

Abstract
Cerebellar Purkinje cells (PCs) encode afferent information in the rate and temporal structure of their spike trains. Both spontaneous firing in these neurons and its modulation by synaptic inputs depend on Ca2+ current carried by Cav2.1 (P/Q) type channels. Previous studies have described how loss-of-function Cav2.1 mutations affect intrinsic excitability and excitatory transmission in PCs. This study examines the effects of the leaner mutation on fast GABAergic transmission and its modulation of spontaneous firing in PCs. The leaner mutation enhances spontaneous synaptic inhibition of PCs, leading to transitory reductions in PC firing rate and increased spike rate variability. Enhanced inhibition is paralleled by an increase in the frequency and amplitude of spontaneous inhibitory postsynaptic currents (sIPSCs) measured under voltage clamp. These differences are abolished by tetrodotoxin, implicating effects of the mutation on spike-induced GABA release. Elevated sIPSC frequency in leaner PCs is not accompanied by increased mean firing rate in molecular layer interneurons, but IPSCs evoked in PCs by direct stimulation of these neurons exhibit larger amplitude, slower decay rate, and a higher burst probability compared to wild-type PCs. Ca2+ release from internal stores appears to be required for enhanced inhibition since differences in sIPSC frequency and amplitude in leaner and wild-type PCs are abolished by thapsigargin, an ER Ca2+ pump inhibitor. These findings represent the first account of the functional consequences of a loss-of-function P/Q channel mutation on PC firing properties through altered GABAergic transmission. Gain in synaptic inhibition shown here would compromise the fidelity of information coding in these neurons and may contribute to impaired cerebellar function resulting from loss-of function mutations in the CaV2.1 channel gene.
Keywords: P/Q-type Ca2+ channels, Ca2+ channelopathies, Cerebellum, Ataxia, Purkinje neurons, Leaner mutation
Introduction
Cerebellar Purkinje cells (PCs) are spontaneously active and encode afferent information through time-dependent synaptic modulation of their intrinsic firing rate. These cells communicate executive commands to various levels of the neural axis and utilize coordination and timing information to refine motor functions [1]. Over 175,000 excitatory and inhibitory synapses relay multimodal afferent signals to each PC, shaping intrinsically generated regular firing of these neurons into a highly structured efferent code [2–4]. The extensive synaptic modulation of PC firing rate along with the key network position and the complex membrane voltage dynamics of these cells [5–8] are suggestive of multifaceted integrative mechanisms for information processing. Yet, despite the well-characterized synaptic inputs and intrinsic properties of PCs, it remains unclear how time-related signals and motor coordination commands are represented in the rate and firing pattern of these neurons.
The required accuracy of temporal information coding in the output of PCs appears to be high as the cerebellum participates in the control of fine movement [9–11]. Damage and disorders to this brain region lead to a breakdown in the timing of muscular events with increased temporal variability in motor function and ataxia [12–16]. Earlier theoretical and experimental studies have discussed the representation of timing information in PC output primarily in relation to excitatory synaptic inputs [17–19]. More recently, however, the significance of intrinsic pacemaker mechanisms and inhibitory synaptic inputs in determining how temporal information is encoded within the temporal structure of PC spike trains has been recognized [3, 7, 20, 21]. A direct link between cerebellar motor deficits and altered temporal properties of PC spiking has been shown recently in several strains of CaV2.1 channel mutant mice [22–24]. Regarding the effects of these mutations that might contribute to deficits in cerebellar function, recent studies [23, 25–29] have revealed that P/Q-type calcium channel mutations significantly alter excitatory synaptic inputs and intrinsic excitability of PCs. However, little information is available as to how these mutations affect synaptic inhibition and modulation of PC output by inhibitory inputs.
In the present study, we compared fast GABAergic synaptic currents and their impact on the PC efferent code in cerebellar slices from mice expressing the leaner CaV2.1 mutation with age-matched wild-type controls. The leaner mutation leads to a ~60% reduction of whole cell Ca2+ currents in dissociated PCs [30–32]. Given the powerful GABAergic synaptic inputs received by PCs from local interneurons and recurrent PC collaterals [33–37] and the dominant role of P/Q-type calcium currents in triggering inhibitory neurotransmitter release at inhibitory synaptic inputs [33–39], attenuation of the inhibitory drive and GABAergic modulation of PC output was expected. However, we found that inhibitory synaptic drive in leaner PCs is enhanced, leading to pronounced changes in the timing of spontaneous action potentials in these neurons. This anomalous gain in inhibitory synaptic drive would be expected to reduce the fidelity of temporal information encoding in PCs and may contribute to impaired cerebellar function in loss-of-function CaV2.1 channel mutant mice.
Methods
Animals and Slice Preparation
C57BL/6J wild-type (++/++) and mice heterozygous for the leaner mutation (Os+/+tgla) with the same background were purchased from Jackson Laboratory (Bar Harbor, ME) and maintained at the Animal Resource Center at Case Western Reserve University. Animals were provided with food and water ad libitum and kept at 22±2°C with a 12/12-h light/dark cycle. Mice that were homozygous for the tgla allele (+tgla/+tgla, referred in this study as leaner) were obtained by brother–sister mating of the mixed heterozygotes (Os+/+tgla) as described previously [40]. Since Os+/Os+ homozygousmice die early in embryogenesis [41], only mice with Os+/+tgla and tgla+/tgla+ genotypes are born. tgla+/tgla+ mice were distinguished from their Os+/+tgla littermates based on their normal paw structure and motor disability, which is evident by roughly postnatal day 10 (P10).
Experimental procedures conformed to guidelines approved by the Institutional Animal Care and Use Committee at Case Western Reserve University. Mice (P16–21) were deeply anesthetized with ketamine (150 mg/kg) and killed by decapitation. The cerebellum was rapidly removed and placed in ice-cold bubbled (95% O2, 5% CO2) low Na+, low Ca2+, high Mg2+ artificial cerebrospinal fluid (ACSF) with a composition (in mM): 75 sucrose, 85 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 0.5 CaCl2, 4 MgCl2, 25 glucose. Sagittal slices (300 µm thick) were cut from the cerebellar vermis on a Vibrotome Series 1,000 (St. Louis, MO) in the same medium and then transferred to a solution with the same composition except that sucrose was omitted and the concentration of NaCl was increased to 125 mM, followed by incubation for 30 min at 32°C with continuous bubbling (95% O2, 5% CO2). Slices were then transferred to ACSF (mM): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2 CaCl2, 2 MgCl2, and 25 glucose, where they were kept at room temperature with continuous bubbling until used.
Electrophysiology
Individual slices were transferred to a recording chamber fixed to the stage of an upright microscope (Leica DM-LFSA) where they were superfused with bubbled ACSF at a rate of ~2–3 ml/min throughout the experiment. Whole cell current- and voltage-clamp and loose seal cell-attached patch recordings were made at 32– 34°C from PCs visually identified under IR-DIC based on their location, size, and morphology. Molecular layer interneurons (basket/stellate cells) were identified visually within the cerebellar cortex for cell-attached patch recordings based on their location, distinct morphology (smaller non-polarized soma and multiple primary dendrites), and lower spontaneous firing rate compared to PCs [42]. Interneurons within the innermost one third of the molecular layer (<50 µm from PC layer) were identified as basket cells [42] and were stimulated to elicit IPSCs that were recorded in PCs under voltage clamp (see below).
All current-clamp recordings were made with an Axoclamp 2B (Axon Instruments, Foster City, CA) using patch pipettes filled with a K-methyl sulfate-based internal solution containing (mM): 140 KCH3O3S, 5 KCl, 5 NaCl, 2 MgATP, 0.01 EGTA, and 10 HEPES, pH 7.3, with a resistance of 3–7 MΩ. Voltage-clamp recordings of spontaneous and evoked IPSCs were made using an Axopatch 200A (Axon Instruments) with symmetrical chloride concentration using a cesium chloride-based internal solution (mM): 150 CsCl, 2 MgCl2, 0.01 EGTA, 10 HEPES, 0.4 NaGTP, and 4 NaATP, pH 7.3, supplemented with 10 mM QX-314. During voltage-clamp recording, the broad-spectrum ionotropic glutamate receptor antagonist kynurenic acid (5 mM) was added to the extracellular recording solution to block fast excitatory synaptic transmission [43]. Capacitance and series resistance compensation was carried out as described previously [28]. Membrane voltage was not corrected for liquid junction potentials. Miniature IPSCs (mIPSCs) were recorded in the presence of kynurenic acid (5 mM) and TTX (1 µM).
Evoked IPSCs (eIPSCs) were elicited in PCs by direct extracellular stimulation of basket cells. After establishing a stable whole cell recording in a given PC, inhibitory inputs from nearby interneurons from the inner third of the molecular layer, identified as basket neurons, were examined with juxtasomatic stimulation using rectangular current pulses with amplitude 30–50 µA and duration 200 µs generated with a stimulus isolator (A-360, WPI) under computer control. After identifying a synaptically connected basket cell/PC pair, the stimulus intensity was adjusted to elicit eIPSCs whose trial-to-trial variability was insensitive to small changes in stimulus strength. In this case, we concluded that the stimuli consistently elicited presynaptic action potentials, with trial-to-trial variability in the measured eIPSCs arising from stochastic properties of release, not AP generation (e.g. see [44]). Monosynaptic IPSCs evoked by single-pulse stimulation were identified based on their short and stable synaptic delay (<4 ms) and insensitivity to small changes in stimulus intensity. Multi-component eIPSCs were defined as eIPSCs in which the first IPSC peak was followed by one or more peaks with inter-event intervals (IEIs) ≤12 ms. This interval was chosen in an effort to characterize secondary and higher order IPSCs that appeared to be correlated with the initial peak. However, this analysis may include some uncorrelated spontaneous events, particularly in cases where the spontaneous IPSC rate is high, and exclude some correlated events with especially large IEIs.
The cell-attached patch recording configuration was established by positioning the patch pipette in close proximity to the neuronal (PC, basket cell, stellate cell) soma followed by application of slight negative pressure to the electrode, which facilitated loose seal formation (<50 MΩ). For these recordings, patch pipettes were filled with external solution, except when comparing PC firing under cell-attached and whole cell conditions where pipettes contained internal solution (see Electronic Supplementary Material (ESM) Fig. 1).
Data Acquisition and Analysis
Analog signals were filtered at 5 kHz, digitized at 10–20 kHz and analyzed off-line using a software written in IgorPro (Wavemetrics, Lake Oswego, OR). Spikes were detected based on a threshold crossing routine, with spike times defined based on the time at which membrane voltage reached a maximum between successive threshold crossings. Inter-spike intervals (ISI) were calculated as the difference between successive spike times. Mean spike frequency (F) was defined as the number of spikes per second. The ISI coefficient of variation (CV) was defined as the ratio of the standard deviation to the mean of the tabulated ISIs.
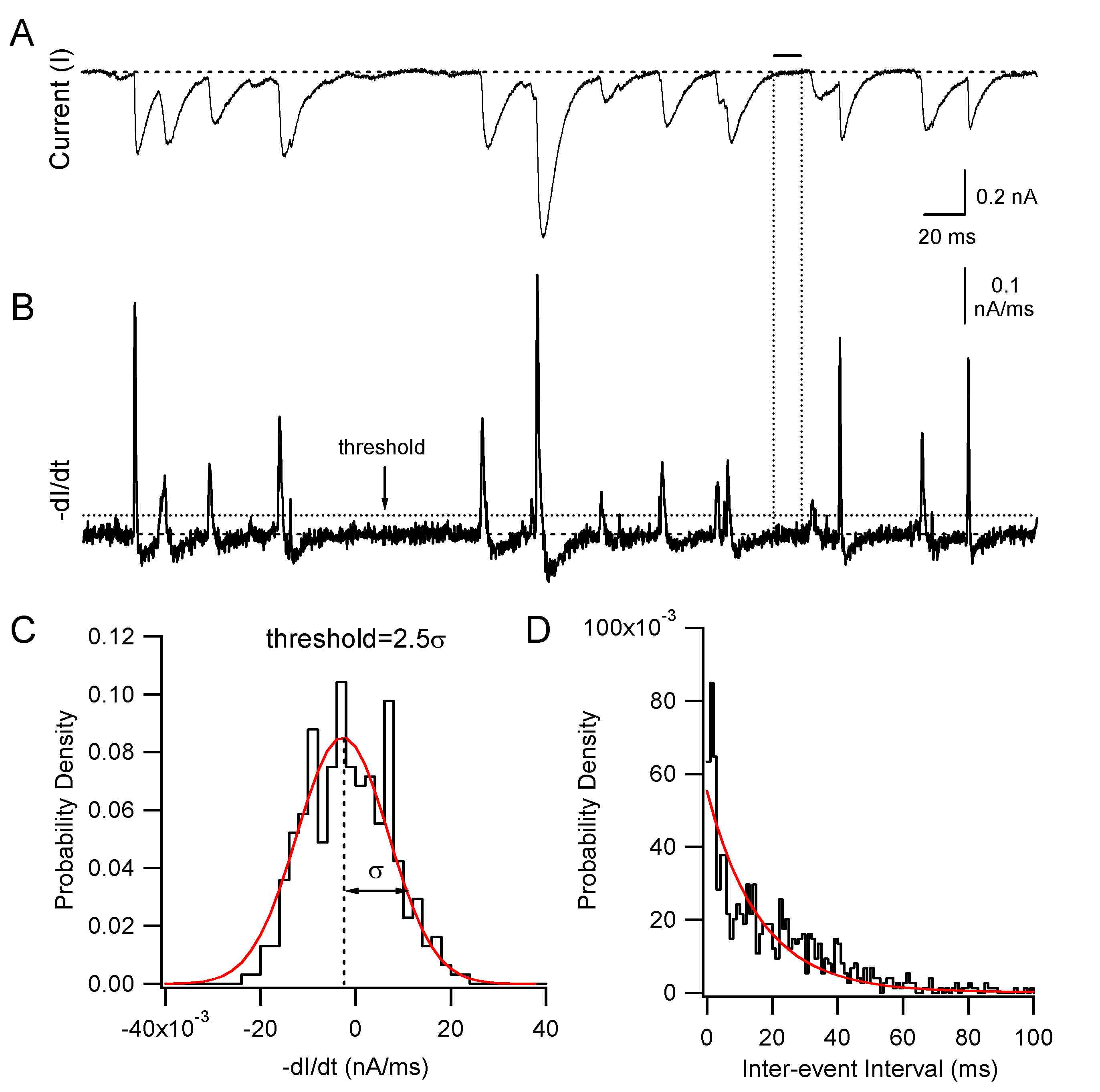
To detect spontaneous synaptic currents recorded under voltage clamp, a method was used that is illustrated in ESM Fig. 2. Raw currents (ESM Fig. 2a) were smoothed (×20, binomial filter), digitally differentiated, and inverted (ESM Fig. 2b). The times at which the inverted derivative exceeded a threshold of 2.5 times the standard deviation of the same quantity measured between synaptic events were then determined; these times corresponded to the rising phase of individual spontaneous IPSCs (sIPSCs). This approach made it possible to detect the vast majority of events that could be identified visually as synaptic currents (ESM Fig. 2a, b) and could be blocked by picrotoxin (see “Results”). After identifying synaptic events, IPSC peaks were located where the current reached a maximum following the threshold crossing of the first derivative. The first current minimum preceding the peak was used to define the pre-IPSC trough, and IPSC amplitude was calculated as the difference between the current peak and trough. IPSC frequency statistics were determined with the same methods used for spike frequency analysis (see above). Inter-event interval distributions were constructed as shown in ESM Fig. 2d. IPSC bursts expected to have an especially pronounced effect on PC spike timing were operationally defined as sequences of three or more IPSCs in which consecutive peaks were spaced by ≤12 ms. Data are reported as mean ± SE and statistical significance is assessed using paired or unpaired Student’s t test, with P<0.05 defining a significant difference.
Chemicals and Drugs
All chemicals and drugs were obtained from Sigma (St. Louis, MO).
Results
Spontaneous Synaptic Inhibition Transiently Reduces Firing Frequency and Increases Spike Rate Variability in Leaner PCs
Previous studies have shown that cerebellar PCs are spontaneously active and that the rate of firing is modulated by fast excitatory and inhibitory synaptic inputs [3]. Given the role of P/Q type Ca2+ channels in regulating intrinsic firing and synaptic inputs to these cells, loss-of-function mutations in P/Q Ca2+ channels could potentially affect both processes. Walter et al. [23] showed that the leaner mutation increases the temporal variability of intrinsic spike firing in PCs, quantified based on the CV of ISI. These measurements were made after blocking fast excitatory and inhibitory synaptic transmission, representing effects of the mutation on intrinsic spike rate variability. To investigate the impact of the mutation on extrinsic contributions to spike rate variability arising from spontaneous synaptic inputs, we compared firing in leaner and wild-type (WT) PCs in the absence of synaptic transmission blockers. Given that WT and leaner PCs show more than one pattern of spontaneous activity [45–47], for simplicity, we restrict analysis here to tonically firing cells.
Figure 1 (top) shows representative recordings of firing in WT (panel a) and leaner (panel b) PCs in the absence of synaptic transmission blockers. The main difference we observed under these conditions is that leaner PCs have a lower firing rate (F) and higher coefficient of variability of interspike intervals (CV) than WT cells: (WT: F=41.4±3.2 Hz; CV=0.08±0.01, n=19; leaner: F=26.3±2.6 Hz; CV=0.35±0.06, n=16). The differences in F and CV between leaner and WT PCs were both significant (P<0.05, unpaired t test). These differences are reflected in the broader and more skewed ISI distributions and less periodic autocorrelograms in leaner compared to WT PCs (see panels a and b, middle and bottom). Distinct hyperpolarizing events during prolonged interspike intervals in leaner PCs (panel b, asterisks) suggest the involvement of inhibitory synaptic transmission. To test this, the effect of picrotoxin (200 µM) on spontaneous firing in leaner and WT PCs was examined and compared (Fig. 1c, d). The combined effect of picrotoxin and kynurenic acid (5 mM) on spontaneous firing was also compared between the two cell groups; since no difference was found between the effects of picrotoxin alone and the effects of picrotoxin and kynurenic acid applied together (results not shown), measurements made under the two pharmacological conditions were pooled. Blockade of inhibitory synaptic transmission significantly reduced CV in leaner PCs to 0.19±0.03 (n=12) compared to the same parameter measured prior to drug application (P<0.03, paired t test). Following blockade of inhibitory synaptic transmission, CV in leaner PCs more closely resembled that measured under the same conditions in WT cells (CV=0.08±0.01, N=13), but was still significantly different from WT (P<0.05). These observations indicate that in addition to the previously described effect of the leaner mutation on intrinsic spike rate variability in PCs, there is a stronger extrinsic contribution to variability that is sensitive to picrotoxin, presumably reflecting changes in the spontaneous inhibitory synaptic inputs received by PCs.
Fig. 1.

Effects of spontaneous synaptic inputs on tonic firing in WT and leaner PCs. a, b Top Representative recordings of spontaneous tonic firing in wild-type and leaner PCs (no injected current). Spike-free gaps of variable duration in leaner PCs accompanied by spontaneous IPSPs (see b, asterisks) increase ISI variability, leading to a broader ISI distribution (middle) and reduced periodicity in the ISI autocorrelogram (bottom). c, d Top Spontaneous spike trains recorded in the same cells in the presence of picrotoxin (200 µM), which greatly reduced CV compared to control, with corresponding modifications of the ISI distribution and autocorrelagram. Note the more pronounced effect of picrotoxin in leaner (b, d) compared to WT (a, c) PCs
Comparison of Spontaneous IPSCs in WT and Leaner PCs
To investigate the basis of enhanced inhibitory modulation of spontaneous firing in leaner PCs, we compared sIPSCs in PCs under voltage clamp. sIPSCs were defined as synaptic currents observed in the presence of kynurenic acid (5 mM). Inhibitory synaptic currents were recorded using symmetrical chloride concentrations at a holding potential of −75 mV, where Cl− currents are inward (Fig. 2a, b; see “Methods”). The average amplitude of sIPSCs varied linearly with holding potential and reversed near the chloride equilibrium potential (Fig. 2a, b), consistent with the expected properties of current carried by GABA-sensitive Cl− channels. Blockade of inhibitory synaptic transmission with picrotoxin (200 µM, WT: n=5, leaner: n=6; Fig. 2c, d) or bicuculline (20 µM, leaner: n=4, not shown) greatly reduced spontaneous synaptic activity in both PC groups (Fig. 2c, d, compare upper and lower traces). In contrast, blockade of excitatory transmission with kynurenic acid alone had little or no effect on either the mean amplitude or frequency of spontaneous synaptic currents in WT or leaner PCs (results not shown). Analysis of sIPSCs in the presence of kynurenic acid indicates that sIPSCs occur at higher frequency in leaner PCs (leaner, 50.3±7.0 Hz, n=6; WT, 28.6±4.7 Hz, n=6, P<0.05, unpaired t test) and have larger amplitude (leaner, 107.6±13.8 pA, n=6; WT, 42.6±7.0 pA, n=6, P<0.05, unpaired t test) compared to WT controls. Figure 2e, f compares normalized distributions of sIPSC amplitude from the same WT and leaner cells illustrated in panels c and d. Figure 2g, h compares IEI distributions in the two cell types, illustrating the finding that IEI distributions can be approximated by single exponential functions. We also characterized sIPSC bursts, defined as sequences of three or more consecutive unitary IPSCs separated by <12 ms (see “Methods”). sIPSC bursts were investigated because they would be expected to have especially pronounced effects on PC firing rate compared to unitary sIPSCs. In addition, the properties of sIPSC bursts can be used to address whether they are a cause, or consequence, of the overall increase in sIPSC frequency observed in leaner PCs (see “Discussion”). Two examples of such bursts from the recordings in Fig. 2d (see 1 and 2) are illustrated on an expanded scale in Fig. 2f (see inset). We found that burst frequency is higher in leaner PCs (2.6±0.8 Hz, n=6) than in WT cells (0.5±0.2 Hz, n=6, P<0.05, unpaired t test). On average, sIPSC bursts consisted of a comparable number of unitary events in WT and mutant PCs (WT, 4.3±0.1; leaner, 4.5±0.2). Effects of the leaner mutation on sIPSC frequency, mean amplitude, and burst frequency are summarized in Fig. 2i. These results demonstrate that compared to WT PCs, leaner PCs receive stronger spontaneous inhibitory synaptic input, characterized by higher mean frequency and amplitude of sIPSCs, along with an increased probability of sIPSC bursts.
Fig. 2.

Spontaneous inhibitory synaptic currents in leaner PCs are more frequent and have larger amplitude than in wild-type cells. a, b Voltage-clamp recordings of sIPSCs in the presence of kynurenic acid at different holding potentials. Mean sIPSC amplitude varies linearly with holding potential and reverses near ECl. c, d Comparison between sIPSCs in WT and leaner PCs under control conditions (upper set of three traces) and in the presence of picrotoxin (200 µM, lower traces). Picrotoxin abolishes the majority of the spontaneous postsynaptic currents. e, f Normalized sIPSC amplitude histograms from the cells shown in c, d. Inset to f shows two examples of sIPSC bursts (from first and second traces of d) on expanded scale. g, h Normalized interevent interval (IEI) histograms from the cells are shown in c, d. Smooth traces show exponential fits to the respective distributions. i Summary data comparing sIPSC frequency, amplitude, and burst frequency in WT and leaner PCs. *P<0.05
TTX Abolishes Differences Between sIPSCs in Leaner and WT PCs
To investigate the basis for enhanced synaptic inhibition in leaner PCs, it was asked whether enhanced inhibition is due to an increase in the number of inhibitory synaptic inputs and/or the postsynaptic sensitivity to these inputs. For this purpose, we compared mIPSCs in WT and leaner PCs in the presence of kynurenic acid (5 mM) and TTX (1 µ;M). TTX treatment reduced the frequency of spontaneous inhibitory synaptic currents in both WT and leaner PCs (Fig. 3, compare panels a, c and b, d), suggesting that many of the sIPSCs are triggered by presynaptic TTX-sensitive action potentials. Moreover, comparing mIPSCs between the two groups (eight leaner and seven WT cells) revealed little or no difference, either in terms of their amplitude (wild type, 31.8±5.3 pA; leaner, 29.1±4.1 pA, P>0.05, unpaired t test) or frequency (wild type, 15.3±4.0 Hz; leaner, 15.1±2.3 Hz, P>0.05, unpaired t test; Fig. 3e). The similarity between mIPSC frequency in leaner and WT PCs is consistent with the idea that neither the number of inhibitory inputs to PCs nor the mechanism of spontaneous GABA release is modified by the leaner mutation. Furthermore, the similarity in mIPSC amplitude, which provides a rough estimate of quantal size in the two cell populations, is consistent with unchanged postsynaptic sensitivity at GABAergic synapses. These results suggest that the higher frequency and amplitude of sIPSC observed in leaner compared to wild-type PCs results specifically from changes in presynaptic, spike-induced, GABA release. Moreover, given the close similarity between mIPSC and sIPSC amplitudes in WT PCs, in contrast to the large difference between the corresponding quantities in leaner PCs (Fig. 3e, central panel), it would appear that the leaner mutation promotes multivesicular release at inhibitory neuron–PC synapses. Notably, TTX also eliminated IPSC bursts in both cell populations (Fig. 3e, right panel), arguing that sIPSC bursts depend on the generation of presynaptic action potentials.
Fig. 3.

Comparison between spontaneous miniature IPSCs (mIPSCs) in wild-type and leaner PCs. a, b Representative recordings of spontaneous IPSCs in wild-type and leaner PCs. c, d Recordings from the same cells after exposure to 1 µM TTX, which reduces sIPSC frequency, leaving mIPSCs that are indistinguishable in the two cell groups both in terms of their frequency and amplitude. TTX also reduces the frequency of bursts to undetectable levels. e Summary of mean mIPSC frequency, amplitude, and burst frequency in WT and leaner PCs. Recordings were made in the presence of 5 mM kynurenic acid. Dotted bars indicate mean values of the respective parameters under control conditions (in the absence of TTX)
Differences in Firing Rate of Presynaptic Gabaergic Neurons Does Not Account for Enhanced Synaptic Inhibition in Leaner PCs
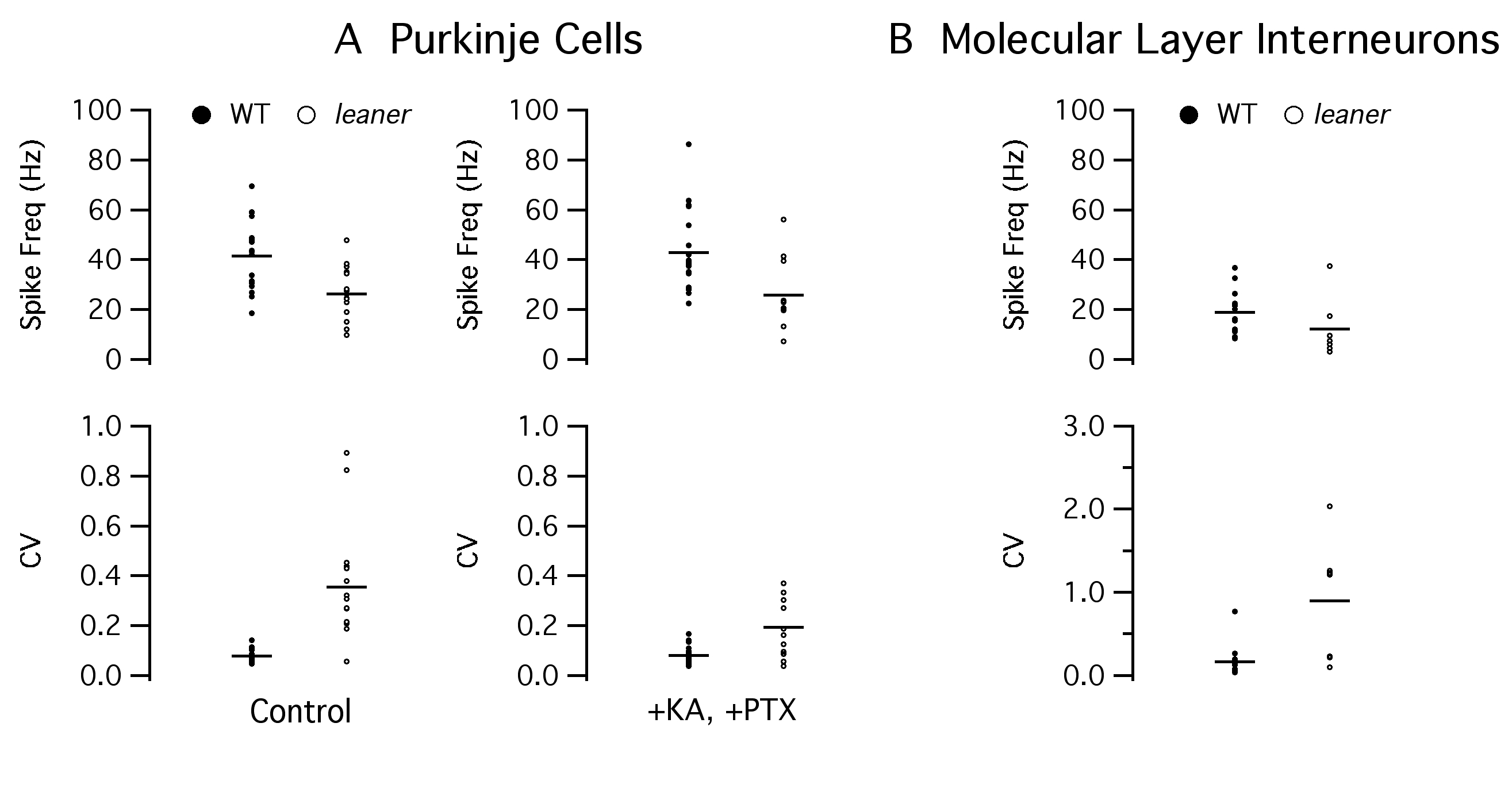
A potential source of increased TTX-sensitive inhibitory synaptic drive in leaner PCs is an increase in the frequency of spontaneously generated action potentials in GABAergic neurons innervating PCs. Purkinje cells receive monosynaptic inhibitory input from interneurons of the molecular layer (basket and stellate cells) and from nearby PCs. Therefore, spontaneous firing activity of these cells was investigated using noninvasive somatic cell-attached patch recording techniques (see also ESM Fig. 1). Note that in most of these experiments, we did not rigorously distinguish between basket and stellate cells and, unless noted otherwise, simply refer to molecular layer interneurons. Figure 4 shows representative interneuronal (panels a–c, putative basket cell) and PC (panels d–f) firing activity from WT and leaner slices under cell-attached recording conditions, along with electrophysiological confirmation of each cell type based on evoked spiking under whole cell current clamp (Fig. 4a, d, bottom). Unlike PCs, which fire at a steady rate during a weak suprathreshold depolarizing current injection, interneurons under the same conditions show spike frequency adaptation (Fig. 4a, d) [36]. While no differences in average firing rate between WT and leaner interneurons was found (WT: F=18.7±2.5 Hz, n=13; leaner: F=12.1±4.5 Hz, n=7, P>0.05, unpaired t test), ISI variability in mutants was significantly higher than in WT cells (WT: CV=0.17±0.05; leaner: CV=0.90±0.28, P<0.038, unpaired t test), which is reflected in the broader ISI distribution and poorly defined autocorrelogram peaks (Fig. 4b, c, middle, bottom). CV appears to be increased in leaner interneurons due to prolonged spike-free gaps (Fig. 4c, top), reminiscent of those seen in leaner PCs under whole cell conditions, which are rarely encountered in WT cells (Fig. 4b, top)
Fig. 4.

Comparison between cell-attached recordings of spontaneous activity in WT and leaner basket and Purkinje neurons. a IR-DIC image of a basket neuron (white arrowhead) with adjacent PC in a WT cerebellar slice. ML molecular layer, PCL Purkinje cell layer, GCL granular cell layer. Recording below shows evoked firing under whole cell current-clamp conditions, illustrating spike frequency adaptation during depolarizing current pulse. b, c Representative cell-attached patch recordings of spontaneous activity in wild-type and leaner basket neurons, with ISI histograms and autocorrelograms below. Mean firing frequency in leaner molecular layer interneurons is similar to WT cells, but ISI variability is more pronounced as indicated by the broader ISI distribution and less defined autocorrelogram peaks, indicative of increased spike rate variability. d IR-DIC image of PC (top, white arrowhead) with recording of evoked firing activity under whole cell current-clamp (bottom). Unlike basket neurons, PCs respond to steady weak depolarizing current injection with sustained repetitive firing without spike frequency adaptation. e, f Representative cell-attached recordings of spontaneous activity from wild-type and leaner PCs with corresponding ISI histograms and autocorrelograms (below). Note higher discharge rate of PCs compared to basket neurons in both WT (compare b and e) and leaner (compare c and e) mice. Also note that cell-attached recordings from WT and leaner PCs confirm differences in firing rate and ISI variability observed during whole cell recordings (see Fig. 1 and ESM Fig. 1)
In a similar way, activity was compared in WT and leaner PCs under cell-attached recording conditions (Fig. 4e, f). Confirming observations made under whole cell current-clamp conditions (Fig. 1), cell-attached recordings indicate that leaner PCs fire at lower rate with higher ISI variability than WT PCs. The differences in frequency and CV between WT and leaner PCs under cell-attached conditions were significant (WT: F=37.1±3.1 Hz; CV=0.10 ± 0.07, n=24; leaner: F=20.1 ± 3.2 Hz; CV=0.37±0.02, n=13, P<0.05, unpaired t test). These results argue that the increase in sIPSC frequency found in leaner PCs is not due to a higher average spontaneous firing rate in presynaptic inhibitory neurons and point to enhanced action potential-induced transmitter release at synapses conveying inhibitory input to mutant PCs.
Analysis of IPSCs Evoked by Direct Basket Cell Stimulation
To test for changes in the strength of spike-induced inhibitory synaptic transmission in leaner PCs, we compared IPSCs in WT and leaner PCs evoked by direct basket cell stimulation (see “Methods”, Fig. 5a). It was found that eIPSCs in leaner PCs have larger amplitude compared to WT cells (leaner, 546.9±78.3 pA; WT, 311.0±50.4 pA, P<0.05) and a slower decay time constant (leaner, 4.5±0.4 ms, n=7; WT, 2.9±0.4 ms, n=7, P<0.05; see Fig. 5e). We also found that in leaner, a larger fraction of eIPSCs consisted of multiple components (leaner, 16.2±3.9%; WT, 4.7±2.2%, P<0.05, unpaired t test) without a difference in the average number of events comprising the multicomponent IPSCs (leaner, 2.5; WT, 2.3); examples of such multicomponent eIPSCs from five consecutive stimuli in a leaner PC are shown in Fig. 5d. These observations point to enhanced action potential-evoked GABA release at leaner basket cell–PC synapses, along with an increased probability of asynchronous IPSC bursts.
Fig. 5.

Comparison between IPSCs evoked in leaner and WT PCs by stimulation of basket cells. a Diagram illustrating placement of stimulating (stim.) and recording (rec.) electrodes. ML molecular layer, PCL Purkinje cell layer, GCL granular cell layer. eIPSCs were recorded in the presence of kynurenic acid (5 mM) and were abolished by picrotoxin (200 µM, data not shown). b, c Superimposed IPSCs in WT and leaner PCs elicited by single pulse basket cell stimulation. Note higher level of spontaneous activity in leaner (c) compared to WT (b). d Examples of multicomponent eIPSCs elicited in a leaner PC from consecutive stimuli. e Summary comparison of eISPC amplitude, decay time constant, and the proportion of eIPSCs consisting of multiple components in WT and leaner PCs. *P<0.05
Enhanced Inhibition in Leaner PCs Requires Ca2+ Loading by Internal Stores
Previous studies have shown that Ca2+ release from presynaptic calcium stores contributes to evoked neurotransmitter release at synapses between inhibitory interneurons and PCs [48–50]; for reviews, see [50, 51]. Ca2+ release triggered by voltage-sensitive Ca2+ entry during a presynaptic action potential would be expected to modify the presynaptic [Ca2+] transient, thereby influencing the rate and ultimate quantity of neurotransmitter release. Under certain conditions, this could include the generation of multiple asynchronous release events that together elicit a postsynaptic IPSC burst. As a first step toward determining if such a mechanism contributes to enhanced inhibitory synaptic drive in leaner PCs, we compared sIPSCs in the two cell groups after a 10- to 15-min pretreatment with the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) inhibitor thapsigargin (Fig. 6) using a concentration (10 µM) like that employed in previous cerebellar slice studies [52, 53]. With this concentration and exposure time, thapsigargin effectively and irreversibly blocks SERCA pumps, leading to the depletion of ER stores through a background Ca2+ leak [54, 55]. We found that thapsigargin reduced both sIPSC frequency and amplitude in leaner PCs (Fig. 6b, d), without significantly altering these parameters in WT cells (Fig. 6a, c). Moreover, after treatment with thapsigargin, WT and leaner PCs were no longer distinguishable in terms of these parameters (frequency: wild type, 27.3±4.5 Hz; leaner, 32.2±4.5 Hz, P>0.05, unpaired t test; amplitude: wild type, 39.6±6.5 pA; leaner, 52.3±5.0 pA, P>0.05, unpaired t test, n=6, 10 for wild type and leaner, respectively; Fig. 6c–e). Thapsigargin also reduced the frequency of sIPSC bursts in leaner PCs without detectably changing the corresponding parameter in WT cells (Fig. 6e). These results indicate that increases in sIPSC frequency and amplitude that contribute to increased inhibitory drive in leaner PCs are sensitive to thapsigargin. This raises the possibility that changes in Ca2+ release from thapsigargin-sensitive presynaptic Ca2+ stores play an important role in amplifying spike-evoked GABA release from molecular layer interneurons that mediate synaptic inhibition in leaner PCs.
Fig. 6.

Effect of thapsigargin on spontaneous IPSCs in WT and leaner PCs. a, b Representative recordings of sIPSCs from control WT and leaner PCs. c, d Representative recordings of sIPSCs from another group of cells that was pretreated for 10–15 min with thapsigargin (10 µM) followed by washout in the recording chamber. e Summary of the effects of thapsigargin on mean sIPSC frequency, amplitude, and burst frequency. Note that thapsigargin specifically reduces mean sIPSC frequency, amplitude, and burst frequency in leaner PCs, but has little effect on WT cells. Moreover, after treatment with thapsigargin, there was little or no difference between WT and leaner PCs in terms of sIPSC frequency, amplitude, and burst frequency. Dotted bars indicate mean values of the respective parameters under control conditions (without thapsigargin treatment). All recordings were made in the presence of kynurenic acid (5 mM)
Discussion
Summary of Main Results
This study demonstrates that the leaner P/Q Ca2+ channel mutation causes a pronounced enhancement in inhibitory synaptic drive in cerebellar Purkinje neurons. This leads to a modification of PC firing properties, most notably transitory depressions in firing rate and increased spike rate variability. Indications of enhanced inhibition include increases in both the frequency and amplitude of sIPSCs measured under voltage clamp and in the frequency of sIPSC bursts. Enhanced spontaneous synaptic inhibitory input in leaner PCs specifically reflects changes in TTX-sensitive (spike-triggered) IPSCs without detectable modifications of mIPSCs either in terms of their mean frequency or amplitude, pointing to changes in both the frequency and amplitude of synaptic currents triggered by spontaneous action potentials in presynaptic GABAergic neurons. Differences in mean firing rate in molecular layer interneurons in WT and leaner mice were not observed, but significant increases were found in the amplitude and decay time constant of evoked IPSCs, as well as in the proportion of evoked responses that consist of more than one distinguishable synaptic current. Taken together, these results suggest that the leaner mutation influences the mechanism by which presynaptic action potentials trigger GABA release. Initial experiments indicate that depletion of thapsigargin-sensitive Ca2+ stores, previously reported to influence evoked presynaptic [Ca2+]i elevations in GABAergic interneurons, eliminates the observed differences between sIPSC in leaner and WT PCs. This raises the possibility that the leaner mutation perturbs presynaptic Ca2+ handling in inhibitory interneurons in a way that overcompensates for reduced voltage-sensitive Ca2+ entry at axonal terminals, ultimately leading to enhanced GABA release (see below).
Loss-of-Function P/Q Channel Mutations and Their Effect on Inhibitory Synaptic Transmission in the Cerebellar Cortex
The leaner mutation is caused by a mutation in the Cacna1a gene which encodes the pore-forming subunit of P/Q-type Ca2+ channels [6, 14]. This leads to the expression of channels with altered intracellular C-terminal sequences [32] (see also [56]) and a ~60% reduction in whole cell (somatic) Ca2+ current in cerebellar PCs [30–32]. While the functional impact of this and other loss-of-function CaV2.1 mouse mutations (e.g., rocker, tottering, and rolling Nagoya) on intrinsic excitability [23, 25, 28] and excitatory synaptic inputs to PCs [26, 27] has been investigated, little information has been available regarding their effects on inhibitory synaptic transmission in these neurons. With emerging evidence for a causal link between abnormal firing in PCs and cerebellar ataxia in CaV2.1 mutant mice [22, 23] and the key role played by GABAergic inputs in shaping PC output [3, 21], there is a strong rationale for the analysis of inhibitory synaptic modulation of PC activity and its sensitivity to P/Q channel mutations.
The Leaner Mutation Disrupts the Efferent Code in Cerebellar Purkinje Neurons
We found that with inhibitory synaptic transmission enabled, action potential generation in leaner Purkinje neurons is punctuated by long interspike intervals, leading to transitory reductions in firing rate and an overall increase in spike rate variability. Since synaptic modulation of the spontaneous firing rate of these neurons is thought to be important for information processing in the cerebellar cortex (see [57] and references therein), such an increase in firing rate variability would be expected to disrupt cerebellar function. Previous work has shown that even in the absence of synaptic transmission, tonic firing in PCs from loss-of-function P/Q channel mutant mice is less regular than in WT animals, a difference that can be attributed to a reduced calcium entry through P/Q-type Ca2+ channels [23]. This finding identifies an intrinsic property of PCs that influences spike rate variability and is sensitive to loss-of function P/Q channel mutations. Our results identify another, extrinsic, property involving spontaneous inhibitory synaptic transmission that influences spike rate variability and is influenced by P/Q channel mutations. While the results demonstrate that this component of variability requires GABAergic transmission, it may also be influenced by changes in intrinsic properties of PCs [28] that modify the impact of inhibitory synaptic transmission on membrane potential dynamics in PCs. Thus, both intrinsic and extrinsic mechanisms must be considered when investigating the effects of P/Q channel mutations on PC firing properties and more generally in other neurons comprising neuronal circuits where these channels play a role.
sIPSC Bursts and Their Role in Disrupting the Temporal Pattern of PC Firing
Pauses in tonic firing seen in leaner PCs were often accompanied by multiple inhibitory postsynaptic potentials (IPSPs; Fig. 1b). Since the sIPSCs that cause such pauses would be most effective in perturbing PC firing rate when they occur in clusters, we measured the frequency of sIPSC bursts, operationally defined as sequences of three or more IPSCs in which consecutive events are separated by no more than 12 ms. It was found that the frequency of such sIPSC bursts was higher in leaner PCs (2.6 Hz) than in WT cells (0.5 Hz). In considering what might be responsible for this increase, the finding that thapsigargin reduced burst frequency in leaner PCs, while having little or no effect in WT cells, raised the possibility that the leaner mutation specifically influences a thapsigargin-sensitive mechanism that generates sIPSC bursts. However, thapsigargin also reduced the mean sIPSC frequency in leaner PCs, questioning whether the higher burst frequency observed in leaner PCs is a specific functional consequence of the leaner mutation or simply results from an elevation in sIPSC frequency.
The finding that the distribution of intervals between consecutive sIPSCs is approximately exponential in both cell types (Fig. 2g, h) is consistent with the idea that sIPSC timing in each cell population is controlled by a homogeneous Poisson process, but with different rates (RWT, Rle). For such processes, it is possible to calculate the probability PBurst(R) that events cluster together in a way that satisfies our definition of a burst:
| (1) |
where the first term in brackets within the sum describes the probability of a sequence of (n − 1) consecutive inter-event intervals ≤12 ms, the second term is the probability of an interval >12 ms, and 1/n is the fraction of interval sequences that end with a long interval. Using a numerical approach to evaluate this equation in the case of WT (RWT=28.6 Hz) and leaner (Rle=50.3 Hz) PCs gives values for PBurst of 0.026 and 0.058, respectively. Multiplying by the respective event frequencies converts these values to burst frequencies (0.74 and 2.92 Hz) which are close to the measured values (0.49 and 2.63 Hz; Fig. 2i, right). Thus, the increase in sIPSC frequency observed in leaner PCs can largely account for the observed increase in burst frequency, directing attention to mechanisms by which the mutation might influence the underlying event frequency.
Potential Mechanisms for Augmented GABAergic Transmission in Leaner PCs
Enhanced inhibitory neurotransmission resulting from a loss-of-function CaV2.1 mutation is surprising given that P/Q-type Ca2+ channels carry a large fraction of the presynaptic Ca2+ entry that triggers GABA release from inhibitory interneurons in the cerebellar cortex [38, 39]. However, recent studies suggest several factors that could contribute to this unanticipated result. For example, PC inhibitory inputs may be tolerant to CaV2.1 channel dysfunction due to a high Ca2+ sensitivity of the vesicular fusion complex at PC inhibitory inputs [58]. Notably, neurotransmission at the only inhibitory synapses (onto thalamic neurons) investigated to date in CaV2.1 mutants is also preserved despite the fact that P/Q-type Ca2+ channels mediate a significant fraction of the presynaptic Ca2+ entry at these synapses [59]. Partial compensation for reduced Ca2+ entry through P/Q-type channels by increased N-type Ca2+ current, previously described at central synapses in CaV2.1 mutants [29, 60, 61], could also reduce the impact of the leaner mutation on GABAergic synaptic drive in PCs. Finally, previous work suggests that leaner Purkinje neurons have weaker cytoplasmic Ca2+ buffering [62, 63] such that despite having smaller Ca2+ currents, depolarization-induced [Ca2+]ielevations are comparable to those seen in WT cells. If a similar change in buffering strength were to occur in terminals of molecular layer interneurons that exert inhibitory control over PCs, it would lessen the impact of reduced Ca2+ entry on presynaptic [Ca2+]i elevations, particularly given that the level of Ca2+ buffering directly regulates PC inhibitory inputs [64]
It is clear that none of the effects described above, alone, can easily account for enhanced synaptic inhibition, specifically involving spike-induced GABA release. Here, the observed effects of thapsigargin suggest a testable hypothesis. Thapsigargin reduced mean sIPSC frequency and amplitude in leaner PCs, but had little or no effect on these parameters in WT cells, implicating Ca2+ uptake and/or release from internal stores in the enhancement of GABA release from leaner inhibitory interneurons; it also argues against nonspecific effects of thapsigargin, e.g., on voltage-gated Ca2+ channels [65], as causes of the observed effects in leaner PCs since such effects would be expected to alter sIPSC properties in wild-type PCs as well. Previous reports have shown that Ca2+ release from presynaptic internal stores influences inhibitory transmission in wild-type Purkinje neurons [48, 66]. Together with ryanodine receptor expression in cerebellar GABAergic synapses [66] and presynaptic specializations with multiple GABA release sites [67], reduced Ca2+ buffering could potentially increase the likelihood of regenerative Ca2+ release in response to action potential-induced Ca2+ entry at inhibitory presynaptic terminals, leading to enhanced GABA release (Fig. 7). In the context of reduced P/Q Ca2+ channel function, weaker cytoplasmic Ca2+ buffering could compensate for diminished presynaptic Ca2+ entry, thereby normalizing evoked GABA release and inhibitory modulation of PC activity. Moreover, weaker Ca2+ buffering could lead to qualitative changes in calcium-induced calcium release (CICR), for example, from a low-gain form that is graded with [Ca2+]i (mode 2 CICR) to a regenerative, high-gain form (mode 3 CICR) [68] that leads to overcompensation for reduced Ca2+ entry. Depending on the spatial relationship between sites of presynaptic Ca2+ entry and release, diminished Ca2+ buffering could reduce spatial and temporal control of [Ca2+]i during action potential-induced Ca2+ entry, leading to larger, less synchronous, and more spatially distributed [Ca2+]i elevations, with consequences for the amount and temporal profile of vesicular GABA release.
Fig. 7.

Hypothetical model of enhanced GABA release at synapses between cerebellar cortical interneurons and PCs in leaner mice. a In WT mice, Ca2+ influx through P/Q type Ca2+ channels in presynaptic terminals of inhibitory interneurons triggers a fast rise in intracellular Ca2+ concentration ([Ca2+]i) that causes synchronous GABA release, opening of postsynaptic chloride channels, and Cl− efflux, which is detected as an inward postsynaptic current (symmetrical Cl−, Vh=−75 mV). b If cytosolic Ca2+ buffering is weaker in leaner compared to WT GABAergic terminals, as has been reported for somatic Ca2+ buffering in leaner PCs [62, 63], evoked [Ca2+]i elevations may be large enough, in spite of reduced Ca2+ entry, to trigger regenerative CICR, leading to enhanced, asynchronous GABA release
Could this hypothesis account for the observations described in this study? If the leaner mutation reduced the strength of presynaptic Ca2+ buffering to the point where action potential-evoked Ca2+ entry and release together produce even larger presynaptic [Ca2+]i elevations than in wild-type terminals, an expected consequence would be enhanced GABA release, leading to eIPSCs with larger mean amplitude (Fig. 5e, left). We observed ~76% larger eIPSCs in leaner compared to wild-type PCs; the proportionally larger increase in mean sIPSC amplitude (153%, Fig. 2i, center) possibly reflects contributions from multiple, nearly simultaneous inputs that are not distinguished by the algorithm we used to detect spontaneous synaptic currents. Weaker presynaptic Ca2+ buffering that favors regenerative CICR could promote asynchronous release from single or multiple release sites and increase the probability that presynaptic action potentials trigger multicomponent IPSCs (Fig. 5d, e, middle), which in the absence of a change in mean firing rate of inhibitory neurons would elevate sIPSC frequency (Fig. 2i, left). We observed an approximately threefold increase in the probability (PMulticomp) of multicomponent eIPSCs from 0.05 (WT) to 0.16 (leaner), with a mean number (N) of events per multicomponent eIPSC of ~2.4 which was not significantly different in two mouse strains.
Can the higher PMulticomp seen in leaner mice account for the observed increase in sIPSC frequency? Given that inter-event sIPSC interval distributions are approximately exponential, this question can be restated as follows: Can the increase in multicomponent IPSC probability in leaner PCs account quantitatively for the higher sIPCS frequency observed in these cells, with the same underlying event rate for the two cell populations? To answer this question, the following equations were solved for the mean event rates in WT and leaner mice (FWT, Fle):
| (2) |
where FObs,WT (28.6 Hz) and FObs,le (50.3 Hz) are the observed sIPSC frequencies and PMulticom,WT (0.05) and PMulticomp,le (0.16) are the probabilities of multicomponent IPSCs with mean number of components (2.4). The deduced event rates in WT and leaner mice are quite different, FWT=26.8 Hz and Fle=41.0 Hz, with the increase in PMulticom accounting for only an ~6 Hz rise in sIPSC frequency, pointing to a larger role for an increase in the underlying rate of sIPSC generation.
How might the leaner mutation increase this rate? We found that the distribution of interspike intervals in leaner molecular layer interneurons was broader than in wild-type neurons, with an increased probability of both large and small interspike intervals but little or no change in the mean. While the increased probability of small intervals would be expected to occasionally raise sIPSC frequency, the results would not predict an increase in mean sIPSC frequency in PCs. The observed increase in mean sIPSC amplitude could be relevant since it would cause a greater proportion of events to surpass a given threshold used for event detection. In any case, reduced presynaptic Ca2+ buffering is only one possible mechanism, and other changes may play a role, such as increased Ca2+ sensitivity of the GABA release machinery as well as altered membrane excitability that promotes action potential invasion beyond axonal branch points, which has been proposed to explain enhanced inhibition in Kv1.1 null mice [69].
Implications for Local Information Processing and Cerebellar Function
The mechanism by which the cerebellar cortex conveys time-varying information to the deep nuclei is likely to require precision of spike timing in individual PCs and in the temporal relationship between firing in different PCs. There is ample evidence indicating behavior-induced changes in PC synchronization and spiking [70–72] that are regulated by local inhibitory networks [3, 73, 74]. The present study shows that GABAergic synaptic drive and properties of IPSCs in leaner PCs are profoundly altered, resulting in an increase in spike rate variability in these cells. In addition to elevating the level of noise in the PC output, increased variability in the rate of tonic firing in PCs would be expected to increase the trial-to-trial variability of PC responses to repeated presentation of afferent stimuli, diminishing the information encoding/processing capabilities of these neurons [75]. Direct experimental evidence supporting this notion has been presented based on studies of P/Q channel mutant tottering mice where an enhanced level of noise in PC spiking leads to the disruption of cerebellar sensorimotor processing and severe oculomotor deficits [22]. Overall, a significant increase in the GABAergic drive in leaner PCs described here would be expected to interfere with temporal coding of sensory information by these cells and communication with downstream targets involved in motor control. The enhanced spontaneous inhibitory input to leaner PCs shown here would shift the synaptic balance toward greater inhibition, potentially disrupting synaptic plasticity and long-term motor adaptation in PCs [20, 76]. While detailed in vivo analysis will be needed to understand how amplified GABAergic inputs affect cerebellar network dynamics and PC function in situ, the experimental data described here complement earlier studies [23, 27, 28] and contribute to a more complete in vitro electrophysiological account of PC intrinsic properties and synaptic functions in CaV2.1 channel mutant leaner mice.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
David Friel would like to thank Dr. Roberto F. Galán for discussions regarding sIPSC burst statistics. This work was supported by a grant from NIH/NINDS (NS 33514) to DDF.
Abbreviations
- PC
Purkinje cell
- BC
Basket cell
- IPSC
Inhibitory postsynaptic current
- sIPSC
Spontaneous IPSC
- eIPSC
Evoked IPSC
- mIPSC
Miniature IPSC
- TTX
Tetrodotoxin
- SERCA
Sarcoplasmic/endoplasmic reticulum Ca/ATPase
- ISI
Inter-spike interval
- IEI
Inter-event interval
- CV
Coefficient of variation
- CICR
Calcium-induced calcium release
- [Ca2+]i
Cytosolic free calcium concentration
- WT
Wild type
- tgla
Leaner
Footnotes
Electronic supplementary material The online version of this article, (doi:10.1007/s12311-010-0210-9) contains supplementary material, which is available to authorized users.
Conflict of interest
The authors report no conflicts of interest regarding the contents of this manuscript.
Contributor Information
Saak V. Ovsepian, Department of Neurosciences, School of Medicine, Case Western Reserve University, 10900 Euclid Avenue, Cleveland, OH 44106, USA
David D. Friel, Email: ddf2@case.edu, Department of Neurosciences, School of Medicine, Case Western Reserve University, 10900 Euclid Avenue, Cleveland, OH 44106, USA; Department of Neurosciences, Case Western Reserve University, 10900 Euclid Ave., Cleveland, OH 44106-4975, USA.
References
- 1.Ito M. The cerebellum and neural control. New York: Raven; 1984. [Google Scholar]
- 2.Jaeger D, Bower JM. Prolonged responses in rat cerebellar Purkinje cells following activation of the granule cell layer: an intracellular in vitro and in vivo investigation. Exp Brain Res. 1994;100(2):200–214. doi: 10.1007/BF00227191. [DOI] [PubMed] [Google Scholar]
- 3.Hausser M, Clark BA. Tonic synaptic inhibition modulates neuronal output pattern and spatiotemporal synaptic integration. Neuron. 1997;19(3):665–678. doi: 10.1016/s0896-6273(00)80379-7. [DOI] [PubMed] [Google Scholar]
- 4.De Schutter E, Steuber V. Patterns and pauses in Purkinje cell simple spike trains: experiments, modeling and theory. Neuroscience. 2009;162(3):816–826. doi: 10.1016/j.neuroscience.2009.02.040. [DOI] [PubMed] [Google Scholar]
- 5.Llinas R, Sugimori M. Electrophysiological properties of in vitro Purkinje cell somata in mammalian cerebellar slices. J Physiol. 1980;305:171–195. doi: 10.1113/jphysiol.1980.sp013357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Llinas R, et al. Blocking and isolation of a calcium channel from neurons in mammals and cephalopods utilizing a toxin fraction (FTX) from funnel-web spider poison. Proc Natl Acad Sci USA. 1989;86(5):1689–1693. doi: 10.1073/pnas.86.5.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jaeger D, De Schutter E, Bower JM. The role of synaptic and voltage-gated currents in the control of Purkinje cell spiking: a modeling study. J Neurosci. 1997;17(1):91–106. doi: 10.1523/JNEUROSCI.17-01-00091.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raman IM, Bean BP. Ionic currents underlying spontaneous action potentials in isolated cerebellar Purkinje neurons. J Neurosci. 1999;19(5):1663–1674. doi: 10.1523/JNEUROSCI.19-05-01663.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diener HC. Cerebellar dysfunction of movement and perception. Can J Neurol Sci. 1993;20(Suppl 3):S62–S69. doi: 10.1017/s031716710004854x. [DOI] [PubMed] [Google Scholar]
- 10.Ivry RB, Keele SW, Diener HC. Dissociation of the lateral and medial cerebellum in movement timing and movement execution. Exp Brain Res. 1988;73(1):167–180. doi: 10.1007/BF00279670. [DOI] [PubMed] [Google Scholar]
- 11.Braitenberg V. Is the cerebellar cortex a biological clock in the millisecond range? Prog Brain Res. 1967;25:334–346. doi: 10.1016/S0079-6123(08)60971-1. [DOI] [PubMed] [Google Scholar]
- 12.Zhuchenko O, et al. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat Genet. 1997;15(1):62–69. doi: 10.1038/ng0197-62. [DOI] [PubMed] [Google Scholar]
- 13.Ophoff RA, et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;87(3):543–552. doi: 10.1016/s0092-8674(00)81373-2. [DOI] [PubMed] [Google Scholar]
- 14.Fletcher CF, et al. Absence epilepsy in tottering mutant mice is associated with calcium channel defects. Cell. 1996;87(4):607–617. doi: 10.1016/s0092-8674(00)81381-1. [DOI] [PubMed] [Google Scholar]
- 15.Harrington DL, et al. Does the representation of time depend on the cerebellum? Effect of cerebellar stroke. Brain. 2004;127(Pt3):561–574. doi: 10.1093/brain/awh065. [DOI] [PubMed] [Google Scholar]
- 16.Timmann D, Watts S, Hore J. Failure of cerebellar patients to time finger opening precisely causes ball high-low inaccuracy in overarm throws. J Neurophysiol. 1999;82(1):103–114. doi: 10.1152/jn.1999.82.1.103. [DOI] [PubMed] [Google Scholar]
- 17.Welsh JP, Llinas R. Some organizing principles for the control of movement based on olivocerebellar physiology. Prog Brain Res. 1997;114:449–461. doi: 10.1016/s0079-6123(08)63380-4. [DOI] [PubMed] [Google Scholar]
- 18.Marr D. A theory of cerebellar cortex. J Physiol. 1969;202(2):437–470. doi: 10.1113/jphysiol.1969.sp008820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Braitenberg V, Atwood RP. Morphological observations on the cerebellar cortex. J Comp Neurol. 1958;109(1):1–33. doi: 10.1002/cne.901090102. [DOI] [PubMed] [Google Scholar]
- 20.Mittmann W, Koch U, Hausser M. Feed-forward inhibition shapes the spike output of cerebellar Purkinje cells. J Physiol. 2005;563(Pt 2):369–378. doi: 10.1113/jphysiol.2004.075028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jaeger D, Bower JM. Synaptic control of spiking in cerebellar Purkinje cells: dynamic current clamp based on model conductances. J Neurosci. 1999;19(14):6090–6101. doi: 10.1523/JNEUROSCI.19-14-06090.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoebeek FE, et al. Increased noise level of Purkinje cell activities minimizes impact of their modulation during sensorimotor control. Neuron. 2005;45(6):953–965. doi: 10.1016/j.neuron.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 23.Walter JT, et al. Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat Neurosci. 2006;9(3):389–397. doi: 10.1038/nn1648. [DOI] [PubMed] [Google Scholar]
- 24.Pietrobon D. Ca(V) 2.1 channnelopathies. Pflungers Archiv-European Journal of Physiology. 2010;460(2):375–393. doi: 10.1007/s00424-010-0802-8. [DOI] [PubMed] [Google Scholar]
- 25.Mori Y, et al. Reduced voltage sensitivity of activation of P/Q-type Ca2+ channels is associated with the ataxic mouse mutation rolling Nagoya (tg(rol)) J Neurosci. 2000;20(15):5654–5662. doi: 10.1523/JNEUROSCI.20-15-05654.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsushita K, et al. Bidirectional alterations in cerebellar synaptic transmission of tottering and rolling Ca2+ channel mutant mice. J Neurosci. 2002;22(11):4388–4398. doi: 10.1523/JNEUROSCI.22-11-04388.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu S, Friel DD. Impact of the leaner P/Q-type Ca2+ channel mutation on excitatory synaptic transmission in cerebellar Purkinje cells. J Physiol. 2008;586(Pt 18):4501–4515. doi: 10.1113/jphysiol.2008.156232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ovsepian SV, Friel DD. The leaner P/Q-type calcium channel mutation renders cerebellar Purkinje neurons hyper-excitable and eliminates Ca2+-Na+ spike bursts. Eur J Neurosci. 2008;27(1):93–103. doi: 10.1111/j.1460-9568.2007.05998.x. [DOI] [PubMed] [Google Scholar]
- 29.Zhou YD, Turner TJ, Dunlap K. Enhanced G protein-dependent modulation of excitatory synaptic transmission in the cerebellum of the Ca2+ channel-mutant mouse, tottering. J Physiol. 2003;547(Pt 2):497–507. doi: 10.1113/jphysiol.2002.033415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dove LS, Abbott LC, Griffith WH. Whole-cell and single-channel analysis of P-type calcium currents in cerebellar Purkinje cells of leaner mutant mice. J Neurosci. 1998;18(19):7687–7699. doi: 10.1523/JNEUROSCI.18-19-07687.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lorenzon NM, et al. Altered calcium channel currents in Purkinje cells of the neurological mutant mouse leaner. J Neurosci. 1998;18(12):4482–4489. doi: 10.1523/JNEUROSCI.18-12-04482.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wakamori M, et al. Single tottering mutations responsible for the neuropathic phenotype of the P-type calcium channel. J Biol Chem. 1998;273(52):34857–34867. doi: 10.1074/jbc.273.52.34857. [DOI] [PubMed] [Google Scholar]
- 33.Andersen P, Eccles JC, Voorhoeve PE. Postsynaptic inhibition of cerebellar Purkinje cells. J Neurophysiol. 1964;27:1138–1153. doi: 10.1152/jn.1964.27.6.1138. [DOI] [PubMed] [Google Scholar]
- 34.Chan-Palay V. The recurrent collaterals of Purkinje cell axons: a correlated study of the rat’s cerebellar cortex with electron microscopy and the Golgi method. Z Anat Entwicklungsgesch. 1971;134(2):200–234. doi: 10.1007/BF00519300. [DOI] [PubMed] [Google Scholar]
- 35.Eccles JC, Llinas R, Sasaki K. The inhibitory interneurones within the cerebellar cortex. Exp Brain Res. 1966;1(1):1–16. doi: 10.1007/BF00235206. [DOI] [PubMed] [Google Scholar]
- 36.Midtgaard J. Stellate cell inhibition of Purkinje cells in the turtle cerebellum in vitro. J Physiol. 1992;457:355–367. doi: 10.1113/jphysiol.1992.sp019382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Orduz D, Llano I. Recurrent axon collaterals underlie facilitating synapses between cerebellar Purkinje cells. Proc Natl Acad Sci USA. 2007;104(45):17831–17836. doi: 10.1073/pnas.0707489104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Forti L, Pouzat C, Llano I. Action potential-evoked Ca2+ signals and calcium channels in axons of developing rat cerebellar interneurones. J Physiol. 2000;527(Pt 1):33–48. doi: 10.1111/j.1469-7793.2000.00033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stephens GJ, et al. The Cav2.1/alpha1A (P/Q-type) voltage-dependent calcium channel mediates inhibitory neurotransmission onto mouse cerebellar Purkinje cells. Eur J Neurosci. 2001;13(10):1902–1912. doi: 10.1046/j.0953-816x.2001.01566.x. [DOI] [PubMed] [Google Scholar]
- 40.Herrup K, Wilczynski SL. Cerebellar cell degeneration in the leaner mutant mouse. Neuroscience. 1982;7(9):2185–2196. doi: 10.1016/0306-4522(82)90129-4. [DOI] [PubMed] [Google Scholar]
- 41.Sidman RL, Green MC, Appel SH. Catalog of the neurological mutants of the mouse. Cambridge: Harvard University Press; 1965. [Google Scholar]
- 42.Vincent P, Marty A. Fluctuations of inhibitory postsynaptic currents in Purkinje cells from rat cerebellar slices. J Physiol. 1996;494(Pt 1):183–199. doi: 10.1113/jphysiol.1996.sp021484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stone TW. Neuropharmacology of quinolinic and kynurenic acids. Pharmacol Rev. 1993;45(3):309–379. [PubMed] [Google Scholar]
- 44.Allen C, Stevens CF. An evaluation of causes for unreliability of synaptic transmission. Proc Natl Acad Sci USA. 1994;91(22):10380–10383. doi: 10.1073/pnas.91.22.10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McKay BE, Turner RW. Physiological and morphological development of the rat cerebellar Purkinje cell. J Physiol. 2005;567(Pt 3):829–850. doi: 10.1113/jphysiol.2005.089383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Womack M, Khodakhah K. Active contribution of dendrites to the tonic and trimodal patterns of activity in cerebellar Purkinje neurons. J Neurosci. 2002;22(24):10603–10612. doi: 10.1523/JNEUROSCI.22-24-10603.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rokni D, et al. Regularity, variability and bi-stability in the activity of cerebellar purkinje cells. Front Cell Neurosci. 2009;3:12. doi: 10.3389/neuro.03.012.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Galante M, Marty A. Presynaptic ryanodine-sensitive calcium stores contribute to evoked neurotransmitter release at the basket cell-Purkinje cell synapse. J Neurosci. 2003;23(35):11229–11234. doi: 10.1523/JNEUROSCI.23-35-11229.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Conti R, Tan YP, Llano I. Action potential-evoked and ryanodine-sensitive spontaneous Ca2+ transients at the presynaptic terminal of a developing CNS inhibitory synapse. J Neurosci. 2004;24(31):6946–6957. doi: 10.1523/JNEUROSCI.1397-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bouchard R, Pattarini R, Geiger JD. Presence and functional significance of presynaptic ryanodine receptors. Prog Neurobiol. 2003;69(6):391–418. doi: 10.1016/s0301-0082(03)00053-4. [DOI] [PubMed] [Google Scholar]
- 51.Collin T, Marty A, Llano I. Presynaptic calcium stores and synaptic transmission. Curr Opin Neurobiol. 2005;15(3):275–281. doi: 10.1016/j.conb.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 52.Fierro L, DiPolo R, Llano I. Intracellular calcium clearance in Purkinje cell somata from rat cerebellar slices. J Physiol. 1998;510(Pt 2):499–512. doi: 10.1111/j.1469-7793.1998.499bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carter AG, et al. Assessing the role of calcium-induced calcium release in short-term presynaptic plasticity at excitatory central synapses. J Neurosci. 2002;22(1):21–28. doi: 10.1523/JNEUROSCI.22-01-00021.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hongpaisan J, et al. Multiple modes of calcium-induced calcium release in sympathetic neurons II: a [Ca2+](i)- and location-dependent transition from endoplasmic reticulum Ca accumulation to net Ca release. J Gen Physiol. 2001;118(1):101–112. doi: 10.1085/jgp.118.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thastrup O, et al. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2(+)-ATPase. Proc Natl Acad Sci USA. 1990;87(7):2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gillard SE, et al. Identification of pore-forming subunit of P-type calcium channels: an antisense study on rat cerebellar Purkinje cells in culture. Neuropharmacology. 1997;36(3):405–409. doi: 10.1016/s0028-3908(97)00046-4. [DOI] [PubMed] [Google Scholar]
- 57.Walter JT, Khodakhah K. The linear computational algorithm of cerebellar Purkinje cells. J Neurosci. 2006;26(50):12861–12872. doi: 10.1523/JNEUROSCI.4507-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sakaba T. Two Ca(2+)-dependent steps controlling synaptic vesicle fusion and replenishment at the cerebellar basket cell terminal. Neuron. 2008;57(3):406–419. doi: 10.1016/j.neuron.2007.11.029. [DOI] [PubMed] [Google Scholar]
- 59.Caddick SJ, et al. Excitatory but not inhibitory synaptic transmission is reduced in lethargic (Cacnb4(lh)) and tottering (Cacna1atg) mouse thalami. J Neurophysiol. 1999;81(5):2066–2074. doi: 10.1152/jn.1999.81.5.2066. [DOI] [PubMed] [Google Scholar]
- 60.Jun K, et al. Ablation of P/Q-type Ca(2+) channel currents, altered synaptic transmission, and progressive ataxia in mice lacking the alpha(1A)-subunit. Proc Natl Acad Sci USA. 1999;96(26):15245–15250. doi: 10.1073/pnas.96.26.15245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Qian J, Noebels JL. Presynaptic Ca(2+) influx at a mouse central synapse with Ca(2+) channel subunit mutations. J Neurosci. 2000;20(1):163–170. doi: 10.1523/JNEUROSCI.20-01-00163.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dove LS, et al. Altered calcium homeostasis in cerebellar Purkinje cells of leaner mutant mice. J Neurophysiol. 2000;84(1):513–524. doi: 10.1152/jn.2000.84.1.513. [DOI] [PubMed] [Google Scholar]
- 63.Murchison D, et al. Homeostatic compensation maintains Ca2+ signaling functions in Purkinje neurons in the leaner mutant mouse. Cerebellum. 2002;1(2):119–127. doi: 10.1080/147342202753671259. [DOI] [PubMed] [Google Scholar]
- 64.Collin T, et al. Developmental changes in parvalbumin regulate presynaptic Ca2+ signaling. J Neurosci. 2005;25(1):96–107. doi: 10.1523/JNEUROSCI.3748-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shmigol A, Kostyuk P, Verkhratsky A. Dual action of thapsigargin on calcium mobilization in sensory neurons: inhibition of Ca2+ uptake by caffeine-sensitive pools and blockade of plasmalemmal Ca2+ channels. Neuroscience. 1995;65(4):1109–1118. doi: 10.1016/0306-4522(94)00553-h. [DOI] [PubMed] [Google Scholar]
- 66.Llano I, et al. Presynaptic calcium stores underlie large-amplitude miniature IPSCs and spontaneous calcium transients. Nat Neurosci. 2000;3(12):1256–1265. doi: 10.1038/81781. [DOI] [PubMed] [Google Scholar]
- 67.Palay SL, Chan-Palay V. Cerebellar cortex: cytology and organization. New York: Springer; 1974. [Google Scholar]
- 68.Albrecht MA, et al. Multiple modes of calcium-induced calcium release in sympathetic neurons I: attenuation of endoplasmic reticulum Ca2+ accumulation at low [Ca2+](i) during weak depolarization. J Gen Physiol. 2001;118(1):83–100. doi: 10.1085/jgp.118.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang CL, Messing A, Chiu SY. Specific alteration of spontaneous GABAergic inhibition in cerebellar purkinje cells in mice lacking the potassium channel Kv1.1. J Neurosci. 1999;19(8):2852–2864. doi: 10.1523/JNEUROSCI.19-08-02852.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fortier PA, Smith AM, Kalaska JF. Comparison of cerebellar and motor cortex activity during reaching: directional tuning and response variability. J Neurophysiol. 1993;69(4):1136–1149. doi: 10.1152/jn.1993.69.4.1136. [DOI] [PubMed] [Google Scholar]
- 71.Krauzlis RJ, Lisberger SG. Directional organization of eye movement and visual signals in the floccular lobe of the monkey cerebellum. Exp Brain Res. 1996;109(2):289–302. doi: 10.1007/BF00231788. [DOI] [PubMed] [Google Scholar]
- 72.Mano N, Ito Y, Shibutani H. Saccade-related Purkinje cells in the cerebellar hemispheres of the monkey. Exp Brain Res. 1991;84(3):465–470. doi: 10.1007/BF00230957. [DOI] [PubMed] [Google Scholar]
- 73.de Solages C, et al. High-frequency organization and synchrony of activity in the purkinje cell layer of the cerebellum. Neuron. 2008;58(5):775–788. doi: 10.1016/j.neuron.2008.05.008. [DOI] [PubMed] [Google Scholar]
- 74.Vincent P, Marty A. Neighboring cerebellar Purkinje cells communicate via retrograde inhibition of common presynaptic interneurons. Neuron. 1993;11(5):885–893. doi: 10.1016/0896-6273(93)90118-b. [DOI] [PubMed] [Google Scholar]
- 75.Chacron MJ, Pakdaman K, Longtin A. Interspike interval correlations, memory, adaptation, and refractoriness in a leaky integrate-and-fire model with threshold fatigue. Neural Comput. 2003;15(2):253–278. doi: 10.1162/089976603762552915. [DOI] [PubMed] [Google Scholar]
- 76.Callaway JC, Lasser-Ross N, Ross WN. IPSPs strongly inhibit climbing fiber-activated [Ca2+]i increases in the dendrites of cerebellar Purkinje neurons. J Neurosci. 1995;15(4):2777–2787. doi: 10.1523/JNEUROSCI.15-04-02777.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.