

Abstract
The abundant axonal microtubule-associated protein tau regulates microtubule and actin dynamics, thereby contributing to normal neuronal function. We examined whether mice deficient in tau (Tau−/−) or with high levels of human tau differ from wild-type (WT) mice in their susceptibility to neuroaxonal injury in experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis. After sensitization with MOG35–55, there was no difference in clinical disease course between human tau and WT mice, but Tau−/− mice had more severe clinical disease and significantly more axonal damage in spinal cord white matter than those in WT mice. Axonal damage in gray matter correlated with clinical severity in individual mice. By immunoblot analysis, the early microtubule-associated protein-1b was increased 2-fold in the spinal cords of Tau−/− mice with chronic experimental autoimmune encephalomyelitis versus naive Tau−/− mice. This difference was not detected in comparable WT animals, which suggests that there was compensation for the loss of tau in the deficient mice. In addition, levels of the growth arrest–specific protein 7b, a tau-binding protein that is stabilized when bound to tau, were higher in WT than those in Tau−/− spinal cord samples. These data indicate that loss of tau exacerbates experimental autoimmune encephalomyelitis and suggest that maintaining tau integrity might reduce the axonal damage that occurs in inflammatory neurodegenerative diseases such as multiple sclerosis.
Keywords: Axonal damage, Experimental autoimmune encephalomyelitis, Growth arrest–specific protein 7b, Microtubule-associated protein 1b, Multiple sclerosis, Myelin oligodendrocyte glycoprotein (MOG), Tau
INTRODUCTION
Disruption of the blood-brain barrier in multiple sclerosis (MS) is associated with an influx of inflammatory cells that compromise normal cell signaling, leading to oligodendrocyte loss, demyelination, axonal damage, and neurodegeneration (1-13). Inflammation is associated with cytokine and chemokine secretion and activation of signaling molecules, including phosphatases and kinases that can post-translationally modify proteins essential for normal cellular functions. Post-translational alterations in neuronal proteins can result in destabilization of the axonal cytoskeleton, thereby negatively affecting neuronal physiology (14). There is an incomplete understanding of how inflammatory changes affect CNS function and contribute to neurodegeneration in MS. To address the role of integral proteins that regulate neuronal cytoskeletal dynamics, we focused on the microtubule-associated protein (MAP) tau in myelin oligoden-drocyte glycoprotein (MOG)– induced experimental autoimmune encephalomyelitis (EAE), a mouse CNS inflammatory disease model of MS (15).
Tau, the major MAP in axons, is important for microtubule dynamics (14). In addition to binding to tubulin and actin (16), it directly interacts with several cytosolic proteins that are involved in axonal transport, myelination, regulation of tau phosphorylation, and neurite outgrowth. These include dynactin (17), protein phosphatase 2a (18), Fyn (19), Pin1 (20-22), forkhead-binding protein 51 (FKBP-51) (23), FKBP-52 (24), and growth arrest–specific protein 7b (Gas7b) (25). Aberrantly phosphorylated tau cannot effectively bind to microtubules, which results in abnormal microtubule dynamics, disassembly of microtubules, and anomalous axonal transport. Whereas hyperphosphorylation and aggregation of tau contribute to pathology in Alzheimer disease (AD), a recent study demonstrated that neuronal c-Abl overexpression led to aberrantly phosphorylated soluble tau, neuronal loss, and neuroinflammation in the forebrain of mice (26). Data from the brainstem of rats showed axonal damage in EAE that was linked to tau pathology (27). Furthermore, soluble and abnormal tau phosphorylation was found in pathologic glia in secondary progressive MS and primary progressive MS (7, 28, 29).
In this study, we examined the contribution of tau to the pathogenesis of EAE by assessing disease progression and recovery in C57Bl/6J mice expressing murine tau (WT), no tau (Tau−/− mice) (30), or excess human tau (hTau) (31, 32). Our goal was to determine whether changes in tau during EAE-induced inflammation affects EAE in these mice and whether other proteins involved in neuritogenesis are differentially expressed in Tau−/− versus WT mice.
MATERIALS AND METHODS
Mice
Tau−/− and hTau male mice were obtained from Dr Peter Davies in collaboration with Dr Karen Duff at Columbia University Medical Center, New York, NY (30-32) and were maintained in conventional housing. The human tau gene was inserted into Tau−/− mice under the control of the tau promoter (31, 32). The mice were backcrossed more than 15 times in the Davies and Shafit-Zagardo Laboratories. Wild-type littermates were used as controls. All experiments were performed with male mice aged 8 weeks. All animal procedures were approved by the Institute of Animal Care Committee at Albert Einstein College of Medicine in complete compliance with the National Institutes of Health Guide for Care and Use of Laboratory Animals.
MOG35–55–Induced EAE
C57Bl/6J WT control mice, hTau mice, and Tau−/− mice were immunized with MOG35–55 peptide at 8 weeks of age (15). The MOG35–55 (3 mg/mL; Peptides International, Cleveland, OH) was emulsified in an equal volume of complete Freund’s adjuvant (CFA) composed of Mycobacterium tuberculosis (10 mg/mL; Difco Laboratories, Detroit, MI) in incomplete Freund’s adjuvant (Difco Laboratories). Mice were anesthetized with isoflurane, and 100 μL of emulsion was injected subcutaneously on each flank (200 μL total/mouse) on day 0. In addition, 200 μL of pertussis toxin (2.5 μg/mL, List Biochemical Laboratories, Campbell, CA) was injected into the tail vein on days 0 and 2. Mice were monitored and graded daily for clinical signs of disease as follows: 0, no disease; 1, limp tail; 2, limp tail and hind limb weakness; 3, hind limb paralysis; 4, hind limb and front limb paralysis; and 5, moribund.
Statistical Analysis
The Student t-test was performed on clinical indices combined from multiple EAE experiments for 2 groups.
Spinal Cord Dissection and Tissue Preparation
Mice were anesthetized with ethyl ether (Fisher Scientific, Pittsburgh, PA) and killed by total body perfusion with 4% paraformaldehyde (Fisher Scientific), Trump fixative (2% glutaraldehyde (Polysciences, Inc, Warrington, PA) + 4% paraformaldehyde in cacodylate buffer), or 1× PBS, pH 7.3. Spinal cords were removed and dissected into cervical, thoracic, and lumbar regions. Sections were placed in fixative for immuno-histochemistry and toluidine blue studies or sonicated on ice with a tissue master 125 sonicator (Omni International, Marietta, GA) in protein buffer (140 mM NaCl, 1 mM Tris, pH 7.4) containing 0.5% Triton X-100 and protease inhibitors (2 μg/mL of leupeptin, 2 mM of ethylene glycol-bis[β-aminoethylether]-N,N,N′,N′-tetraacetic acid, 4 μg/mL of pepstatin, 5 mM of sodium pyrophosphate, 30 mM of β-glycerophosphate, 30 mM of sodium fluoride, 100 mM of sodium orthovanadate, 100 mM of 4-[2-aminoethyl] benzenesulfonyl fluoride hydrochloride) to yield total protein homogenates. The homogenates were cleared by centrifugation at 4°C at 7,000 × g for 10 minutes. Aliquots were frozen at −80°C.
Western Blot Analysis
Protein homogenates were loaded in 1× final concentration loading buffer containing 2% sodium dodecyl sulfate, 0.017% bromophenol blue, and 0.28 M β-mercaptoethanol and separated by electrophoresis in a 10% sodium dodecyl sulfate polyacrylamide gel (33). After electrophoresis, proteins were transferred to nitrocellulose (34). Blots were incubated with 5% nonfat dry milk and 5% goat serum in 1× Tris-buffered saline (TBS) for 1 hour at room temperature. After blocking, membranes were incubated with the respective primary antibodies followed by horseradish peroxidase–conjugated secondary antibodies (1:20,000; Jackson ImmunoResearch Laboratories, West Grove, PA). Visualization of all secondary antibodies was by enhanced chemiluminescence (GE Healthcare, Piscataway, NJ). Bands were analyzed by calculating the relative densitometric intensity (rdi) of each protein normalized to β-actin using ImageJ software (National Institutes of Health). Changes in tau phosphorylation after EAE induction were evaluated using immunoblot and relative densitometric analysis of protein homogenates prepared from the lumbar spinal cord of naive, EAE-induced WT, and hTau mice with chronic EAE.
Immunohistochemistry
Paraformaldehyde-fixed sections were stored overnight at 4-C and processed for paraffin embedding. Ten-micrometer-thick sections were dewaxed with xylenes and rehydrated through descending alcohols and brought to 1× TBS pH 7.4. All sections were incubated for 30 minutes with 1× TBS containing 0.25% Triton X-100, followed by a 1-hour incubation in 5% goat serum and 5% nonfat dry milk in 1× TBS, then incubated with antibodies diluted in 5% nonfat dry milk in 1× TBS, overnight at 4°C. Sections were washed three times in 1× TBS and incubated with secondary antibody followed by incubation with the appropriate staining kit (Vector Laboratories, Inc, Burlingame, CA) and visualized by diaminobenzidine (Sigma). For triple-label immunofluorescence, sections were incubated for 1 hour with directly conjugated, fluorescently labeled, isotype-specific secondary antibodies and visualized on an Olympus digital fluorescent microscope. In addition, 1-μm-thick sections were prepared from Trump fixed sections and stained with toluidine blue stain in the Analytical Imaging Facility. Sections of spinal cord were cut and stained for hematoxylin and eosin and Luxol fast blue in the Histopathology Core Facility.
Antibodies
Unless otherwise noted, all anti-tau antibodies were generated in the Davies Laboratory. Monoclonal antibodies (mAbs) DA9 and Tau46 (Chemicon, Billerica, MA) recognize nonphosphorylated sites on native tau. Phosphotau sites studied included sites recognized by mAbs-CP13 (phospho202), CP9 (phospho231), and PHF1 (phospho396). The microtubule-associated protein 2 (MAP2) (clone HM-2, M9942) and β-actin antibodies were purchased from Sigma. The neurofilament mAb SMI32 and antibody to myelin basic protein (MBP) were purchased from Covance, Emeryville, CA. The anti-CD45 mAb was from BD Biosciences (San Jose, CA); antibody to amyloid precursor protein (APP) was purchased from Invitrogen; anti-Iba1 (019–19741) was from Wako Chemicals (Richmond, VA). The polyclonal antibody (pAb) to high-molecular-weight (HMW) tau (Big Tau) (35) was generated in the laboratory of Dr Itzak Fischer at Drexel University College of Medicine, Philadelphia, PA. Dr Lester Binder at Northwestern University Medical School, Chicago, IL, generously provided the MAP1b mAb 3G5. The anti-Gas7 antibody was purchased from Novus Biologicals, Littleton, CO. For semiquantitative analysis, Iba1-positive CD45-positive cells, SMI32-positive and APP-positive axons/lesion in 40× fields were counted.
RESULTS
EAE in hTau and WT Mice
The average day of EAE onset was approximately day 10 in both WT C57Bl/6J and hTau mice, and there were no differences in the clinical courses between hTau and WT mice (Fig. 1a). Because there is ascending paralysis beginning in the lumbar spinal cord in EAE, our immunohistochemical and immunoblot analysis focused on neurofilament and tau phosphorylation in the lumbar region. Changes in the state of neurofilament phosphorylation were assessed by comparing the abundance of nonphosphorylated neurofilament proteins to phosphorylated neurofilament proteins using the SMI32 and SMI31 mAbs, respectively. At the time of death, the mean clinical scores indicated recovery (clinical index [CI] <1.5); the mice had higher clinical scores during the acute phase (Fig. 1A). The WT mice (Fig. 1bA) and the hTau mice (Fig. 1bE) had similar SMI32-positive staining in the lumbar spinal cord white matter. The SMI31 immunoreactivity in WT and hTau mice also appeared similar (Figs. 1b: B, F, respectively), indicating that overexpression of hTau did not appear to affect the extent of axonal damage in MOG-induced EAE. Hematoxylin and eosin sections also showed no overt differences in the extent of inflammatory cells in WT and hTau spinal cord (Figs. 1b: C and G, respectively); and Luxol fast blue staining showed no obvious differences in demyelination at the chronic stage (Figs. 1b: D, H).
FIGURE 1.
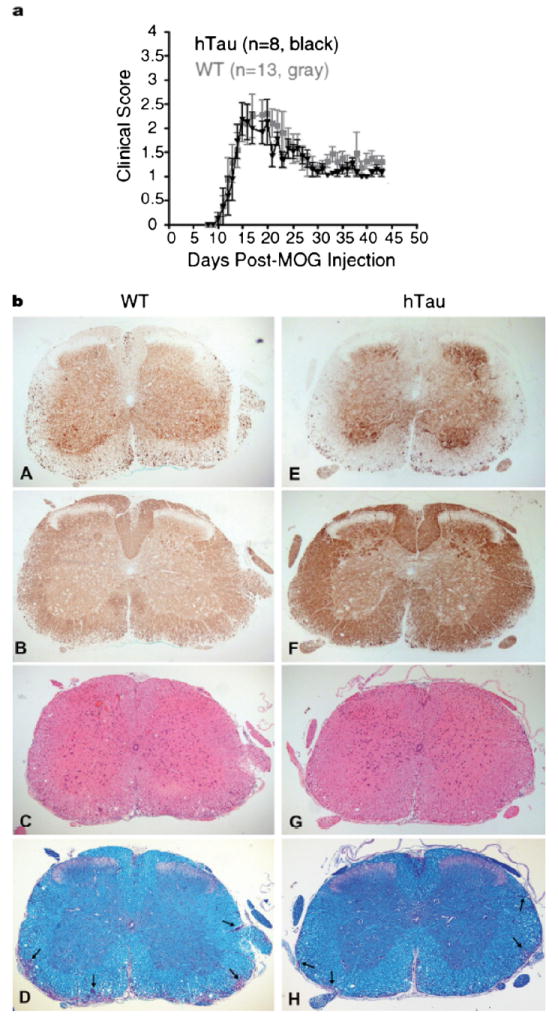
Tau overexpression does not alter disease severity in myelin oligodendrocyte glycoprotein (MOG)–induced experimental autoimmune encephalomyelitis (EAE). In (a) mice were scored daily for clinical signs of disease, and the clinical scores were charted up to day 43 after immunization. The incidence of disease was 93% for both groups of mice. Gray squares represent the mean daily clinical score ± SEM of wild-type (WT) mice (n = 13); black triangles represent the mean ± SEM of mice with high levels of human tau (hTau) (n = 8). (b) Histochemical (C, D, G, H) and immunohistochemical (A, B, E, F) stainings of the spinal cord of hTau mice (E–H) at the chronic stage of disease show that hTau mice are not more severely affected than WT mice (A–D). Nonphosphorylated neurofilaments were detected using antibody SMI32 (A, E); phosphorylated neurofilament staining was detected using SMI31 (B, F). Arrows (D, H) indicate areas of demyelination. (C, G) Hematoxylin and eosin; (D, H) Luxol fast blue stain. Original magnification: 50×.
Changes in Tau Phosphorylation
The ratio of phosphotau normalized to total tau in MOG-sensitized mice relative to naive mice was examined. Total tau was determined using the mAb DA9 that recognizes all non-phosphorylated tau isoforms. Although hTau mice expressed more tau protein than WT mice did, there was no marked difference in the ratio of DA9-positive tau in lumbar spinal cord homogenates of mice with EAE versus that from naive mice (Fig. 2). For WT mice, the ratio was 1.2; for hTau mice, the ratio was 1.0. MAP2, another major MAP predominantly expressed in the dendrites and cell body of neurons and recognized by the anti-HM2 mAb, was marginally changed in EAE versus the naive mouse homogenates. For WT mice, the ratio was 1.4; for hTau mice, the ratio was 1.6. Phosphotau immunoreactivity was observed in both naive mice and mice with EAE using several anti-tau mAbs. Phosphotau epitopes, including phosphotau231, phosphotau202, and phosphotau396 (recognized by CP9, CP13, and PHF1 mAbs, respectively) were detected in lumbar spinal cord protein homogenates isolated from both naive mice and mice with chronic EAE. After normalization to DA9, the ratio of phosphotau in MOG-sensitized lumbar spinal cord to phosphotau in naive lumbar spinal cord homogenate was negligibly altered in hTau mice (0.98–1.5), suggesting that the hTau overexpression did not negatively contribute to enhancement of abnormal tau phosphorylation or further impact axonal damage in the samples tested.
FIGURE 2.
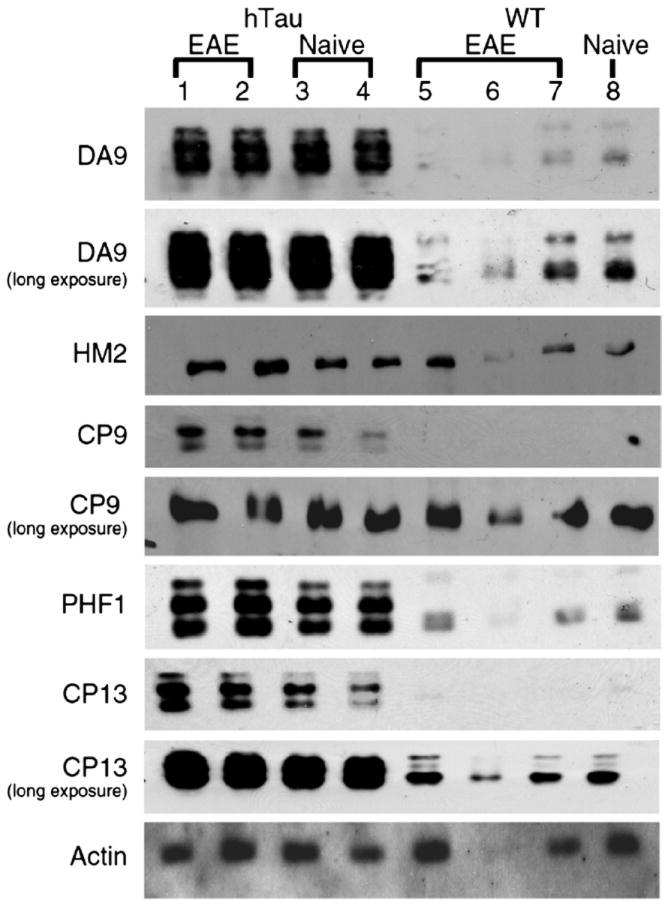
Minimal relative changes in tau phosphorylation in lumbar spinal cords of mice with high levels of human tau (hTau) and wild-type (WT) mice during chronic experimental auto-immune encephalomyelitis (EAE). Monoclonal antibody (mAb) DA9 recognizes nonphosphorylated tau. The mAb clone HM2 recognizes high-molecular-weight microtubule-associated protein 2 (HMW MAP2). The mAbs CP9, PHF1, CP13 recognize phosphorylated tau epitopes. Visualization was by enhanced chemiluminescence. To visualize tau in hTau or WT mice, both short and long exposures are shown. Lanes 1 and 2, hTau spinal cord chronic EAE; lanes 3 and 4, naive hTau spinal cord; lanes 5 to 7, WT spinal cord chronic EAE; lane 8, naive WT spinal cord. Fifty micrograms of protein was loaded in each lane.
The expression of HMW tau was studied in naive mice at 2 and 9 months of age with a generic tau antibody tau46 (1:1000) and a polyclonal antibody specific for HMW tau (Fig. 3a). The HMW tau is expressed in murine spinal cord at both ages. Using this specific antibody, 110-kDa tau immunohistochemical staining of WT and hTau spinal cord was examined. No significant difference in the pattern of staining was observed between the naive spinal cord of hTau mouse (Fig. 3b) or WT spinal cord (not shown). The HMW staining was detected predominantly in the substantia gelatinosa of Rolando, with some staining of neuron cell bodies. Figure 3c shows hematoxylin and eosin and HMW tau immunostaining for WT (Fig. 3c: A, C, E, G) and hTau (Fig. 3c: B, D, F, H) mice, focusing on the substantia gelatinosa and adjacent gray matter. During both the acute stage of EAE (day 15 post-MOG injection; Figs. 3c: A–D) and at the chronic stage (day 47 postimmunization; Figs. 3c: E–H), there was no apparent difference in 110-kDa tau immunostaining between hTau and WT mice.
FIGURE 3.
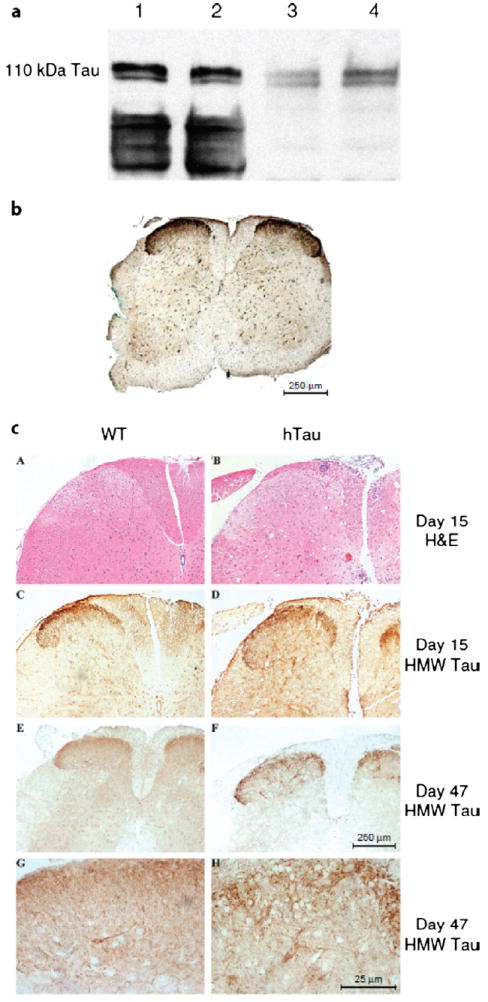
The 110-kDa high-molecular-weight (HMW) tau is expressed in the spinal cord during experimental autoimmune encephalomyelitis (EAE). (a) Immunoblot analysis for HMW tau in mouse spinal cord. Twenty micrograms of protein homogenate from the lumbar spinal cord of wild-type (WT) mice at 2 months (lanes 1, 3) and 9 months (lanes 2, 4) of age was loaded per lane. Lanes 1 and 2 were incubated with mAb tau 46 (1:1000); lanes 3 and 4 were incubated with HMW tau polyclonal antibody (pAb) (1:20,000). (b) Spinal cord section from a mouse with high levels of human tau (hTau) stained with HMW tau pAb (1:10,000). (c) Paraffin-embedded sections of WT (A, C, E, G) and hTau (B, D, F, H) mice stained with hematoxylin and eosin (A, B) or immunostained with anti-HMW tau polyclonal antibody (C–H). Scale bars = (b, c [A–F]) 250 μm; (c [G, H]) 25 μm.
Thus, our combined data indicate that overexpression of tau does not contribute to a more severe course of disease in hTau mice in acute or chronic EAE. Although immunoblot analysis showed increased phosphotau, we did not observe tau inclusions in gray matter neurons or glia within the spinal cord in chronic EAE by immunohistochemistry.
EAE in Tau−/− Mice
To evaluate the consequence of the loss of tau expression during EAE, Tau−/− mice and WT mice were compared during acute and chronic diseases. A representative experiment is shown in Figure 4a, and cumulative data for 4 independent experiments are shown in Figure 4b. Because the day of onset was the same for both the Tau−/− and WT mice, we normalized all data to the first day of clinical signs for each individual mouse. The Tau−/− mice had significantly higher clinical scores during the acute phase of disease, that is, day 14 through day 20, with the exception of day 17 (days 14, 18, 19, 20, p < 0.05; days 15, 16, p < 0.02 by unpaired Student t-test). The mean clinical scores ± SEM from days 14 to 20 for the WT mice was 1.9 ± 0.05 versus 2.4 ± 0.06 for the Tau−/− mice (p = 0.0001). Five (11%) of 43 of the Tau−/− mice became moribund, whereas only 1 (2%) of 46 WT mice became moribund. During chronic disease, the Tau−/− mice tended to have higher clinical scores, but differences from WT were not significant.
FIGURE 4.
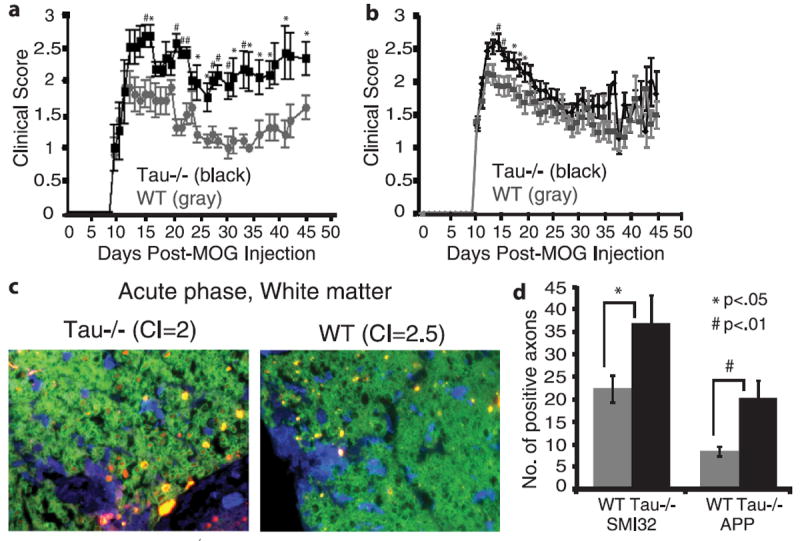
Tau-deficient (Tau−/−) mice have more severe clinical disease and neuronal injury than wild-type (WT) mice during experimental autoimmune encephalomyelitis (EAE). (a) Mice were scored for clinical disease beginning on day 10 postsensitization. (b) Data combined from 4 experiments show that Tau−/− mice have higher clinical scores during acute disease. (c) At day 19, Tau−/− mice have more SMI32 staining than WT mice. Paraffin-embedded sections were stained with an antibody to myelin basic protein ([MBP] 1:1000; green), Iba1 (1:400; blue), and SMI32 (1:20,000; red). (d) The numbers of SMI32-positive or amyloid precursor protein (APP)–positive axons/lesion in a 40× field were counted. The disease incidence was 94% in both groups. CI = clinical index.
To determine if clinical score differences between WT and Tau−/− mice during the acute phase was caused by increased inflammatory cells in the spinal cords, WT (n = 4) and Tau−/− (n = 6) spinal cords were immunostained for Iba1 or CD45. There was no significant difference in the mean number of inflammatory cells/lesion (40×); all lesions in each section were analyzed (data not shown). The mean number of Iba1-positive cells was 81.8 ± 13.1 for WT mice and 94.5 ± 14.2 for Tau−/− mice (p > 0.05). The mean number of CD45-positive cells was 20.2 ± 3.0 for WT mice and 15.9 ± 2.7 for Tau−/− mice (p > 0.05) (Table).
TABLE.
Summary of Clinical Severity and Immunohistochemical Findings in Tau−/− and Wild-Type Mice in Acute Experimental Autoimmune Encephalomyelitis
| Tau−/− Mice | Wild-Type Mice | p | |
|---|---|---|---|
| Average CI (days 14–20) | 2.4 ± 0.06 | 1.9 ± 0.05 | <0.0001 |
| CD45-positive cells/lesion | 15.9 ± 2.7 | 20.2 ± 3.0 | >0.05 |
| Iba1-positive cells/lesion | 94.5 ± 14.2 | 81.8 ± 13.1 | >0.05 |
| SMI32-positive axons/lesion | 37.0 ± 6.2 | 22.4 ± 3.0 | <0.05 |
| APP-positive axons/lesion | 20.3 ± 4.0 | 8.5 ± 1.0 | <0.01 |
All data are mean ± SEM. Counts were done in 40× fields.
APP, amyloid precursor protein; CI, clinical index.
Axonal Damage in White and Gray Matter in Tau−/− Mice
In contrast to the number of inflammatory cells, the number of SMI32-positive and APP-positive axons/lesion (40×) was significantly higher in Tau−/− mice versus WT mice during the acute phase at day 19. In lumbar spinal cord sections incubated with antibodies to MBP (green), SMI32 (red), and Iba1 (blue; Fig. 4c), there were fewer SMI32-positive axons in WT than those in Tau−/− mice (22.4 ± 3.0 vs 37.0 ± 6.2, p < 0.05). There were also fewer APP-positive swollen axons in WT versus Tau−/− mice (8.5 ± 1.0 vs 20.3 ± 4.0, p = 0.002; Fig. 4d).
Several Tau−/− mice had hind limb and front limb paralysis during the acute phase of EAE. In sections prepared from these mice, neurons in the gray matter of lumbar spinal cord showed neuritic swellings. In Figure 5, the upper panels show gray matter regions from the lumbar spinal cord of a naive Tau−/− mouse stained with SMI32, Iba1, and APP. There were no axonal neuritic swellings observed with either APP or SMI32 staining and no Iba1-positive activated microglia in naive mice. By contrast, the lower panels of Figure 5 show that, in Tau−/− mice with hind limb and front limb paralysis, there are focal axonal swellings that stain with SMI32 and APP within the gray matter. This damage correlated with the presence of activated microglia near large motor neurons. The WT mice did not show SMI32-positive or APP-positive axonal swellings in the gray matter, and there was less extensive Iba1-positive immunostaining.
FIGURE 5.
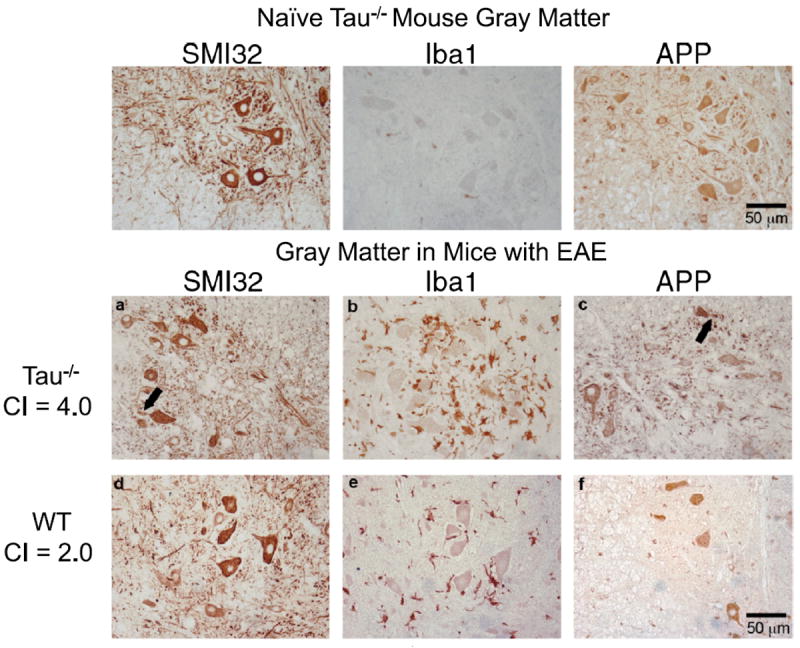
Neuronal damage in gray matter of tau-deficient (Tau−/−) mice in acute experimental autoimmune encephalomyelitis (EAE). Paraffin-embedded sections of lumbar spinal cord from a naive Tau−/− mouse (upper row), a Tau−/− mouse with EAE (day 19) (middle row), and a wild-type (WT) mouse with EAE (day 19) (bottom row) were immunostained with antibodies to amyloid precursor protein (APP), SMI32 and Iba1. (a–c) In Tau−/− mice, neurons with SMI32-positive (a) and APP-positive (c) aberrant neurites (arrows) were near Iba1-positive activated microglia (b). (d–f) The WT mouse with EAE has no detectable SMI32-positive (d) or APP-positive (f) swellings and fewer immunostained microglia (e). CI = clinical index.
Thus, the significant difference in mean clinical scores during acute disease correlated with more axonal damage in the spinal cords of Tau−/− mice than in the spinal cords of WT mice. In addition to the detectable differences in the mean number of SMI32-positive and APP-positive swellings/lesion in the white matter of Tau−/− mice, there was evidence of motor neuron injury in their gray matter.
Cytoskeletal Proteins in WT and Tau−/− Mice in Chronic EAE
We next examined tau and MAP1b expression in protein homogenates from lumbar spinal cord of naive mice and WT and Tau−/− mice during chronic EAE to determine whether MAP1b, a MAP expressed during neural development, was upregulated in Tau−/− mice (Fig. 6a). As expected, tau was not expressed in lumbar spinal cord homogenates from naive Tau−/− mice (lane 1) or from Tau−/− with chronic EAE (lanes 2–9). Tau expression was detected in spinal cord homogenates from naive WT (lane 13) and WT mice with chronic EAE (lanes 10–12). When normalized to β-actin, the total tau expression increased less than 1.5-fold during chronic EAE versus naive WT mice. MAP1b was previously shown to be reexpressed during EAE and to correlate with a reparative process. With the exception of 1 WT sample that did not have detectable MAP1b (lane 10), MAP1b was present in all WT and Tau−/− spinal cord homogenates. The expression of MAP1b was not elevated in spinal cord homogenates from chronic WT mice relative to naive WT homogenates; the mean increase was 1.2-fold after normalization to β-actin. Additional WT samples did not show a significant increase in MAP1b during chronic disease. By contrast, relative to naive Tau−/− mice, MAP1b was increased in homogenates from Tau−/− mice during chronic EAE. Of the 8 homogenates examined, the mean increase of MAP1b was 2.2 ± 3.4-fold, with a range of 1.4- to 3.9-fold (p < 0.05) (Fig. 6b).
FIGURE 6.
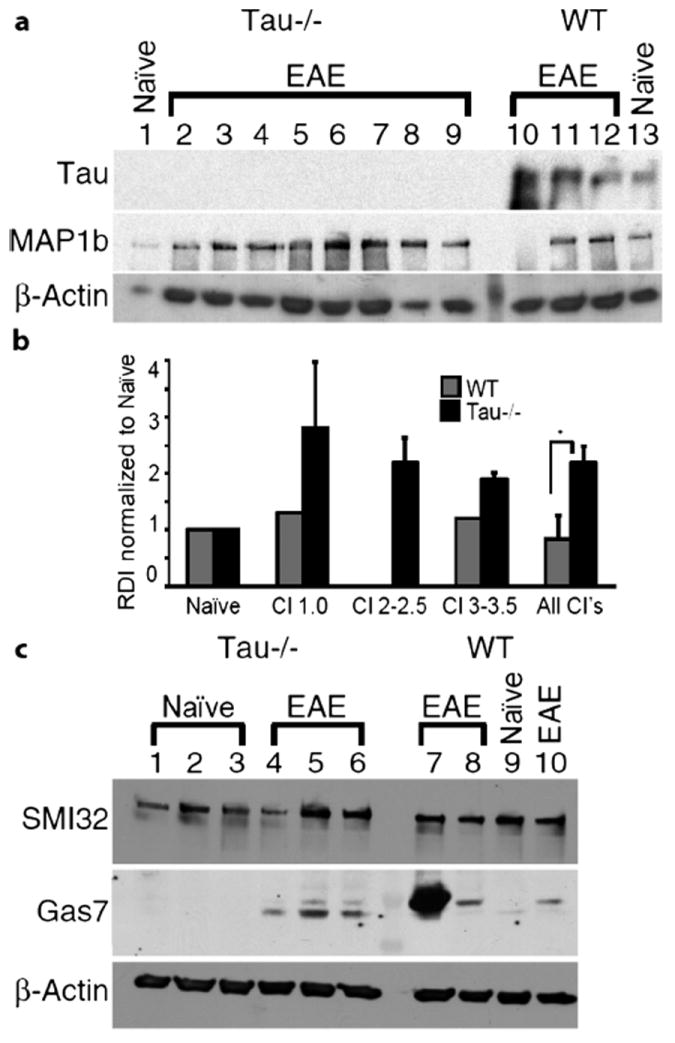
Expression of cytoskeletal proteins in wild-type (WT) and tau-deficient (Tau−/−) mice during chronic experimental autoimmune encephalomyelitis (EAE). (a) Comparison of microtubule-associated protein 1b (MAP1b) and tau in naive and chronic Tau−/− and WT mice. Blot was cut, and the upper portion was incubated with the MAP1b monoclonal antibody (mAb) 3G5 (1:10). The middle section was incubated with tau mAb DA9 (1:5000), and the lower panel was incubated with A-actin, mAb (1:5000). Lane 1, naive Tau−/− mouse; lanes 2 to 9, chronic Tau−/− mice; lanes 10 to 12, chronic WT mice; lane 13, naive WT mouse. Clinical index (CI) for individual mice were as follows: lanes 2 and 3, CI = 3.5; lane 4, CI = 3; lane 5 and 6, CI = 2.5; lane 7, CI = 2; lanes 8 and 9, CI = 1; lanes 10 and 11, CI = 3.0; lane 12, CI = 1. The lane between lanes 9 and 10 is the molecular weight marker. (b) Quantification of microtubule-associated protein 1b (MAP1b). Relative densitometric intensity (RDI) was normalized to naive for WT and Tau−/−; MAP1b was significantly increased in Tau−/− mice versus WT mice (p < 0.05) (c) Tau−/− mice express less Gas7b during chronic EAE. Three naive Tau−/− mice (lanes 1–3) and 3 chronic Tau−/− mice (lanes 4–6) were compared with chronic WT mice (lanes 7, 8, 10) and a WT mouse injected with complete Freund’s adjuvant (CFA) and pertussis toxin (no myelin oligodendrocyte glycoprotein [MOG]) (lane 9). Two Gas7 bands were detected; Gas7b is the upper band migrating at approximately 50 kDa, and Gas7a is the lower band migrating at approximately 45 kDa. The CI for the mice were as follows: chronic Tau−/− lane 4, CI = 2; chronic Tau−/− lane 5, CI = 1.5; chronic Tau−/− lane 6, CI = 1; chronic WT lane 7, CI = 2.5; chronic WT lane 8, CI = 1; chronic WT lane 10, CI = 1. CFA without MOG plus pertussis toxin WT, lane 9, CI = 0. Gas7 mAb was diluted 1:400. The lane between lanes 6 and 7 is the molecular weight marker. Twenty micrograms of total protein was loaded in each lane in all blots.
Although we determined that, during the chronic phase, there were more SMI32-positive axons within the anterior column in Tau−/− versus WT mice, we did not detect increased expression by immunoblot analysis (Fig. 6c). This is likely caused by the abundance of nonphosphorylated neurofilament proteins normally expressed within neuronal cell bodies and dendrites. Lumbar spinal cord homogenates probed for SMI32 from 3 naive Tau−/− mice (lanes 1–3), 3 Tau−/− mice with chronic EAE (lanes 4–6), 3 chronic WT mice with chronic EAE (lanes 7, 8, 10), and a WT mouse that received CFA plus pertussis toxin but no MOG peptide (lane 9) are shown in Figure 6c. In addition, we examined Gas7, another protein predominantly expressed in neurons and associated with neuritogenesis and growth arrest. Two Gas7 isoforms, Gas7a and Gas7b, 45 kDa and 50 kDa, respectively, interact with the cytoskeleton during process outgrowth. Gas7a colocalizes with the actin network and physically binds to N-WASP to enhance neurite outgrowth. Gas7b binds to tau and enhances microtubule polymerization and neurite outgrowth. The physical association of Gas7b with tau stabilizes Gas7b; therefore, we examined whether Gas7b expression was altered in Tau−/− mice. As shown in Figure 6c, there was no detectable Gas7a (lower band) or Gas7b (upper band) in homogenates from naive Tau−/− mice (lanes 1–3); we detected faint Gas7a in WT mice sensitized with CFA (lane 9). As shown in lanes 4 to 6, Gas7a (lower band) was expressed at higher amounts than Gas7b in spinal cord homogenates from Tau−/− mice with chronic disease. By contrast, in protein homogenates isolated from chronic WT mice, most of the Gas7 expression was Gas7b, with minimal Gas7a detected. During chronic disease, the observed changes in cytoskeletal proteins MAP1b, Gas7a, and Gas7b in Tau−/− mice suggest a delay in recovery with altered expression of proteins associated with the cytoskeleton and neurite outgrowth. In combination with increased axonal damage early in disease in Tau−/− mice, these data suggest a protective role for tau in axons during EAE.
DISCUSSION
The Role of Tau in the CNS
The primary cause of disability in MS is the result of neuronal damage and loss. Because tau controls neuronal cytoskeletal dynamics and is the major MAP in the axon interacting with and fine tuning microtubule and actin dynamics, we investigated its role in neuroprotection in EAE. Tau is a major MAP in the vertebrate nervous system consisting of 6 low-molecular-weight isoforms and 110-kDa HMW isoforms with 3 or 4 repeats of the microtubule-binding domain (MTBD). All tau isoforms are generated by alternative splicing from a single gene. The 6 low-molecular-weight isoforms differ by having none, one, or two N-terminal insertions at the N-terminus of the protein as well as 3 or 4 repeats of the MTBD (14).
Several tau-deficient mouse strains develop normally, are healthy, and their fertility and viability are comparable to those of WT age-matched mice, suggesting that during development, there is redundancy and compensation by other MAPs. By contrast, abnormal tau hyperphosphorylation is a major biochemical and pathologic hallmark of AD and other tauopathies. Hyperphosphorylated tau results in tau dissociation from microtubules, with disruption of normal neuronal function, axonal transport, and synaptogenesis. Neurofibrillary tangles, the hallmark of AD, are composed of hyperphosphorylated tau. Although our study examined young adult hTau mice and the effect of tau overexpression during MOG-induced EAE, by 5 months of age, hTau mice have abnormal tau phosphorylation, with neurofibrillary tangles similar to that observed in AD (31).
Robertson et al (36) determined that reducing endogenous tau was beneficial and protected against excitotoxin-induced neuronal dysfunction in a mouse model of AD. Compared with WT mice, Tau−/− mice were resistant to seizures induced by intraperitoneal injection of kainate. Moreover, a transgenic mouse expressing APP with loss of tau expression ameliorated β-amyloid–induced behavioral deficits. However, the EAE model differs from the β-amyloid transgenic models in that there are no paired helical filament formations resulting in neurofibrillary tangles during EAE. Instead, there is breakdown of the blood-brain barrier, an influx of immune cells from the periphery, glial activation, demyelination, and axonal damage that requires a rapid response and remodeling of the axonal cytoskeleton.
In addition to binding to tubulin, tau has multiple binding partners that regulate cellular dynamics. Known tau binding/interacting partners include Fyn, Abl, protein phosphatase 2a, the prolylpeptidyl isomerases Pin1, FKBP-51, FKBP-52, Gas7b, and actin (17-26). While tau functions on microtubules to regulate neurite outgrowth, tau also affects actin dynamics, and independently binds to the neural plasma membrane via its amino-terminal projection domain (37-39).
Effects of Tau Overexpression in MOG-Induced EAE
Because inflammation and cytokine production may correlate with abnormal tau phosphorylation, we expected to observe increases in tau hyperphosphorylation during EAE and worsening of clinical scores in the hTau mice. However, tau overexpression and the higher level of tau phosphorylation versus controls were not associated with effects on clinical disease in hTau mice. Both WT and hTau mice sensitized with MOG33–55 had an approximately 2-fold increase in abnormal tau phosphorylation in lumbar spinal cord by immunoblotting, but there was no relative difference in the extent of phosphorylation in mice expressing hTau versus WT mice. The WT and hTau mice also succumbed to disease by the same day. Thus, the expression of tau with different numbers of MTBP repeats did not adversely affect disease severity during the course of the disease. The hTau mice express human tau with both 3 and 4 repeats of the MTBD, whereas WT mice express predominantly tau with 4 repeats of the MTBD. Whereas 4R tau is purported to enhance microtubule stability, hTau mice did not have higher mean clinical scores or greater axonal damage as assessed by SMI32-positive axonal staining. The pathologic loss of SMI31 immuno-reactivity in white matter correlates with axonal swellings that are dephosphorylated and immunoreactive for SMI32. The extent of axonal damage and spheroids induced in the hTau and WT mice during chronic EAE as assessed by SMI32 immunoreactivity was not increased in hTau versus WT spinal cord.
Examination of spinal cord sections with an antibody specific to HMW tau determined that axons were immuno-positive throughout the cord particularly in the substantia gelatinosa of Rolando. This region, located at the dorsal surface of the dorsal column, extends the entire length of the spinal cord into the medulla oblongata. This region functions in the transmission of sensory pain information and pain management caused by injury to the spinal cord, peripheral nerve lesions, and inflammation associated with MS (40). The substantia gelatinosa contains small nerve cells with unmyelinated axons and few thinly myelinated fibers and does not stain for phosphorylated neurofilament proteins. The paucity of myelinated fibers is consistent with the finding that phosphorylated neurofilament expression correlates with myelination, and expression of abundant HMW tau in the substantia gelatinosa of Rolando suggests that HMW tau may stabilize these small axons.
EAE in Tau−/− Mice
Tau−/− mice are relatively normal during neurodevelopment and have uncompromised viability, but in EAE, the absence of tau negatively affected the clinical course and axonal integrity of these mice compared with WT mice. Spinal cord lesions from Tau−/− mice had more SMI32-positive and APP-positive axons than were observed in WT spinal cord. Paraplegic Tau−/− mice with clinical scores of 4.0 had gray matter abnormalities consisting of APP-positive and SMI32-positive axons surrounded by Iba1-positive glia that were not observed in the WT mice.
We considered the possibility that the loss of tau during EAE would induce the reexpression of MAPs expressed during early development to compensate for the loss of tau. MAP1b is the most abundant and earliest MAP expressed in neurons and oligodendrocytes in the developing nervous system (41-43). In neurons, MAP1b is downregulated during maturation when MAP1a is upregulated. In oligodendrocytes, MAP1b expression precedes morphological differentiation (44). MAP1b binds to microtubules and actin in a phosphorylation-dependent manner, where it functions in growing axons and is the major MAP associated with microtubules early in development. The expression of MAP1b is down-regulated 10-fold in maturing myelinating brain. In EAE, the increased MAP1b expression observed in the spinal cords of Tau−/− mice likely was the result of immature oligodendrocytes, and compromised axons undergoing axonal outgrowth and elongation. The upregulation of MAP1b is likely protective and aids in process stability and outgrowth caused by the loss of tau. Based on the higher clinical scores during acute and chronic EAE and the continued expression of MAP1b in Tau−/− mice during the chronic phase of disease, we suggest that axonal repair is ongoing in the Tau−/− mice. In addition, we considered how the loss of tau expression might influence tau protein interactions. Gas7b binds Tau, and a Gas7b/Tau complex interacts with the microtubules to promote tubulin polymerization (25, 45). Studies determined that tau stabilizes Gas7b and that the brains from patients with AD had decreased levels of Gas7b. Other studies showed that Gas7b has a role in microtubule organization (45). We did not detect Gas7b expression in naive WT and Tau−/− mice, but in mice with chronic EAE, we observed more Gas7b expression in WT spinal cord homogenates than that in Tau−/− spinal cord homogenates. Although all Gas7b values for WT homogenates were higher than the values for the Tau−/− homogenates, the differences were not significant because of the high standard deviation resulting from 1 WT homogenate with high Gas7b values. Our data are consistent with a previous report demonstrating that tau stabilizes Gas7b and loss of tau reduces Gas7b expression in the CNS (25). Although we did not show a direct interaction between tau and Gas7b in axonal cytoskeleton stabilization, it is likely that the reduced expression of Gas7b combined with increased MAP1b expression and increased SMI32-positive and APP-positive immunoreactivity all contribute to the delayed recovery in spinal cord of Tau−/− mice during EAE. In addition, the increase in Gas7a in Tau−/− is suggestive of neurite outgrowth as Gas7a colocalizes with the actin network and, by physically binding to N-WASP, enhances neurite outgrowth (46).
In summary, the combined data show that Tau−/− mice have significantly higher clinical scores, more axonal damage early in disease, and delayed recovery detected by changes in the cytoskeleton during chronic disease. The higher mean clinical scores and corresponding increase in MAP1b and Gas7a and decrease in Gas7b in Tau−/− mice suggest that tau may help limit axon lesion size during EAE either directly or in combination with microtubule-associated binding partners.
Acknowledgments
We thank Drs Karen Duff and Peter Davies for the hTau and Tau−/− mice. We thank Dr Itzak Fischer Department of Neurobiology and Anatomy, Drexel University College of Medicine, Philadelphia, PA, for providing us with multiple bleeds of his HMW Big Tau antibodies. We thank Ms Lauren Bayer for her help with statistical analysis. Ashrei Bayewitz and Lauren Bayer were participants in the summer undergraduate research program at Albert Einstein College of Medicine. We thank Dr Cedric Raine for helpful discussions.
This work was supported by Grant No. RG4046-A-6 from the National Multiple Sclerosis Society and Grant No. MH038623-27 from the National Institutes of Health.
References
- 1.Alvarez JI, Cayrol R, Prat A. Disruption of central nervous system barriers in multiple sclerosis. Biochim Biophys Acta. 2011;1812:252–64. doi: 10.1016/j.bbadis.2010.06.017. [DOI] [PubMed] [Google Scholar]
- 2.Barnett MH, Parratt JD, Pollard JD, et al. MS: Is it one disease? Int MS J. 2009;16:57–65. [PubMed] [Google Scholar]
- 3.Frohman EM, Racke MK, Raine CS. Multiple sclerosis—The plaque and its pathogenesis. N Engl J Med. 2006;354:942–55. doi: 10.1056/NEJMra052130. [DOI] [PubMed] [Google Scholar]
- 4.Davie CA, Barker GJ, Webb S, et al. Persistent functional deficit in multiple sclerosis and autosomal dominant cerebellar ataxia is associated with axon loss. Brain. 1995;118:1583–92. doi: 10.1093/brain/118.6.1583. [DOI] [PubMed] [Google Scholar]
- 5.De Stefano N, Matthews PM, Fu L, et al. Axonal damage correlates with disability in patients with relapsing-remitting multiple sclerosis. Results of a longitudinal magnetic resonance spectroscopy study. Brain. 1998;21:1469–77. doi: 10.1093/brain/121.8.1469. [DOI] [PubMed] [Google Scholar]
- 6.Kornek B, Lassmann H. Axonal pathology in multiple sclerosis. A historical note. Brain Pathol. 1999;9:651–56. doi: 10.1111/j.1750-3639.1999.tb00547.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferguson B, Matyszak MK, Esiri MM, et al. Axonal damage in acute multiple sclerosis lesions. Brain. 1997;120:393–99. doi: 10.1093/brain/120.3.393. [DOI] [PubMed] [Google Scholar]
- 8.Trapp BD, Peterson J, Ransohoff RM, et al. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–85. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- 9.Bitsch A, Schuchardt J, Bunkowski S, et al. Acute axonal injury in multiple sclerosis. Correlation with demyelination and inflammation. Brain. 2000;123:117–83. doi: 10.1093/brain/123.6.1174. [DOI] [PubMed] [Google Scholar]
- 10.Compston A. Making progress on the natural history of multiple sclerosis. Brain. 2006;129:561–63. doi: 10.1093/brain/awl034. [DOI] [PubMed] [Google Scholar]
- 11.O’Neill JK, Baker D, Morris MM, et al. Optic neuritis in chronic relapsing experimental allergic encephalomyelitis in Biozzi ABH mice: Demyelination and fast axonal transport changes in disease. J Neuroimmunol. 1998;82:210–18. doi: 10.1016/s0165-5728(97)00203-8. [DOI] [PubMed] [Google Scholar]
- 12.Aboul-Enein F, Weiser P, Hoftberger R, et al. Transient axonal injury in the absence of demyelination: A correlate of clinical disease in acute experimental autoimmune encephalomyelitis. Acta Neuropathol. 2006;111:539–47. doi: 10.1007/s00401-006-0047-y. [DOI] [PubMed] [Google Scholar]
- 13.Shriver LP, Dittel BN. T cell–mediated disruption of the neuronal microtubule network: Correlation with early reversible axonal dysfunction in acute experimental autoimmune encephalomyelitis. Am J Pathol. 2006;169:999–1011. doi: 10.2353/ajpath.2006.050791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goedert M. Tau protein and the neurofibrillary pathology of Alzheimer’s disease. Trends Neurosci. 1993;16:460–65. doi: 10.1016/0166-2236(93)90078-z. [DOI] [PubMed] [Google Scholar]
- 15.Pluchino S, Quattrini A, Brambilla E, et al. Injection of adult neurospheres induces recovery in a chronic model of multiple sclerosis. Nature. 2003;422:688–94. doi: 10.1038/nature01552. [DOI] [PubMed] [Google Scholar]
- 16.Kempf M, Clement A, Faissner A, et al. Tau binds to the distal axon early in development of polarity in a microtubule- and microfilament-dependent manner. J Neurosci. 1996;16:5583–92. doi: 10.1523/JNEUROSCI.16-18-05583.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Magnani E, Fan J, Gasparini L, et al. Interaction of tau protein with the dynactin complex. EMBO J. 2007;26:4546–54. doi: 10.1038/sj.emboj.7601878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sontag E, Nunbhakdi-Craig V, Lee G, et al. Regulation of the phosphorylation state and microtubule-binding activity of Tau by protein phosphatase 2A. Neuron. 1996;17:1201–7. doi: 10.1016/s0896-6273(00)80250-0. [DOI] [PubMed] [Google Scholar]
- 19.Lee G, Newman ST, Gard DL, et al. Tau interacts with src-family non-receptor tyrosine kinases. J Cell Sci. 1998;111:3167–77. doi: 10.1242/jcs.111.21.3167. [DOI] [PubMed] [Google Scholar]
- 20.Lu PJ, Wulf G, Zhou XZ, et al. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature. 1999;399:784–88. doi: 10.1038/21650. [DOI] [PubMed] [Google Scholar]
- 21.Lim J, Lu KP. Pinning down phosphorylated tau and tauopathies. Biochim Biophys Acta. 2005;1739:311–22. doi: 10.1016/j.bbadis.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 22.Liou YC, Sun A, Ryo A, et al. Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature. 2003;424:556–61. doi: 10.1038/nature01832. [DOI] [PubMed] [Google Scholar]
- 23.Borysov SI, Schmid AB, Abisambra JF, et al. The Hsp90 cochaperone, FKBP51, increases Tau stability and polymerizes microtubules. J Neurosci. 2010;30:591–9. doi: 10.1523/JNEUROSCI.4815-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chambraud B, Sardin E, Giustiniani J, et al. A role for FKBP52 in Tau protein function. Proc Natl Acad Sci USA. 2010;107:2658–63. doi: 10.1073/pnas.0914957107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akiyama H, Gotoh A, Shin RW, et al. A novel role for hGas7b in microtubular maintenance: Possible implication in tau-associated pathology in Alzheimer disease. J Biol Chem. 2009;284:32695–99. doi: 10.1074/jbc.M109.035998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schlatterer SD, Tremblay MA, Acker CM, et al. c-Abl in neuro-degenerative disease. J Mol Neurosci. 2011;25:119–33. doi: 10.3233/JAD-2011-102025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schneider A, Araújo GW, Trajkovic K, et al. Hyperphosphorylation and aggregation of tau in experimental autoimmune encephalomyelitis. J Biol Chem. 2004;279:55833–39. doi: 10.1074/jbc.M409954200. [DOI] [PubMed] [Google Scholar]
- 28.Anderson JM, Hampton JM, Patani R, et al. Abnormally phosphorylated tau is associated with neuronal and axonal loss in experimental auto-immune encephalomyelitis and multiple sclerosis. Brain. 2008;131:1736–48. doi: 10.1093/brain/awn119. [DOI] [PubMed] [Google Scholar]
- 29.Anderson JM, Patani R, Reynolds R, et al. Evidence for abnormal tau phosphorylation in early aggressive multiple sclerosis. Acta Neuropathol. 2009;117:583–89. doi: 10.1007/s00401-009-0515-2. [DOI] [PubMed] [Google Scholar]
- 30.Tucker KL, Meyer M, Barde YA. Neurotrophins are required for nerve growth during development. Nat Neurosci. 2001;4:29–37. doi: 10.1038/82868. [DOI] [PubMed] [Google Scholar]
- 31.Andorfer C, Kress Y, Espinoza M, et al. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem. 2003;86:582–90. doi: 10.1046/j.1471-4159.2003.01879.x. [DOI] [PubMed] [Google Scholar]
- 32.Duff K, Knight H, Refolo LM, et al. Characterization of pathology in transgenic mice overexpressing human genomic and cDNA tau transgenes. Neurobiol Dis. 2000;7:87–98. doi: 10.1006/nbdi.1999.0279. [DOI] [PubMed] [Google Scholar]
- 33.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–85. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 34.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–54. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boyne LJ, Tessler A, Murray M, et al. Distribution of Big tau in the central nervous system of the adult and developing rat. J Comp Neurol. 1995;358:279–93. doi: 10.1002/cne.903580209. [DOI] [PubMed] [Google Scholar]
- 36.Roberson ED, Scearce-Levie K, Palop JJ, et al. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316:750–54. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 37.Sharma VM, Litersky JM, Bhaskar K, et al. Tau impacts on growth factor–stimulated actin remodeling. J Cell Sci. 2007;120:748–57. doi: 10.1242/jcs.03378. [DOI] [PubMed] [Google Scholar]
- 38.Leugers CJ, Lee G. Tau potentiates nerve growth factor-induced mitogen-activated protein kinase signaling and neurite initiation without a requirement for microtubule binding. J Biol Chem. 2010;285:19125–34. doi: 10.1074/jbc.M110.105387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brandt R, Léger J, Lee G. Interaction of tau with the neural plasma membrane mediated by tau’s amino-terminal projection domain. J Cell Biol. 1995;131:1327–40. doi: 10.1083/jcb.131.5.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hantman AW, van den Pol AN, Perl ER. Morphological and physiological features of a set of spinal substantia gelatinosa neurons defined by green fluorescent protein expression. J Neurosci. 2004;24:836–42. doi: 10.1523/JNEUROSCI.4221-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tucker RP, Matus A. Microtubule-associated proteins characteristic of embryonic brain are found in the adult mammalian retina. Dev Biol. 1998;130:423–34. doi: 10.1016/0012-1606(88)90338-7. [DOI] [PubMed] [Google Scholar]
- 42.Tucker RP. The roles of MAPs in brain morphognesis: A review. Brain Res Brain Res Rev. 1990;15:101–20. doi: 10.1016/0165-0173(90)90013-e. [DOI] [PubMed] [Google Scholar]
- 43.Tint I, Fischer I, Black M. Acute inactivation of MAP1b in growing sympathetic neurons destabilizes axonal microtubules. Cell Motil Cytoskeleton. 2005;60:48–65. doi: 10.1002/cm.20045. [DOI] [PubMed] [Google Scholar]
- 44.Vouyiouklis DA, Brophy PJ. MAP-1b expression precedes the morphological differentiation of oligodendrocytes. J Neurosci Res. 1993;35:257–67. doi: 10.1002/jnr.490350305. [DOI] [PubMed] [Google Scholar]
- 45.Uchida T, Akiyama H, Sakamoto W, et al. Direct optical microscopic observation of the microtubule polymerization intermediate sheet structure in the presence of gas7. J Mol Biol. 2009;391:849–57. doi: 10.1016/j.jmb.2009.06.081. [DOI] [PubMed] [Google Scholar]
- 46.You JJ, Lin-Chao S. Gas7 functions with N-WASP to regulate the enurite outgrowth of hippocampal neurons. J Biol Chem. 2010;285:11652–66. doi: 10.1074/jbc.M109.051094. [DOI] [PMC free article] [PubMed] [Google Scholar]