

Abstract
Myelodysplastic syndromes (MDS) are a heterogeneous group of clonal hematopoeitic disorders characterized by ineffective hematopoiesis and potential transformation to acute myeloid leukemia (AML). For decades, the mainstay of treatment for MDS was supportive care, including transfusion of blood products and growth factors. Further understanding of disease biology led to the discovery of a high prevalence of hypermethylation of tumor suppressor genes in high-risk MDS and secondary leukemias. Hence, the role of irreversible DNA methlytransferase inhibitors such as azacitidine was investigated with promising outcomes in the treatment of MDS. Azacitidine was initially approved in the USA by the Food and Drug Administration (FDA) in 2004 for the treatment of all subtypes of MDS and was granted expanded approval in 2009 to reflect new overall survival data demonstrated in the AZA-001 study of patients with higher-risk MDS. Azacitidine has demonstrated significant and clinically meaningful prolongation of survival in higher-risk patients with MDS and has changed the natural history of these disorders. The agent maintains a relatively safe toxicity profile, even in older patients. The role of azacitidine has been explored in the treatment of AML and chronic myelomonocytic leukemia and has also been studied in the peritransplant setting. Azacitidine has been combined with other novel agents such as lenalidomide, histone deacetylase inhibitors and growth factors in the hope of achieving improved outcomes. Currently, both intravenous and subcutaneous forms of azacitidine are approved for use in the USA with the oral form being granted fast track status by the FDA.
Keywords: acute myeloid leukemia, azacitidine, methylation, myelodysplastic syndromes
Introduction
Myelodysplastic syndromes (MDS) comprise a heterogeneous group of clonal hematological disorders, characterized by ineffective hematopoiesis with subsequent cytopenia and potential transformation to acute myeloid leukemia (AML) [de Angelo and Stone, 2005].
MDS may originate de novo or may be secondary to exposure to DNA-damaging agents such as ionizing radiation, alkylating agents or topoisomerase-2 inhibitors. In the USA, about five new cases per 100,000 of the general population are diagnosed with MDS each year. The incidence increases with age and reaches about 20–50 cases per 100,000 individuals above the age of 60. Age is a primary factor, with the median age at diagnosis being 70 years.
Classification and subsequent prognostic models have helped hematologists fine-tune patient selection and treatment strategies to a great extent. The 2008 World Health Organization (WHO) classification of MDS represents a minor modification of the third edition WHO classification which was originally published in 2001, building on the basis of the 1982 French–American–British (FAB) classification. The current WHO classification divides these syndromes into 10 different groups based on key features seen in the peripheral blood and bone marrow, as well as specific cytogenetic abnormalities (Table 1). In 1997, in an attempt to develop a consensus prognostic risk-based analysis system, an international MDS risk analysis workshop was convened and the International Prognostic Scoring System (IPSS) was developed [Greenberg et al. 1997]. The IPSS stratifies patients with MDS into four risk categories: low, intermediate-1, intermediate-2 and high risk based on the sum of three variable subscores: number of cytopenias, percentage of bone marrow blasts and cytogenetic abnormalities. Patients over age 60 with a low IPSS score have a median survival of 4.8 years, whereas patients with a high IPSS score have a median survival of only 4 months. While the IPSS defines critical prognostic features of MDS, shows the importance of age-related stratification, and provides improved prognostic evaluation, it does not distinguish between patients with severe and moderate degrees of cytopenias. Similarly, it also fails to identify patients who may face a poor prognosis despite having lower-risk disease (low and intermediate-1 risk).
Table 1.
2008 World Health Organization classification of myelodysplastic syndromes/neoplasms
| Name | Abbreviation | Peripheral blood: key features | Bone marrow: key features | WHO estimated proportion of patients with MDS |
|---|---|---|---|---|
| Refractory cytopenias with unilineage dysplasia (RCUD): | ||||
| refractory anemia | RA | Anemia <1% blasts |
Unilineage erythroid dysplasia (in 10% of cells) <5% blasts |
10–20% |
| refractory neutropenia | RN | Neutropenia <1% blasts |
Unilineage granulocytic dysplasia <5% blasts |
<1% |
| refractory thrombocytopenia | RT | Thrombocytopenia <1% blasts |
Unilineage megakaryocytic dysplasia <5% blasts |
<1% |
| Refractory anemia with ring sideroblasts | RARS | Anemia No blasts |
Unilineage erythroid dysplasia 15% of erythroid precursors are ring sideroblasts <5% blasts |
3–11% |
| Refractory cytopenias with multilineage dysplasia | RCMD | Cytopenia(s) <1% blasts No Auer rods |
Multilineage dysplasia ± ring sideroblasts <5% blasts No Auer rods |
30% |
| Refractory anemia with excess blasts, type 1 | RAEB-1 | Cytopenia(s) <5% blasts No Auer rods |
Unilineage or multilineage dysplasia 5–9% blasts No Auer rods |
40% |
| Refractory anemia with excess blasts, type 2 | RAEB-2 | Cytopenia(s) 5–19% blasts ±Auer rods |
Unilineage or multilineage dysplasia 10–19% blasts ±Auer rods |
|
| MDS associated with isolated del(5q) | Del(5q) | Anemia Normal or high platelet count <1% blasts |
Isolated 5q31 chromosome deletion Anemia, hypolobated megakaryocytes <5% blasts |
Uncommon |
| Childhood MDS, including refractory cytopenia of childhood (provisional) | RCC | Pancytopenia | <5% marrow blasts for RCC Marrow usually hypocellular |
<1% |
| MDS, unclassifiable | MDS-U | Cytopenias 1% blasts |
Does not fit other categories Dysplasia and <5% blasts If no dysplasia, MDS-associated karyotype |
? |
Adapted from Steensma et al. [2009].
If peripheral blood blasts are 2–4%, the diagnosis is RAEB-1 even if marrow blasts are less than 5%. If Auer rods are present, the WHO considers the diagnosis RAEB-2 if the blast proportion is less than 20% (even if less than 10%), AML if at least 20% blasts. For all subtypes, peripheral blood monocytes are less than 1 × 109/liter. Bicytopenia may be observed in RCUD subtypes, but pancytopenia with unilineage marrow dysplasia should be classified as MDS-U. Therapy-related MDS (t-MDS), whether due to alkylating agents, topoisomerase II inhibitors, or radiation, is classified together with therapy-related acute myeloid leukemia (t-MDS/t-AML) in the WHO classification of AML and precursor lesions. The listing in this table excludes MDS/myeloproliferative neoplasm overlap categories, such as chronic myelomonocytic leukemia, juvenile myelomonocytic leukemia, and the provisional entity RARS with thrombocytosis.
MDS, myelodysplastic syndromes; WHO, World Health Organization.
Taking these flaws into account, various newer scoring systems have been developed. The MD Anderson Cancer Center score (MDACC) was developed to provide insight into which patients may benefit from more aggressive treatment [Garcia-Manero et al. 2008b]. The MDACC scoring system may find its utility in better identifying a subset of patients with lower-risk disease who would benefit from early therapeutic intervention. The WHO prognostic scoring system is another risk model that takes into account the severity of anemia as assessed by transfusion requirements in addition to the blast percentage and karyotype [Malcovati et al. 2007]. It uses a six-category system and allows for more precise prognostication of overall survival (OS) duration and risk of progression to AML. More recently, the IPSS has also been revised in order to better stratify patients into risk categories (IPSS-R) [Greenberg et al. 2012]. Data from a total of 7012 patients from multiple centers in 11 countries were reviewed. Major changes include incorporation of five cytogenetic prognostic subgroups instead of three. Depth of cytopenia is also considered in the new version. In addition, the IPSS-R score established for each patient can now assess the individual prognostic risk in five categories instead of four. According to the groups, the estimated median survival ranges from 8.8 years in the very low-risk group to 0.8 years in the very high-risk group (Table 2).
Table 2.
IPSS-R Classification system for myelodysplastic syndromes
| Prognostic variable | 0 | 0.5 | 1 | 1.5 | 2 | 3 | 4 |
|---|---|---|---|---|---|---|---|
| Cytogenetics | Very good | Good | Intermediate | Poor | Very poor | ||
| Bone marrow blast (%) | 2 | >2 to <5 | 5–10 | >10 | |||
| Hemoglobin | 10 | 8 to <10 | <8 | ||||
| Platelets | 100 | 50 to <100 | <50 | ||||
| ANC | 0.8 | <0.8 | |||||
|
| |||||||
| Risk category | Risk score | ||||||
|
| |||||||
| Very low | 1.5 | ||||||
| Low | >1.5–3 | ||||||
| Intermediate | >3–4.5 | ||||||
| High | >4.5–6 | ||||||
| Very high | >6 | ||||||
Adapted from Greenberg et al. [2012].
ANC, absolute neutrophil count; IPSS-R, International Prognostic Scoring System Risk categories.
Pathophysiology of myelodysplastic syndromes
Steps associated with the pathogenesis of MDS include enhanced self-renewal of a hematopoietic stem cell or acquisition of self-renewal in a progenitor cell; increased proliferative capacity in the disease-sustaining clone and/or in its more differentiated progeny; impaired or blocked differentiation; genetic and epigenetic instability; antiapoptotic mechanisms in the disease-sustaining cell; evasion of the immune system; and suppression of normal hematopoiesis.
The capacity for self-renewal must be present in the MDS disease-initiating cell [Nimer, 2008]. This cell may arise from a self-renewing hematopoietic stem cell or it may come from a more differentiated myeloid progenitor that acquires the ability to self-renew. Further clonal expansion may occur through increased proliferation or resistance to apoptosis, and an abnormal bone marrow microenvironment could favor the development of a neoplastic clone. MDS occur when at least one of the molecular lesions present in the dominant clone or in its microenvironment also causes dysplastic differentiation of one or more myeloid lineages giving rise to ineffective hematopoiesis. The degree to which each step is affected can determine how the disease manifests clinically, including the types and degree of cytopenia present and whether the disease is indolent or rapidly progressive.
Advances in our knowledge of epigenetics have also led to better understanding of the specifics of the pathogenesis of MDS. DNA methylation provides a major epigenetic code of the lineage and development-specific genes that control expression of normal cells [Ruter et al. 2004]. The most relevant molecular mediators of the epigenetic state in MDS are gene-expression patterns maintained by methylation of cytosine residues in DNA and covalent modification of histones.
DNA methyl-transferases convert cytosine bases into 5-methylcytosines, particularly when they form the first base in a CpG dinucleotide. These CpG islands are commonly found clustered in and around gene promoters, consistent with their role in the regulation of gene expression. Methylation of CpG islands can alter their interaction with DNA-binding proteins, such as transcription factors and histone-modifying enzymes. Typically, methylation of CpG islands in promoters leads to silencing of neighboring genes and represents a mechanism for loss of tumor suppressor gene expression. In MDS and AML, several genes have been described as targets of DNA hypermethylation. These include the cell cycle regulators CDKN2A (p14 and p16) and CDKN2B (p15), CTNNA1, E-cadherin (CDH1), and many others. Genome-wide increases in promoter hypermethylation predict survival, even after taking into consideration age, sex, and IPSS risk group, and are seen during progression to AML [Bejar et al. 2011; Jiang et al. 2009; Shen et al. 2010]. These observations provide a rationale for the use of hypomethylating agents in MDS.
Our knowledge of various other genes that play a role in the pathophysiology of MDS is also rapidly advancing and studies continue to investigate the biological and clinical consequences of each genetic lesion. For example, one recent study analyzed the impact of ten-eleven-translocation 2 (TET2) mutations on response to azacitidine in MDS [Itzykson et al. 2011]. It was noted that the response rate (including HI) was 82% in patients carrying the mutated gene versus 45% in patients carrying the wild type (p = 0.007). Thus, TET2 status may be a genetic predictor of response to azacitidine, independently of karyotype, and holds promise as one of the tools available to help in better patient selection.
Azacitidine chemistry and pharmacodynamic properties
Azacitidine is a pyrimidine nucleoside analog that was chemically synthesized and characterized in Czechoslovakia by Frantisek Sorm and his fellow investigators in the 1960s (Figure 1) [Vogler et al. 1976]. Shortly afterwards, azacitidine was also microbiologically isolated from the fermentation beer of Streptoverticillium ladakanus. Azacitidine differs from cytosine primarily by the presence of a nitrogen at position 5. The hypomethylating effect appears to primarily depend upon the presence of this altered C5 position [Leone et al. 2002].
Figure 1.
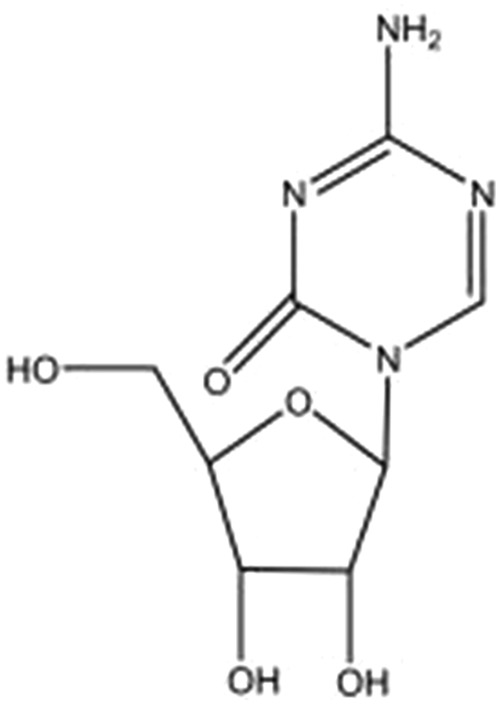
Chemical structure of azacitidine.
The cytotoxicity of azacitidine is a consequence of its incorporation into RNA as well as DNA. Azacitidine is transported into the mammalian cell by the same facilitated nucleoside transport system as operates for uridine and cytidine. Intracellular azacitidine undergoes three sequential phosphorylation reactions to reach its presumed active form, azacitidine triphosphate [Glover et al. 1987].
Being a ribonucleoside, azacitidine is incorporated in the RNA and to a lesser degree in the DNA. Incorporation into RNA results in interference in the synthesis of nucleic acids and proteins. Although azacitidine is incorporated to a lesser extent into DNA compared with RNA, its inhibitory effect on DNA synthesis is much greater than on RNA synthesis. After incorporation into DNA, azacitidine noncompetitively inhibits the enzyme DNA methyltransferase (DNMT1), which is responsible for DNA methylation [Glover et al. 1987]. Azacitidine thereby causes a block in cytosine methylation in newly replicated DNA but not in resting, nondividing cells. In summary, azacitidine causes depletion of DNA methyltransferases, hypomethylation of DNA, and induction of DNA damage [Hagemann et al. 2011; Hollenbach et al. 2010]. Other cytidine nucleoside analogs have also been studied in preclinical studies to better understand their mechanism of action, mainly decitabine and zebularine. The main difference of decitabine from azacitidine is that it is phosphorylated by different kinases and is incorporated solely in the DNA. In addition, it has also been shown that decitabine and azacitidine have different effects in their actions on cell viability, protein synthesis, cell cycle, and gene expression [Hollenbach et al. 2010].
Zebularine, a cytidine lacking the 4-amino group, is the most recent addition to the nucleoside analogues. Similar to azacitidine, it is a DNMT inhibitor, inhibiting DNA methylation and reactivating silenced genes. Moreover, it also enhances tumor cell chemosensitivity and radiosensitivity and has antimitogenic and angiostatic activities. In addition, zebularine is also very stable. Recent studies suggest differences in its mechanism of action from azacitidine and decitabine and other nucleosides [Champion et al. 2010]. An increase is seen in the DNA binding when zebularine is incorporated into the DNA compared with deoxycytidine and 5-fluorodeoxycytidine, together with a strong decrease in the dissociation rate. The intermediate covalent complex between the enzyme and the DNA is reversible, differing thus from 5-fluorodeoxycytidine. No methylation reaction occurs when zebularine is present in the DNA. Zebularine exerts its hypomethylation activity by stabilizing the binding of DNMTs to DNA, hindering the methylation and decreasing the dissociation, thereby trapping the enzyme and preventing turnover even at other sites.
Preclinical activity
Preclinical studies involving human promyelocytic leukemia HL-60 cells suggest a difference in the mechanism of action of azacitidine depending on drug concentration. At low doses (2–8 mmol/liter), azacitidine seems to preferentially target RNA and induces cytotoxicity in G1 cells [Murakami et al. 1995]. Higher concentrations (16 mmol/liter) are associated with disturbances in RNA and DNA metabolism and trigger cell death in both G1 and S phases of the cell cycle. Azacitidine also demonstrates differentiation-inducing activity at low doses and strong antileukemic effects at higher concentrations in cell line models. Azacitidine hence exerts its antineoplastic effect by two distinct mechanisms: cytotoxicity and induction of hypomethylation leading to cellular effects that are distinct from immediate cytotoxicity. DNA methylation profiles done 2 weeks after the first cycle of azacitidine in one study showed marked differences from the baseline DNA methylation profiles of the patients and in some predicted eventual hematologic responses [Fandy et al. 2009].
Pharmacokinetic properties, dosing, formulations and safety
The intravenous pharmacokinetic properties of azacitidine were first studied in 1972 utilizing radioactive labeling of the drug [Troetel et al. 1972]. The half life of azacitidine was 3.5 h with peak concentrations reached within 90 min. Most of the drug was found to be cleared renally. The bioavailability and pharmacokinetics of subcutaneously administered azacitidine was compared with intravenous administration in six patients in a randomized study [Marcucci et al. 2005]. Patients were administered a single dose of azacitidine 75 mg/m2 either subcutaneously or intravenously with a minimum of 7 days and maximum of 28 days were permitted between treatments. Subcutaneous azacitidine was found to have good bioavailability compared with intravenous administration, with area under the curve values within 89% of that measured following intravenous administration. Mean intravenous half life was approximately 22 ± 1 min and mean subcutaneous half life was 41 ± 8 min. The higher mean half life of the subcutaneous form has been attributed to the additional transition time required for azacitidine to move from the subcutaneous compartment into the circulation. Another explanation is that azacitidine remains both stable and bioavailable at the subcutaneous depot site until entering the plasma compartment. Following subcutaneous administration of azacitidine, maximum plasma concentration was observed at 0.5 h in all six patients. The primary route of excretion for azacitidine and its metabolites is through the urine.
Although the drug is mainly excreted renally (50–85%), renal complications are rare [Peterson et al. 1981]. Since azacitidine has not been studied in patients with concurrent renal dysfunction and MDS, caution is advised when dealing with such situations and dose reductions may be necessary. Azacitidine is also contraindicated in patients with advanced liver tumors. Hepatotoxicity may also occur in patients with severe preexisting hepatic impairment. [Vogler et al. 1976].
The current standard dosing schedule of azacitidine is 75 mg/m2 subcutaneously for 7 consecutive days (525 mg/m2 total), every 4 weeks, and is certainly feasible. In the AZA-001 study, 86% of patients had no dose reduction, and 80% of cycles were given at 4- or 5-week intervals, without prophylactic myeloid growth factor use [Fenaux et al. 2009]. Moreover, patients treated with azacitidine did not experience higher rates of infection or bleeding relative to the observation/conventional therapy groups.
Oral formulations of azacitidine have also been recently studied with the premise being that this would facilitate consistent dosing, reduce side effects, and favor compliance. A phase 0 pilot study demonstrated that plasma concentrations of orally administered azacitidine were comparable to those achieved by subcutaneous injection [Garcia-Manero et al. 2008c]. Subsequently, a phase I study of 41 patients receiving the first 75 mg/m2 dose of azacitidine subcutaneously and then escalating doses (from 120 to 600 mg) of oral azacitidine over the next 7 days demonstrated that oral azacitidine was well tolerated and had a low toxicity profile [Garcia-Manero et al. 2011]. The maximum tolerated oral dose was 480 mg, with grade 3 and 4 diarrhea observed in two of the three patients in the 600 mg cohort. Mean relative oral bioavailability ranged from 6.3% to 20%. Overall response rate (complete remission, hematologic improvement, or red blood cell or platelet transfusion independence) was 35% in previously treated patients and 73% in previously untreated patients.
Azacitidine has generally been found to be a well tolerated drug, with the most common grade 3 or 4 events being cytopenia [Vogler et al. 1976]. The most common nonhematological complications related to the subcutaneous route of administration are injection site reactions such as erythema and ecchymosis, followed by nausea and vomiting. Myelosuppression can sometimes be severe, but is usually transient, and most patients recover before their next treatment or require a dose delay (23–29%) [Santini et al. 2010]. Most of the adverse events are seen in the first two cycles and diminish subsequently. Infectious complications are not statistically different when compared with best supportive care. It has also been noted that comorbid conditions in older patients do seem to have an impact on the tolerability of azacitidine [Seymour et al. 2010].
Women of childbearing age should be warned of the potential harm of azacitidine on pregnancy. Women should be instructed not to breastfeed and to avoid pregnancy while on therapy. The safety profile of azacitidine is not known in children.
Clinical efficacy
We have already discussed the genetic and epigenetic pathways that may lead to the development of MDS. These changes are reversible, and hypomethylating agents have been shown to alter the methylation patterns seen in these diseases, making them the most active agents available for the treatment of high-risk MDS [Bejar et al. 2011].
Before hypomethylating agents became the mainstay of therapy for patients with MDS, treatment usually consisted only of supportive care for older patients and many other different treatment strategies for younger patients. These ranged from single-agent ara-c to the more aggressive multiagent chemotherapy similar to that employed in the treatment of AML as well as stem-cell transplantation. Cure rates as well as survival rates were low and patient selection for different therapeutic interventions was often difficult because of the limited prognostic information available. Advances in prognostic information gleaned from our discovery of the various genetic and epigenetic factors that play a role in the development of MDS in recent years and the introduction of hypomethylating agents has changed the landscape of how we treat patients with MDS.
Azacitidine was first evaluated in a series of phase I and II trials in the late 1960s and early 1970s as a cytotoxic agent and found to be efficacious in patients with myeloid malignancies [Cataldo et al. 2009; Goldberg et al. 1993; McCredie et al. 1973; Saiki et al. 1981; Vigil et al. 2010; Vogler et al. 1976]. Most of the patients in these studies were suffering from relapsed AML and azacitadine was mainly used as part of combination regimens with varying doses ranging from 100 to 750 mg/m2 with response rates anywhere from 0% to 58%. However, there was a trend towards better remission rates with a lower dosing schedule of azacitidine in one of the phase I studies [Saiki et al. 1981]. These initial studies then led to the larger phase II and phase III studies by Cancer and Leukemia Group B (CALGB), establishing azacitidine as frontline therapy for patients with MDS.
Establishment of azacitidine as frontline therapy in MDS
CALGB conducted a phase I/II study (CALGB 8421) of patients with largely refractory anemia with excess blasts (RAEB) and RAEB in transformation (RAEB-t), using azacitidine at 75 mg/m2/day continuous intravenous infusion for 7 days every 28 days [Silverman et al. 1993]. Responses were seen in 49% of evaluable patients with 12% of patients achieving complete response (CR), 25% partial response (PR), and 12% hematological improvement (HI). Median survival for all patients was 13.3 months, whereas median remission duration was 14.7 months.
This was followed by a second phase II study by the same group (CALGB 8921), using a subcutaneous bolus injection of azacitidine at the same dose [Silverman et al. 2006]. Responses were once again seen in close to 50% of patients with 27% achieving CR/PR and 13% demonstrating HI.
With these encouraging results, a phase III trial was conducted (CALGB 9221) which randomized 191 patients to subcutaneous azacitidine at 75 mg/m2/day for 7 days every 28 days versus supportive care only [Silverman et al. 2002]. Patients in the supportive care arm with progressive disease were allowed to crossover to the azacitidine arm which occurred in nearly half of the patients. The FAB classification was used at study entry as the IPSS had not yet been developed. CALGB response criteria were used as patient enrollment predated the establishment of the International Working Group (IWG) criteria [Cheson et al. 2000]. Responses occurred in 60% of patients in the azacitidine arm (7% CR, 16% PR, 37% HI) compared with 5% receiving supportive care (p < 0.001). As seen in previous studies, most of the responses were seen in the third and fourth month of treatment. Median time to leukemic transformation or death was 21 months for azacitidine versus 13 months for supportive care (p = 0.007). Enhancement in quality of life was also found to be significant in patients initially randomized to azacitidine with an improvement in fatigue, dyspnea, physical functioning and psychological distress compared with those in the supportive care arm [Kornblith et al. 2002]. This study provided conclusive evidence that azacitidine provided significantly higher response rates, improved quality of life, and reduced risk of leukemic transformation compared with supportive care. The crossover design of the study precluded demonstration of a survival advantage for azacitidine-treated patients, although subset analysis of high-risk patients suggested a survival benefit.
As a result of this study, subcutaneous azacitidine became the first drug to be approved for the treatment of MDS of all FAB subtypes in May 2004. The intravenous form of azacitidine was also approved by the US Food and Drug Administration (FDA) in January 2007 with a similar dosing schedule to that of the subcutaneous route.
A further analysis of the three CALGB studies was published in 2006 using the WHO classification system and the IWG response criteria [Silverman et al. 2006]. Response rates in azacitidine-treated patients were consistent across studies, with 40–47% of patients demonstrating a response. CR was achieved in 10–17% of patients, PR was rare, and 23–36% of patients had HI. Median duration of response was 13.1 months. In all three studies, the median number of cycles from first treatment with azacitidine to any response (CR, PR, or HI) was three, with the majority of responders (90%) achieving a response by the sixth cycle.
A second randomized trial, AZA-001, was conducted to evaluate the impact on OS of hypomethylating therapy (HMT) in MDS. Efficacy of azacitidine was compared with conventional care regimens in patients with high-risk MDS [Fenaux et al. 2009]. A total of 358 patients were randomized 1:1 to receive azacitidine or a conventional care regimen that could include supportive care, low-dose cytarabine, or induction-type chemotherapy. The conventional care regimen was chosen by the investigator prior to randomization. Azacitidine was administered subcutaneously at 75 mg/m2 daily for 7 consecutive days every 28 days for at least six cycles. A median of nine cycles of azacitidine was administered (range 4–15 cycles). The primary endpoint in the AZA-001 study was OS. Patients treated with azacitidine had median OS of 24.5 months, while patients receiving conventional care had a median OS of 15.0 months (Figure 2). The estimated 2-year survival rates were 50.8% for patients receiving azacitidine and 26.2% for patients receiving conventional care; patients in the azacitidine group also had higher rates of CR (17% versus 8%, p = 0.015) and PR (12% versus 4%, p = 0.0094). Likewise, the median time to disease progression, relapse after CR or PR, and death were significantly longer in the azacitidine group than in the conventional care group (14.1 versus 8.8 months, p = 0.047). The median duration of hematological response (CR, PR, and HI) was significantly longer in the azacitidine group than in the conventional care group (13.6 versus 5.2 months, p = 0.0002). The rate of transformation to AML was lower in the azacitidine group than in the conventional care group, and the median time to AML transformation was 17.8 months in the azacitidine group compared with 11.5 months in the conventional care group. Although statistical significance was not reached because of lack of power, this study showed for the first time that azacitidine prolonged survival and decreased the risk of transformation to AML in patients with high-risk MDS compared with conventional therapies. This benefit favoring azacitidine was seen across groups regardless of which conventional care regimen the investigator thought was the most appropriate comparator.
Figure 2.

Overall survival of patients with myelodysplastic syndromes treated with azacitidine and conventional care.
Adapted from Fenaux et al. [2009].
A secondary analysis of the AZA-001 study was recently published which evaluated the potential benefit of continuing azacitidine therapy beyond first response [Silverman et al. 2011]. Overall, 91 of 179 patients achieved a response to azacitidine; responding patients received a median of 14 treatment cycles. Median time to first response was two cycles. A total of 91% of first responses occurred by six cycles, with the remaining 9% achieving their first response by cycle 12. Although the first response was the best response for 52% of patients, a median of three additional cycles were needed for the rest of the patients to achieve their best response, suggesting that prolonged treatment with azacitidine may maximize response.
An interesting aspect of the CALGB and the AZA-001 trials to note is the relatively low CR rate. Although conventional wisdom would suggest that achieving CR is paramount for survival benefit, a retrospective subset analysis examining the survival impact of azacitidine excluding patients who achieved CR suggests otherwise [List et al. 2008]. One-year survival rates were superior for azacitidine treatment versus conventional treatment, 68% versus 56% (p = 0.15) respectively. Thus, even a response less than CR trends towards improved survival.
Investigators at the Western Pennsylvania Cancer Institute (WPCI) have had considerable experience with azacitidine in MDS. In this group’s earliest experience, 48 patients were evaluated for efficacy in the WPCI azacitidine program; 46 were transfusion dependent before treatment [Gryn et al. 2002]. Eighteen of these patients (39%) became transfusion independent. The majority of this group required two or more cycles to respond. The median duration of response was 7 months, with three responses continuing beyond 2 years. The FAB classification and the IPSS did not predict response to azacitidine. However, a decrease in the white blood cell count during the initial cycle of azacitidine correlated with a higher response rate. Further studies of 86 patients at the WPCI revealed improvement in hematologic response to azacitidine if given in combination with granulocyte colony-stimulating factor (84% versus 51%, p = 0.003) [Abdulhaq and Rossetti, 2007].
Other studies analyzing the response to azacitidine with regards to karyotype as well as the potential utility in patients with marrow fibrosis or with secondary MDS have also been carried out at the WPCI. A retrospective analysis of 80 patients with MDS treated with azacitidine at the WPCI showed no statistical difference in response rate between patients with simple (less than two cytogenetic abnormalities) and complex karyotype (more than three cytogenetic abnormalities) (38% versus 68%, p = 0.15)[Abdulhaq and Rossetti, 2007]. In another study, it was observed that azacitidine does better than conventional chemotherapy for patients with chromosome 5 or 7 abnormalities [Ravandi et al. 2009]. In a retrospective review of 41 patients with MDS with marrow fibrosis treated with azacitidine, the overall response rate was found to be 63% (7% CR, 20% PR, and 36% HI) [Juvvadi et al. 2007]. Treatment was well tolerated with the most common side effects being nausea, vomiting, diarrhea, and cytopenia. This study demonstrated that azacitidine is effective in treating patients with MDS regardless of marrow histology and has equal efficacy in patients with marrow fibrosis compared with those without fibrosis. This group has also studied the utility of azacitidine in chronic myelomonocytic leukemia (CMML) with results being discussed later in this paper.
Alternative dosing and administration schedule
Although studies have reported on alternative dosing schedules of azacitidine having similar response rates to the published regimen from the AZA-001 trial, none of them have been shown to be superior [Lyons et al. 2009]. One such study concluded that for patients with lower risk disease for whom the treatment goal of improved quality of life and reduced transfusion requirement was sought, the use of 75 mg/m2 subcutaneously for 5 days every 4 weeks was reasonable if a 7-day regimen could not be administered, although there are no data to support an OS benefit over the standard dosing schedule [Lyons et al. 2009]. In this randomized trial, patients with MDS were given azacitidine subcutaneously in one of three schedules every 4 weeks for six cycles: AZA 5-2-2 (75 mg/m2 daily for 5 days, followed by 2 days of no treatment, and then 75 mg/m2 daily for 2 more days for a total dose of 525 mg/m2 per cycle), AZA 5-2-5 (50 mg/m2 daily for 5 days followed by 2 days of no treatment, and then 50 mg/m2 for 5 days for a total dose of 500 mg/m2 per cycle), or AZA 5 (75 mg/m2 for 5 days for a total dose of 375 mg/m2). After six cycles of treatment, HI was reported in 44%, 45%, and 56% of the patients in the AZA 5-2-2, AZA 5-2-5, and AZA 5 arms respectively. Packed red blood cell transfusion independence was achieved by 50%, 55%, and 64% in the AZA 5-2-2, AZA 5-2-5, and AZA 5 arms respectively. More than one grade 3 or 4 adverse event occurred in 84%, 77%, and 58% of patients in the AZA 5-2-2, AZA 5-2-5, and AZA 5 arms respectively.
Another retrospective Spanish registry study of azacitidine reported on 144 patients who received azacitidine 75 mg/m2 for either 5 days, 5 days–weekend off–2 days (5/2/2), or 7 consecutive days [Garcia et al. 2009]. Those receiving 7 total days of azacitidine had a higher CR rate, 22% versus 12%. Patients who received 7 consecutive days had a higher overall response rate of 74%, compared with 65% for 5/2/2, or 58% for 5 days.
Prolonged administration of azacitidine over a 10-day period has also been studied in a randomized phase II study from the US Leukemia Intergroup (E1905 study) [ClinicalTrials.gov identifier: NCT00313586] testing 10 days of azacitidine (50 mg/m2/day subcutaneously) versus 10 days of azacitidine plus the histone deacetylase (HDAC) inhibitor entinostat (4 mg/m2/day orally on days 3 and 10) [Prebet et al. 2010b]. Of 150 patients, 70 demonstrated baseline cytogenetic abnormalities. A total of 40 patients (27 MDS and 13 AML) were evaluable. The clinical response rates (CR plus PR plus trilineage HI) according to IPSS cytogenetic risk stratification were 20%, 33%, and 35% for favorable, intermediate, and poor cytogenetic risk groups respectively (p = nonsignificant). A complete cytogenetic response (CyR) of 13% and a partial CyR of 23% as a proportion of all treated patients with initial cytogenetic abnormalities was seen. A total of 65 azacitidine-treated patients from AZA-001 had comparable cytogenetic results available. Only three patients (4.6%) experienced complete CyR with 22 patients (33.8%) achieving partial CyR. In contrast, the reported rate of clinical response was similar in the two studies. Complete CyR was more frequent in the E1905 study 10-day AZA schedule compared with the 7-day schedule of the AZA study (p = 0.007). There was no difference in overall CyR between the two studies. Further studies are warranted comparing this longer dosing schedule with the more conventional 7-day schedule to better evaluate these results.
Another study utilizing a lower dose of azacitidine (15 mg/m2 per day for 14 days) resulted in modest activity without significant myelosuppression [Chitambar et al. 1991].
Additional studies at WPCI have also looked at alternative dosing schedules. In a retrospective analysis of eight patients who received azacitidine 100 mg/m2 for 5 days every 4 weeks, the overall response was found to be 63% with average response duration of 6.2 months [Haq et al. 2006]. Myelosuppression leading to discontinuation of treatment was only seen in two patients. It was an otherwise well tolerated regimen.
Thus, while the 7-day subcutaneous or intravenous regimens of azacitidine remain the FDA-approved standards, alternative schedules seem feasible for carefully selected patients who can otherwise not tolerate the 7-day standard of care approach.
Azacitidine as maintenance therapy
In a Nordic MDS study, 23 patients in CR after induction chemotherapy received azacitidine as consolidative therapy because of their ineligibility for allogeneic transplantation. Azacitidine was given at 60 mg/m2 subcutaneously daily for 5 days in a 28-day cycle until relapse or unacceptable toxicity occurred. The median duration of response was 13.5 months (range 2–49 months) with 30% of the cases remaining in CR beyond 20 months [Grovdal et al. 2010].
Another study evaluated 51 patients with AML or MDS who were treated with azacitidine after receiving intensive chemotherapy (IC) [Gardin et al. 2009]. A total of 46 evaluable patients had achieved CR (n = 33), CR with incomplete platelet recovery (CRi) (n = 11) and PR (n = 2) before study entry. Diagnosis at IC onset was MDS (N = 13) and AML (N = 33). The median number of azacitidine maintenance cycles was 5.5 in CR patients and 2 in CRi or PR patients. Median follow up was 24 months. Median disease-free survival (DFS) was 6.5 months. Median OS from response was 15 months. 18-month DFS and OS were 30% and 56% in the 33 CR patients (median OS 24 months) compared with 0% and 17% in the 11 PR/CRi patients (p = 0.0001 and 0.04 respectively). A total of 21 of the 33 CR patients had relapsed and 3 had CR duration greater than 24 months. None of the patients in CRi/PR after IC improved their response with azacitidine.
These studies suggest that there might be a role for azacitidine as maintenance therapy in patients who have achieved a response with standard induction therapy to help with prolongation of response duration while minimizing excess toxicity.
Role of azacitidine in the transplant setting
Although allogeneic hematopoietic stem-cell transplantation (HSCT) is the only known curative option for MDS, because of age restrictions, concomitant medical conditions, and donor availability, fewer than 5% of patients with MDS are transplant candidates. For those who do undergo HSCT, results from several large centers indicate DFS rates of approximately 30–50%, depending on MDS type [Runde et al. 1998]. Failure is due primarily to transplant-related mortality in patients with low-risk MDS and disease recurrence in patients with high-risk MDS. Long-term follow-up data demonstrate an overall DFS rate of 40%, similar to rates in patients who received intensive chemotherapy [Appelbaum and Anderson, 1998]. Azacitidine has been investigated extensively in the peritransplant setting.
In a retrospective study the outcomes of 34 patients with MDS who underwent HSCT were analyzed, 14 of whom had received azacitidine at standard doses before transplantation. The Kaplan–Meier estimates for OS and progression-free survival between the two groups did not show clear evidence of a favorable outcome for either group, and there were no marked differences in toxic effects and other complications between the two groups [Vigil et al. 2010].
Another study analyzed the post-transplant outcomes of 54 consecutive patients with MDS or CMML who received HSCT from HLA-compatible donors according to pretransplant azacitidine exposure [Field et al. 2010]. A total of 30 patients received a median of four (range, one to seven) cycles of azacitidine, and 24 patients did not receive azacitidine before HCT. The 1-year estimates of OS, relapse-free survival, and cumulative incidence of relapse were 47%, 41%, and 20%, for azacitidine patients and 60%, 51%, and 32% for nonazacitidine patients respectively. These observations suggest that outcomes are similar in both groups, with a trend towards decreased early relapse in patients receiving azacitidine.
A more recent study included 19 patients who received HMT followed by allogeneic HSCT [Kim et al. 2012]. HMT consisted of decitabine in 9 patients and azacitidine in 10. After HMT, two patients achieved CR, six marrow CR (mCR), three HI alone, and six stable disease (SD) in terms of best response. In all, 2-year OS rates were 68%, and the overall outcomes of those who achieved CR/mCR with HMT tended to be superior to those without CR/mCR.
Azacitidine maintenance therapy after transplantation has also been explored with a goal of decreasing relapse rates. In one study, 45 patients with high-risk MDS or AML in first complete remission (CR1) after allogeneic HSCT were treated with maintenance azacitidine [de Lima et al. 2010]. By using a Bayesian adaptive method to determine the best dose/schedule combination based on time to toxicity, the authors investigated combinations of five daily azacitidine doses: 8, 16, 24, 32, and 40 mg/m2, and four schedules: one, two, three, or four cycles, each with 5 days of drug and 25 days of rest. Reversible thrombocytopenia was the dose-limiting toxicity. The optimal combination was found to be 32 mg/m2 given for four cycles. Median follow up was 20.5 months. One-year event-free survival and OS were 58% and 77%, respectively. The authors concluded that azacitidine at 32 mg/m2 given for 5 days is safe and can be administered after allogeneic transplant for at least four cycles to heavily pretreated patients with AML/MDS. The trial suggests that this treatment may prolong event-free survival and OS, and that more cycles may be associated with greater benefit.
Few treatment options are available for patients whose disease relapses after transplantation. The WPCI group studied the treatment of patients who have relapsed after unrelated donor peripheral stem-cell transplantation [Rossetti et al. 2007]. This study included six patients with high-risk myeloid malignancies and cytogenetic relapse after transplantation who received azacitidine at a minimum dose of 25 mg/m2 for 5 days. A reduction of cytogenetic abnormalities was observed in 83% of the patients shortly after one cycle of therapy, with one patient remaining in CR 4 months after the completion of therapy. The remaining patients relapsed 30 days after the completion of therapy, reflecting activity but a short-lived response.
Another prospective, multicenter, single-arm phase-II trial studies the utility of azacitidine and donor lymphocyte infusion (DLI) as first salvage therapy in 30 patients with MDS or AML relapsing after HSCT [Schroeder et al. 2010]. A total of 25 patients were evaluable. Of these, 23 had AML (15 de novo/8 secondary following MDS), 1 had MDS (RAEB type 1) and one had a myelodysplastic/myeloproliferative syndrome (CMML type 1). The median age was 54 years. Relapse occurred in all patients after a median of 160 days following HSCT. Patients received a median of three courses of azacitidine and 18 of 25 patients received DLI. Following treatment, overall response rate was 64% with five patients (20%) achieving a CR or CRi, three (12%) a PR, and eight (32%) SD. Median response duration was 266 days. After a median follow up of 100 days, 15 of 25 patients (60%) were alive at the time of publication of the abstract. Median OS of all patients was 184 days. All patients who achieved a CR/CRi remained in ongoing remission for a median time of 229 days.
These studies suggest that there might be a role for azacitidine in the treatment of patients who relapse after HSCT, whether as monotherapy or in combination with other therapies.
Efficacy of azacitidine in lower-risk myelodysplastic syndromes
The vast majority of patients who are initially diagnosed with MDS present with anemia and eventually become transfusion dependent. Although growth factors, such as the erythropoiesis-stimulating agents, with or without granulocyte colony-stimulating factors, can be used in the initial management of patients with low-risk MDS, a considerable number of patients will need alternative therapy. As described before, although the IPSS can predict the natural history of MDS, it has certain limitations, one of which is the identification of patients who may face a poorer prognosis despite having lower-risk disease (low and intermediate-1). Although there is a paucity of data with regards to the use of azacitidine in low-risk MDS, some studies have shown that azacitidine can certainly be effectively used in such patients.
The CALGB 9221 trial included 44 patients with low-risk disease in its analysis. The overall response observed in patients with low-risk MDS receiving azacitidine was 59% (9% CR, 18% PR, and 32% HI) with an OS of 44 months compared with 27 months for the control group [Silverman et al. 2006]. There has also been a report published on a series of 52 low-risk transfusion-dependent patients by a multicenter prospective community-based study (AVIDA) [Grinblatt et al. 2008]. In total, 42% achieved transfusion independence while on azacitidine; 67% of the patients who achieved transfusion independence did so after the second cycle of treatment. A significant 62% of patients were able to reach platelet transfusion independence; 88% of the patients who achieved platelet transfusion independence did so after the second treatment course, with minimal side effects. Another retrospective Italian study evaluated 74 patients with low-risk MDS who received azacitidine at 75 or 100 mg/m2 in monthly schedules subcutaneously [Musto et al. 2010]. The overall response in these patients was 45% (10% CR, 9.5% PR, and 20.3% HI). HIs were not as strong as those reported in higher-risk populations.
These data suggest that azacitidine is a viable option in a subset of patients with even lower-risk disease, although there remains absence of data on survival. Certainly, the lineage specificity of azacitidine with its utility in reducing platelet transfusion requirements in patients can be taken advantage of and help improve the quality of life of patients. As described before, the newer scoring systems can provide better guidance to physicians in identifying patients with low-risk MDS who would benefit from more aggressive treatment.
Azacitidine use in older patients
There are limited treatment options for patients older than 75 years or fragile patients who cannot otherwise tolerate cytotoxic therapy. An important goal of therapy in this subgroup of patients is to reduce transfusion dependence and delay progression of disease while limiting toxicities of any therapeutic intervention. A recent subset analysis of the AZA-001 trial was published looking at patients older than 75 years [Seymour et al. 2010]. There was a higher OS rate demonstrated at 2 years (55%) than in the conventional care group (15%). Azacitidine was also generally well tolerated in patients older than 75 years and produced transfusion independence in 44% of the patients who received it, compared with 22% in the conventional care group. Although there might have been a bias towards including relatively fit patients in this study, this analysis demonstrates that the use of azacitidine in patients older than 75 years has better response rates compared with conventional care with an acceptable safety profile.
Use of azacitidine in chronic myelomonocytic leukemia and other myeloproliferative disorders
Although azacitidine has received FDA approval for the treatment of CMML, phase II and III studies that assessed the utility of azacitidine in MDS included only a small number of patients with this particular entity. A retrospective analysis was recently published analyzing 38 patients with CMML treated with azacitidine at the Western Pennsylvania Cancer Institute [Costa et al. 2011]. Azacitidine was administered at 75 mg/m2 per day for 7 days or 100 mg/m2 per day for 5 days every 4 weeks. The modified IWG criteria were used to assess response. The overall response rate was 39% (CR 11%, PR 3%, and HI 25%). The median OS was 12 months. There was a statistically significant OS advantage in responders compared with nonresponders: 15.5 versus 9 months (p = 0.04). Treatment was found to be generally well tolerated with acceptable therapy-associated toxicity.
Utility of azacitidine has also been reported in the treatment of patients with Philadelphia (Ph)-negative myeloproliferative neoplasm (MPN) who have progressed to MDS or AML by the Groupe Francophone des Myelodysplasies recently [Thepot et al. 2010]. The study included 54 patients with a median age of 69.5 years. The initial MPN was polycythemia vera (PV) in 21 patients (39%), essential thrombocytosis (ET) in 21 patients (39%), primary myelofibrosis (PMF) in 7 patients (13%), and unclassified MPN in 5 patients (9%). At inclusion in the azacitidine program, 26 patients (48%) had AML and 28 patients (52%) had MDS. Most of the patients (65%) received azacitidine at the FDA approved dose (75 mg/m2/day during 7 days every 28 days) and the median number of cycles administered was six. The overall response rate was 52% (24% CR, 11% PR, 8% with marrow CR or CR with incomplete recovery of cytopenias, 9% HI) and median response duration was 9 months. Patients who evolved from ET did better than patients who evolved from PV as far as OS (71% versus 33% responses in ET and PV respectively) and achievement of CR (14% CR for PV versus 43% for ET) were concerned. The median OS was 11 months. Interestingly, recurrence of chronic phase features of the initial MPN was observed in 38% of the responders. These results suggest that azacitidine can certainly induce a substantial response rate with relatively low toxicity in Ph-negative MPN having progressed to AML/MDS.
Azacitidine in combination with other agents
Azacitidine has been combined with other agents to augment the response rates and to prolong response duration while maintaining low toxicity. The synergistic effects of azacitidine and HDAC inhibitors in reactivating silent genes encouraged clinical studies of this combination in MDS [Garcia-Manero et al. 2006]. One such agent is valproic acid, which is a short-chain fatty acid HDAC inhibitor with modest activity when used alone. However, it has shown enhanced activity when combined with hypomethylating agents [Garcia-Manero et al. 2006; Voso et al. 2009]. The median OS for patients receiving valproic acid plus azacitidine was 14.4 months with disease progression in 32% of patients. Another phase I study combining azacitidine with the HDAC inhibitor phenylbutyrate showed a 44% overall response rate, with 7 of 13 patients with MDS achieving a response [Gore et al. 2006].
More recently, data on the combination of azacitidine and the HDAC inhibitor entinostat (MS-275) [Prebet et al. 2010a]. This phase II study randomized 150 patients into two arms. Arm B received azacitidine 50 mg/m2 for 10 days plus entinostat, while arm A received azacitidine 50 mg/m2 for 10 days alone. Following six cycles of treatment, patients with documented clinical response (IWG 2000: CR, PR, or trilineage HI) continued for the lesser of a total of 24 cycles or disease progression. Overall hematologic normalization (HN) rate was 28%: 10% CR, 8% PR, and 10% trilineage HI. The HN rates were 31% in arm A and 24% in arm B (p = nonsignificant). Nontrilineage HI was achieved in an additional 12% of patients in arm A and 19% of patients in arm B; thus, total hematologic response was 43% and 44%. The median time to best response was 6 months in both arms (range 1–14); median duration of response was 11 months (range 1–18) in arm A and 10 months (range 2–19) in arm B (p = nonsignificant). With a median follow up of 17 months, median OS was 18 months in arm A and 13 months in arm B (p = 0.15). The study concluded that although prolonged administration of azacitidine improved hematologic normalization compared indirectly with the current standard of care, the addition of entinostat did not improve the response rate.
MGCD0103 is a selective HDAC inhibitor that demonstrated promising activity as a single agent in patients with MDS. In a phase I/II study, patients with relapsed/refractory MDS or AML received a standard dose of subcutaneous azacitidine plus MGCD0103 in escalating doses from 35 mg to 135 mg three times per week starting on day 5 of azacitidine [Garcia-Manero et al. 2008a]. A total of 30% of the patients responded, with four achieving a CR, five achieving an incomplete response, and two achieving a PR. The maximum tolerated dose of MGCD0103 was fixed at 90 mg because of severe nausea, vomiting, and dehydration at higher doses. Further trials are underway using both broad and specific HDAC inhibitors.
Azacitidine has also been combined with thalidomide and lenolidamide. In one study, thalidomide, which is an effective modulator of immune response with antiangiogenic activity, was administered in escalating doses with a standard dose of azacitidine for 5 days [Raza et al. 2008]. Of the 40 patients enrolled, 15% experienced a CR and 42% had HI. In another phase I study of 18 patients, azacitidine was tested in combination with lenolidamide, which had demonstrated activity in patients with MDS with the 5q deletion [Sekeres et al. 2010]. The treatment regimen was well tolerated, and the overall response rate for the 17 evaluable patients was 71%, with 41% of patients achieving a CR. The combination was well tolerated with significant clinical activity.
One phase II study has also evaluated the efficacy of combining azacitidine with etanercept, which is a tumor necrosis factor α blocker [Holsinger et al. 2007]. This was based on preclinical studies of mechanisms that implicated tumor necrosis factor α2 receptors in the development of MDS. In 23 patients, azacitidine was given in the standard 7-day dose and schedule, while etanercept was administered at 25 mg subcutaneously twice a week for 2 weeks in a 28-day cycle. A total of 14 patients responded, with 28% achieving a CR and 44% achieving a PR.
Gemtuzumab ozogamicin (GO), which is a previously available anti-CD 33 antibody/toxin conjugate, has demonstrated a good response in hematological malignancies. The combination of GO and azacitidine has also been evaluated in early clinical trials in patients with refractory or relapsed AML or MDS [Michaelis et al. 2009]. Median OS was 21 weeks, and 27% of patients achieved a CR. It is of note that 26% of patients with refractory disease had a documented CR with a median OS of 40 weeks. Despite these encouraging data, GO is no longer available commercially.
Azacitidine versus decitabine
Decitabine is another azanucleoside that was approved by the FDA on the basis of a randomized phase III trial versus best supportive care [Kantarjian et al. 2006]. An overall response rate of 30% with a CR rate of 9% was seen in this trial. Another multicenter study of 99 patients with MDS using a different dose and schedule showed a CR rate of 17%, with an overall response rate of 51% including HI [Steensma et al. 2009].
The results reported from the European Organization for Research and Treatment of Cancer/German MDS trial (06011), which randomly assigned 223 patients with MDS to best supportive care with or without decitabine, showed that at a median follow up of 2.5 years, the median OS was 8.5 months for best supportive care versus 10.1 months for decitabine, a difference that was not statistically significant (p = 0.38) [Lubbert et al. 2011].
Although there have been two meta-analyses comparing azacitidine with decitabine, there has not yet been a head-to-head comparison between azacitidine and decitabine in a prospective trial. A comparative trial of azacitidine versus decitabine was opened for recruitment [ClinicalTrials.gov identifier: NCT01011283] but had to be terminated because of poor accrual.
One meta-analysis and systematic review included four trials with 952 patients that examined the effect of azacitidine and decitabine [Gurion et al. 2010]. When OS was analyzed, there was an advantage of azacitidine [hazard ratio (HR) 0.67, 95% confidence interval (CI) 0.54–0.83; two trials, 549 patients] over decitabine (HR 0.88, 95% CI 0.66–1.17; one trial, 233 patients). There was also an advantage of azacitidine over decitabine in prolonging the time to AML transformation or death.
Another meta-analysis/systematic review was published which included the same randomized controlled trials but used slightly different statistical methods [Kumar et al. 2010]. The authors found that an indirect comparison of azacitidine to decitabine showed a statistically significant benefit for the outcome of OS with azacitidine (HR 0.63, 95% CI 0.46–0.85; p = 0.003) but there was no difference between azacitidine and decitabine for time to AML transformation or death.
Although it would appear that azacitidine has an advantage over decitabine based on these studies, caution should be exercised in interpreting the results. There were a limited number of trials and patients treated with decitabine included in the meta-analyses. Moreover, there has also been a difference in the duration of treatment between azacitidine and decitabine, with decitabine administered for a median of only three to four cycles compared with nine cycles for azacitidine.
Directions
Recent years have witnessed remarkable advances in our understanding of the biology of MDS. Many novel agents have been introduced into clinical trials with encouraging results. Azacitidine, the first hypomethylating agent to be approved by the FDA for treatment of MDS, was the first drug to prove that a change in the natural history of these diseases could be accomplished. It has risen as a keystone in the treatment of MDS as it prolongs survival and has a relatively safe toxicity profile. Azacitidine has also been shown to impact all three cell lines, delay leukemic transformation, and improve quality of life. With a better understanding of the biology of MDS and the ever-changing landscape of cytogenetics and molecular studies, we may be able to refine further the selection of patients who would benefit most from the different types of treatments becoming available.
Numerous challenges still remain, however, one of the more important ones being the clinical course of patients following azacitidine failure. Recent studies have shown that the prognosis for these patients is quite poor. One study analyzed data from 151 patients whose condition failed to respond to azacitidine therapy and showed a 12-month OS of only 20% [Lin et al. 2010]. Similar results were reported in another analysis of 435 patients post azacitidine failure, showing a median OS of 5.6 months and a 2-year survival probability of only 15% [Prebet et al. 2011].
The availability of azacitidine has significantly impacted the way clinicians approach the treatment of MDS and has given much hope to patients with these diseases. We continue to learn important information on alternative dosing and combination therapy with other agents. The promise of azacitidine treatment in MDS (and AML) may result in future trials studying the utility of this agent in the treatment of other malignant conditions.
Footnotes
Funding: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement: The authors declare no conflicts of interest in preparing this article.
Contributor Information
Cyrus Khan, Western Pennsylvania Cancer Institute, The Western Pennsylvania Hospital, Pittsburgh, PA, USA.
Neeta Pathe, Western Pennsylvania Cancer Institute, The Western Pennsylvania Hospital, Pittsburgh, PA, USA.
Salman Fazal, Western Pennsylvania Cancer Institute, The Western Pennsylvania Hospital, Pittsburgh, PA, USA.
John Lister, Western Pennsylvania Cancer Institute, The Western Pennsylvania Hospital, Pittsburgh, PA, USA.
James M. Rossetti, Western Pennsylvania Cancer Institute, The Western Pennsylvania Hospital, 4800 Friendship Avenue, Suite 2303, Pittsburgh, PA 15224, USA
References
- Abdulhaq H., Rossetti J. (2007) The role of azacitidine in the treatment of myelodysplastic syndromes. Expert Opin Invest Drugs 16: 1967–1975 [DOI] [PubMed] [Google Scholar]
- Appelbaum F., Anderson J. (1998) Allogeneic bone marrow transplantation for myelodysplastic syndrome: outcomes analysis according to IPSS score. Leukemia 12(Suppl. 1): 25–29 [PubMed] [Google Scholar]
- Bejar R., Levine R., Ebert B. (2011) Unraveling the molecular pathophysiology of myelodysplastic syndromes. J Clin Oncol 29: 504–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo V., Cortes J., Quintas-Cardama A. (2009) Azacitidine for the treatment of myelodysplastic syndrome. Exp Rev Anticancer Ther 9: 875–884 [DOI] [PubMed] [Google Scholar]
- Champion C., Guianvarc’h D., Senamaud-Beaufort C., Jurkowska R., Jeltsch A., Ponger L., et al. (2010) Mechanistic insights on the inhibition of c5 DNA methyltransferases by zebularine. PloS One 5: e12388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheson B., Bennett J., Kantarjian H., Pinto A., Schiffer C., Nimer S., et al. (2000) Report of an international working group to standardize response criteria for myelodysplastic syndromes. Blood 96: 3671–3674 [PubMed] [Google Scholar]
- Chitambar C., Libnoch J., Matthaeus W., Ash R., Ritch P., Anderson T. (1991) Evaluation of continuous infusion low-dose 5-azacytidine in the treatment of myelodysplastic syndromes. Am J Hematol 37: 100–104 [DOI] [PubMed] [Google Scholar]
- Costa R., Abdulhaq H., Haq B., Shadduck R., Latsko J., Zenati M., et al. (2011) Activity of azacitidine in chronic myelomonocytic leukemia. Cancer 117: 2690–2696 [DOI] [PubMed] [Google Scholar]
- de Angelo D., Stone R. (2005) Myelodysplastic Syndromes: Biology and Treatment, Vol. 1, 4th ed. Philadelphia, PA: Churchill Livingstone [Google Scholar]
- de Lima M., Giralt S., Thall P., de Padua Silva L., Jones R., Komanduri K., et al. (2010) Maintenance therapy with low-dose azacitidine after allogeneic hematopoietic stem cell transplantation for recurrent acute myelogenous leukemia or myelodysplastic syndrome. Cancer 116: 5420–5431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fandy T., Herman J., Kerns P., Jiemjit A., Sugar E., Choi S., et al. (2009) Early epigenetic changes and DNA damage do not predict clinical response in an overlapping schedule of 5-azacytidine and entinostat in patients with myeloid malignancies. Blood 114: 2764–2773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenaux P., Mufti G., Hellstrom-Lindberg E., Santini V., Finelli C., Giagounidis A., et al. (2009) Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol 10: 223–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field T., Perkins J., Huang Y., Kharfan-Dabaja M., Alsina M., Ayala E., et al. (2010) 5-Azacitidine for myelodysplasia before allogeneic hematopoietic cell transplantation. Bone Marrow Transplant 45: 255–260 [DOI] [PubMed] [Google Scholar]
- Garcia R., de Miguel D., Bailen A., González J., Sanz G., Falantes J., et al. (2009) Different clinical results with the use of different dosing schedules of azacitidine in patients with myelodysplastic syndrome managed in community-based practice: effectiveness and safety data from the Spanish Azacitidine Compassionate Use Registry. Blood (ASH Annual Meeting Abstracts) 114: abstract 2773 [Google Scholar]
- Garcia-Manero G., Assouline S., Cortes J., Estrov Z., Kantarjian H., Yang H., et al. (2008a) Phase 1 study of the oral isotype specific histone deacetylase inhibitor MGCD0103 in leukemia. Blood 112: 981–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Manero G., Gore S., Cogle C., Ward R., Shi T., Macbeth K., et al. (2011) Phase I study of oral azacitidine in myelodysplastic syndromes, chronic myelomonocytic leukemia, and acute myeloid leukemia. J Clin Oncol 29: 2521–2527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Manero G., Kantarjian H., Sanchez-Gonzalez B., Yang H., Rosner G., Verstovsek S., et al. (2006) Phase 1/2 study of the combination of 5-aza-2’-deoxycytidine with valproic acid in patients with leukemia. Blood 108: 3271–3279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Manero G., Shan J., Faderl S., Cortes J., Ravandi F., Borthakur G., et al. (2008b) A prognostic score for patients with lower risk myelodysplastic syndrome. Leukemia 22: 538–543 [DOI] [PubMed] [Google Scholar]
- Garcia-Manero G., Stoltz M., Ward M., Kantarjian H., Sharma S. (2008c) A pilot pharmacokinetic study of oral azacitidine. Leukemia 22: 1680–1684 [DOI] [PubMed] [Google Scholar]
- Gardin C., Prebet T., Bouabdallah K., Caillot D., Guerci A., Raffoux E., et al. (2009) A phase II study of post-remission therapy with azacitidine (AZA) in patients with AML post-MDS and high-risk MDS: a GFM group study. Blood (ASH Annual Meeting Abstracts) 114: abstract 844 [Google Scholar]
- Glover A., Leyland-Jones B., Chun H., Davies B., Hoth D. (1987) Azacitidine: 10 years later. Cancer Treat Rep 71: 737–746 [PubMed] [Google Scholar]
- Goldberg J., Gryn J., Raza A., Bennett J., Browman G., Bryant J., et al. (1993) Mitoxantrone and 5-azacytidine for refractory/relapsed ANLL or CML in blast crisis: a leukemia intergroup study. Am J Hematol 43: 286–290 [DOI] [PubMed] [Google Scholar]
- Gore S., Baylin S., Sugar E., Carraway H., Miller C., Carducci M., et al. (2006) Combined DNA methyltransferase and histone deacetylase inhibition in the treatment of myeloid neoplasms. Cancer Res 66: 6361–6369 [DOI] [PubMed] [Google Scholar]
- Greenberg P., Cox C., LeBeau M., Fenaux P., Morel P., Sanz G., et al. (1997) International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 89: 2079–2088 [PubMed] [Google Scholar]
- Greenberg P., Tuechler H., Schanz J., Sanz G., Garcia-Manero G., Sole F., et al. (2012) Revised International Prognostic Scoring System (IPSS-R) for myelodysplastic syndromes. Blood 120: 2454–2465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinblatt D., Narang M., Malone J. (2008) Treatment of patients with low-risk myelodysplastic syndromes receiving azacitidine who are enrolled in AVIDA, a longitudinal patient registry. Blood (ASH Annual Meeting Abstracts) 112: abstract 1646 [Google Scholar]
- Grovdal M., Karimi M., Khan R., Aggerholm A., Antunovic P., Astermark J., et al. (2010) Maintenance treatment with azacytidine for patients with high-risk myelodysplastic syndromes (MDS) or acute myeloid leukaemia following MDS in complete remission after induction chemotherapy. Br J Haematol 150: 293–302 [DOI] [PubMed] [Google Scholar]
- Gryn J., Zeigler Z., Shadduck R., Lister J., Raymond J., Sbeitan I., et al. (2002) Treatment of myelodysplastic syndromes with 5-azacytidine. Leuk Res 26: 893–897 [DOI] [PubMed] [Google Scholar]
- Gurion R., Vidal L., Gafter-Gvili A., Belnik Y., Yeshurun M., Raanani P., et al. (2010) 5-Azacitidine prolongs overall survival in patients with myelodysplastic syndrome – a systematic review and meta-analysis. Haematologica 95: 303–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemann S., Heil O., Lyko F., Brueckner B. (2011) Azacytidine and decitabine induce gene-specific and non-random DNA demethylation in human cancer cell lines. PloS One 6: e17388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haq B., Rossetti J., Kramer W., Latsko J., Jasthy S., Lister J., et al. (2006) Response and tolerability of a 5-day azacytidine schedule in patients with myelodysplastic syndromes. J Clin Oncol (Meeting Abstracts) 24(18 Suppl.): 16532 [Google Scholar]
- Hollenbach P., Nguyen A., Brady H., Williams M., Ning Y., Richard N., et al. (2010) A comparison of azacitidine and decitabine activities in acute myeloid leukemia cell lines. PloS One 5: e9001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holsinger A., Ramakishnan A., Storer B. (2007) Therapy of myelodysplastic syndrome with azacitidine given in combination with etanercept: a phase II study. Blood (ASH Annual Meeting Abstracts) 110: 1452 [Google Scholar]
- Itzykson R., Kosmider O., Cluzeau T., Mansat-De Mas V., Dreyfus F., Beyne-Rauzy O., et al. (2011) Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia 25: 1147–1152 [DOI] [PubMed] [Google Scholar]
- Jiang Y., Dunbar A., Gondek L., Mohan S., Rataul M., O’Keefe C., et al. (2009) Aberrant DNA methylation is a dominant mechanism in MDS progression to AML. Blood 113: 1315–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juvvadi R., Rossetti J., Shadduck R., Kaplan R., Kennedy M., Kramer W., et al. (2007) Response to azacitidine in patients with myelodysplastic syndrome with marrow fibrosis. J Clin Oncol (Meeting Abstracts) 25(18 Suppl.): 7089 [Google Scholar]
- Kantarjian H., Issa J., Rosenfeld C., Bennett J., Albitar M., DiPersio J., et al. (2006) Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer 106: 1794–1803 [DOI] [PubMed] [Google Scholar]
- Kim D., Lee J., Park Y., Kim S., Choi Y., Lee S., et al. (2012) Feasibility of hypomethylating agents followed by allogeneic hematopoietic cell transplantation in patients with myelodysplastic syndrome. Bone Marrow Transplant 47: 374–379 [DOI] [PubMed] [Google Scholar]
- Kornblith A., Herndon J., Silverman L., Demakos E., Odchimar-Reissig R., Holland J., et al. (2002) Impact of azacytidine on the quality of life of patients with myelodysplastic syndrome treated in a randomized phase III trial: a Cancer and Leukemia Group B study. J Clin Oncol 20: 2441–2452 [DOI] [PubMed] [Google Scholar]
- Kumar A., List A., Hozo I., Komrokji R., Djulbegovic B. (2010) Decitabine versus 5-azacitidine for the treatment of myelodysplastic syndrome: adjusted indirect meta-analysis. Haematologica 95: 340–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone G., Teofili L., Voso M., Lubbert M. (2002) DNA methylation and demethylating drugs in myelodysplastic syndromes and secondary leukemias. Haematologica 87: 1324–1341 [PubMed] [Google Scholar]
- Lin K., Reljic T., Kumar A., Lancet J., List A., Komrokji R. (2010) Poor outcome of patients with myelodysplastic syndrome (MDS) after azacitidine treatment failure. Blood (ASH Annual Meeting Abstracts) 116: abstract 2913 [DOI] [PubMed] [Google Scholar]
- List A., Fenaux P., Mufti G., Hellstrom-Lindberg E., Gore S., Bennett J. (2008) Effect of azacitidine on overall survival in higher-risk myelodysplastic syndromes (MDS) without complete remission. J Clin Oncol 26: 7007 [Google Scholar]
- Lubbert M., Suciu S., Baila L., Ruter B., Platzbecker U., Giagounidis A., et al. (2011) Low-dose decitabine versus best supportive care in elderly patients with intermediate- or high-risk myelodysplastic syndrome (MDS) ineligible for intensive chemotherapy: final results of the randomized phase III study of the European Organisation for Research and Treatment of Cancer Leukemia Group and the German MDS Study Group. J Clin Oncol 29: 1987–1996 [DOI] [PubMed] [Google Scholar]
- Lyons R., Cosgriff T., Modi S., Gersh R., Hainsworth J., Cohn A., et al. (2009) Hematologic response to three alternative dosing schedules of azacitidine in patients with myelodysplastic syndromes. J Clin Oncol 27: 1850–1856 [DOI] [PubMed] [Google Scholar]
- Malcovati L., Germing U., Kuendgen A., Della Porta M., Pascutto C., Invernizzi R., et al. (2007) Time-dependent prognostic scoring system for predicting survival and leukemic evolution in myelodysplastic syndromes. J Clin Oncol 25: 3503–3510 [DOI] [PubMed] [Google Scholar]
- Marcucci G., Silverman L., Eller M., Lintz L., Beach C. (2005) Bioavailability of azacitidine subcutaneous versus intravenous in patients with the myelodysplastic syndromes. J Clin Pharmacol 45: 597–602 [DOI] [PubMed] [Google Scholar]
- McCredie K., Bodey G., Burgess M., Gutterman J., Rodriguez V., Sullivan M., et al. (1973) Treatment of acute leukemia with 5-azacytidine (NSC-102816). Cancer Chemother Rep 57: 319–323 [PubMed] [Google Scholar]
- Michaelis L., Shafer D., Barton K. (2009) Azacitidine and low-dose gemtuzumab ozogamicin for the treatment of poor-risk AML and MDS including relapsed refractory disease. Blood (ASH Annual Meeting Abstracts) 114: 1034 [Google Scholar]
- Murakami T., Li X., Gong J., Bhatia U., Traganos F., Darzynkiewicz Z. (1995) Induction of apoptosis by 5-azacytidine: drug concentration-dependent differences in cell cycle specificity. Cancer Res 55: 3093–3098 [PubMed] [Google Scholar]
- Musto P., Maurillo L., Spagnoli A., Gozzini A., Rivellini F., Lunghi M., et al. (2010) Azacitidine for the treatment of lower risk myelodysplastic syndromes : a retrospective study of 74 patients enrolled in an Italian named patient program. Cancer 116: 1485–1494 [DOI] [PubMed] [Google Scholar]
- Nimer S. (2008) MDS: a stem cell disorder – but what exactly is wrong with the primitive hematopoietic cells in this disease? Hematol Am Soc Hematol Educ Program 43–51 [DOI] [PubMed] [Google Scholar]
- Peterson B., Collins A., Vogelzang N., Bloomfield C. (1981) 5-Azacytidine and renal tubular dysfunction. Blood 57: 182–185 [PubMed] [Google Scholar]
- Prebet T., Gore S., Esterni B., Gardin C., Itzykson R., Thepot S., et al. (2011) Outcome of high-risk myelodysplastic syndrome after azacitidine treatment failure. J Clin Oncol 29: 3322–3327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prebet T., Gore S., Sun Z., Greenberg P., Juckett M., Malick L., et al. (2010a) Prolonged administration of azacitidine with or without entinostat increases rate of hematologic normalization for myelodysplastic syndrome and acute myeloid leukemia with myelodysplasia-related changes: results of the US Leukemia Intergroup Trial E1905. Blood (ASH Annual Meeting Abstracts) 116: 601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prebet T., Sun Z., Ketterling R., Hicks G., Beach C., Greenberg P., et al. (2010b) A 10 day schedule of azacitidine induces more complete cytogenetic remissions than the standard schedule in myelodysplasia and acute myeloid leukemia with myelodysplasia-related changes: results of the E1905 US Leukemia Intergroup Study. Blood (ASH Annual Meeting Abstracts) 116: 4013 [Google Scholar]
- Ravandi F., Issa J., Garcia-Manero G., O’Brien S., Pierce S., Shan J., et al. (2009) Superior outcome with hypomethylating therapy in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome and chromosome 5 and 7 abnormalities. Cancer 115: 5746–5751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raza A., Mehdi M., Mumtaz M., Ali F., Lascher S., Galili N. (2008) Combination of 5-azacytidine and thalidomide for the treatment of myelodysplastic syndromes and acute myeloid leukemia. Cancer 113: 1596–1604 [DOI] [PubMed] [Google Scholar]
- Rossetti J., Shadduck R., Thatikinda C. (2007) Low dose azacitidine for relapse of MDS/AML after unrelated donor blood peripheral stem cell transplantation. Blood (ASH Annual Meeting Abstracts) 110: 5034 [Google Scholar]
- Runde V., de Witte T., Arnold R., Gratwohl A., Hermans J., van Biezen A., et al. (1998) Bone marrow transplantation from HLA-identical siblings as first-line treatment in patients with myelodysplastic syndromes: early transplantation is associated with improved outcome. Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation. Bone Marrow Transplant 21: 255–261 [DOI] [PubMed] [Google Scholar]
- Ruter B., Wijermans P., Lubbert M. (2004) DNA methylation as a therapeutic target in hematologic disorders: recent results in older patients with myelodysplasia and acute myeloid leukemia. Int J Hematol 80: 128–135 [DOI] [PubMed] [Google Scholar]
- Saiki J., Bodey G., Hewlett J., Amare M., Morrison F., Wilson H., et al. (1981) Effect of schedule on activity and toxicity of 5-azacytidine in acute leukemia: a Southwest Oncology Group Study. Cancer 47: 1739–1742 [DOI] [PubMed] [Google Scholar]
- Santini V., Fenaux P., Mufti G., Hellstrom-Lindberg E., Silverman L., List A., et al. (2010) Management and supportive care measures for adverse events in patients with myelodysplastic syndromes treated with azacitidine. Eur J Haematol 85: 130–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder T., Czibere A., Kroger N., Platzbecker U., Bug G., Rieger K., et al. (2010) Azacitidine (Vidaza(R), Aza) and donor lymphocyte infusions (DLI) in patients with acute myeloid leukemia (AML) or myelodysplastic syndromes (MDS) relapsing after allogeneic hematopoietic stem cell transplantation (HSCT): interim-analysis from the AZARELA-Trial (NCT-00795548). Blood (ASH Annual Meeting Abstracts) 116: 1294 [Google Scholar]
- Sekeres M., List A., Cuthbertson D., Paquette R., Ganetzky R., Latham D., et al. (2010) Phase I combination trial of lenalidomide and azacitidine in patients with higher-risk myelodysplastic syndromes. J Clin Oncol 28: 2253–2258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seymour J., Fenaux P., Silverman L., Mufti G., Hellstrom-Lindberg E., Santini V., et al. (2010) Effects of azacitidine compared with conventional care regimens in elderly (>/= 75 years) patients with higher-risk myelodysplastic syndromes. Crit Rev Oncol Hematol 76: 218–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L., Kantarjian H., Guo Y., Lin E., Shan J., Huang X., et al. (2010) DNA methylation predicts survival and response to therapy in patients with myelodysplastic syndromes. J Clin Oncol 28: 605–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman L., Demakos E., Peterson B., Kornblith A., Holland J., Odchimar-Reissig R., et al. (2002) Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol 20: 2429–2440 [DOI] [PubMed] [Google Scholar]
- Silverman L., Fenaux P., Mufti G., Santini V., Hellstrom-Lindberg E., Gattermann N., et al. (2011) Continued azacitidine therapy beyond time of first response improves quality of response in patients with higher-risk myelodysplastic syndromes. Cancer 117: 2697–2702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman L., Holland J., Weinberg R., Alter B., Davis R., Ellison R., et al. (1993) Effects of treatment with 5-azacytidine on the in vivo and in vitro hematopoiesis in patients with myelodysplastic syndromes. Leukemia 7(Suppl. 1): 21–29 [PubMed] [Google Scholar]
- Silverman L., McKenzie D., Peterson B., Holland J., Backstrom J., Beach C., et al. (2006) Further analysis of trials with azacitidine in patients with myelodysplastic syndrome: studies 8421, 8921, and 9221 by the Cancer and Leukemia Group B. J Clin Oncol 24: 3895–3903 [DOI] [PubMed] [Google Scholar]
- Steensma D. (2009) The changing classification of myelodysplastic syndromes: what’s in a name? Hematol Am Soc Hematol Educ Prog 645–655 [DOI] [PubMed] [Google Scholar]
- Steensma D., Baer M., Slack J., Buckstein R., Godley L., Garcia-Manero G., et al. (2009) Multicenter study of decitabine administered daily for 5 days every 4 weeks to adults with myelodysplastic syndromes: the alternative dosing for outpatient treatment (ADOPT) trial. J Clin Oncol 27: 3842–3848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thepot S., Itzykson R., Seegers V., Raffoux E., Quesnel B., Chait Y., et al. (2010) Treatment of progression of Philadelphia-negative myeloproliferative neoplasms to myelodysplastic syndrome or acute myeloid leukemia by azacitidine: a report on 54 cases on the behalf of the Groupe Francophone des Myelodysplasies (GFM). Blood 116: 3735–3742 [DOI] [PubMed] [Google Scholar]
- Troetel W., Weiss A., Stambaugh J., Laucius J., Manthei R. (1972) Absorption, distribution, and excretion of 5-azacytidine (NSC-102816) in man. Cancer Chemother Rep 56: 405–411 [PubMed] [Google Scholar]
- Vigil C., Martin-Santos T., Garcia-Manero G. (2010) Safety and efficacy of azacitidine in myelodysplastic syndromes. Drug Des Dev Ther 4: 221–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogler W., Miller D., Keller J. (1976) 5-Azacytidine (NSC 102816): a new drug for the treatment of myeloblastic leukemia. Blood 48: 331–337 [PubMed] [Google Scholar]
- Voso M., Santini V., Finelli C., Musto P., Pogliani E., Angelucci E., et al. (2009) Valproic acid at therapeutic plasma levels may increase 5-azacytidine efficacy in higher risk myelodysplastic syndromes. Clin Cancer Res 15: 5002–5007 [DOI] [PubMed] [Google Scholar]