0000244681.72738.bc. [DOI] [PubMed] [Google Scholar]
- 25.Mallat Z, Lambeau G, Tedgui A. Lipoprotein-associated and secreted phospholipases A in cardiovascular disease: Roles as biological effectors and biomarkers. Circulation. 2010;122:2183–200. doi: 10.1161/CIRCULATIONAHA.110.936393. [DOI] [PubMed] [Google Scholar]
- 26.Ryu SK, Mallat Z, Benessiano J, et al. Phospholipase A2 enzymes, high-dose atorvastatin, and prediction of ischemic events after acute coronary syndromes. Circulation. 2012;125:757–66. doi: 10.1161/CIRCULATIONAHA.111.063487. [DOI] [PubMed] [Google Scholar]
- 27.Nicholls SJ, Cavender MA, Kastelein JJ, et al. Inhibition of secretory phospholipase A(2) in patients with acute coronary syndromes: Rationale and design of the Vascular Inflammation Suppression to Treat Acute Coronary Syndrome for 16 Weeks (VISTA-16) trial. Cardiovasc Drugs Ther. 2012;26:71–5. doi: 10.1007/s10557-011-6358-9. [DOI] [PubMed] [Google Scholar]
- 28.Cybulsky MI, Iiyama K, Li H, et al. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J Clin Invest. 2001;107:1255–62. doi: 10.1172/JCI11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gu L, Okada Y, Clinton SK, et al. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol Cell. 1998;2:275–81. doi: 10.1016/s1097-2765(00)80139-2. [DOI] [PubMed] [Google Scholar]
- 30.Plutzky J. Inflammatory pathways in atherosclerosis and acute coronary syndromes. Am J Cardiol. 2001;88:10K–5K. doi: 10.1016/s0002-9149(01)01924-5. [DOI] [PubMed] [Google Scholar]
- 31.Kadar A, Glasz T. Development of atherosclerosis and plaque biology. Cardiovasc Surg. 2001;9:109–21. doi: 10.1016/s0967-2109(00)00097-1. [DOI] [PubMed] [Google Scholar]
- 32.Oemar BS, Tschudi MR, Godoy N, Brovkovich V, Malinski T, Luscher TF. Reduced endothelial nitric oxide synthase expression and production in human atherosclerosis. Circulation. 1998;97:2494–8. doi: 10.1161/01.cir.97.25.2494. [DOI] [PubMed] [Google Scholar]
- 33.Braganza DM, Bennett MR. New insights into atherosclerotic plaque rupture. Postgrad Med J. 2001;77:94–8. doi: 10.1136/pmj.77.904.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Libby P. Changing concepts of atherogenesis. J Intern Med. 2000;247:349–58. doi: 10.1046/j.1365-2796.2000.00654.x. [DOI] [PubMed] [Google Scholar]
- 35.Pinderski LJ, Fischbein MP, Subbanagounder G, et al. Overexpression of interleukin-10 by activated T lymphocytes inhibits atherosclerosis in LDL receptor-deficient mice by altering lymphocyte and macrophage phenotypes. Circ Res. 2002;90:1064–71. doi: 10.1161/01.res.0000018941.10726.fa. [DOI] [PubMed] [Google Scholar]
- 36.Jones P, Kafonek S, Laurora I, Hunninghake D. Comparative dose efficacy study of atorvastatin versus simvastatin, pravastatin, lovastatin, and fluvastatin in patients with hypercholesterolemia (the CURVES study) Am J Cardiol. 1998;81:582–7. doi: 10.1016/s0002-9149(97)00965-x. [DOI] [PubMed] [Google Scholar]
- 37.Insull W, Kafonek S, Goldner D, Zieve F. Comparison of efficacy and safety of atorvastatin (10mg) with simvastatin (10mg) at six weeks. ASSET Investigators. Am J Cardiol. 2001;87:554–9. doi: 10.1016/s0002-9149(00)01430-2. [DOI] [PubMed] [Google Scholar]
- 38.Albert MA, Danielson E, Rifai N, Ridker PM. Effect of statin therapy on C-reactive protein levels: The pravastatin inflammation/CRP evaluation (PRINCE): A randomized trial and cohort study. JAMA. 2001;286:64–70. doi: 10.1001/jama.286.1.64. [DOI] [PubMed] [Google Scholar]
- 39.Kinlay S, Schwartz GG, Olsson AG, et al. High-dose atorvastatin enhances the decline in inflammatory markers in patients with acute coronary syndromes in the MIRACL study. Circulation. 2003;108:1560–6. doi: 10.1161/01.CIR.0000091404.09558.AF. [DOI] [PubMed] [Google Scholar]
- 40.Ridker PM, Rifai N, Lowenthal SP. Rapid reduction in C-reactive protein with cerivastatin among 785 patients with primary hypercholesterolemia. Circulation. 2001;103:1191–3. doi: 10.1161/01.cir.103.9.1191. [DOI] [PubMed] [Google Scholar]
- 41.Jialal I, Stein D, Balis D, Grundy SM, Adams-Huet B, Devaraj S. Effect of hydroxymethyl glutaryl coenzyme a reductase inhibitor therapy on high sensitive C-reactive protein levels. Circulation. 2001;103:1933–5. doi: 10.1161/01.cir.103.15.1933. [DOI] [PubMed] [Google Scholar]
- 42.Riesen WF, Engler H, Risch M, Korte W, Noseda G. Short-term effects of atorvastatin on C-reactive protein. Eur Heart J. 2002;23:794–9. doi: 10.1053/euhj.2001.2967. [DOI] [PubMed] [Google Scholar]
- 43.van de Ree MA, Huisman MV, Princen HM, Meinders AE, Kluft C. Strong decrease of high sensitivity C-reactive protein with high-dose atorvastatin in patients with type 2 diabetes mellitus. Atherosclerosis. 2003;166:129–35. doi: 10.1016/s0021-9150(02)00316-7. [DOI] [PubMed] [Google Scholar]
- 44.Koh KK, Schenke WH, Waclawiw MA, Csako G, Cannon RO., III Statin attenuates increase in C-reactive protein during estrogen replacement therapy in postmenopausal women. Circulation. 2002;105:1531–3. doi: 10.1161/01.cir.0000013837.81710.da. [DOI] [PubMed] [Google Scholar]
- 45.Kent SM, Flaherty PJ, Coyle LC, Markwood TT, Taylor AJ. Effect of atorvastatin and pravastatin on serum C-reactive protein. Am Heart J. 2003;145:e8. doi: 10.1067/mhj.2003.34. [DOI] [PubMed] [Google Scholar]
- 46.Ansell BJ, Watson KE, Weiss RE, Fonarow GC. hsCRP and HDL effects of statins trial (CHEST): Rapid effect of statin therapy on C-reactive protein and high-density lipoprotein levels: A clinical investigation. Heart Dis. 2003;5:2–7. doi: 10.1097/01.HDX.0000050407.62572.DE. [DOI] [PubMed] [Google Scholar]
- 47.Kleemann R, Verschuren L, de Rooij BJ, et al. Evidence for anti-inflammatory activity of statins and PPARalpha activators in human C-reactive protein transgenic mice in vivo and in cultured human hepatocytes in vitro. Blood. 2004;103:4188–94. doi: 10.1182/blood-2003-11-3791. [DOI] [PubMed] [Google Scholar]
- 48.Romano M, Diomede L, Sironi M, et al. Inhibition of monocyte chemotactic protein-1 synthesis by statins. Lab Invest. 2000;80:1095–100. doi: 10.1038/labinvest.3780115. [DOI] [PubMed] [Google Scholar]
- 49.Blake GJ, Ridker PM. Novel clinical markers of vascular wall inflammation. Circ Res. 2001;89:763–71. doi: 10.1161/hh2101.099270. [DOI] [PubMed] [Google Scholar]
- 50.Koh KK, Son JW, Ahn JY, et al. Non-lipid effects of statin on hypercholesterolemic patients established to have coronary artery disease who remained hypercholesterolemic while eating a step-II diet. Coron Artery Dis. 2001;12:305–11. doi: 10.1097/00019501-200106000-00006. [DOI] [PubMed] [Google Scholar]
- 51.Rezaie-Majd A, Maca T, Bucek RA, et al. Simvastatin reduces expression of cytokines interleukin-6, interleukin-8, and monocyte chemoattractant protein-1 in circulating monocytes from hypercholesterolemic patients. Arterioscler Thromb Vasc Biol. 2002;22:1194–9. doi: 10.1161/01.atv.0000022694.16328.cc. [DOI] [PubMed] [Google Scholar]
- 52.Brull DJ, Sanders J, Rumley A, Lowe GD, Humphries SE, Montgomery HE. Statin therapy and the acute inflammatory response after coronary artery bypass grafting. Am J Cardiol. 2001;88:431–3. doi: 10.1016/s0002-9149(01)01696-4. [DOI] [PubMed] [Google Scholar]
- 53.Ikeda U, Shimada K. Statins and monocytes. Lancet. 1999;353:2070. doi: 10.1016/S0140-6736(05)77885-5. [DOI] [PubMed] [Google Scholar]
- 54.Ferro D, Parrotto S, Basili S, Alessandri C, Violi F. Simvastatin inhibits the monocyte expression of proinflammatory cytokines in patients with hypercholesterolemia. J Am Coll Cardiol. 2000;36:427–31. doi: 10.1016/s0735-1097(00)00771-3. [DOI] [PubMed] [Google Scholar]
- 55.Bustos C, Hernandez-Presa MA, Ortego M, et al. HMG-CoA reductase inhibition by atorvastatin reduces neointimal inflammation in a rabbit model of atherosclerosis. J Am Coll Cardiol. 1998;32:2057–64. doi: 10.1016/s0735-1097(98)00487-2. [DOI] [PubMed] [Google Scholar]
- 56.Ikeda U, Shimpo M, Ohki R, et al. Fluvastatin inhibits matrix metalloproteinase-1 expression in human vascular endothelial cells. Hypertension. 2000;36:325–9. doi: 10.1161/01.hyp.36.3.325. [DOI] [PubMed] [Google Scholar]
- 57.Aikawa M, Rabkin E, Sugiyama S, et al. An HMG-CoA reductase inhibitor, cerivastatin, suppresses growth of macrophages expressing matrix metalloproteinases and tissue factor in vivo and in vitro. Circulation. 2001;103:276–83. doi: 10.1161/01.cir.103.2.276. [DOI] [PubMed] [Google Scholar]
- 58.Crisby M, Nordin-Fredriksson G, Shah PK, Yano J, Zhu J, Nilsson J. Pravastatin treatment increases collagen content and decreases lipid content, inflammation, metalloproteinases, and cell death in human carotid plaques: implications for plaque stabilization. Circulation. 2001;103:926–33. doi: 10.1161/01.cir.103.7.926. [DOI] [PubMed] [Google Scholar]
- 59.Seljeflot I, Tonstad S, Hjermann I, Arnesen H. Reduced expression of endothelial cell markers after 1 year treatment with simvastatin and atorvastatin in patients with coronary heart disease. Atherosclerosis. 2002;162:179–85. doi: 10.1016/s0021-9150(01)00696-7. [DOI] [PubMed] [Google Scholar]
- 60.Sardo MA, Castaldo M, Cinquegrani M, et al. Effects of atorvastatin treatment on sICAM-1 and plasma nitric oxide levels in hypercholesterolemic subjects. Clin Appl Thromb Hemost. 2002;8:257–63. doi: 10.1177/107602960200800310. [DOI] [PubMed] [Google Scholar]
- 61.Niwa S, Totsuka T, Hayashi S. Inhibitory effect of fluvastatin, an HMG-CoA reductase inhibitor, on the expression of adhesion molecules on human monocyte cell line. Int J Immunopharmacol. 1996;18:669–75. doi: 10.1016/s0192-0561(96)00068-9. [DOI] [PubMed] [Google Scholar]
- 62.O’Donoghue M, Morrow DA, Sabatine MS, et al. Lipoprotein-associated phospholipase A2 and its association with cardiovascular outcomes in patients with acute coronary syndromes in the PROVE IT-TIMI 22 (PRavastatin Or atorVastatin Evaluation and Infection Therapy-Thrombolysis In Myocardial Infarction) trial. Circulation. 2006;113:1745–52. doi: 10.1161/CIRCULATIONAHA.105.612630. [DOI] [PubMed] [Google Scholar]
- 63.Tsiara S, Elisaf M, Mikhailidis DP. Early vascular benefits of statin therapy. Curr Med Res Opin. 2003;19:540–56. doi: 10.1185/030079903125002225. [DOI] [PubMed] [Google Scholar]
- 64.Suzumura K, Yasuhara M, Narita H. Superoxide anion scavenging properties of fluvastatin and its metabolites. Chem Pharm Bull (Tokyo) 1999;47:1477–80. doi: 10.1248/cpb.47.1477. [DOI] [PubMed] [Google Scholar]
- 65.Suzumura K, Yasuhara M, Tanaka K, Odawara A, Narita H, Suzuki T. An in vitro study of the hydroxyl radical scavenging property of fluvastatin, and HMG-CoA reductase inhibitor. Chem Pharm Bull (Tokyo) 1999;47:1010–2. doi: 10.1248/cpb.47.1010. [DOI] [PubMed] [Google Scholar]
- 66.Suzumura K, Yasuhara M, Tanaka K, Suzuki T. Protective effect of fluvastatin sodium (XU-62-320), a 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitor, on oxidative modification of human low-density lipoprotein in vitro. Biochem Pharmacol. 1999;57:697–703. doi: 10.1016/s0006-2952(98)00341-4. [DOI] [PubMed] [Google Scholar]
- 67.Hussein O, Schlezinger S, Rosenblat M, Keidar S, Aviram M. Reduced susceptibility of low density lipoprotein (LDL) to lipid peroxidation after fluvastatin therapy is associated with the hypocholesterolemic effect of the drug and its binding to the LDL. Atherosclerosis. 1997;128:11–8. doi: 10.1016/s0021-9150(96)05972-2. [DOI] [PubMed] [Google Scholar]
- 68.Kalinowski L, Dobrucki IT, Malinski T. Cerivastatin potentiates nitric oxide release and enos expression through inhibition of isoprenoids synthesis. J Physiol Pharmacol. 2002;53:585–95. [PubMed] [Google Scholar]
- 69.Kalinowski L, Dobrucki LW, Brovkovych V, Malinski T. Increased nitric oxide bioavailability in endothelial cells contributes to the pleiotropic effect of cerivastatin. Circulation. 2002;105:933–8. doi: 10.1161/hc0802.104283. 26; [DOI] [PubMed] [Google Scholar]
- 70.Fuhrman B, Koren L, Volkova N, Keidar S, Hayek T, Aviram M. Atorvastatin therapy in hypercholesterolemic patients suppresses cellular uptake of oxidized-LDL by differentiating monocytes. Atherosclerosis. 2002;164:179–85. doi: 10.1016/s0021-9150(02)00063-1. [DOI] [PubMed] [Google Scholar]
- 71.Endres M, Laufs U. Effects of statins on endothelium and signaling mechanisms. Stroke. 2004;35(11 Suppl 1):2708–11. doi: 10.1161/01.STR.0000143319.73503.38. [DOI] [PubMed] [Google Scholar]
- 72.Vaughan CJ, Murphy MB, Buckley BM. Statins do more than just lower cholesterol. Lancet. 1996;348:1079–82. doi: 10.1016/S0140-6736(96)05190-2. [DOI] [PubMed] [Google Scholar]
- 73.Jeremy JY, Rowe D, Emsley AM, Newby AC. Nitric oxide and the proliferation of vascular smooth muscle cells. Cardiovasc Res. 1999;43:580–94. doi: 10.1016/s0008-6363(99)00171-6. [DOI] [PubMed] [Google Scholar]
- 74.De Caterina R, Libby P, Peng HB, et al. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J Clin Invest. 1995;96:60–8. doi: 10.1172/JCI118074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Laufs U, La Fata V, Plutzky J, Liao JK. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation. 1998;97:1129–35. doi: 10.1161/01.cir.97.12.1129. [DOI] [PubMed] [Google Scholar]
- 76.Morikawa S, Takabe W, Mataki C, et al. The effect of statins on mRNA levels of genes related to inflammation, coagulation, and vascular constriction in HUVEC. Human umbilical vein endothelial cells. J Atheroscler Thromb. 2002;9:178–83. doi: 10.5551/jat.9.178. [DOI] [PubMed] [Google Scholar]
- 77.Laufs U, Gertz K, Dirnagl U, Bohm M, Nickenig G, Endres M. Rosuvastatin, a new HMG-CoA reductase inhibitor, upregulates endothelial nitric oxide synthase and protects from ischemic stroke in mice. Brain Res. 2002;942:23–30. doi: 10.1016/s0006-8993(02)02649-5. [DOI] [PubMed] [Google Scholar]
- 78.Laufs U, Endres M, Custodis F, et al. Suppression of endothelial nitric oxide production after withdrawal of statin treatment is mediated by negative feedback regulation of rho GTPase gene transcription. Circulation. 2000;102:3104–10. doi: 10.1161/01.cir.102.25.3104. [DOI] [PubMed] [Google Scholar]
- 79.Laufs U, Liao JK. Targeting Rho in cardiovascular disease. Circ Res. 2000;87:526–8. doi: 10.1161/01.res.87.7.526. [DOI] [PubMed] [Google Scholar]
- 80.Jacobson JR. Statins in endothelial signaling and activation. Antioxid Redox Signal. 2009;11:811–21. doi: 10.1089/ars.2008.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wagner AH, Kohler T, Ruckschloss U, Just I, Hecker M. Improvement of nitric oxide-dependent vasodilatation by HMG-CoA reductase inhibitors through attenuation of endothelial superoxide anion formation. Arterioscler Thromb Vasc Biol. 2000;20:61–9. doi: 10.1161/01.atv.20.1.61. [DOI] [PubMed] [Google Scholar]
- 82.Jagroop IA, Mikhailidis DP. Effect of endothelin-1 on human platelet shape change: Reversal of activation by naftidrofuryl. Platelets. 2000;11:272–7. doi: 10.1080/09537100050129288. [DOI] [PubMed] [Google Scholar]
- 83.Barton M, Lattmann T, d’Uscio LV, Luscher TF, Shaw S. Inverse regulation of endothelin-1 and nitric oxide metabolites in tissue with aging: Implications for the age-dependent increase of cardiorenal disease. J Cardiovasc Pharmacol. 2000;36(5 Suppl 1):S153–6. doi: 10.1097/00005344-200036051-00048. [DOI] [PubMed] [Google Scholar]
- 84.Hernandez-Perera O, Perez-Sala D, Navarro-Antolin J, et al. Effects of the 3-hydroxy-3-methylglutaryl-CoA reductase inhibitors, atorvastatin and simvastatin, on the expression of endothelin-1 and endothelial nitric oxide synthase in vascular endothelial cells. J Clin Invest. 1998;101:2711–9. doi: 10.1172/JCI1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dupuis J, Tardif JC, Cernacek P, Theroux P. Cholesterol reduction rapidly improves endothelial function after acute coronary syndromes. The RECIFE (ReductIon of Cholesterol in Ischemia and Function of the Endothelium) trial. Circulation. 1999;99:3227–33. doi: 10.1161/01.cir.99.25.3227. [DOI] [PubMed] [Google Scholar]
- 86.de Jongh S, Lilien MR, op’t Roodt J, Stroes ES, Bakker HD, Kastelein JJ. Early statin therapy restores endothelial function in children with familial hypercholesterolemia. J Am Coll Cardiol. 2002;40:2117–21. doi: 10.1016/s0735-1097(02)02593-7. [DOI] [PubMed] [Google Scholar]
- 87.Marchesi S, Lupattelli G, Siepi D, et al. Short-term atorvastatin treatment improves endothelial function in hypercholesterolemic women. J Cardiovasc Pharmacol. 2000;36:617–21. doi: 10.1097/00005344-200011000-00011. [DOI] [PubMed] [Google Scholar]
- 88.Tsunekawa T, Hayashi T, Kano H, et al. Cerivastatin, a hydroxymethylglutaryl coenzyme a reductase inhibitor, improves endothelial function in elderly diabetic patients within 3 days. Circulation. 2001;104:376–9. doi: 10.1161/hc2901.094094. [DOI] [PubMed] [Google Scholar]
- 89.Omori H, Nagashima H, Tsurumi Y, et al. Direct in vivo evidence of a vascular statin: A single dose of cerivastatin rapidly increases vascular endothelial responsiveness in healthy normocholesterolaemic subjects. Br J Clin Pharmacol. 2002;54:395–9. doi: 10.1046/j.1365-2125.2002.01677.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Landmesser U, Bahlmann F, Mueller M, et al. Simvastatin versus ezetimibe: pleiotropic and lipid-lowering effects on endothelial function in humans. Circulation. 2005;111:2356–63. doi: 10.1161/01.CIR.0000164260.82417.3F. [DOI] [PubMed] [Google Scholar]
- 91.Colli S, Eligini S, Lalli M, Camera M, Paoletti R, Tremoli E. Vastatins inhibit tissue factor in cultured human macrophages. A novel mechanism of protection against atherothrombosis. Arterioscler Thromb Vasc Biol. 1997;17:265–72. doi: 10.1161/01.atv.17.2.265. [DOI] [PubMed] [Google Scholar]
- 92.Ferro D, Basili S, Alessandri C, Cara D, Violi F. Inhibition of tissue-factor-mediated thrombin generation by simvastatin. Atherosclerosis. 2000;149:111–6. doi: 10.1016/s0021-9150(99)00291-9. [DOI] [PubMed] [Google Scholar]
- 93.Nagata K, Ishibashi T, Sakamoto T, et al. Rho/Rho-kinase is involved in the synthesis of tissue factor in human monocytes. Atherosclerosis. 2002;163:39–47. doi: 10.1016/s0021-9150(01)00750-x. [DOI] [PubMed] [Google Scholar]
- 94.Eto M, Kozai T, Cosentino F, Joch H, Luscher TF. Statin prevents tissue factor expression in human endothelial cells: Role of Rho/Rho-kinase and Akt pathways. Circulation. 2002;105:1756–9. doi: 10.1161/01.cir.0000015465.73933.3b. [DOI] [PubMed] [Google Scholar]
- 95.Camera M, Toschi V, Comparato C, et al. Cholesterol-induced thrombogenicity of the vessel wall: Inhibitory effect of fluvastatin. Thromb Haemost. 2002;87:748–55. [PubMed] [Google Scholar]
- 96.Baetta R, Camera M, Comparato C, Altana C, Ezekowitz MD, Tremoli E. Fluvastatin reduces tissue factor expression and macrophage accumulation in carotid lesions of cholesterol-fed rabbits in the absence of lipid lowering. Arterioscler Thromb Vasc Biol. 2002;22:692–8. doi: 10.1161/01.atv.0000012802.69414.a8. [DOI] [PubMed] [Google Scholar]
- 97.Ferro D, Basili S, Alessandri C, Mantovani B, Cordova C, Violi F. Simvastatin reduces monocyte-tissue-factor expression type IIa hypercholesterolaemia. Lancet. 1997;350:1222. doi: 10.1016/S0140-6736(05)63452-6. [DOI] [PubMed] [Google Scholar]
- 98.Holschermann H, Hilgendorff A, Kemkes-Matthes B, et al. Simvastatin attenuates vascular hypercoagulability in cardiac transplant recipients. Transplantation. 2000;69:1830–6. doi: 10.1097/00007890-200005150-00017. [DOI] [PubMed] [Google Scholar]
- 99.Cortellaro M, Cofrancesco E, Arbustini E, et al. Atorvastatin and thrombogenicity of the carotid atherosclerotic plaque: The ATROCAP study. Thromb Haemost. 2002;88:41–7. [PubMed] [Google Scholar]
- 100.Hansen JB, Huseby KR, Huseby NE, Sandset PM, Hanssen TA, Nordoy A. Effect of cholesterol lowering on intravascular pools of TFPI and its anticoagulant potential in type II hyperlipoproteinemia. Arterioscler Thromb Vasc Biol. 1995;15:879–85. doi: 10.1161/01.atv.15.7.879. [DOI] [PubMed] [Google Scholar]
- 101.Lorena M, Perolini S, Casazza F, Milani M, Cimminiello C. Fluvastatin and tissue factor pathway inhibitor in type IIA and IIB hyperlipidemia and in acute myocardial infarction. Thromb Res. 1997;87:397–403. doi: 10.1016/s0049-3848(97)00143-6. [DOI] [PubMed] [Google Scholar]
- 102.Nordoy A, Bonaa KH, Sandset PM, Hansen JB, Nilsen H. Effect of omega-3 fatty acids and simvastatin on hemostatic risk factors and postprandial hyperlipemia in patients with combined hyperlipemia. Arterioscler Thromb Vasc Biol. 2000;20:259–65. doi: 10.1161/01.atv.20.1.259. [DOI] [PubMed] [Google Scholar]
- 103.Morishita E, Asakura H, Saito M, et al. Elevated plasma levels of free-form of TFPI antigen in hypercholesterolemic patients. Atherosclerosis. 2001;154:203–12. doi: 10.1016/s0021-9150(00)00463-9. [DOI] [PubMed] [Google Scholar]
- 104.Krysiak R, Okopien B, Herman Z. Effects of HMG-CoA reductase inhibitors on coagulation and fibrinolysis processes. Drugs. 2003;63:1821–54. doi: 10.2165/00003495-200363170-00005. [DOI] [PubMed] [Google Scholar]
- 105.Szczeklik A, Musial J, Undas A, et al. Inhibition of thrombin generation by simvastatin and lack of additive effects of aspirin in patients with marked hypercholesterolemia. J Am Coll Cardiol. 1999;33:1286–93. doi: 10.1016/s0735-1097(99)00023-6. [DOI] [PubMed] [Google Scholar]
- 106.Musial J, Undas A, Undas R, Brozek J, Szczeklik A. Treatment with simvastatin and low-dose aspirin depresses thrombin generation in patients with coronary heart disease and borderline-high cholesterol levels. Thromb Haemost. 2001;85:221–5. [PubMed] [Google Scholar]
- 107.Aoki I, Aoki N, Kawano K, et al. Platelet-dependent thrombin generation in patients with hyperlipidemia. J Am Coll Cardiol. 1997;30:91–6. doi: 10.1016/s0735-1097(97)00129-0. [DOI] [PubMed] [Google Scholar]
- 108.Alessandri C, Basili S, Maurelli M, et al. Relationship between prothrombin activation fragment F1 + 2 and serum cholesterol. Haemostasis. 1996;26:214–9. doi: 10.1159/000217210. [DOI] [PubMed] [Google Scholar]
- 109.Puccetti L, Bruni F, Di Renzo M, et al. Hypercoagulable state in hypercholesterolemic subjects assessed by platelet-dependent thrombin generation: In vitro effect of cerivastatin. Eur Rev Med Pharmacol Sci. 1999;3:197–204. [PubMed] [Google Scholar]
- 110.Puccetti L, Bruni F, Bova G, et al. Effect of diet and treatment with statins on platelet-dependent thrombin generation in hypercholesterolemic subjects. Nutr Metab Cardiovasc Dis. 2001;11:378–87. [PubMed] [Google Scholar]
- 111.Meade TW, Mellows S, Brozovic M, et al. Haemostatic function and ischaemic heart disease: Principal results of the Northwick Park Heart Study. Lancet. 1986;2:533–7. doi: 10.1016/s0140-6736(86)90111-x. [DOI] [PubMed] [Google Scholar]
- 112.Wilhelmsen L, Svardsudd K, Korsan-Bengtsen K, Larsson B, Welin L, Tibblin G. Fibrinogen as a risk factor for stroke and myocardial infarction. N Engl J Med. 1984;311:501–5. doi: 10.1056/NEJM198408233110804. [DOI] [PubMed] [Google Scholar]
- 113.Kannel WB, Wolf PA, Castelli WP, D’Agostino RB. Fibrinogen and risk of cardiovascular disease. The Framingham Study. JAMA. 1987;258:1183–6. [PubMed] [Google Scholar]
- 114.Davidson M, McKenney J, Stein E, et al. Comparison of one-year efficacy and safety of atorvastatin versus lovastatin in primary hypercholesterolemia. Atorvastatin Study Group I. Am J Cardiol. 1997;79:1475–81. doi: 10.1016/s0002-9149(97)00174-4. [DOI] [PubMed] [Google Scholar]
- 115.Seeger H, Wallwiener D, Mueck AO. Lipid-independent effects of an estrogen-statin combination: Inhibition of expression of adhesion molecules and plasminogen activator inhibitor-I in human endothelial cell cultures. Climacteric. 2001;4:209–14. [PubMed] [Google Scholar]
- 116.Warkentin TE. Hemostasis and atherosclerosis. Can J Cardiol. 1995;11(Suppl C):29C–34C. [PubMed] [Google Scholar]
- 117.Essig M, Nguyen G, Prie D, Escoubet B, Sraer JD, Friedlander G. 3-Hydroxy-3-methylglutaryl coenzyme A reductase inhibitors increase fibrinolytic activity in rat aortic endothelial cells. Role of geranylgeranylation and Rho proteins. Circ Res. 1998;83:683–90. doi: 10.1161/01.res.83.7.683. [DOI] [PubMed] [Google Scholar]
- 118.Mussoni L, Banfi C, Sironi L, Arpaia M, Tremoli E. Fluvastatin inhibits basal and stimulated plasminogen activator inhibitor 1, but induces tissue type plasminogen activator in cultured human endothelial cells. Thromb Haemost. 2000;84:59–64. [PubMed] [Google Scholar]
- 119.Haslinger B, Goedde MF, Toet KH, Kooistra T. Simvastatin increases fibrinolytic activity in human peritoneal mesothelial cells independent of cholesterol lowering. Kidney Int. 2002;62:1611–9. doi: 10.1046/j.1523-1755.2002.00601.x. [DOI] [PubMed] [Google Scholar]
- 120.Lopez S, Peiretti F, Bonardo B, Juhan-Vague I, Nalbone G. Effect of atorvastatin and fluvastatin on the expression of plasminogen activator inhibitor type-1 in cultured human endothelial cells. Atherosclerosis. 2000;152:359–66. doi: 10.1016/s0021-9150(00)00454-8. [DOI] [PubMed] [Google Scholar]
- 121.Bourcier T, Libby P. HMG CoA reductase inhibitors reduce plasminogen activator inhibitor-1 expression by human vascular smooth muscle and endothelial cells. Arterioscler Thromb Vasc Biol. 2000;20:556–62. doi: 10.1161/01.atv.20.2.556. [DOI] [PubMed] [Google Scholar]
- 122.Ishibashi T, Nagata K, Ohkawara H, et al. Inhibition of Rho/Rho-kinase signaling downregulates plasminogen activator inhibitor-1 synthesis in cultured human monocytes. Biochim Biophys Acta. 2002;1590:123–30. doi: 10.1016/s0167-4889(02)00201-x. [DOI] [PubMed] [Google Scholar]
- 123.Wiesbauer F, Kaun C, Zorn G, Maurer G, Huber K, Wojta J. HMG CoA reductase inhibitors affect the fibrinolytic system of human vascular cells in vitro: A comparative study using different statins. Br J Pharmacol. 2002;135:284–92. doi: 10.1038/sj.bjp.0704454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: The Scandinavian Simvastatin Survival Study (4S) Lancet. 1994;344:1383–9. [PubMed] [Google Scholar]
- 125.Sacks FM, Pfeffer MA, Moye LA, et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial investigators. N Engl J Med. 1996;335:1001–9. doi: 10.1056/NEJM199610033351401. [DOI] [PubMed] [Google Scholar]
- 126.Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. N Engl J Med. 1998;339:1349–57. doi: 10.1056/NEJM199811053391902. [DOI] [PubMed] [Google Scholar]
- 127.Stenestrand U, Wallentin L. Early statin treatment following acute myocardial infarction and 1-year survival. JAMA. 2001;285:430–6. doi: 10.1001/jama.285.4.430. [DOI] [PubMed] [Google Scholar]
- 128.Aronow HD, Topol EJ, Roe MT, et al. Effect of lipid-lowering therapy on early mortality after acute coronary syndromes: An observational study. Lancet. 2001;357:1063–8. doi: 10.1016/S0140-6736(00)04257-4. [DOI] [PubMed] [Google Scholar]
- 129.Cannon CP, McCabe CH, Bentley J, Braunwald E. Early statin therapy is associated with markedly lower mortality in patients with acute coronary syndromes: Observations from OPUS-TIMI 16. J Am Coll Cardiol. 2001;37(Suppl A):334A. (Absts 831-2). [Google Scholar]
- 130.Newby LK, Kristinsson A, Bhapkar MV, et al. Early statin initiation and outcomes in patients with acute coronary syndromes. JAMA. 2002;287:3087–95. doi: 10.1001/jama.287.23.3087. [DOI] [PubMed] [Google Scholar]
- 131.Heeschen C, Hamm CW, Laufs U, Snapinn S, Bohm M, White HD. Withdrawal of statins increases event rates in patients with acute coronary syndromes. Circulation. 2002;105:1446–52. doi: 10.1161/01.cir.0000012530.68333.c8. [DOI] [PubMed] [Google Scholar]
- 132.Schiele R, Gitt AK, Heer T. Early statin use in acute myocardial is associated with a reduced hospital mortality: Results of the Mitra-2. Circulation [Abstract] 2000;102(Suppl 2):II-435. (Abst 2117). [Google Scholar]
- 133.Briguori C, Colombo A, Airoldi F, et al. Statin administration before percutaneous coronary intervention: Impact on periprocedural myocardial infarction. Eur Heart J. 2004;25:1822–8. doi: 10.1016/j.ehj.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 134.Schwartz GG, Olsson AG, Ezekowitz MD, et al. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: The MIRACL study: A randomized controlled trial. JAMA. 2001;285:1711–8. doi: 10.1001/jama.285.13.1711. [DOI] [PubMed] [Google Scholar]
- 135.Thompson PL, Meredith I, Amerena J, Campbell TJ, Sloman JG, Harris PJ. Effect of pravastatin compared with placebo initiated within 24 hours of onset of acute myocardial infarction or unstable angina: the Pravastatin in Acute Coronary Treatment (PACT) trial. Am Heart J. 2004;148:e2. doi: 10.1016/j.ahj.2003.10.052. [DOI] [PubMed] [Google Scholar]
- 136.LaBlanche JM, Jukema JW, Charbonneau F, et al., editors. PRINCESS: Prevention of ischemic events by early treatment of cerivastatin after acute myocardial infarction. ESC Congress; Munich. August 28 to Septenmber 1, 2004. [Google Scholar]
- 137.Serruys PW, de Feyter P, Macaya C, et al. Fluvastatin for prevention of cardiac events following successful first percutaneous coronary intervention: a randomized controlled trial. JAMA. 2002;287:3215–22. doi: 10.1001/jama.287.24.3215. [DOI] [PubMed] [Google Scholar]
- 138.Liem AH, van Boven AJ, Veeger NJ, Withagen AJ, Robles de Medina RM, Tijssen JG, et al. Effect of fluvastatin on ischaemia following acute myocardial infarction: a randomized trial. Eur Heart J. 2002;23:1931–7. doi: 10.1053/euhj.2002.3291. [DOI] [PubMed] [Google Scholar]
- 139.Kayikcioglu M, Turkoglu C, Kultursay H, Evrengul H, Can L. The short term results of combined use of pravastatin with thrombolytic therapy in acute myocardial infarction. Circulation. 1999;100(Suppl 1):I–303. (Abst 1586). [Google Scholar]
- 140.Arntz HR, Agrawal R, Wunderlich W, et al. Beneficial effects of pravastatin (+/−colestyramine/niacin) initiated immediately after a coronary event (the randomized Lipid-Coronary Artery Disease [L-CAD] Study) Am J Cardiol. 2000;86:1293–8. doi: 10.1016/s0002-9149(00)01230-3. [DOI] [PubMed] [Google Scholar]
- 141.de Lemos JA, Blazing MA, Wiviott SD, et al. Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes: Phase Z of the A to Z trial. JAMA. 2004;292:1307–16. doi: 10.1001/jama.292.11.1307. [DOI] [PubMed] [Google Scholar]
- 142.Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med. 2004;350:1495–504. doi: 10.1056/NEJMoa040583. [DOI] [PubMed] [Google Scholar]
- 143.Briel M, Vale N, Schwartz GG, et al. Updated evidence on early statin therapy for acute coronary syndromes: Meta-analysis of 18 randomized trials involving over 14,000 patients. Int J Cardiol. 2012;158:93–100. doi: 10.1016/j.ijcard.2011.01.033. [DOI] [PubMed] [Google Scholar]
- 144.Briel M, Schwartz GG, Thompson PL, et al. Effects of early treatment with statins on short-term clinical outcomes in acute coronary syndromes: A meta-analysis of randomized controlled trials. JAMA. 2006;295:2046–56. doi: 10.1001/jama.295.17.2046. [DOI] [PubMed] [Google Scholar]
- 145.Hulten E, Jackson JL, Douglas K, George S, Villines TC. The effect of early, intensive statin therapy on acute coronary syndrome: A meta-analysis of randomized controlled trials. Arch Intern Med. 2006;166:1814–21. doi: 10.1001/archinte.166.17.1814. [DOI] [PubMed] [Google Scholar]
- 146.Vale N, Nordmann AJ, Schwartz GG, et al. Statins for acute coronary syndrome. Cochrane Database Syst Rev. 2011;(6):CD006870. doi: 10.1002/14651858.CD006870.pub2. [DOI] [PubMed] [Google Scholar]
- 147.Shiu JR, Pearson GJ, Charrois TL, Gyenes G, Koshman SL. Frequency of intensive statin therapy in patients with acute coronary syndrome admitted to a tertiary care center. Am J Cardiol. 2012;109:1–5. doi: 10.1016/j.amjcard.2011.07.064. [DOI] [PubMed] [Google Scholar]
- 148.Ostadal P, Alan D, Vejvoda J. Statins in the first-line therapy of acute coronary syndrome – similar to aspirin? Exp Clin Cardiol. 2005;10:9–16. [PMC free article] [PubMed] [Google Scholar]
- 149.Lefer AM, Campbell B, Shin YK, Scalia R, Hayward R, Lefer DJ. Simvastatin preserves the ischemic-reperfused myocardium in normocholesterolemic rat hearts. Circulation. 1999;100:178–84. doi: 10.1161/01.cir.100.2.178. [DOI] [PubMed] [Google Scholar]
- 150.Ueda Y, Kitakaze M, Komamura K, et al. Pravastatin restored the infarct size-limiting effect of ischemic preconditioning blunted by hypercholesterolemia in the rabbit model of myocardial infarction. J Am Coll Cardiol. 1999;34:2120–5. doi: 10.1016/s0735-1097(99)00440-4. [DOI] [PubMed] [Google Scholar]
- 151.Lefer DJ, Scalia R, Jones SP, et al. HMG-CoA reductase inhibition protects the diabetic myocardium from ischemia-reperfusion injury. Faseb J. 2001;15:1454–6. doi: 10.1096/fj.00-0819fje. [DOI] [PubMed] [Google Scholar]
- 152.Di Napoli P, Antonio Taccardi A, et al. Simvastatin reduces reperfusion injury by modulating nitric oxide synthase expression: An ex vivo study in isolated working rat hearts. Cardiovasc Res. 2001;51:283–93. doi: 10.1016/s0008-6363(01)00306-6. [DOI] [PubMed] [Google Scholar]
- 153.Bauersachs J, Galuppo P, Fraccarollo D, Christ M, Ertl G. Improvement of left ventricular remodeling and function by hydroxymethylglutaryl coenzyme a reductase inhibition with cerivastatin in rats with heart failure after myocardial infarction. Circulation. 2001;104:982–5. doi: 10.1161/hc3401.095946. [DOI] [PubMed] [Google Scholar]
- 154.Hayashidani S, Tsutsui H, Shiomi T, et al. Fluvastatin, a 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitor, attenuates left ventricular remodeling and failure after experimental myocardial infarction. Circulation. 2002;105:868–73. doi: 10.1161/hc0702.104164. [DOI] [PubMed] [Google Scholar]
- 155.Bell RM, Yellon DM. Atorvastatin, administered at the onset of reperfusion, and independent of lipid lowering, protects the myocardium by up-regulating a pro-survival pathway. J Am Coll Cardiol. 2003;41:508–15. doi: 10.1016/s0735-1097(02)02816-4. [DOI] [PubMed] [Google Scholar]
- 156.Wolfrum S, Dendorfer A, Schutt M, et al. Simvastatin acutely reduces myocardial reperfusion injury in vivo by activating the phosphatidylinositide 3-kinase/Akt pathway. J Cardiovasc Pharmacol. 2004;44:348–55. doi: 10.1097/01.fjc.0000137162.14735.30. [DOI] [PubMed] [Google Scholar]
- 157.Zheng X, Hu SJ. Effects of simvastatin on cardiohemodynamic responses to ischemia-reperfusion in isolated rat hearts. Heart Vessels. 2006;21:116–23. doi: 10.1007/s00380-005-0868-y. [DOI] [PubMed] [Google Scholar]
- 158.Szarszoi O, Maly J, Ostadal P, et al. Effect of acute and chronic simvastatin treatment on post-ischemic contractile dysfunction in isolated rat heart. Physiol Res. 2008;57:793–6. doi: 10.33549/physiolres.931559. [DOI] [PubMed] [Google Scholar]
- 159.Ostadal P, Alan D, Hajek P, et al. The effect of early treatment by cerivastatin on the serum level of C-reactive protein, interleukin-6, and interleukin-8 in the patients with unstable angina and non-Q-wave myocardial infarction. Mol Cell Biochem. 2003;246:45–50. [PubMed] [Google Scholar]
- 160.Luo Y, Jiang D, Wen D, Yang J, Li L. Changes in serum interleukin-6 and high-sensitivity C-reactive protein levels in patients with acute coronary syndrome and their response to simvastatin. Heart Vessels. 2004;19:257–62. doi: 10.1007/s00380-004-0776-6. [DOI] [PubMed] [Google Scholar]
- 161.Macin SM, Perna ER, Farias EF, et al. Atorvastatin has an important acute anti-inflammatory effect in patients with acute coronary syndrome: Results of a randomized, double-blind, placebo-controlled study. Am Heart J. 2005;149:451–7. doi: 10.1016/j.ahj.2004.07.041. [DOI] [PubMed] [Google Scholar]
- 162.Pitt B, Loscalzo J, Ycas J, Raichlen JS. Lipid levels after acute coronary syndromes. J Am Coll Cardiol. 2008;51:1440–5. doi: 10.1016/j.jacc.2007.11.075. [DOI] [PubMed] [Google Scholar]
- 163.Ostadal P, Alan D, Vejvoda J, et al. Immediate effect of fluvastatin on lipid levels in acute coronary syndrome. Mol Cell Biochem. 2007;306:19–23. doi: 10.1007/s11010-007-9549-8. [DOI] [PubMed] [Google Scholar]
- 164.Vondrakova D, Ostadal P, Kruger A. Immediate effect of intensive atorvastatin therapy on lipid parameters in patients with acute coronary syndrome. Lipids Health Dis. 2010;9:71. doi: 10.1186/1476-511X-9-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Michelena HI, Osorio LA, Citkowitz E. Cholesterol levels after 3 days of high-dose simvastatin in patients at moderate to high risk for coronary events. Int J Cardiol. 2005;101:111–4. doi: 10.1016/j.ijcard.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 166.Ostadal P, Alan D, Vejvoda J, et al. Fluvastatin in the first-line therapy of acute coronary syndrome: Results of the multicenter, randomized, double-blind, placebo-controlled trial (the FACS-trial) Trials. 2010;11:61. doi: 10.1186/1745-6215-11-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kim JS, Kim J, Choi D, et al. Efficacy of high-dose atorvastatin loading before primary percutaneous coronary intervention in ST-segment elevation myocardial infarction: The STATIN STEMI trial. JACC Cardiovasc Interv. 2010;3:332–9. doi: 10.1016/j.jcin.2009.11.021. [DOI] [PubMed] [Google Scholar]