

Abstract
Epigenetic alterations may mechanistically explain the developmental origins of adult disease, namely the hypothesis that many complex adult chronic diseases originate as a result of conditions encountered in utero. If true, epigenetically regulated imprinted genes, critical to normal growth and development, may partially mediate these outcomes. We determine the influence of in utero exposure to cigarette smoking on methylation at two differentially methylated regions (DMRs) regulating Insulin-like Growth Factor 2 (IGF2) and H19, and how this might relate to birth weight of infants born to 418 pregnant women. Smoking status was ascertained through self-report and medical records. Bisulfite pyrosequencing was used to measure methylation in umbilical cord blood DNAs. Least squares DNA methylation means at each DMR and birth weight were compared between infants of smokers and non-smokers, using generalized linear models. While there were no significant differences at the H19 DMR, infants born to smokers had higher methylation at the IGF2 DMR than those born to never smokers or those who quit during pregnancy (49.5%, SD=8.0 versus 46.6%, SD=5.6 and 45.8%, SD=6.3, respectively; p=0.0002). The smoking-related increase in methylation was most pronounced in male offspring (p for sex interaction=0.03), for whom approximately 20% of smoking-related low birth weight was mediated by DNA methylation at the IGF2 DMR. Our findings suggest that IGF2 DMR plasticity is an important mechanism by which in utero adjustments to environmental toxicants are conferred. Larger studies to replicate these findings are required.
Keywords: Sex-specific methylation effects, imprinted gene, pyrosequencing, early origins hypothesis
1. Introduction
Despite repeated demonstrations of adverse birth outcomes resulting from in utero exposure to cigarette smoking, the prevalence of smoking during pregnancy remains at around 14% in the United States (CDC, 2009). The mechanisms that link exposure to adverse outcomes are poorly understood but appear to be mediated by non-genotoxic adaptations to the gestational environment and/or exposure to toxicants (Barker, 1990; Gluckman et al., 2005). Epidemiologic evidence strongly supports links between conditions encountered during embryonic or fetal development and adult-onset diseases, including atherosclerosis, coronary heart disease, type 2 diabetes mellitus, obesity, and cancer [for review, see (Godfrey and Barker, 2001)]. These chronic diseases and conditions have also been linked to low birth weight resulting from exposure to both active (Stillman et al., 1986) and passive cigarette smoking during pregnancy (Martin and Bracken, 1986).
Commonly studied non-genotoxic effects are epigenetic mechanisms, including modification of histone proteins and DNA methylation, which have also been hypothesized to explain how in utero exposure to cigarette smoking affects adult-onset diseases. However, the ability to test this hypothesis has been stymied due to the restricted type of biological specimens obtainable from otherwise healthy populations (e.g., peripheral blood, buccal cells) coupled with the lack of identified mitotically stable epigenetic targets that are not substantially influenced by inherent tissue specific differences in methylation.
DNA methylation, the most studied epigenetic mechanism in epidemiologic studies, is typically associated with chromatin condensation through the binding of methyl-CpG binding proteins and recruitment of proteins involved in chromatin modification that ultimately lead to transcriptional silencing (Portela and Esteller, 2010). DNA methylation regulates genomically imprinted genes through differential marking of the parental alleles during two epigenetic reprogramming events in early pregnancy; in gametogenesis, when methylation is erased and re-established to reflect the sex of the individual in which the developing gametes reside, and after fertilization, when the maternal and paternal pronuclei undergo demethylation and subsequent remethylation that will help guide germ layer specification and tissue differentiation (Feng et al., 2010; Kota and Feil, 2010). We have previously postulated that the mitotic stability of the methylation marks associated with imprinted genes coupled with establishment pre-gastrulation makes them especially suited for epidemiologic study of in utero environmental effects on the epigenome (Hoyo et al., 2009). The inherent plasticity of the epigenome during this period of epigenetic remodeling may allow for adaptive epigenetic profiles to be established at the sequences regulating imprinted genes, modifying gene expression patterns in response to perceived in utero environmental conditions.
The well-characterized imprinted domain at human chromosome 11p15.5 contains paternally expressed Insulin-like Growth Factor II (IGF2) and the maternally expressed H19 gene, from which is transcribed a non-coding RNA. Imprinted expression and transcription of IGF2 are regulated in large part through the patterns of DNA methylation of at least two differentially methylated regions (DMRs), one of which is located near the H19 promoter (H19 DMR) and the other upstream of the three IGF2 promoters that are subject to imprinting (IGF2 DMR). Both DMRs have been shown to exhibit altered methylation in cigarette smoking-related malignancies (Cui et al., 2002; Cruz-Correa et al., 2004). In the present study, we examined the influence of in utero exposure to cigarette smoke on DNA methylation at the IGF2/H19 imprinted domain using umbilical cord blood specimens naïve to the ex utero environment. We furthermore determined the extent to which methylation profiles mediate the link between maternal smoking and low birth weight.
2. Materials and methods
2.1. Study participants
Study participants were enrolled as part of the Newborn Epigenetics STudy (NEST), a multiethnic birth cohort designed to identify the effects of early exposures on epigenetic profiles and phenotypic outcomes. These studies were approved by the Duke University Institutional Review Board and written consent was obtained from all mothers participating as study subjects. A detailed description of identification and enrollment procedures has been described elsewhere (Hoyo et al., 2011). Briefly, pregnant women were recruited from prenatal clinics serving Duke University Hospital and Durham Regional Hospital Obstetrics facilities in Durham, North Carolina from April 2005 to June 2008. Eligibility criteria were: age ≥18 years, English speaking, pregnant, and intention to use one of the two obstetrics facilities for the index pregnancy to enable access to labor and birth outcome data as well as umbilical cord blood. To ensure an adequate sample size for cigarette exposure, smoking women were targeted for the first ~200 women recruited. Gestational age at enrollment ranged from 19 to 42 weeks. Of the 1101 women approached, 940 (85%) were enrolled. Cord blood samples were collected from 720 infants. Bisulfite pyrosequencing has been completed on the first 428 cord blood samples for the H19 and IGF2 DMRs. The 428 participants in whom specimens were available were similar to the 940 enrolled in the study, with respect to maternal age (p=0.55) maternal education (p=0.94), race/ethnicity (p=0.39) and infant sex (p=0.81). Six of the 428 were excluded because smoking status could not be defined with certainty; four additional mothers gave conflicting answers to smoking-related questions and were also excluded. Final analyses included the 418 samples for which smoking and methylation data were available.
2.2. Data collection
At enrollment, a standardized questionnaire was administered that included maternal demographic characteristics, health status, reproductive factors, anthropometric measurements, and lifestyle factors such as tobacco use, alcohol intake and use of dietary supplements. We abstracted medical charts to verify responses to our main exposure and outcomes, cigarette smoking and birth weight.
2.3. Ascertainment of cigarette smoking status
To improve the accuracy of self-reported smoking status, cigarette smoking was ascertained by responses to four questions. First, women were asked to answer “Yes” or “No” to the question, “Before the year you found out you were pregnant, did you smoke?” They were also asked, “After you found out you were pregnant, which of the following best describes your behavior?” The four possible responses were, “I continue to smoke,” or “I stopped during the first/second/third trimester.” To further verify smoking behavior, in two different parts of the questionnaire women were also asked, “Have you ever smoked 100 cigarettes or more in your lifetime?” (Yes/No), followed by, “Do you smoke now?” (Yes/No). Three maternal smoking categories were created as follows: Women were considered “smokers during pregnancy” if they answered affirmatively to ever having smoked 100 cigarettes or more, being a current smoker, and smoking during pregnancy; women were considered “quitters during pregnancy” if they had responded affirmatively to ever smoking 100 cigarettes or more, smoking during the year of pregnancy, and stopping smoking any time during the pregnancy; “non-smokers” were women who responded negatively to ever smoking 100 cigarettes or more, and negatively to current smoking.
2.4. Specimen collection
Immediately following newborn delivery, the umbilical vein was punctured and umbilical cord blood was collected into K3EDTA-containing vacutainer tubes. The leukocyte-containing buffy coat was isolated following centrifugation at 3,500 × g for 20m at 4°C. Aliquots were prepared and stored at −80°C.
2.5. Other variables
At enrollment, a standardized questionnaire was used to solicit information on maternal age, race/ethnicity, marital status, education, pre-pregnancy weight, and height. Medical records were used to obtain gestational age at enrollment and at delivery, birth weight and infant gender. Body mass index (BMI) was computed by dividing weight by height squared.
2.6. DNA methylation analysis
Genomic DNA from buffy coat specimens was extracted using Gentra Puregene Reagents (Qiagen, Valencia, CA). The IGF2 and H19 DMRs were analyzed by bisulfite pyrosequencing. Bisulfite modification was performed as previously described (Huang et al., 2006). Pyrosequencing assays were designed using PSQ Assay Design Software and duplicate reactions run on a Pyromark Q96 MD Pyrosequencer (Qiagen). The percent methylation for each of the CpGs within the target sequence was calculated using PyroQ CpG Software (Qiagen). Bisulfite conversion efficiency was assessed using the PyroQ CpG Software and averaged 97.3% (SD=0.98%) for the H19 DMR and 99.5% (SD=2.7%) for the IGF2 DMR.
The region analyzed for the IGF2 DMR includes three CpG dinucleotides upstream of exon 3 (chr 11p15.5; CpG site 1: 2,169,518; CpG site 2: 2,169,515; and CpG site 3: 2,169,499; NCBI Human Genome Build 37/hg19) (Cui et al., 2002). The region studied for the H19 DMR encompasses four dinucleotides located upstream of the H19 gene (chr 11p15.5; CpG site 1: 2,024,261, CpG site 2: 2,024,259, CpG site 3: 2,024,257, and CpG site 4: 2,024,254; NCBI Human Genome Build 37/hg19), which are located within one of the six CTCF binding sites of this region (Cui et al., 2001).
For the IGF2 DMR, 50 ng of bisulfite-modified DNA was amplified using 2U Platinum Taq DNA polymerase (Invitrogen, Carlsbad, CA) and 0.15 µM each forward (5’-GGA GGG GGT TTA TTT TTT TAG GAA G-3’) and reverse (5’-[Biotin]-AAC CCC AAC AAA AAC CAC TAA ACA C-3’) primers in a 40 µl reaction volume with 0.2 mM dNTPs and 3 mM MgCl2. PCR conditions were 94°C for 3m, then 5 cycles of 94°C for 30s, 70°C for 30s, 72°C for 30s; then 5 cycles of 94°C for 30s, 68°C for 30s, 72°C for 30s; then 50 cycles of 94°C for 30s, 66°C for 30s, 72°C for 30s; followed by a 5m extension at 72°C. Biotin-labeled single-stranded amplicons were isolated according to protocol using the Qiagen Pyromark Q96 Work Station and underwent pyrosequencing with 0.5 µM primer 5’-GGG GTT TAT TTT TTT AGG A-3’.
For the H19 DMR, 40 ng of bisulfite-modified DNA was amplified using 1U Platinum Taq DNA polymerase and 0.15 µM each forward (5’-TTT GTT GAT TTT ATT AAG GGA G-3’) and reverse (5’-[Biotin]CTA TAA ATA AAC CCC AAC CAA AC-3’) primers in a 20 µl reaction volume with 0.1 mM dNTPs and 3 mM MgCl2. PCR conditions were 94°C for 3m, then 5 cycles of 94°C for 30s, 64°C for 30s, 72°C for 30s; then 5 cycles of 94°C for 30s, 61°C for 30s, 72°C for 30s; then 35 cycles of 94°C for 30s, 58°C for 30s, 72°C for 30s; followed by a 5m extension at 72°C. The biotin-labeled single-stranded amplicons were isolated as described above and underwent pyrosequencing using 0.5 µM of primer 5’-GTG TGG AAT TAG AAG T-3’.
Assay validation was performed by analysis of defined ratios of plasmids that contain inserts derived from the bisulfite modified version of the methylated and unmethylated sequences, as previously described (Wong et al., 2006).
2.7. Statistical analysis
Covariates considered as potential confounders or effect modifiers are included in Table 1 and included maternal age (< 30 years, 30–39 years, > 40+ years), body mass index (BMI; kg/m2) before pregnancy, cigarette smoking (never smoked, smoked during early pregnancy but stopped, or current smoker), gestational age (< 37 weeks or ≥ 37 weeks), birth weight (< 2500 grams [low] or ≥ 2500 grams [normal]), gender of the offspring (male or female), mother’s education (less than high school, high school/GED, some college, college graduate, or graduate education), and mother’s race/ethnicity (African American, Caucasian or other).
Table 1.
Maternal and Infant Characteristics by Cigarette-smoking Behavior During Pregnancy
| Characteristics | Smokers (N=75) # (%) |
Quit in pregnancy (N=110) # (%) |
Never smoked (N=222) # (%) |
Fisher’s Exact p- value |
|---|---|---|---|---|
| Maternal age | ||||
| < 30 years | 49 (65.3) | 64 (58.2) | 113 (50.9) | 0.03 |
| 30–39 years | 23 (30.7) | 39 (35.4) | 101 (45.5) | |
| 40 years | 3 (4.0) | 7 (6.4) | 8 (3.6) | |
| Education | <0.0001 | |||
| < High school | 50 (66.7) | 49 (44.5) | 55 (24.9) | |
| High school or < college | 19 (25.3) | 29 (26.4) | 63 (28.5) | |
| College or more | 6 (8.0) | 32 (29.1) | 103 (46.6) | |
| Missing | 0 | 0 | 1 | |
| Race | 0.2 | |||
| African American | 41 (54.7) | 51 (46.4) | 110 (49.5) | |
| White | 33 (44.0) | 49 (44.5) | 93 (41.9) | |
| Asian/Hispanic/other/missing | 1 (1.3) | 10 (9.1) | 19 (8.6) | |
| Marital status | 0.05 | |||
| Single | 30 (40.0) | 26 (23.6) | 67 (30.2) | |
| Married/partner | 36 (48.0) | 70 (63.6) | 148 (66.7) | |
| Divorce/widowed | 9 (12.0) | 14 (12.7) | 7 (3.2) | |
| BMI | 0.4 | |||
| <25 | 29 (46.8) | 50 (50.0) | 110 (53.1) | |
| 25–29 | 17 (27.4) | 18 (18.0) | 41(19.8) | |
| 30–34 | 10 (16.1) | 13 (13.0) | 25(12.1) | |
| 35+ | 6 (9.7) | 19 (19.0) | 31(15.0) | |
| Missing | 13 | 10 | 15 | |
| Gestational age at birth | 0.03 | |||
| ≤ 36 weeks | 14 (18.7) | 15 (13.6) | 29 (13.1) | |
| > 36 weeks | 61 (81.3) | 95 (86.4) | 193 (86.9) | |
| Birth weight | <0.0001 | |||
| < 2500 grams | 18 (24.3) | 16 (14.7) | 23 (10.4) | |
| ≥ 2500 grams | 56 (75.7) | 93 (85.3) | 197 (89.6) | |
| Missing | 1 | 1 | 2 | |
| Offspring sex | 0.8 | |||
| Female | 34 (47.2) | 52 (48.6) | 107 (48.6) | |
| Male | 38 (52.8) | 55 (51.4) | 113 (51.4) | |
| Missing | 3 | 3 | 2 | |
| 5-minute Apgar score | 0.7 | |||
| 0–8 | 5 (6.9) | 16 (15.8) | 27 (12.9) | |
| 9–10 | 68 (93.1) | 85 (84.2) | 182 (87.1) | |
| Missing | 2 | 9 | 13 | |
DMR methylation percentages at each CpG dinucleotide were evaluated for normal distribution using Kolmogorov-Smirnov tests. Principal components and confirmatory factor analyses were then used to determine whether methylation at the individual CpGs analyzed at the IGF2 and H19 DMRs were correlated enough for each region to be represented by a single mean. The values for Cronbach’s alpha, a measure of internal consistency, for the CpGs were 86% and 87% at the IGF2 DMR and H19 DMR, respectively. These high correlations at each DMR justified the use of a single mean for each DMR.
We used chi-square tests to determine whether categorical maternal and offspring characteristics varied by maternal cigarette smoking. F-tests from generalized linear models (PROC GLM) were used to compare unadjusted and adjusted least squares mean methylation levels of infants born to women who reported continuing to smoke during pregnancy to those of women who 1) never smoked and 2) stopped smoking during pregnancy. We also employed logistic regression models to confirm the association between cigarette smoking and low birth weight, and to estimate the percent of smoking-related low birth weight mediated by aberrant IGF2 methylation. In such analyses, aberrant methylation at the IGF2 DMR was dichotomized as the methylation in the lowest quartile of the distribution, given that hypomethylation at this DMR has been linked to loss of genomic imprinting (Cui et al., 2003) and periconceptional caloric restriction (Heijmans et al., 2008). Although we considered for potential confounding factors that were associated with birth weight, rapid growth or childhood obesity in the population and in our data set in Table 1 (-value<0.25) one at a time, we retained only maternal education, gestational age, birth weight, race/ethnicity and BMI before pregnancy entered as continuous and categorical variables (Table 1), as these factors contributed significantly to the model (p<0.05).
Because early exposures have been shown to influence the IGF2 methylation profiles differently by sex (Heijmans et al., 2008), and because epigenetic response may vary by genetic factors (Waterland et al., 2010), we repeated these analyses stratified by sex and race. To allow for unrestrained model entry of individual CpGs into statistical models, we repeated the above using mixed effects models. Finally, we conducted mediation analyses to determine the extent to which the association between maternal cigarette smoking and low birth weight was mediated by methylation at either the H19 or IGF2 DMR using standard methods described (Baron and Kenny, 1986).
3. Results
The average gestational age at enrollment was 22 weeks (range 19 to 42 weeks). Although smokers were targeted for the first ~200 recruited, the majority of pregnant women (55%) had never smoked, 27% reported having stopped smoking during the first trimester, and 18% were smokers at enrollment. Of the 110 women who quit smoking during pregnancy, 52 (47%) quit during the first trimester, 14 (13%) quit during the second trimester, and 7 (6%) quit during the third trimester. The remaining 37 women (33%) did not report the time during pregnancy that they quit smoking. Women who smoked during pregnancy and those who stopped smoking early in pregnancy did not significantly differ from never smokers by maternal age, alcohol intake, race, obesity status, gestational age at birth, sex or Apgar score of the offspring (Table 1). However, only 48% (n=36) of women who smoked compared to 67% (n=148) of women who never smoked were married or lived with a partner (p<0.001). Smokers and non-smokers also differed by educational attainment; just 25% (n=55) of non-smokers but 67% (n=50) of smokers had less than a high school education (p<0.001). As expected, infants born to smokers were two times more likely to weigh less than 2500 grams at birth compared to offspring of women who reported never smoking (p=0.001). Hence, in subsequent analysis, we assessed potential confounding by maternal education, marital status, gestational age and birth weight, as were the infant’s sex, maternal BMI before pregnancy and race/ethnicity; the latter factors have been associated with methylation patterns and/or birth weight in other studies (Adkins et al., 2011).
Overall, DNA methylation means were 61.2% (SD=4.2%) and 47.3% (SD=4.5%) at the H19 and IGF2 DMRs, respectively, and were similar at each CpG dinucleotide. Table 2 shows methylation percentages for each CpG evaluated, and the average methylation levels at each DMR in offspring of smokers and non-smokers. Infants born to women who never smoked had a slightly lower methylation fraction on average (46.6%) when compared to infants of women who either quit during pregnancy or continued to smoke (47.8%). However, when pregnant smokers were categorized into two groups including those who quit smoking during pregnancy and those who continued to smoke, we found that infants born to women who continued to smoke during pregnancy had a 3.6% (p=0.001) and 2.9% (p=0.002) higher mean methylation fraction at the IGF2 DMR (49.5%, SD=8.0; n=63) than infants born to women who stopped smoking in pregnancy (45.8%, SD=6.3; n=86) or infants born to women who never smoked (46.6, SD=5.6%; n=178; p=0.002; Table 2). These differences were adjusted for maternal BMI before pregnancy, race/ethnicity, education, gestational age, sex, and birth weight. This significant difference persisted at each of the three CpG dinucleotides evaluated. Neither modeling these relationships using mixed effects models to allow unconstrained model entry of each CpG dinucleotide, nor estimating unadjusted DNA methylation differences altered these findings. Removal of the 10 observations with very low or high DNA methylation also did not alter these findings. However, at the H19 DMR methylation differences were not apparent among the 75 infants born to women who continued to smoke during pregnancy and the 110 born to those who stopped in early pregnancy (p=0.8) and those born to 222 women who never smoked (p=0.8).
Table 2.
Methylation fractions at the IGF2/H19 Imprinted domain by maternal cigarette smoking during pregnancy
| Pregnancy-related smoking behavior | Methylation differences between current smokers and those who |
||||
|---|---|---|---|---|---|
| Smokers (n=75) |
Quit in pregnancy (n=110) |
Never smoked (n=222) |
Quit in pregnancy (p-value) |
Never smoked (p-value) |
|
| H19_CG1 | 59.84 | 60.13 | 59.82 | 0.29 (0.8) | −0.01(1.0) |
| H19_CG2 | 56.81 | 57.50 | 57.58 | 0.68 (0.6) | 0.77 (0.5) |
| H19_CG3 | 59.27 | 60.08 | 59.43 | 0.80 (0.5) | 0.16 (0.9) |
| H19_CG4 | 58.68 | 59.53 | 58.91 | 0.85 (0.5) | 0.23 (0.9) |
| H19 DMR mean | 58.65 | 59.31 | 58.94 | 0.66 (0.6) | 0.29 (0.8) |
|
Smokers (n=63) |
Quit in pregnancy (n=86) |
Never smoked (n=178) |
Quit in pregnancy (p-value) |
Never smoked (p-value) |
|
| IGF2_CG1 | 45.61 | 40.56 | 41.13 | −5.05 (0.0003) | −4.48 (0.001) |
| IGF2_CG2 | 54.17 | 50.74 | 51.44 | −3.43 (0.0006) | −2.73 (0.01) |
| IGF2_CG3 | 53.90 | 49.14 | 49.31 | −4.76 (0.0004) | −4.59 (0.0005) |
| IGF2 DMR mean | 49.50 | 45.86 | 46.64 | −3.63 (0.001) | −2.86 (0.002) |
Means adjusted for maternal education, gestational age, maternal body mass index before pregnancy, race/ethnicity, offspring’s sex, birth weight
As noted previously, several lines of evidence suggest that adverse environmental effects such as exposure to cadmium (Tellez-Plaza et al., 2010) or severe caloric restriction (Heijmans et al., 2008) are gender-dependent. We therefore stratified our analyses by sex of infant. At the IGF2 DMR, we found no methylation differences in female infants born to women who continued to smoke during pregnancy (n=34) and those who either never smoked (n=107) (0.6%, p=0.7) or those who stopped smoking after pregnancy began (n=52) (0.9%, p=0.5). However, among males, those born to women who stopped smoking during pregnancy had 6.5% lower methylation (n=55; p=0.0001) while those born to women who never smoked had 4.7% lower methylation (n=117; p=0.0002), than those born to women who continued to smoke during pregnancy (n=38; Fig. 1). Neither analyses unadjusted for potential confounders nor mixed models altered these findings (data not shown). The interaction term for infant sex and maternal smoking during pregnancy that was included in the final models was statistically significant (p=0.03).
Fig. 1.
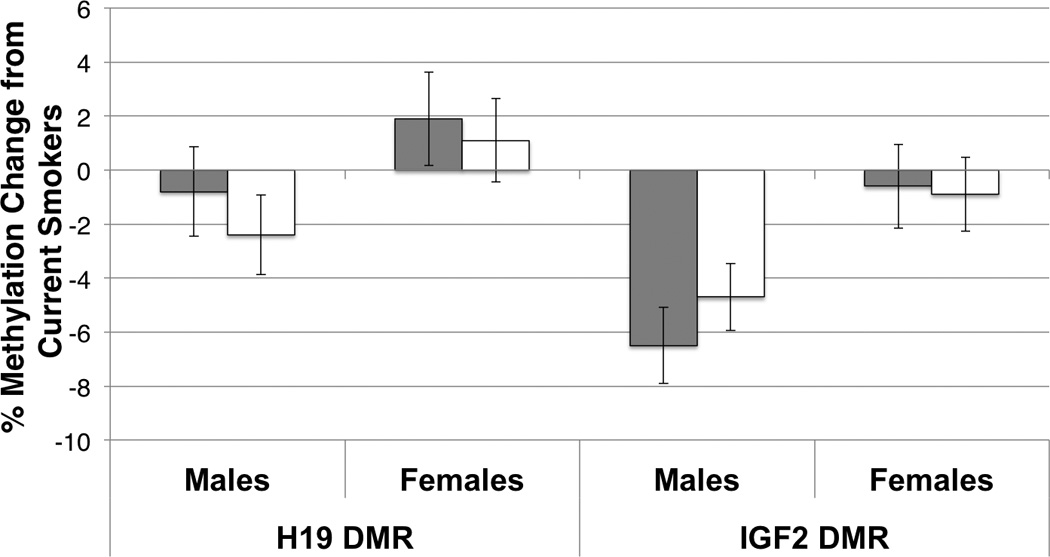
Percent differences in methylation at the H19 and IGF2 DMRs in male and female infants in relation to maternal smoking status during pregnancy. For each DMR, the average difference in methylation is shown (+/− SEM) on the y-axis for male and female infants born to mothers who quit smoking during pregnancy (grey bars) or never smoked cigarettes (white bars) as compared to infants born to mothers who smoked throughout pregnancy (current smokers; baseline methylation). There was no significant difference at the H19 DMR for n=206 males (quit: −0.8% methylation difference, p=0.61; never: −2.4% methylation difference, p=0.13) or n=193 females (quit: +1.9% methylation difference, p=0.32; never: +1.1% methylation difference, p=0.54). The IGF2 DMR also showed no significant difference for n=151 females (quit: −0.6% methylation difference, p=0.73; never: −0.9% methylation difference, p=0.51) while male infants (n=169) born to current smokers showed significantly elevated methylation relative to those born to mothers who quit smoking during pregnancy (−6.5% difference, p<0.0001) and those born to mothers who never smoked (−4.7% methylation difference, p=0.0002).
We also computed odds ratios for the association between low birth weight and cigarette smoking, and estimated the proportion of smoking-related low birth weight that is mediated by variation in IGF2 DMR methylation. After adjusting for potential confounding factors, we found an over four-fold increase in the risk of low birth weight associated with continued maternal cigarette smoking (OR=4.16, 95% CI=1.44–11.99). These analyses revealed that 21% of the association of maternal cigarette smoking and low birth weight in male infants was mediated by aberrant methylation the IGF2 DMR, compared to 2.2% in female infants (Table 3).
Table 3.
*Odds Ratios for the association between maternal smoking and birth weight among 323 newborns
| Characteristic | OR (95% CI) |
|---|---|
| Smoking v. never | 4.16 (1.44, 11.99) |
| Quit v. never | 1.70 (0.60, 4.85) |
| IGF2 DMR mean methylation (<25th percentile) | 0.82 (0.30,2.24) |
| Race | |
| African Americans v. others | 6.52 (0.48, 88.59) |
| Whites v. others | 1.80 (0.14, 23.45) |
| Sex (male v. females) | 0.61 (0.25,1.50) |
| Gestational age <36 wks v. ≥36 wks | 27.05 (9.48,77.18) |
| Marital status (single v. widow) | 1.52 (0.18,13.17) |
| Not single v. widow | 2.23 (0.30,16.50) |
Factors mutually adjusted for each other
4. Discussion
In the present study, we tested the hypothesis that maternal cigarette smoking is associated with changes in DNA methylation at two DMRs regulating the expression of H19 and IGF2, and that methylation profiles mediate the association between maternal cigarette smoking and low birth weight. While no associations were found between H19 DMR methylation and maternal cigarette smoking, we found significantly higher IGF2 DMR methylation in infants born to women who smoked throughout pregnancy when compared to those who either stopped smoking early in pregnancy or those who never smoked. Methylation differences were most apparent in male infants. Approximately 21% of smoking-related low birth weight (with smoking defined as either continued to smoke during pregnancy or quit in early pregnancy) was mediated by aberrant IGF2 methylation in males compared to 2% in female infants.
Our findings support earlier studies showing that the effects of in utero tobacco smoke exposure may at least partially be mediated through alterations in DNA methylation (Breton et al., 2009). These results therefore support the interpretation that epigenetic plasticity is an important mechanism by which early adaptations to environmental cues such as cigarette smoking are realized. Some (Heijmans et al., 2008) but not all (Waterland et al., 2010) have found methylation differences at the IGF2 DMR between individuals exposed and not exposed to environmental stimuli such as nutrient challenges, suggesting that this DMR is a reasonable epigenetic biosensor for evaluating early environmental exposures (Hoyo et al., 2009). Deregulation of IGF2 expression has been linked to overgrowth disorders (Morison and Reeve, 1998; Delaval et al., 2006; Riccio et al., 2009), obesity (Roth et al., 2002; Gomes et al., 2005) and cancer (Taniguchi et al., 1995; Sullivan et al., 1999; Ravenel et al., 2001; Cui et al., 2003; Murphy et al., 2006; Xu et al., 2006; Byun et al., 2007; Chao and D'Amore, 2008; Ito et al., 2008; Pollak, 2008; Paradowska et al., 2009; Shetty et al., 2010; Zuo et al., 2011). We (Hoyo et al., 2009) and others (Mathers, 2007; Heijmans et al., 2009) have proposed that methylation shifts at sequences regulating genomic imprinting can serve as archives of the periconceptional and prenatal environment.
Methylation differences observed in this study are small when compared to those found in animal exposure (Dolinoy et al., 2007) or human pathology (Sullivan et al., 1999; Murphy et al., 2006; Xu et al., 2006; Cui, 2007; Ito et al., 2008; Murrell et al., 2008a). However, this should be expected, given the exposure heterogeneity of human populations combined with an apparent intolerance for sustained dramatic shifts in methylation, especially at this locus, which are often associated with malignancy. To gain insights into the functional significance of methylation differences observed, we explored our unpublished gene expression microarray data (Affymetrix U133 HTA platform) for 41 infants included in this study to determine the relationship between IGF2 expression (IGF2 probe 202409_at) and methylation at the IGF2 DMR. Although the correlation coefficient was in the expected direction (rho = −0.20), it was not statistically significant (p=0.22), likely owing to small sample size. Nevertheless, these data suggest that for every 1% decrease in methylation, there is an approximate two-fold increase in IGF2 transcription (Supplementary Figure 1), theoretically equivalent to what would be observed if the maternal allele was aberrantly activated, as is seen with loss of imprinting. Thus, a 5% decrease in methylation at the IGF2 DMR is associated with an approximate 32-fold increase in IGF2 expression levels.
The magnitude of the effect on aberrant methylation in our study is strikingly similar to the 4.5% methylation difference reported at the IGF2 DMR in 17-month old Dutch children exposed to FA during periconception versus those unexposed to FA, as well as the differences that were found in 60 year old adults who were periconceptionally exposed to the Dutch famine of 1944–45. As adults, Dutch famine survivors are at significantly higher risk of neurological disorders, (1998) obesity in early and late adulthood, (Ravelli et al., 1976; Ravelli et al., 1999) breast cancer, (Elias et al., 2004b) infertility, (Elias et al., 2004a) and metabolic disorders that include type-2 diabetes and dyslipidemia. (Barker, 2004; Yajnik, 2004; Painter et al., 2005; Gluckman et al., 2007). Modest aberrant methylation changes at the H19 or IGF2 DMR have been associated with increased expression of IGF2 (Sullivan et al., 1999; Takano et al., 2000; Cui et al., 2001; Nakagawa et al., 2001; Takai et al., 2001; Cui et al., 2002; Murphy and Jirtle, 2003; Murphy et al., 2006; Xu et al., 2006; Murrell et al., 2008b) and increased susceptibility to several chronic diseases (Cui et al., 2003; Cruz-Correa et al., 2004; Murphy et al., 2006; Feinberg, 2007; Jirtle and Skinner, 2007). Equally small effect sizes have been observed in abused children (McGowan et al., 2009) and in individuals conceived using assisted reproductive technologies (Katari et al., 2009). Because these epigenetic shifts correlated significantly with gene expression, together these studies with the idea that while major epigenetic shifts at imprint regulatory elements may be lethal, adaptive response to perceived environment is likely to be subtle epigenetic changes.
Our findings do not support the hypothesis that methylation changes at the IGF2 DMR are limited to the periconceptional period. In our sample, methylation levels in the offspring of non-smokers were indistinguishable from those of women who quit smoking upon learning they were pregnant, although this may also reflect unmeasured environmental tobacco smoke among women who never smoked. Although the exact mechanism by which cigarette smoking induces alterations in methylation profiles remains unknown, the nicotine in inhaled tobacco smoke has been shown to be vasoconstrictive in humans (Xiao et al., 2007). Vasoconstriction may limit nutrient flow to the growing fetus, mimicking the effects of caloric restriction previously linked to epigenetic perturbation at this locus (Heijmans et al., 2008).
Our study results suggest, for the first time, that the epigenetic effects of maternal cigarette smoking during pregnancy may be more pronounced in males than in females. The finding that males may be more susceptible to environmental toxicants is supported by recent studies of human conceptuses in abortions and stillbirths showing that the male embryo may be more vulnerable than the female to abnormal methylation (Pliushch et al., 2010). Sex differences in DNA methylation patterns in response to caloric restriction have also been previously reported among Dutch famine victims (Heijmans et al., 2007). Other studies of population-wide human disasters have revealed excess phenotypic response and risk among males (Catalano et al., 2005; Khashan et al., 2011). Although the mechanisms that give rise to these effects are unknown, sex effects could reflect differences in epigenetic mechanisms that are responsive to environmental cues. Alternatively, early adaptations resulting from the effects of maternal cigarette smoke may be similar in male and female fetuses, but malleability following changes in exposure (such as quitting) may be male-specific. While speculative, these hypotheses may be testable in other birth cohorts.
Although our findings support the hypothesis that maternal cigarette smoking is associated with epigenetic changes at DMRs regulating the imprinted expression of IGF2, the direction of the association is dissimilar to that of the Dutch famine studies. Whereas exposure to caloric restriction was associated with hypomethylation at the IGF2 DMR (Heijmans et al., 2008), we found increases in methylation at this DMR in relation to maternal cigarette smoking. Similarly, in a study of 17-month-old children, investigators reported an increase in methylation in response to folic acid taken during the periconceptional period, and suggested that folic acid improved a hypomethylated state at the IGF2 DMR, presumably restoring genomic imprinting (Steegers-Theunissen et al., 2009). These findings could not be confirmed in a study of individuals conceived during the nutritionally challenging rainy season in a Gambian rural community (Waterland et al., 2010). We contend that the periconceptional period represents a particularly vulnerable time period, not only for establishment of the epigenome through epigenetic reprogramming, but also for beneficial or deleterious epigenetic responses to exposures such as cigarette smoking. For example, the in utero environment for infants of smokers and quitters during pregnancy is similar with regard to tobacco smoke exposure at the time of periconception, giving rise to similar DNA methylation profiles at IGF2 and perhaps other imprinted DMRs. However, epigenetic profiles in infants born to quitters may trigger compensatory measures at the cessation of exposure which counter increased methylation, and this may lead to over-compensation and a lower methylation profile than that observed in the women who did not smoke. In contrast, infants born to those who continued to smoke throughout pregnancy sustained the high level of methylation due to the continued effects of the exposure, and at some point during gestation these patterns become stable and may always be maintained in the offspring. Alternatively, DNA demethylation pathways involving TET-mediated conversion of 5-methylcytosine to 5-hydroxymethylcytosine (5-hmC) and AID/TDG-mediated base excision repair to replace 5-hmC with cytosine, combined with an inability to distinguish 5-mC from 5-hmC using current technologies, may miss smoking status-related differences in the abundance of these modified bases at these regions. For example, the continuous smoking group may have increased methylation at the IGF2 DMR based on pyrosequencing, but this could represent an increase in 5-hmC and/or lack of ability to undergo TDG-mediated base excision repair. While speculative, technological advances should clarify the underlying mechanisms.
Major strengths of our study include the large sample size and the use of quantitative methods of analysis for DNA methylation (Heijmans et al., 2007). Pyrosequencing methodology has the capacity to detect subtle differences that have the potential to increase susceptibility to chronic disease in later life. Another strength of our analysis is the focus on the DMR regulating IGF2, one of the best-characterized imprinted genes to date, and one with important roles in early embryonic and fetal growth. Finally, because cigarette smoking is a relatively common prenatal exposure, with approximately 10–20% of pregnant women reporting smoking in the United States (Tong et al., 2009) the finding that prenatal cigarette smoke exposure may be an important in utero effector of epigenetic profiles is significant. If our findings are confirmed by others, interventions aimed at smoking cessation during pregnancy, as monitored by methylation profiles in the offspring, offers the potential for identifying nutrients that can negate these effects. Such findings could have a dramatic public health and therapeutic impact on preventing adverse long-term health consequences of these epigenetic alterations.
A limitation of our study is the evaluation of DNA methylation at only two DMRs in the IGF2/H19 imprinted domain. We chose to focus specifically on this region because it is clear that IGF2 plays a fundamental role in regulating growth and expression of IGF2 is deregulated in many disorders and diseases. As compared to other human imprinted domains, methylation at the IGF2/H19 imprinted domain is very well characterized as is involvement of methylation changes at these DMRs in regulating imprint status and levels of IGF2 expression. Another limitation relates to our ascertainment of maternal smoking status. Although women were asked four different questions to determine past and current smoking behavior, and medical records were used to verify smoking status, it is possible that some smokers were misclassified. Cigarette smoking is particularly vulnerable to misclassification because smoking while pregnant is a socially undesirable behavior. However, because women were unaware of the study hypothesis and we verified exposure using medical records, we expect misclassification in this cohort to be minimal. Moreover, women are unlikely to affirm smoking status if they do not smoke. In the event that some cigarette smoking was misclassified, it is unlikely that such misclassification would differ by methylation fraction or other newborn characteristics. Non-differential misclassification, if it exists, would likely attenuate the differences observed. This vulnerability to misclassification also resulted in the suboptimal response to questions on the timing when women stopped smoking, relative to when the pregnancy began, and the number of cigarettes smoked per day. These limitations in smoking ascertainment precluded evaluation of the effects of quitting smoking before the pregnancy began separately from never smoking, as well as intensities of smoking on the epigenetic profile.
4.1. Conclusions
Despite these limitations, we provide the first evidence for smoking-related, subtle but stable methylation shifts in utero, offering a potential explanation for the increased frequency of low birth weight infants born to women who smoke during pregnancy. We found that maternal cigarette smoking differentially alters IGF2–regulating methylation in a manner depending on infant sex, and that up to 20% of smoking-related low birth weight is mediated by methylation shifts at this DMR. This initial evidence for IGF2 DMR plasticity provides a potential explanation for higher susceptibility to toxins in males. The methylation target identified in this study could be further developed as a biosensor for public health or therapeutic interventions aimed at ameliorating the phenotypic consequences of in utero exposure to cigarette smoking.
Supplementary Material
Acknowledgments
This research was supported by National Institutes of Health grants R21ES014947, R01ES016772, R01DK085173 and by funding from the Duke Comprehensive Cancer Center.
Abbreviations
- BMI
body mass index
- CI
confidence interval
- CpG
cytosine-phosphate-guanine dinucleotide pair
- DMR
differentially methylated region
- DNA
deoxyribonucleic acid
- IGF2
insulin-like growth factor II
- GED
general education development diploma
- H19
maternally expressed non-coding RNA H19
- NEST
Newborn Epigenetics STudy
- OR
odds ratio
- PCR
polymerase chain reaction
- SD
standard deviation
- SEM
standard error of the mean
Footnotes
Author’s contributions
C.H. and S.K.M. conceived and designed the study, C.H., S.K.M., Z.H., F.O. and A.P.M. acquired and/or generated data, C.H., F.W., S.K.M., Z.H., J.S.S., R.L.J. and E.SI. contributed to data analysis and interpretation, A.A., S.K.M. and C.H. drafted the manuscript, C.H., S.K.M., A.A., Z.H., J.S.S., R.L.J., A.P.M. and E.S.I helped to revise the manuscript, and all authors provided final approval of the manuscript.
REFERENCES
- Clinical guidelines on the identification, evaluation, and treatment of overweight and obesity in adults: executive summary. Expert Panel on the Identification, Evaluation, and Treatment of Overweight in Adults. Am J Clin Nutr. 1998;68:899–917. doi: 10.1093/ajcn/68.4.899. [DOI] [PubMed] [Google Scholar]
- Adkins RM, Thomas F, Tylavsky FA, Krushkal J. Parental ages and levels of DNA methylation in the newborn are correlated. BMC medical genetics. 2011;12:47. doi: 10.1186/1471-2350-12-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJ. The fetal and infant origins of adult disease. BMJ. 1990;301:1111. doi: 10.1136/bmj.301.6761.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJ. The developmental origins of adult disease. J Am Coll Nutr. 2004;23:588S–595S. doi: 10.1080/07315724.2004.10719428. [DOI] [PubMed] [Google Scholar]
- Baron RM, Kenny DA. The moderator-mediator variable distinction in social psychological research: conceptual, strategic, and statistical considerations. Journal of personality and social psychology. 1986;51:1173–1182. doi: 10.1037//0022-3514.51.6.1173. [DOI] [PubMed] [Google Scholar]
- Breton CV, Byun HM, Wenten M, Pan F, Yang A, Gilliland FD. Prenatal tobacco smoke exposure affects global and gene-specific DNA methylation. Am J Respir Crit Care Med. 2009;180:462–467. doi: 10.1164/rccm.200901-0135OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun HM, Wong HL, Birnstein EA, Wolff EM, Liang G, Yang AS. Examination of IGF2 and H19 loss of imprinting in bladder cancer. Cancer Res. 2007;67:10753–10758. doi: 10.1158/0008-5472.CAN-07-0329. [DOI] [PubMed] [Google Scholar]
- Catalano R, Bruckner T, Gould J, Eskenazi B, Anderson E. Sex ratios in California following the terrorist attacks of September 11, 2001. Hum Reprod. 2005;20:1221–1227. doi: 10.1093/humrep/deh763. [DOI] [PubMed] [Google Scholar]
- CDC: Trends in Smoking Before, During, and After Pregnancy — Pregnancy Risk Assessment Monitoring System (PRAMS), United States, 31 Sites, 2000–2005. Surveillance Summaries. 2009 No. SS-4. [PubMed] [Google Scholar]
- Chao W, D'Amore PA. IGF2: epigenetic regulation and role in development and disease. Cytokine Growth Factor Rev. 2008;19:111–120. doi: 10.1016/j.cytogfr.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Correa M, Cui H, Giardiello FM, Powe NR, Hylind L, Robinson A, Hutcheon DF, Kafonek DR, Brandenburg S, Wu Y, He X, Feinberg AP. Loss of imprinting of insulin growth factor II gene: a potential heritable biomarker for colon neoplasia predisposition. Gastroenterology. 2004;126:964–970. doi: 10.1053/j.gastro.2003.12.051. [DOI] [PubMed] [Google Scholar]
- Cui H. Loss of imprinting of IGF2 as an epigenetic marker for the risk of human cancer. Dis Markers. 2007;23:105–112. doi: 10.1155/2007/363464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui H, Cruz-Correa M, Giardiello FM, Hutcheon DF, Kafonek DR, Brandenburg S, Wu Y, He X, Powe NR, Feinberg AP. Loss of IGF2 imprinting: a potential marker of colorectal cancer risk. Science. 2003;299:1753–1755. doi: 10.1126/science.1080902. [DOI] [PubMed] [Google Scholar]
- Cui H, Niemitz EL, Ravenel JD, Onyango P, Brandenburg SA, Lobanenkov VV, Feinberg AP. Loss of imprinting of insulin-like growth factor-II in Wilms' tumor commonly involves altered methylation but not mutations of CTCF or its binding site. Cancer Res. 2001;61:4947–4950. [PubMed] [Google Scholar]
- Cui H, Onyango P, Brandenburg S, Wu Y, Hsieh CL, Feinberg AP. Loss of imprinting in colorectal cancer linked to hypomethylation of H19 and IGF2. Cancer Res. 2002;62:6442–6446. [PubMed] [Google Scholar]
- Delaval K, Wagschal A, Feil R. Epigenetic deregulation of imprinting in congenital diseases of aberrant growth. Bioessays. 2006;28:453–459. doi: 10.1002/bies.20407. [DOI] [PubMed] [Google Scholar]
- Dolinoy DC, Huang D, Jirtle RL. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci U S A. 2007;104:13056–13061. doi: 10.1073/pnas.0703739104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias SG, Onland-Moret NC, Peeters PH, Rinaldi S, Kaaks R, Grobbee DE, Van Noord PA. Urinary endogenous sex hormone levels in postmenopausal women after caloric restriction in young adulthood. Br J Cancer. 2004a;90:115–117. doi: 10.1038/sj.bjc.6601513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias SG, Peeters PH, Grobbee DE, van Noord PA. Breast cancer risk after caloric restriction during the 1944–1945 Dutch famine. J Natl Cancer Inst. 2004b;96:539–546. doi: 10.1093/jnci/djh087. [DOI] [PubMed] [Google Scholar]
- Feinberg AP. An epigenetic approach to cancer etiology. Cancer J. 2007;13:70–74. doi: 10.1097/PPO.0b013e31803c6e3b. [DOI] [PubMed] [Google Scholar]
- Feng S, Jacobsen SE, Reik W. Epigenetic reprogramming in plant and animal development. Science. 2010;330:622–627. doi: 10.1126/science.1190614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gluckman P, Hanson M, Beedle A. Early life events and their consequences for later disease: A life history and evolutionary perspective. Am J Hum Biol. 2007;19 doi: 10.1002/ajhb.20590. [DOI] [PubMed] [Google Scholar]
- Gluckman PD, Cutfield W, Hofman P, Hanson MA. The fetal, neonatal, and infant environments-the long-term consequences for disease risk. Early Hum Dev. 2005;81:51–59. doi: 10.1016/j.earlhumdev.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Godfrey KM, Barker DJ. Fetal programming and adult health. Public Health Nutr. 2001;4:611–624. doi: 10.1079/phn2001145. [DOI] [PubMed] [Google Scholar]
- Gomes MV, Soares MR, Pasqualim-Neto A, Marcondes CR, Lobo RB, Ramos ES. Association between birth weight, body mass index and IGF2/ApaI polymorphism. Growth Horm IGF Res. 2005;15:360–362. doi: 10.1016/j.ghir.2005.06.016. [DOI] [PubMed] [Google Scholar]
- Heijmans BT, Kremer D, Tobi EW, Boomsma DI, Slagboom PE. Heritable rather than age-related environmental and stochastic factors dominate variation in DNA methylation of the human IGF2/H19 locus. Hum Mol Genet. 2007;16:547–554. doi: 10.1093/hmg/ddm010. [DOI] [PubMed] [Google Scholar]
- Heijmans BT, Tobi EW, Lumey LH, Slagboom PE. The epigenome: archive of the prenatal environment. Epigenetics. 2009;4:526–531. doi: 10.4161/epi.4.8.10265. [DOI] [PubMed] [Google Scholar]
- Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, Slagboom PE, Lumey LH. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A. 2008;105:17046–17049. doi: 10.1073/pnas.0806560105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyo C, Murphy SK, Jirtle RL. Imprint regulatory elements as epigenetic biosensors of exposure in epidemiological studies. J Epidemiol Community Health. 2009;63:683–684. doi: 10.1136/jech.2009.090803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyo C, Murtha AP, Schildkraut JM, Forman MR, Calingaert B, Demark-Wahnefried W, Kurtzberg J, Jirtle RL, Murphy SK. Folic acid supplementation before and during pregnancy in the Newborn Epigenetics Study (NEST) BMC Public Health. 2011;11:46. doi: 10.1186/1471-2458-11-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Wen Y, Shandilya R, Marks JR, Berchuck A, Murphy SK. High throughput detection of M6P/IGF2R intronic hypermethylation and LOH in ovarian cancer. Nucleic Acids Res. 2006;34:555–563. doi: 10.1093/nar/gkj468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y, Koessler T, Ibrahim AE, Rai S, Vowler SL, Abu-Amero S, Silva AL, Maia AT, Huddleston JE, Uribe-Lewis S, Woodfine K, Jagodic M, Nativio R, Dunning A, Moore G, Klenova E, Bingham S, Pharoah PD, Brenton JD, Beck S, Sandhu MS, Murrell A. Somatically acquired hypomethylation of IGF2 in breast and colorectal cancer. Hum Mol Genet. 2008;17:2633–2643. doi: 10.1093/hmg/ddn163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet. 2007;8:253–262. doi: 10.1038/nrg2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katari S, Turan N, Bibikova M, Erinle O, Chalian R, Foster M, Gaughan JP, Coutifaris C, Sapienza C. DNA methylation and gene expression differences in children conceived in vitro or in vivo. Human molecular genetics. 2009;18:3769–3778. doi: 10.1093/hmg/ddp319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khashan AS, McNamee R, Henriksen TB, Pedersen MG, Kenny LC, Abel KM, Mortensen PB. Risk of affective disorders following prenatal exposure to severe life events: A Danish population-based cohort study. J Psychiatr Res. 2011 doi: 10.1016/j.jpsychires.2010.12.005. [DOI] [PubMed] [Google Scholar]
- Kota SK, Feil R. Epigenetic transitions in germ cell development and meiosis. Dev Cell. 2010;19:675–686. doi: 10.1016/j.devcel.2010.10.009. [DOI] [PubMed] [Google Scholar]
- Martin TR, Bracken MB. Association of low birth weight with passive smoke exposure in pregnancy. Am J Epidemiol. 1986;124:633–642. doi: 10.1093/oxfordjournals.aje.a114436. [DOI] [PubMed] [Google Scholar]
- Mathers JC. Early nutrition: impact on epigenetics. Forum Nutr. 2007;60:42–48. doi: 10.1159/000107066. [DOI] [PubMed] [Google Scholar]
- McGowan PO, Sasaki A, D'Alessio AC, Dymov S, Labonte B, Szyf M, Turecki G, Meaney MJ. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nature neuroscience. 2009;12:342–348. doi: 10.1038/nn.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morison IM, Reeve AE. Insulin-like growth factor 2 and overgrowth: molecular biology and clinical implications. Mol Med Today. 1998;4:110–115. doi: 10.1016/s1357-4310(97)01197-0. [DOI] [PubMed] [Google Scholar]
- Murphy SK, Huang Z, Wen Y, Spillman MA, Whitaker RS, Simel LR, Nichols TD, Marks JR, Berchuck A. Frequent IGF2/H19 domain epigenetic alterations and elevated IGF2 expression in epithelial ovarian cancer. Mol Cancer Res. 2006;4:283–292. doi: 10.1158/1541-7786.MCR-05-0138. [DOI] [PubMed] [Google Scholar]
- Murphy SK, Jirtle RL. Imprinting evolution and the price of silence. BioEssays. 2003;25:577–588. doi: 10.1002/bies.10277. [DOI] [PubMed] [Google Scholar]
- Murrell A, Ito Y, Verde G, Huddleston J, Woodfine K, Silengo MC, Spreafico F, Perotti D, De Crescenzo A, Sparago A, Cerrato F, Riccio A. Distinct methylation changes at the IGF2-H19 locus in congenital growth disorders and cancer. PLoS ONE. 2008a;3:e1849. doi: 10.1371/journal.pone.0001849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell A, Ito Y, Verde G, Huddleston J, Woodfine K, Silengo MC, Spreafico F, Perotti D, De Crescenzo A, Sparago A, Cerrato F, Riccio A, Xu W, Fan H, He X, Zhang J, Xie W. Distinct methylation changes at the IGF2-H19 locus in congenital growth disorders and cancer LOI of IGF2 is associated with esophageal cancer and linked to methylation status of IGF2 DMR. PLoS ONE. 2008b;3:e1849. doi: 10.1371/journal.pone.0001849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa H, Chadwick RB, Peltomaki P, Plass C, Nakamura Y, de La Chapelle A. Loss of imprinting of the insulin-like growth factor II gene occurs by biallelic methylation in a core region of H19-associated CTCF-binding sites in colorectal cancer. Proc Natl Acad Sci U S A. 2001;98:591–596. doi: 10.1073/pnas.011528698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter RC, Roseboom TJ, Bleker OP. Prenatal exposure to the Dutch famine and disease in later life: an overview. Reprod Toxicol. 2005;20:345–352. doi: 10.1016/j.reprotox.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Paradowska A, Fenic I, Konrad L, Sturm K, Wagenlehner F, Weidner W, Steger K. Aberrant epigenetic modifications in the CTCF binding domain of the IGF2/H19 gene in prostate cancer compared with benign prostate hyperplasia. Int J Oncol. 2009;35:87–96. doi: 10.3892/ijo_00000316. [DOI] [PubMed] [Google Scholar]
- Pliushch G, Schneider E, Weise D, El Hajj N, Tresch A, Seidmann L, Coerdt W, Muller AM, Zechner U, Haaf T. Extreme methylation values of imprinted genes in human abortions and stillbirths. Am J Pathol. 2010;176:1084–1090. doi: 10.2353/ajpath.2010.090764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. 2008;8:915–928. doi: 10.1038/nrc2536. [DOI] [PubMed] [Google Scholar]
- Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28:1057–1068. doi: 10.1038/nbt.1685. [DOI] [PubMed] [Google Scholar]
- Ravelli AC, van Der Meulen JH, Osmond C, Barker DJ, Bleker OP. Obesity at the age of 50 y in men and women exposed to famine prenatally. Am J Clin Nutr. 1999;70:811–816. doi: 10.1093/ajcn/70.5.811. [DOI] [PubMed] [Google Scholar]
- Ravelli GP, Stein ZA, Susser MW. Obesity in young men after famine exposure in utero and early infancy. N Engl J Med. 1976;295:349–353. doi: 10.1056/NEJM197608122950701. [DOI] [PubMed] [Google Scholar]
- Ravenel JD, Broman KW, Perlman EJ, Niemitz EL, Jayawardena TM, Bell DW, Haber DA, Uejima H, Feinberg AP. Loss of imprinting of insulin-like growth factor-II (IGF2) gene in distinguishing specific biologic subtypes of Wilms tumor. J Natl Cancer Inst. 2001;93:1698–1703. doi: 10.1093/jnci/93.22.1698. [DOI] [PubMed] [Google Scholar]
- Riccio A, Sparago A, Verde G, De Crescenzo A, Citro V, Cubellis MV, Ferrero GB, Silengo MC, Russo S, Larizza L, Cerrato F. Inherited and Sporadic Epimutations at the IGF2-H19 locus in Beckwith-Wiedemann syndrome and Wilms' tumor. Endocr Dev. 2009;14:1–9. doi: 10.1159/000207461. [DOI] [PubMed] [Google Scholar]
- Roth SM, Schrager MA, Metter EJ, Riechman SE, Fleg JL, Hurley BF, Ferrell RE. IGF2 genotype and obesity in men and women across the adult age span. Int J Obes Relat Metab Disord. 2002;26:585–587. doi: 10.1038/sj.ijo.0801927. [DOI] [PubMed] [Google Scholar]
- Shetty PJ, Movva S, Pasupuleti N, Vedicherlla B, Vattam KK, Venkatasubramanian S, Ahuja YR, Hasan Q. Regulation of IGF2 transcript and protein expression by altered methylation in breast cancer. J Cancer Res Clin Oncol. 2010 doi: 10.1007/s00432-010-0890-z. [DOI] [PubMed] [Google Scholar]
- Steegers-Theunissen RP, Obermann-Borst SA, Kremer D, Lindemans J, Siebel C, Steegers EA, Slagboom PE, Heijmans BT. Periconceptional Maternal Folic Acid Use of 400 microg per Day Is Related to Increased Methylation of the IGF2 Gene in the Very Young Child. PLoS One. 2009;4:e7845. doi: 10.1371/journal.pone.0007845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stillman RJ, Rosenberg MJ, Sachs BP. Smoking and reproduction. Fertil Steril. 1986;46:545–566. doi: 10.1016/s0015-0282(16)49628-7. [DOI] [PubMed] [Google Scholar]
- Sullivan MJ, Taniguchi T, Jhee A, Kerr N, Reeve AE. Relaxation of IGF2 imprinting in Wilms tumours associated with specific changes in IGF2 methylation. Oncogene. 1999;18:7527–7534. doi: 10.1038/sj.onc.1203096. [DOI] [PubMed] [Google Scholar]
- Takai D, Gonzales FA, Tsai YC, Thayer MJ, Jones PA. Large scale mapping of methylcytosines in CTCF-binding sites in the human H19 promoter and aberrant hypomethylation in human bladder cancer. Hum Mol Genet. 2001;10:2619–2626. doi: 10.1093/hmg/10.23.2619. [DOI] [PubMed] [Google Scholar]
- Takano Y, Shiota G, Kawasaki H. Analysis of genomic imprinting of insulin-like growth factor 2 in colorectal cancer. Oncology. 2000;59:210–216. doi: 10.1159/000012163. [DOI] [PubMed] [Google Scholar]
- Taniguchi T, Sullivan MJ, Ogawa O, Reeve AE. Epigenetic changes encompassing the IGF2/H19 locus associated with relaxation of IGF2 imprinting and silencing of H19 in Wilms tumor. Proc Natl Acad Sci U S A. 1995;92:2159–2163. doi: 10.1073/pnas.92.6.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellez-Plaza M, Navas-Acien A, Crainiceanu CM, Sharrett AR, Guallar E. Cadmium and peripheral arterial disease: gender differences in the 1999–2004 US National Health and Nutrition Examination Survey. Am J Epidemiol. 2010;172:671–681. doi: 10.1093/aje/kwq172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong VT, Jones JR, Dietz PM, D'Angelo D, Bombard JM. Trends in smoking before, during, and after pregnancy - Pregnancy Risk Assessment Monitoring System (PRAMS), United States, 31 sites, 2000–2005. MMWR. Surveillance summaries : Morbidity and mortality weekly report. Surveillance summaries / CDC. 2009;58:1–29. [PubMed] [Google Scholar]
- Waterland RA, Kellermayer R, Laritsky E, Rayco-Solon P, Harris RA, Travisano M, Zhang W, Torskaya MS, Zhang J, Shen L, Manary MJ, Prentice AM. Season of conception in rural gambia affects DNA methylation at putative human metastable epialleles. PLoS Genet. 2010;6:e1001252. doi: 10.1371/journal.pgen.1001252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong HL, Byun HM, Kwan JM, Campan M, Ingles SA, Laird PW, Yang AS. Rapid and quantitative method of allele-specific DNA methylation analysis. Biotechniques. 2006;41:734–739. doi: 10.2144/000112305. [DOI] [PubMed] [Google Scholar]
- Xiao D, Huang X, Yang S, Zhang L. Direct effects of nicotine on contractility of the uterine artery in pregnancy. J Pharmacol Exp Ther. 2007;322:180–185. doi: 10.1124/jpet.107.119354. [DOI] [PubMed] [Google Scholar]
- Xu W, Fan H, He X, Zhang J, Xie W. LOI of IGF2 is associated with esophageal cancer and linked to methylation status of IGF2 DMR. Exp Clin Cancer Res. 2006;25:543–547. [PubMed] [Google Scholar]
- Yajnik CS. Early life origins of insulin resistance and type 2 diabetes in India and other Asian countries. J Nutr. 2004;134:205–210. doi: 10.1093/jn/134.1.205. [DOI] [PubMed] [Google Scholar]
- Zuo QS, Yan R, Feng DX, Zhao R, Chen C, Jiang YM, Cruz-Correa M, Casson AG, Kang XD, Han F, Chen T. Loss of imprinting and abnormal expression of the insulin-like growth factor 2 gene in gastric cancer. Mol Carcinog. 2011 doi: 10.1002/mc.20731. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.