

Abstract
Human nicotinamide phosphoribosyltransferase (NAMPT) replenishes the NAD pool and controls the activities of sirtuins (SIRT), mono- and poly-(ADP-ribose) polymerases (PARP) and NAD nucleosidase (CD38). The nature of the enzymatic transition-state (TS) is central to understanding the function of NAMPT. We determined the TS structure for pyrophosphorolysis of nicotinamide mononucleotide (NMN) by kinetic isotope effects (KIEs). With the natural substrates, NMN and pyrophosphate (PPi), the intrinsic KIEs of [1′-14C], [1-15N], [1′-3H] and [2′-3H] are 1.047, 1.029, 1.154 and 1.093, respectively. A unique quantum computational approach was used for TS analysis that included structural elements of the catalytic site. Without constraints (e.g. imposed torsion angles), the theoretical and experimental data are in good agreement. The quantum-mechanical calculations incorporated a crucial catalytic site residue (D313), two magnesium atoms and coordinated water molecules. The transition state model predicts primary 14C, α-secondary 3H, β-secondary 3H and primary 15N KIE close to the experimental values. The analysis reveals significant ribocation character at the TS. The attacking PPi nucleophile is weakly interacting (rC-O = 2.60 Å) and the N-ribosidic C1′-N bond is highly elongated at the TS (rC-N = 2.35 Å), consistent with an ANDN mechanism. Together with the crystal structure of the NMN•PPi•Mg2•enzyme complex, the reaction coordinate is defined. The enzyme holds the nucleophile and leaving group in relatively fixed positions to create a reaction coordinate with C1′-anomeric migration from nicotinamide to the PPi. The transition state is reached by a 0.85 Å migration of C1′.
INTRODUCTION
NAD is an important coenzyme for redox reactions as well as the substrate for regulatory NAD-linked enzymes. The poly ADP-ribosyltransferases (PARP) (Fig. 1a) are responsible for post-translational modifications of proteins by transfer of ADP-ribose (ADPR) to an amino acid acceptor of the target.1 Involved in DNA damage detection and repair, their role is decisive for the maintenance of the genomic stability.2 The second family includes the silent information regulator 2 (Sir2), initially characterized in yeast.3 Homologues of Sir2, SIRT1 and six other sirtuins encoded in the human genome, catalyze the NAD-dependent deacetylation of histones and transcription factors (e.g. p53, FOXO; Fig. 1a).4 Enhanced SIRT1 activity, along with high concentrations of NAD, have been correlated with resistance to oxidative stress.5 Deacetylations of FOXO6 and p537 promote lifespan extension through inhibition of apoptosis, reduction of damages from oxidative stress and mediating the up-regulation of manganese superoxide dismutase (MnSOD), catalase8 and thioredoxin9 (Fig. 1a). NAD nucleosidase (CD38) catalyzes the hydrolysis of NAD to ADPR and NAM and is also responsible for the production of cyclic ADPR,10 a signaling molecule with Ca2+-mobilizing activity (cADPR; Fig. 1a).
Fig. 1.

The role of NAMPT in mammalian NAD metabolism. (a) The salvage of NAD (red labels) is predominantly by NAMPT, the rate-limiting formation of NMN from NAM and α-D-5-phosphoribosyl 1-pyrophosphate (PRPP). NMN is converted to NAD by three isoforms of NMN adenylyltransferase (NMNAT, EC 2.7.7.1). NMN can also arise from phosphorylation of nicotinamide riboside (NR), an additional salvageable NAD precursor (nicotinamide riboside kinase; NRK, EC 2.7.1.22). Other ways to generate NAD include its salvage from NA and the de novo synthesis from L-Trp. The NAD pool needs constant replenishment because of NAD use (blue labels). Lifespan extension may be promoted by SIRT1 through FOXO/p53 deacetylation. Stress stimuli (IL-1β) enhances NAD production through NAMPT over-expression. Likewise, STAT 3 (through an IL-6 signaling pathway), NF-κB and TNFα, up-regulate nampt (red DNA). PGE2 is implicated in arthritis (via mPGES-1 up-regulation). FK866 and GMX17778 inhibit NAMPT. (b) The pyrophosphorolysis reaction catalyzed by NAMPT
Cellular NAD consumption is high and constant recycling is required to sustain the activities of these enzymes. In mammals, nicotinate mononucleotide (NAMN) and NMN are precursors of NAD (Fig. 1a). Although NAD can arise from nicotinate (NA) salvage or de novo synthesis from L-tryptophan (Fig. 1a), its biosynthesis is predominantly achieved by NAMPT in recycling NAM from SIRT, PARP and CD38 reactions (Fig. 1). NAMPT catalyzes the rate limiting step in NAD salvage, a documented role in human metabolism.11 It is linked to the function of sirtuins through modulation of NAD synthesis.12 NAMPT inhibition causes reduced levels of NAD and induces premature senescence, while its over-expression inhibits cell death.13 NAMPT is up-regulated in several cancers where NF-κB, IL-6 and nitric oxide synthase promote angiogenesis (Fig. 1a).14–17
NAMPT has been validated as a cancer target by the inhibitor FK866 (Fig. 2).18 Crystallographic studies19,20 and mechanistic insights21 on NAMPT-catalyzed NMN synthesis provide a functional description of this target and have promoted the search for more powerful inhibitors. Despite the description of additional drugs (e.g. GMX1778, Fig. 2), the in vivo inhibition of this enzyme is difficult because (a) modest NAMPT inhibitors are ineffective at physiological concentrations of NAM18,22 and (b) enzyme mutations occur and result in a 20,000-fold loss of efficiency for this class of compounds.23
Fig. 2.

Similarities between NAM and NAMPT inhibitors. In the NAMPT active site, NAM interacts with Tyr*18 and Phe193 in a π-stacking fashion [PDB: 3DKJ].19 FK866 resembles NAM; its pyridyl group (red) also interacts with NAMPT at the Tyr*18 and Phe193 levels. Additional interactions inside a β-barrel (β7, β8, β11 and β12) are provided by the peptidic moiety (black) while hydrophobic interactions are ensured by the “tail” of the inhibitor (blue). No structure for NAMPT-bound GMX1778 has been reported, but its relation to FK866 suggests similar interactions. Parameters such as Km, Kd and Ki are given for a better illustration of these similarities.11,20
Kinetic isotope effects (KIEs) provide the only experimental approach to deliver atomic-level information of the transition-state (TS) structure (bond distances, angles and charges). Here we measured the intrinsic KIEs for the pyrophosphorolysis reaction catalyzed by human NAMPT (Fig. 1b) and used a complementary quantum chemical approach to establish the TS structure from the experimental data. The TS structure and its electrostatic potential surface (ESPS) provide an unprecedented tool for understanding catalysis and to provide a blueprint for the design of transition state analogues.
MATERIALS AND METHODS
Protein expression and purification
Human NAMPT was expressed and purified as previously described.21 A pBAD vector, encoding for the ribokinase from E. coli was kindly provided by Dr. Sherry L. Mowbray (Swedish University of Agricultural Sciences, Uppsala, Sweden); the protein expression and purification were performed as reported.24 A pTEV6 plasmid encoding for Salmonella enterica PncA nicotinamidase (with a N-terminal maltose-binding protein-hexahistidine tag) was the generous gift of Dr. Jorge C. Escalante-Semerena (University of Wisconsin, Madison); the protein was expressed and purified as previously described.25
Isotopic labeling of NMN substrates and purification
[5′-3H]NMN, [5′-14C]NMN, [4′-3H]NMN, [1′-14C]NMN and [1′-3H]NMN were synthesized enzymatically from [6-3H]glucose, [6-14C]glucose, [5-3H]glucose, [1-14C]ribose and [1-3H]ribose, respectively (purchased from American Radiolabeled Chemicals Inc. and Moravek Biochemicals) as shown in Fig. S1a. A detailed protocol is available (Supporting Information Materials and Methods). [2′-3H]NMN and [1-15N, 5′-14C]NMN were synthesized from [2-3H]ribose (Fig. S1b) and [1-15N]NAM (Fig. S1c) whose syntheses are described (Supporting Information Materials and Methods).
Kinetic Isotope Effect measurements – Experimental KIEs
KIEs were measured under competitive conditions using the isotope ratio method.26 The isotopic label of interest was mixed with the appropriate remote label (the [5′-14C]NMN being the remote label used for all 3H KIEs while the [5′-3H] and [4′-3H]NMN were used as the remote labels for all other KIEs). The 3H to 14C cpm ratio was 1:1 with no less than 4 × 105 cpm of 14C per experiment. The reaction mixture (2 mL) contained 50 mM MES (pH = 6.30), 100 mM KCl, 5 mM MgCl2, 1 mM THP, 75 μM PPi, 25 μM NMN (3H and 14C labels included). After 10 min incubation at 37°C, the reaction was initiated by adding HsNAMPT to a final concentration of 230 nM. At 15–20% depletion of the substrate, 1400 μL of the reaction were quenched, and six 200 μL samples were loaded onto charcoal-cellulose columns (1:1 w/w, pre-equilibrated with water). The reaction product was then eluted with 1 mL of water and 5 mL of a 0.5 M solution of ammonium acetate (pH = 6.6). The 100% depletion of NMN was achieved by adding extra PPi and enzyme (final concentration brought to 250 μM and 1.8 μM, respectively) to the 600 μL reaction volume. After completion, six 40 μL fractions were loaded onto similar columns as described above, following the same elution protocol. All samples (twelve per independent experiment) were dried on a speed-vac, dissolved in 200 μL of water, mixed with 12 mL of scintillation fluid (Ultima Gold, PerkinElmer) and their 3H/14C ratios were measured by scintillation counting (30 min/cycle, 10 cycles; dual channel detector, Wallac 1414 LSC, PerkinElmer) after deconvolution of both channels. The channel 1 and 2 were set to have all 3H signal (>99.9%) in channel 1, whereas counts in channel 2 arise only from 14C; after calibration of the ratio (β) between those two channels using a source of 14C (> 1 × 105cpm), the exact amount of 3H and 14C in each of the samples was determined by the following relationships:
The corresponding 3H/14C ratios (R), for the partial (~20 %, Rf) and complete (100%, R0) conversion were used to correct the KIEs to 0% depletion of substrate according to Eq. 1:
| [1] |
Measurements of commitment factor Cf and Cr
The external reverse commitment factor (Cr) for the HsNAMPT-catalyzed reaction was evaluated under conditions where the NMN pyrophosphorolysis was irreversible. Because nicotinic acid is not a NAMPT substrate,19 the [1′-3H] KIE was measured following the same procedure as above, but in the presence of an excess of nicotinamidase. The forward commitment factor (Cf) to catalysis was measured by isotope trapping using rapid-mix pre-steady state conditions.27 NAMPT (50μM) was incubated at 37°C with [5′-3H]NMN (175 μM; 5 × 106 cpm for 100 μL total final volume) in 50 mM MES (pH = 6.30), 100 mM KCl, 5 mM MgCl2 and 1 mM THP. A chase solution (7920 μL; 50 mM MES pH = 6.30, 100 mM KCl, 5 mM MgCl2, 1 mM THP, 1 mM unlabeled NMN and 75 μM PPi), pre-equilibrated at 37°C, was rapidly added to 80 μL of the above radioactive solution. Samples (1 mL, for four aliquots of 200 μL each) were quenched at the indicated time (Fig. 4c) and quantified for product formation (PRPP elution on charcoal-cellulose columns as described above). Five replicates of this experiment were done. Background correction used the same experiment without enzyme. The concentration of labeled PRPP formed following addition of the chase solution was plotted as a function of the time (Fig. 4c). As described in Eq. 2, extrapolation of this concentration to zero time allows determination of the fraction of product formed (PRPP) versus bound NMN (E•14C-NMN) following dilution in excess of unlabeled NMN (cf. NMN Kd determination; Fig. 4a, Supporting Information Materials and Methods). After correction for forward commitment (Eq. 3–4), the intrinsic KIEs were suitable for the computational chemistry study.
Fig. 4.
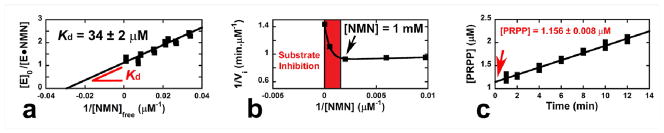
Determination of the forward commitment factor (Cf) by isotope trapping for the pyrophosphorolysis of NMN catalyzed by HsNAMPT. (a) Determination of the NMN dissociation constant at 37 °C and pH 6.30 (MES). The dissociation constant between NMN and NAMPT (Kd = 34 ± 2 μM) allows determination of the E•NMN complex concentration (by mixing 50 μM of enzyme with 175 μM of radioactive NMN, the E•NMN complex concentration becomes equal to 39.95 μM; Supporting Information Materials and Methods). (b) The NMN substrate inhibition. The isotope trapping method, used to determine Cf, is performed at high concentration of NMN; yet, this method is irrelevant in the presence of a substrate inhibition (red zone). Here, a 1 mM NMN concentration is still suitable (Supporting Information Materials and Methods). (c) Isotope trapping experiment. The synthesis of PRPP is monitored over time. After extrapolation to the origin ([PRPP] = 1.156 ± 0.008 μM), Eq. 2–4 allow determination of the Cf value (Y = 1.156 / 39.95 = 0.0289 and Cf = 0.0298 ± 0.0003; see Methods). Plots were made with KaleidaGraph 3.6 (Synergy Software).
| [2] |
| [3] |
| [4] |
Computational modeling of transition-state structure
A previously published crystal structure of NAMPT, with bound NMN and Mg2PPi (PDB: 3DHD) was used as the starting point for the calculations.20 Each of the calculated transition structure is characterized by only one imaginary frequency corresponding to reaction coordinate motion. KIEs for these transition structures are computed using ISOEFF9828,29 and a one-dimensional infinite parabola correction is applied to account for tunneling contributions to the KIEs.30 The electrostatic potential surfaces (ESPS) were calculated by using the CUBE program from Gaussian 09.31 Checkpoints files for CUBE inputs were generated at B3LYP/6-31G* for the optimized geometries at the same level of theory and visualized with GaussView 3.0.
Reactivity of 5P-pNR against NAMPT and inhibition properties
Reactivity assay was performed in TRIS buffer (50 mM, pH = 8.0) supplied with 1 mM PPi, 5 mM MgCl2 and 100 μM 5P-pNR. The reaction started by addition of NAMPT (10 μM final concentration). Signal was monitored at 430 nm and compared against a control sample (same as above without NAMPT). Possible inhibition of the NMN pyrophosphorolysis by 5P-pNR was assessed by monitoring the initial reaction rates at various 5P-pNR concentrations (5 to 100 μM) using 20 μM [5′-3H]NMN, 1 mM PPi, 5 mM MgCl2 and 90 nM NAMPT (50 mM HEPES, pH 7.5). Radiolabeled product was isolated by solid phase extraction and radioactivity was quantified by scintillation counting. Correlation between initial velocities and the corresponding inhibitor concentrations allows determination of the Ki.
RESULTS
Experimental KIEs are suppressed under typical experimental conditions
Intrinsic KIEs are required for the interpretation of enzymatic transition-states.32 The [1′-3H] label was used to evaluate the extent of KIE suppression as a function of experimental conditions. The [1′-3H] position is alpha to the N-ribosidic bond and is expected to display a significant KIE for the NMN pyrophosphorolysis reaction. Under optimum conditions for conversion of NMN to NAM (1 mM PPi, pH = 7.30), this KIE is near unity (1.011 ± 0.002; Fig. 3a). Although HsNAMPT activity is restricted to a narrow pH range,21 the [1′-3H] KIE at pH values (between 8.30 and 5.95; Fig. 3a) revealed that acidic conditions contribute to better expression of the experimental KIE (Fig. 3a). Indeed, NAMPT reveals a pH dependence for kcat (Fig. 3b) while Km is almost constant (1 to 2 μM). Acidic pH (6.30) decreases the kcat/Km by a factor of 15, consistent with the improved expression of KIEs.
Fig. 3.
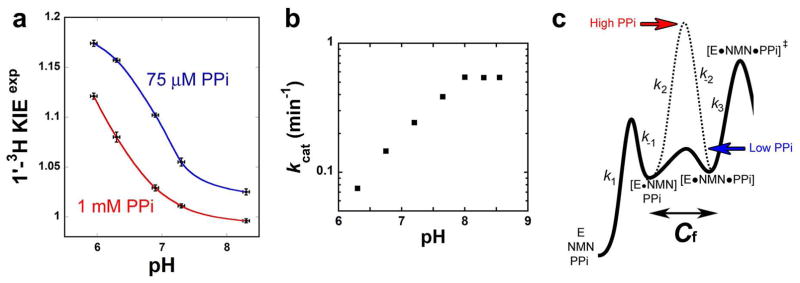
Influence of pH and PPi concentration on the experimental [1′-3H] KIE. (a) Experimental [1′-3H] KIEs were measured at high and low concentrations of PPi (1 mM and 75 μM, red and blue fit, respectively). For each set, a pH dependence profile was also established. Each data is representative of one single experiment (see Methods); the error bars for KIEs are the corresponding standard errors. Plots were made using KaleidaGraph 3.6 (Synergy Software). (b) Evaluation of kcat upon pH variation. (c) Qualitative representation of the PPi concentration effect on forward commitment (Cf). At low PPi concentration, on this hypothetical reaction coordinate diagram, the transition-state [E•14C-NMN•PPi]‡ is bearing the highest energy barrier (step 3, k3 < k-2; plain trace); in this case, the KIEs will be close to intrinsic values. At high PPi concentration (dash trace), after crossing the higher barrier on step 2, it the Michaelis complex evolves toward product formation. In this case, the experimental KIE will be reduced.
Reducing the PPi concentration to 75 μM also increased the isotope effect and at the lowest pH, the experimental KIE value approaches the upper limit of an intrinsic KIE calculated for this position (Fig. 3a). Increased pyrophosphate concentration affects the KIEs of this bi-substrate reaction by increased forward commitment factors.33 At high PPi concentrations the KIEs are abolished. At low PPi concentrations, the isotope effects tend to their intrinsic values (Fig. 3c). These observations are reminiscent from results previously obtained with orotate phopshoribosyltransferase, another pyrophosphate dependent N-ribosyl transferase. The use of natural substrates (i.e. orotidine 5′-monophosphate and PPi) abrogates KIE expression at physiological pH; it is only when the slow substrate analogue phosphonoacetic acid is used that the KIEs can be measured.34 Hence, experimental conditions were varied to reduce the commitment factors and allow determination of KIEs close to their intrinsic values. Optimum KIEs were observed at 37°C, pH 6.30, and in the presence of 75 μM PPi.
Forward and reverse commitment factors (Cf and Cr)
Commitment factors obscure intrinsic KIEs and only the intrinsic KIEs are useful in defining TS structure. Intrinsic KIEs are therefore required for TS determination. Observed, experimental KIEs can be corrected to intrinsic KIEs by determination of kinetic commitment factors.
The forward commitment factor (Cf) reflects the probability of the Michaelis complex [E•14C- NMN•PPi] to dissociate to unreacted substrates relative to generating products (i.e. Cf = kcat/koff where kcat and koff are equivalent to k3 and k-2, respectively; Fig. 3c). We determined Cf by the isotope-trapping method of Rose.27 From a 40 μM preformed [E•14C-NMN] complex (Kd = 32 μM; Fig. 4a), a rapid-mix dilution into excess NMN (i.e. 1 mM; Fig. 4b) and 75 μM PPi allowed the monitoring of radiolabeled PRPP formation (Fig. 4c). With those conditions, only 2.89% of the initial [E•14C-NMN] complex is converted to product (1.156 μM PRPP; Fig. 4c). The corresponding Cf is 0.0298 (Eq. 2–3, Fig. 4c).
Isotope trapping may also be applied to the determination of reverse commitment, the ability of enzyme-bound products to reform the enzyme-bound substrates before product release. However, the chemical instability of PRPP makes this experiment technically challenging. Instead, we measured the [1′-3H] KIE under conditions where NMN pyrophosphorolysis is made externally irreversible by hydrolysis of NMN with excess nicotinamidase. When using nicotinamidase, the experimental KIE was 1.148 ± 0.002 (SD = 0.022), compared to 1.161 ± 0.002 (SD = 0.019) in absence of the deaminase. Although these results are statistically different (T-test, t = 8.85 and p < 0.001) a lower KIE value obtained with nicotinamidase precludes significant Cr. Thus, the [1′-3H] KIE was averaged to 1.154 (Table 1).
Table 1.
Intrinsic and calculated KIEs for the pyrophosphorolysis of NMN catalyzed by HsNAMPT
| substrate | type of KIE
|
intrinsic KIEsa
|
calculated KIEs
|
|
|---|---|---|---|---|
| [1-15N, 5′-14C]NMN | Primary 15N | 1.029 ± 0.002b (8; ± 0.003)c | 1.024d | 1.024e |
| [1′-14C]NMN | Primary 14C | 1.047 ± 0.002 (8; ± 0.004) | 1.043 | 1.046 |
| [1′-3H]NMN | α-secondary 3H | 1.154 ± 0.001 (8; ± 0.003) | 1.154 | 1.163 |
| [2′-3H]NMN | β-secondary 3H | 1.093 ± 0.001 (4; ± 0.003) | 1.142 | 1.090 |
| [4′-3H]NMN | γ-secondary 3H | 0.998 ± 0.001 (4; ± 0.003) | – | |
| [5′-3H]NMN | δ-secondary 3H | 1.019 ± 0.001 (4; ± 0.004) | – | |
Experimental KIEs have been previously corrected to 0% substrate depletion using Eq. 1. Intrinsic KIEs were obtained by correction of the observed KIEs for remote 3H label and for forward commitment factor (Cf = 0.0298 ± 0.0003; Fig. 4) using Eq. 2-4.
Each intrinsic KIE is provided with its standard error; standard errors less than 0.001 were rounded-up to 0.001.
The numbers in parenthesis are the number of independent experiments and the 95% confidence intervals, respectively. Each independent experiment is composed of six samples for 20% substrate depletion and six samples for 100% substrate depletion, which by combination provide a total of thirty six KIEs.
Theoretical KIEs were determined on the final TS model from Fig. 5a using B3LYP/6-31G* gas phase calculations (see Methods).
Theoretical KIEs were determined on the final TS (rC-O = 2.60 Å, rC-N = 2.35 Å) using B3LYP(PCM:diethyl ether)/6-31G* calculations (see Methods); remote 5′- and 4′-positions are used only as remote labels and were not included in the analysis.
Intrinsic KIEs and their significance
Intrinsic KIEs for atoms near the reaction center report on the nature of the TS. The isotope effect from [5′-14C]NMN is three bonds away from the reaction center and is assumed to be unity. The [1-15N]NMN and [1′-14C]NMN experimental KIEs were corrected for their [5′-3H] control labels using Eq. 5. The contributions from forward and reverse commitment are small and the experimental and intrinsic KIEs were within experimental error (Table 1).
| [5] |
The KIE for the C1′ position is sensitive to atomic motion along the reaction coordinate and reflects the interactions of the leaving group (NAM) and nucleophile (PPi) with the anomeric carbon at the TS. This KIE is often critical for defining the reaction mechanism (e.g. SN1 vs. SN2).35 The 1.047 primary [1′-14C]NMN isotope effect for NMN pyrophosphorolysis suggests significant nucleophilic involvement at the transition-state. The accompanying large [1-15N]NMN isotope effect is consistent with full C-N bond cleavage at the TS.
The α-secondary [1′-3H] KIE reflects an increase in the out-of-plane C1′-H1′ bending mode as C1′ rehybridizes from sp3 to sp2. Large KIEs ( > 1.250) at this position are characteristic of steric freedom for the C1′-H bond at the TS relative to substrate, with a limited participation of both nucleophile and leaving group (i.e. highly developed oxacarbenium).36 We measured a 1.154 KIE for the H1′ position; consistent with weak residual bond order to C1′ at the TS (Table 1).
We used [2′-3H]NMN and detected a β-secondary KIE of 1.093. The magnitude of this effect provides information about the ribosyl geometry at the TS, where a large KIE is characteristic of maximum hyperconjugation between the C2′-H2′ σ bond and the 2pz orbital of the anomeric carbon (C1′) at the TS.37 Although this position is crucial to determine the TS geometry, a direct interpretation requires computational chemistry (see below).
The tritium isotope effects at the 5′- and 4′-position were determined for remote control purposes. These two positions are not considered in the determination of the final TS geometry (Table 1).
Modeling the HSNAMPT TS
A TS model consistent with the intrinsic KIEs for pyrophosphorolysis of NMN was determined in B3LYP/6-31G* calculations38 as implemented in Gaussian 09. The TS model includes unmodified substrates NMN and PPi, two Mg2+ ions critical to the reaction, and five water molecules. Also included is a chemical mimic of an active site aspartate (D313), which along with PPi, complete the octahedral coordination of both Mg2+ ions (Fig. 5a).20 A series of TSs were located by fixing the two key distances along the reaction coordinate, i.e. the forming C-O bond (rC-O) and the breaking C-N (rC-N) bond, with no additional restraints placed on other parameters (Supporting Information Materials and Methods). To better simulate the enzyme active site, the geometry that gave the best fit to the experimental KIEs in the gas phase calculations was re-calculated using a polarizable continuum model (PCM) with three different solvents – diethyl ether, acetone and water (with increasing dielectric constants of 4.24, 20.49 and 78.36, respectively). Inclusion of a solvent model had a significant impact on the accurate prediction of the [2′-3H] KIE but did not significantly alter the heavy-atom KIE predictions (Table 1). The closest match to experimental KIEs was obtained with the B3LYP(PCM:diethy ether)/6-31G* calculations. A low dielectric constant (diethyl ether) is often used for PCM calculations when modeling of a protein environment is desired.39,40
Fig. 5.

TS of HsNAMPT – geometry and characteristics. (a) Geometry of the transition-state matching the intrinsic KIEs. The C1′-nucleophile (rC-O) and the C1′-leaving group (rC-N) distances are depicted in red- and blue-dashed lines, respectively. Also represented, and incorporated in the computational model, magnesium atoms (green balls) and their corresponding ligands (grey-dashed lines). (b) Close-up on the electrostatic potential surface (ESPS) of the TS, at the C1′ vicinity. (c) Close-up on the ESPS of the NMN reactant state, at the C1′ vicinity. ESPSs were calculated at B3LYP/6-31G* for the optimized geometries at the same level of theory and visualized with GaussView 3.0 (Supporting Information Materials and Methods); blue coloring corresponds to a relative electron deficiency, while red defines a relative electron enrichment (see color coding and its arbitrary scaling units). The natural charges from NBO calculations for O4′, C1′, H1′ and N1 atoms are also represented (italic values).
The TS that best matches the intrinsic KIEs for the HsNAMPT-catalyzed reaction exhibits a minimal bond order to the attacking pyrophosphate nucleophile (rC-O = 2.60 Å) and weak but significant bond order to the leaving group (rC-N = 2.35 Å; Table 1 and Fig. 5a). There is significant ribocation character at the transition-state, akin to other N-ribosyltransferases (Fig. 5b, 5c).41 We performed a charge analysis of the TS relative to the ground state (Fig. 5b, 5c). Though NMN is often depicted with a positive charge “+” on its nitrogen, the positive charge of NMN resides primarily on the anomeric carbon in the ground state as evidenced by the +0.259 natural charge on C1′ and the −0.326 natural charge on N1. As the C1′-N1 bond strength decreases at the TS, increased electron density appears on N1 (Fig. 5b). The development of ribocation character at the TS is supported by the decreased electron density on O4′ (natural charge increasing from −0.596 to −0.434) and the sustained electron deficiency on C1′ (Fig. 5b). The presence of the attacking oxygen atom from PPi (at 2.60 Å from C1′) has an electrostatic contribution to the net charge of C1′, even though there is no significant bond order to this oxygen atom1.
The pucker of the ribose ring is governed by its contacts with D313 and one of the active site Mg2+ ions. The D313(Oδ2) forms hydrogen bonds to the hydroxyl protons from O2′ and O3′ and the Mg2+ ion coordinates with the same O2′ and O3′ atoms (Fig. 5a). The coordination of the same Mg2+ ion to D313(Oδ1) increases the rigidity of the ribose ring, thus enforcing a “flat” geometry at the TS. The other Mg2+ ion is coordinated to two oxygen atoms of PPi and four water molecules, two of which interact with the 5′-phosphate moiety through hydrogen bonding.
Considering that the transition structure in Fig. 5a was constrained only along the reaction coordinate and was relaxed in all other dimensions, the agreement of experimental and predicted KIEs is remarkable (Table 1). Shown in Table 1 is a comparison of the experimental KIEs and the KIE predictions for the TS geometry in Fig. 5a (for both gas phase and PCM (diethyl ether) structures). The predicted KIEs at the reaction center, i.e. [1′-14C] and [1′-3H] and [1-15N] KIEs, are in close agreement to intrinsic KIEs values in both gas phase and PCM calculations. The [2′-3H] KIE, which reports on the ribose ring pucker, is usually challenging to predict without restraining the H2′-C2′-C1′-H1′ dihedral angle. Since our model includes the active site elements that determine the ring pucker (namely the aspartate mimic and one of the Mg2+ ions), we expected to obtain a good match for the [2′-3H] KIE even without restricting the H2′-C2′-C1′-H1′ dihedral angle. Our gas phase model slightly over-predicts the [2′-3H] KIE (1.142); however, inclusion of a PCM model resulted in an excellent match of experiment (1.093 ± 0.003) and theory (1.090). We attribute this excellent match of theory and experiment to the detailed modeling of the chemically relevant active site environment with full substrates, Mg2+ ions and their ligands, as well as the use of a PCM (diethylether)2 to provide an accurate description of the catalytic site environment for HsNAMPT.
The slight discrepancy between experimental and predicted [1-15N] KIE (1.029 vs. 1.024; Table 1) might suggest to the reader that C–N bond-scission is slightly more advanced than 2.35 Å at the transition state. Geometries with slightly extended rC-N distances (up to 2.80 Å) and slightly shortened rC-N distances (that still gave good match of the [1′-14C] KIE) resulted in a slightly better 15N KIE prediction of up to 1.026; however, the [1′-3H] and [2′-3H] KIE predictions for these geometries departed significantly from experiment (Supporting Information Materials). The use of a higher level of theory with diffuse functions (B3LYP/6-31+G*) did not significantly improve the [1-15N] KIE prediction (Supporting Information Materials). While the predicted 1.024 15N KIE seems reasonably close to the experimental value of 1.029 (especially if one considers the corresponding 95% confidence interval of 0.003; Table 1), we believe that the slightly lower predicted 15N KIE might be a result of not considering the environment around the leaving group in our model (e.g. π-stacking, H-bonding). Finally, in addition to the concerted bimolecular process (ANDN) described above, two stepwise mechanisms were also considered – (a) SN1 mechanism with irreversible leaving group departure (the first step, DN‡*AN) and (b) SN1 mechanism with irreversible nucleophilic attack (the second step, DN*AN‡). Both these possibilities were discounted based on the poor match of experimental and theoretical KIEs (See Supporting Information for full details on these calculations).
Leaving group activation in the NAMPT-catalyzed reaction
The 5-phospho-p-nitrophenyl β-D-ribofuranoside (5P-pNR) is an excellent probe to elucidate the mechanism by which NAMPT achieves its oxacarbenium ion transition state. If the enzyme ionizes a ribosyl hydroxyl to facilitate the departure of nicotinamide, or interacts with the ribosyl group of NMN to stabilize an oxacarbenium ion, the 5P-pNR is expected to be a good substrate for NAMPT; the departure of the p-nitrophenyl moiety does not need assistance from the enzyme (e.g. hydrogen bound, protonation). On the other hand, if nicotinamide activation is required to reach the TS, the 5P-pNR would be a poor substrate. The chemical mechanism by which NAMPT achieves its TS was analyzed with in presence of PPi (see Materials and Methods).34 5P-pNR was not a substrate for NAMPT under conditions that would have detected 10−4 of the rate relative to NMN. However, 5P-pNR was capable of binding to NAMPT to give a Ki of 2.5 μM vs. KmNMN = 0.74 μM. We conclude that leaving-group activation is the major force in reaching the TS for the NAMPT-catalyzed reaction.
DISCUSSION
Kinetic isotope effects (KIEs) provide the one experimental approach capable of providing atomic-level information of the TS geometry (bond distances, angles and charges). Our results provide the most accurate representation of the reaction catalyzed by NAMPT.
Superimposition of the NAMPT crystal structures (NAMPT•NMN•Mg2PPi and NAMPT•benzamide•PRPP, 3DHD and 3DKJ, respectively) supported a reaction mechanism of “nucleophilic displacement by electrophile migration”.20 According to our transition-state model, C1′ “migrates” 0.87 Å (C1′-N = 1.48 Å in NMN and 2.35 Å at the TS) from the leaving group NAM to reach the TS (residual bond order, n = 0.06). At the TS, the bond order to the nucleophile is low (n = 0.02) and C1′ is still 2.60 Å from the nucleophilic oxygen atom of PPi. An additional migration of 1.15 Å is required to complete the reaction coordinate and generate PRPP. The 2.0 Å excursion of C1′ from leaving group to nucleophile is close to the distance from the crystal structures cited above (2.2 Å). At the TS, the C1′-O4′ bond becomes significantly shorter (Δn = +0.65), the C1′-N1 bond elongates (Δn = −0.85).These atomic modifications cause accumulation of negative charge on the nicotinamide ring. A decreased natural charge on the nitrogen atom (Δ = −0.129) matches the formation of a positive charge on the ribosyl ring (Fig. 5). The extra positive charge on C1′ (Δ = +0.194) justifies the term “ribooxacarbenium ion”. Enzymes exclude competitive nucleophiles (i.e. water) from their active sites to promote an efficient substrate/product transformation during catalysis. With NAMPT, the vicinity of C1′ is water-free except for WAT532 positioned 3.7 Å from the reactive center (PDB: 3DHD). Free water diffuses at 1 Å ps−1 and it would take approximately 2 ps for an unfettered water at this position to diffuse and react with the ribocation. However, this structural water participates in hydrogen bonding with Y*18(OH, from the neighboring subunit) and R311(Nη1) at 2.7 and 3.2 Å, respectively, precluding water diffusion and reaction with the ribocation. In human purine nucleoside phosphorylase, a chemistry-related N-ribosyltransferase, atomic motion constituting the reaction coordinate is complete in under 100 fs, faster than diffusion times for even closer waters.42
NAMPT’s active site imposes constraints on atomic motions to promote trajectories that would be less probable in solution chemistry. When comparing NMN hydrolysis (k = 1.04 ± 0.01 × 10−3 hr−1; Fig. S3a) to the enzyme catalyzed NMN pyrophosphorolysis (kcat = 0.48 ± 0.01 min−1; Fig. S3b), we estimate that this reaction is improved 30,000-fold by NAMPT. This modest rate enhancement (ξ; Fig. S3) emphasizes the chemical instability of the N-ribosidic bond in NMN. Rate enhancement occurs by stabilization of the ribocation through electrostatic interactions and by charge delocalization from the ribose ring through hydrogen bonds as proposed for diphtheria toxin (DT).43
Enzymes with nicotinamide N-ribosyltransferase activities similar to NAMPT include DT and sirtuins. X-ray analysis and molecular dynamics studies of DT revealed the crucial role of Glu148 forming a hydrogen bond to the 2′-OH in both enzyme-substrate and enzyme-TS complexes.44,45 This interaction causes charge delocalization in both ground state and TS to facilitate formation of a ribooxocarbenium ion at the TS. Ionization of the 2′-OH in solution (i.e. NAD and nicotinamide riboside) would facilitate the nicotinamide-ribosyl bond hydrolysis at a rate 10,000-fold that for a fully protonated 2′-OH.46 Despite the stabilization of the ribocation near the transition state, the reaction is initiated by leaving-group interactions with the nicotinamide since p-nitrophenyl-β-D-riboside would be a good substrate if reaction initiation occurred via formation of a ribocation.47
Other phosphoribosyltransferases (PRTases) also place aspartatic and/or glutamic residues in the O2′ and O3′ vicinity (e.g. hypoxanthine/guanine/xanthine-, adenine- and orotate PRTases, Fig. S4). Although the position of these conserved residues among PRTases is similar to that in NAMPT structures, the binding modes for substrates and Mg2+ are very different. NAMPT always contains two structural Mg2+ ions and NAMPT D313 coordinates with a bound magnesium ion. The aspartic or glutamic moieties from other PRTases are part of the second coordination sphere and the 2′- and/or 3′-hydroxyls from the respective ribosyl substrates (Fig. S4). According to our calculations, the natural charge at O2′ is −0.779 and −0.797 for NMN and the TS, respectively; such a variation precludes a D313-mediated ionization. Instead, D313 may form a favorable hydrogen bond with NMN (distance Oδ2-O3′ being 2.5 Å; PDB: 3DHD). Its main role in catalysis might be limited to Mg2+ coordination for optimal conformation of substrates (cf. computational model; Fig. 5a).
Leaving group activation (for NAM) is a major driving force in the NAMPT reaction, similar to recent results with OPRTase.48 Unlike related PRTases (APRTase or HGXPRTase), where protonation/deprotonation of the purine ring is ensured by acid/base catalysis (Fig. S4), hydrogen bonding from Arg311(Nε)-Oamide (3.1 Å) and Asp(Oδ2)-Namide (3.2 Å) to NAM provides sufficient stabilization and electrostatic interactions from π-stacking with Phe193/Tyr*18 to activate the leaving group.
The transition-state from NAMPT is concerted: the residual bond order to the leaving group is 0.06, and the bond order to the approaching nucleophile is 0.02 (Table 2). Our TS shares similarities with the TS from the NAD hydrolysis catalyzed by diphtheria toxin.49 Although both reactions follow an ANDN mechanism, the N-ribosidic bond is elongated to a greater extent for the DT catalyzed hydrolysis (Table 2). In both cases, the approaching nucleophile weakly interacts (n = 0.02–0.03; Table 2). This characteristic is very distinct from the NAD hydrolysis catalyzed by Pertussis toxin where participation of the water nucleophile is weak (SN1 reaction, n = 0.001, rC-O > 3.5 Å; Table 2).50 Finally, when compared to the ADP-ribosylations catalyzed by DT and PT toxins, the pyrophosphorolysis of NMN is more concerted. Although the rC-Nu distances are quite similar (~ 2.50–2.60 Å), the loss in bond order of the nicotinamide leaving group is more substantial for the NAMPT catalyzed reaction (n = 0.06; Table 2) than it is for the ADP-ribosylation of eEF-2 catalyzed by DT (n = 0.18; Table 2).43,51,52
Table 2.
Bond order and bond length at the transition-state of the NMN pyrophosphorolysis catalyzed by HsNAMPT and comparison with other reactions catalyzed by NAD-utilizing enzymes
| system |
rC-LG(rC-N)
|
rC-Nu(rC-X)a
|
||
|---|---|---|---|---|
| bond orderb | Å | bond order | Å | |
| DT NAD hydrolysis (O)a | 0.02 | 2.65 | 0.03 | 2.46 |
| NAMPT NMN PPi (O) | 0.06 | 2.35 | 0.02 | 2.60 |
| PT NAD hydrolysis (O) | 0.11 | 2.14 | 0.001 | 3.50 |
| PT NAD Giα1 (S) | 0.13 | 2.10 | 0.09 | 2.55 |
| PT NAD αi3C20 (S) | 0.15 | 2.06 | 0.12 | 2.47 |
| DT NAD eEF-2 (N) | 0.18 | 1.99 | 0.03 | 2.58 |
The nature of the atom nucleophile depends on the system studied. It is an oxygen atom (O) for the NAD hydrolysis catalyzed by both Diphtheria and Pertussis toxins (DT and PT, respectively) or for the pyrophosphorolysis of NMN catalyzed by NAMPT. It is a sulfur atom (S) for the ADP-ribosylation of Giα1 and αi3C20 peptides by PT toxin. Finally, it is a nitrogen atom (N) when the DT toxin ADP-ribosylates eukaryotic elongation factor 2 (eEF-2).
Pauling bond orders are calculated using the relation ni = e(r-ri)/0.3, where r is the bond length of a single bond (i.e. rC-N = 1.48 Å, rC-S = 1.83 Å, rC-O = 1.44 Å) and ri is the interatomic distance between C1′ and the atom of interest (i.e. rC-LG and rC-Nu).
CONCLUSIONS
Intrinsic KIEs were measured with pyrophosphate as the nucleophile in the NAMPT reaction. By incorporating structural elements from the catalytic site into the quantum chemical calculations, an unbiased TS structure was located to match the intrinsic KIEs. This unique computational approach will be useful in TS analysis of other enzymes, especially those involving pyrophosphorolysis. A ribosyl carbocation develops at the TS surrounded by nicotinamide and pyrophosphate, 2.35 Å and 2.60 Å away from the anomeric C1′, respectively. Determination of the TS features permits the creation of an electrostatic potential surface that may assist in the design of TS analogues for NAMPT.
Supplementary Material
Acknowledgments
We thank P.C. Tyler of Industrial Research Laboratory, Inc. for supplying the 5P-pNR; S.L. Mowbray (Swedish University of Agricultural Sciences, Uppsala, Sweden) for providing E. coli ribokinase and J.C. Escalante-Semerena (University of Wisconsin, Madison) for the S. enterica PncA nicotinamidase; and the US National Institutes of Health grant GM41916 for funding.
Abbreviations
- NAMPT
nicotinamide phosphoribosyltransferase
- SIRT
sirtuins
- PARP
poly-(ADP-ribose) polymerase
- CD38
NAD nucleosidase
- PRPP
α-D-5-phosphoribosyl-1-pyrophosphate
- NAM
nicotinamide
- NMN
nicotinamide mononucleotide
- NAMN
nicotinate mononucleotide
- PPi
pyrophosphate
- ADPR
ADP-ribose
- cADPR
cyclic ADP-ribose
- THP
tris(hydroxypropyl)phosphine
- KIE
kinetic isotope effect
- TS
transition-state
- ESPS
electrostatic potential surface
- Cr/Cf
reverse and forward commitment, respectively
- 5P-pNR
5′-phospho-p-nitrophenyl-β-D-ribofuranoside
- PRTase
phosphoribosyltransferase
- PncA
nicotinamidase
- ξ
rate enhancement
Footnotes
NBO analysis of the TS with identical rC-N but without the attacking nucleophile showed an increased positive charge at C1′ when compared to NMN.
A simpler model with only one Mg2+ ion and a 5′-deoxy nicotinamide riboside was unable to simultaneously predict all the experimental KIEs (Fig. S2).
Supporting Materials and Methods including the synthesis of labeled substrates (15N, 14C and 3H), the dissociation constant (Kd) for NMN onto HsNAMPT and the substrate inhibition experiment, the description of computational methodology and corresponding coordinates of calculated structures. Supporting Information Figure S1, synthesis of radiolabeled NMN; Supporting Information Figure S2, first approach to solve HsNAMPT TS by computational chemistry using 5′-deoxy-nicotinamide riboside, MgPPi and acetic acid; Supporting Information Figure S3, determination of the rate enhancement for HsNAMPT; Supporting Information Figure S4, comparison of catalytic features of a few PRTases. This information is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Corda D, Di Girolamo M. EMBO J. 2003;22:1953. doi: 10.1093/emboj/cdg209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Menissier de Murcia J, Ricoul M, Tartier L, Niedergang C, Huber A, Dantzer F, Schreiber V, Ame JC, Dierich A, LeMeur M, Sabatier L, Chambon P, de Murcia G. EMBO J. 2003;22:2255. doi: 10.1093/emboj/cdg206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blander G, Guarente L. Annu Rev Biochem. 2004;73:417. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- 4.Sauve AA, Wolberger C, Schramm VL, Boeke JD. Annu Rev Biochem. 2006;75:435. doi: 10.1146/annurev.biochem.74.082803.133500. [DOI] [PubMed] [Google Scholar]
- 5.van der Veer E, Ho C, O’Neil C, Barbosa N, Scott R, Cregan SP, Pickering JG. J Biol Chem. 2007;282:10841. doi: 10.1074/jbc.C700018200. [DOI] [PubMed] [Google Scholar]
- 6.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. Science. 2004;303:2011. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 7.Vaziri H, Dessain SK, Eaton NgE, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. Cell. 2001;107:149. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 8.Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC, Rabinovitch PS. Science. 2005;308:1909. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 9.Mitsui A, Hamuro J, Nakamura H, Kondo N, Hirabayashi Y, Ishizaki-Koizumi S, Hirakawa T, Inoue T, Yodoi J. Antioxid Redox Signal. 2002;4:693. doi: 10.1089/15230860260220201. [DOI] [PubMed] [Google Scholar]
- 10.Graeff R, Liu Q, Kriksunov IA, Kotaka M, Oppenheimer N, Hoa Q, Lee HC. J Biol Chem. 2009;284:27629. doi: 10.1074/jbc.M109.030965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burgos ES. Curr Med Chem. 2011;18:1947. doi: 10.2174/092986711795590101. [DOI] [PubMed] [Google Scholar]
- 12.Revollo JR, Grimm AA, Imai S. J Biol Chem. 2004;279:50754. doi: 10.1074/jbc.M408388200. [DOI] [PubMed] [Google Scholar]
- 13.Jia SH, Li Y, Parodo J, Kapus A, Fan L, Rotstein OD, Marshall JC. J Clin Invest. 2004;113:1318. doi: 10.1172/JCI19930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van Beijnum JR, Moerkerk PT, Gerbers AJ, De Bruine AP, Arends JW, Hoogenboom HR, Hufton SE. Int J Cancer. 2002;101:118. doi: 10.1002/ijc.10584. [DOI] [PubMed] [Google Scholar]
- 15.Bae YH, Bae MK, Kim SR, Lee JH, Wee HJ, Bae SK. Biochem Biophys Res Commun. 2009;379:206. doi: 10.1016/j.bbrc.2008.12.042. [DOI] [PubMed] [Google Scholar]
- 16.Bauer L, Venz S, Junker H, Brandt R, Radons J. Int J Oncol. 2009;35:97. doi: 10.3892/ijo_00000317. [DOI] [PubMed] [Google Scholar]
- 17.Wang B, Hasan MK, Alvarado E, Yuan H, Wu H, Chen WY. Oncogene. 2010 doi: 10.1038/onc.2010.468. [DOI] [PubMed] [Google Scholar]
- 18.Hasmann M, Schemainda I. Cancer Res. 2003;63:7436. [PubMed] [Google Scholar]
- 19.Khan JA, Tao X, Tong L. Nat Struct Mol Biol. 2006;13:582. doi: 10.1038/nsmb1105. [DOI] [PubMed] [Google Scholar]
- 20.Burgos ES, Ho MC, Almo SC, Schramm VL. Proc Natl Acad Sci U S A. 2009;106:13748. doi: 10.1073/pnas.0903898106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burgos ES, Schramm VL. Biochemistry. 2008;47:11086. doi: 10.1021/bi801198m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Watson M, Roulston A, Belec L, Billot X, Marcellus R, Bedard D, Bernier C, Branchaud S, Chan H, Dairi K, Gilbert K, Goulet D, Gratton MO, Isakau H, Jang A, Khadir A, Koch E, Lavoie M, Lawless M, Nguyen M, Paquette D, Turcotte E, Berger A, Mitchell M, Shore GC, Beauparlant P. Mol Cell Biol. 2009;29:5872. doi: 10.1128/MCB.00112-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olesen UH, Petersen JG, Garten A, Kiess W, Yoshino J, Imai S, Christensen MK, Fristrup P, Thougaard AV, Bjorkling F, Jensen PB, Nielsen SJ, Sehested M. BMC Cancer. 2010;10:677. doi: 10.1186/1471-2407-10-677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andersson CE, Mowbray SL. J Mol Biol. 2002;315:409. doi: 10.1006/jmbi.2001.5248. [DOI] [PubMed] [Google Scholar]
- 25.Garrity J, Gardner JG, Hawse W, Wolberger C, Escalante-Semerena JC. J Biol Chem. 2007;282:30239. doi: 10.1074/jbc.M704409200. [DOI] [PubMed] [Google Scholar]
- 26.Parkin DW. In: Enzyme mechanism from isotope effects. Cook PF, editor. CRC Press, Inc; Boca Raton, Florida 33431: 1991. p. 269. [Google Scholar]
- 27.Rose IA. Methods Enzymol. 1980;64:47. doi: 10.1016/s0076-6879(80)64004-x. [DOI] [PubMed] [Google Scholar]
- 28.Anisimov V, Paneth P. J Math Chem. 1999;26:75. [Google Scholar]
- 29.Scott AP, Radom L. J Phys Chem. 1996;100:16502. [Google Scholar]
- 30.Bell RP. The tunnel effect in chemistry. Chapman & Hall; London: 1980. [Google Scholar]
- 31.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima TE, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian, Inc; Wallingford, CT: 2009. [Google Scholar]
- 32.Northrop DB. Biochemistry. 1975;14:2644. doi: 10.1021/bi00683a013. [DOI] [PubMed] [Google Scholar]
- 33.Northrop DB. Annu Rev Biochem. 1981;50:103. doi: 10.1146/annurev.bi.50.070181.000535. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Y, Luo M, Schramm VL. J Am Chem Soc. 2009;131:4685. doi: 10.1021/ja808346y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berti PJ, Tanaka KSE. Adv Phys Org Chem. 2002;37:239. [Google Scholar]
- 36.Glad SS, Jensen F. J Am Chem Soc. 1997;119:227. [Google Scholar]
- 37.Sunko DE, Szele I, Hehre WJ. J Am Chem Soc. 1977;99:5000. [Google Scholar]
- 38.Becke AD. J Chem Phys. 1993;98:5648. [Google Scholar]
- 39.Himo F, Siegbahn PE. J Am Chem Soc. 2001;123:10280. doi: 10.1021/ja010715h. [DOI] [PubMed] [Google Scholar]
- 40.Kahraman A, Morris RJ, Laskowski RA, Favia AD, Thornton JM. Proteins. 2010;78:1120. doi: 10.1002/prot.22633. [DOI] [PubMed] [Google Scholar]
- 41.Singh V, Evans GB, Lenz DH, Mason JM, Clinch K, Mee S, Painter GF, Tyler PC, Furneaux RH, Lee JE, Howell PL, Schramm VL. J Biol Chem. 2005;280:18265. doi: 10.1074/jbc.M414472200. [DOI] [PubMed] [Google Scholar]
- 42.Saen-Oon S, Quaytman-Machleder S, Schramm VL, Schwartz SD. Proc Natl Acad Sci U S A. 2008;105:16543. doi: 10.1073/pnas.0808413105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parikh SL, Schramm VL. Biochemistry. 2004;43:1204. doi: 10.1021/bi035907z. [DOI] [PubMed] [Google Scholar]
- 44.Bell CE, Eisenberg D. Biochemistry. 1996;35:1137. doi: 10.1021/bi9520848. [DOI] [PubMed] [Google Scholar]
- 45.Kahn K, Bruice TC. J Am Chem Soc. 2001;123:11960. doi: 10.1021/ja0113807. [DOI] [PubMed] [Google Scholar]
- 46.Handlon AL, Xu C, Muller-Steffner HM, Schuber F, Oppenheimer NJ. J Am Chem Soc. 1994;116:12087. [Google Scholar]
- 47.Mazzella LJ, Parkin DW, Tyler PC, Furneaux RH, Schramm VL. J Am Chem Soc. 1996;118:2111. [Google Scholar]
- 48.Zhang Y, Deng H, Schramm VL. J Am Chem Soc. 2010;132:17023. doi: 10.1021/ja107806j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Berti PJ, Blanke SR, Schramm VL. J Am Chem Soc. 1997;119:12079. doi: 10.1021/ja971317a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scheuring J, Schramm VL. Biochemistry. 1997;36:4526. doi: 10.1021/bi962841h. [DOI] [PubMed] [Google Scholar]
- 51.Scheuring J, Berti PJ, Schramm VL. Biochemistry. 1998;37:2748. doi: 10.1021/bi972594x. [DOI] [PubMed] [Google Scholar]
- 52.Scheuring J, Schramm VL. Biochemistry. 1997;36:8215. doi: 10.1021/bi970379a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.