

Abstract
In this Letter, we describe a short, 6-step enantioselective route to spiroaminal lactam model systems reminiscent of marineosins A and B has been developed starting from either (R)- or (S)-hydroxysuccinic acid, respectively, in ~9% overall yield. This route enables late stage incorporation of the pyrrole ring at C5 via nucleophilic displacement of an iminium triflate salt.
Keywords: marineosin, iminium triflate, enantioselective, pyrrole, alkaloid
In 2008, Fenical and co-workers reported the discovery of two novel spiroaminals, marineosins A (1) and B (2), from a marine-derived Streptomyces-related actinomycete (Figure 1),1 and related to the prodigiosin family.2 Both 1 and 2 displayed inhibition of human colon carcinoma cell growth (HCT-116 IC50s of 0.5 μM and 46 μM, respectively).1 Fenical also proposed a biosynthesis of 1 and 2 that proceeded through an inverse-electron demand hetero Diels-Alder reaction with 3 to provide 4, which is then reduced to afford 1 and 2. We evaluated this biosynthetic proposal, and while 3 was accessible in high yield, we were unable to affect the intramolecular inverse-electron demand hetero Diels-Alder reaction under a variety of conditions. Attempts with multiple substrates for intermolecular variants were equally unsuccessful.3
Figure 1.
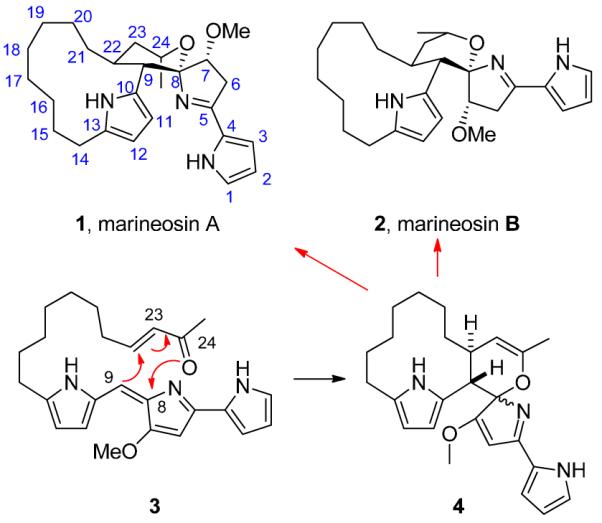
Structures and proposed biosynthesis of marineosins A (1) and B (2).
In 2010, Snider and co-workers proposed an alternative biosynthesis of 1 and 2 from undecylprodigiosin that only requires a single two-electron oxidation.4 Based on this proposal, Snider developed a seven step route to a model system 7 for the spiroiminal moiety from methylvalerolactone 5 (Scheme 1). While an important advance towards the synthesis of 1 and 2, we aimed to avoid long equilibration times, inseparable equilibrium mixtures, and, importantly, early installation of the pyrrole.4
Scheme 1.
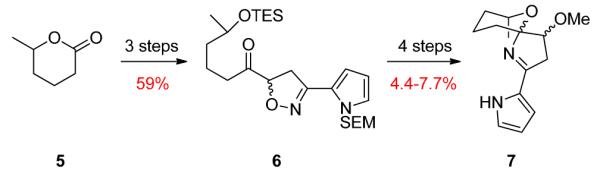
Snider’s Spiroiminal Model System.
After the unsuccessful biosynthetic approach,3 our lab has pursued multiple synthetic strategies en route to a total synthesis of 1 and 2. Uniformly, routes with early incorporation of the C1-C4 pyrrole moiety, led to reactivity/stability issues that forced abandonment of advanced intermediates and strategies. Based on this outcome, we decided to re-design our routes to install the pyrrole moiety as the final step of the synthesis (Scheme 2). To determine the viability of this new approach, we developed a short, enantioselective synthesis of two spiroiminal model systems of 1 and 2 (highlighted in red). This route enables late stage incorporation of the pyrrole ring at C5 via a novel application of nucleophilic displacement of an iminium triflate salt.
Scheme 2.
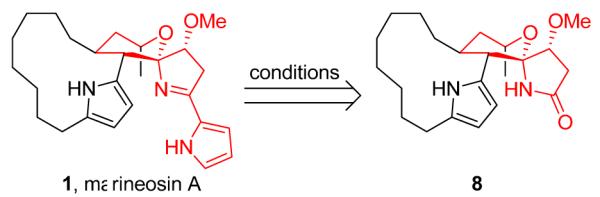
Envisioned Disconnection for the Synthesis of 1.
Our model system was inspired by the work of Huang for the construction of aza-spiropyran derivatives by the addition of functionalized Grignard reagents into maleimides.5 The synthesis of the proposed model system began with the requisite THP-protected bromobutanol 12 (Scheme 3) following a Grieco procedure.6 Here, tetrahydrofuran is opened with HBr to afford 10 in 75% yield. Protection as the THP ether afforded 12 in 90% yield, which is then converted into Grignard reagent 13.6
Scheme 3.
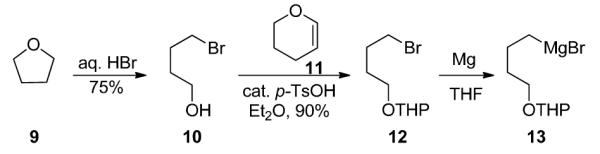
Synthesis of the Key Gringard Reagent 13.
With 13 in hand, we prepared the maleimide fragment relevant for a model system of 1.7,8 Starting from (R)-hydroxy succinic acid 14, refluxing in m-xylenes with p-methoxybenzyl amine 15 affords the desired maleimide 16 in 78% yield (Scheme 4). Silver oxide mediated alkylation with MeI in MeCN affords key coupling partner 17 in 78% yield. Addition of 13 into 17 provided hydroxy aminal 18 in 80% yield (Scheme 5).15
Scheme 4.
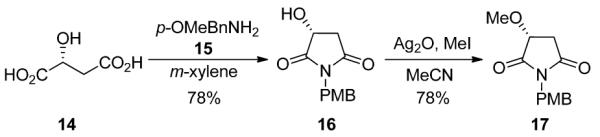
Synthesis of the Key Malimide 17.
Scheme 5.
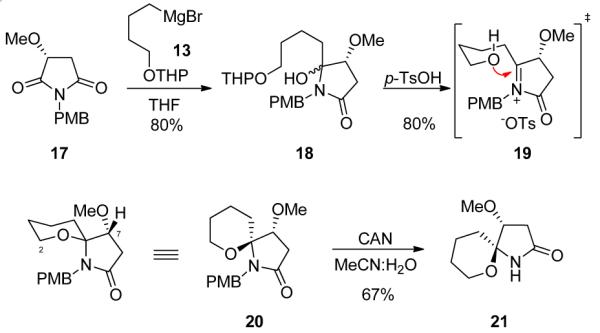
Synthesis of the Spiroaminal Moiety of 1.
Treatment with p-TsOH cleaves the THP ether and generates iminium salt 19 which is attacked by the free hydroxyl leading to formation of the spiroaminal 20 in 80% yield.16 Finally, ceric ammonium nitrate (CAN)-mediated removal of the p-methoxybenzyl (PMB) group provides lactam 21 in 67% yield.17 Model system 21 possessed the correct stereochemistry at C7 for 1, but the opposite absolute stereochemistry at C8. However, 21 is a valuable model from which to develop chemistry for the late stage installation of the pyrrole at C5, and not consume valuable late stage 8.
Stereochemical assignments of 20, with anti O-1,O-7 geometry, was made based on literature precedent and from extensive nOe studies (Figure 2).9
Figure 2.
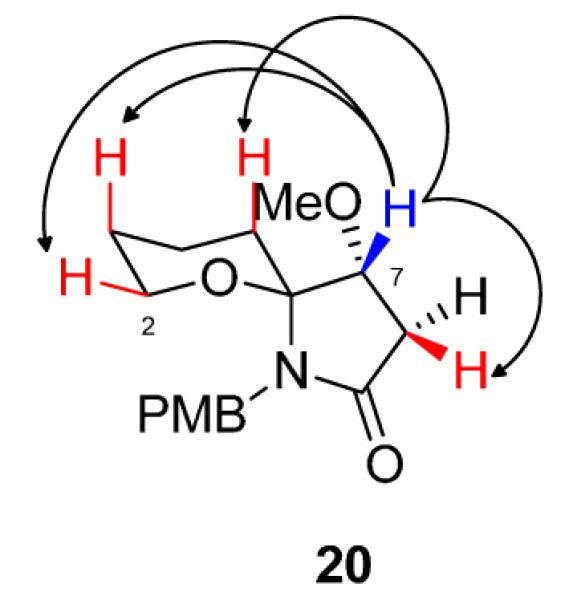
Diagnostic nOe correlations in the (6S,7R)-spiroaminal 20 model system reminiscent of 1.
With 21 in hand, we were poised to evaluate conditions to install the pyrrole moiety at C5 to validate our retrosynthetic approach aimed at accessing 8. Our initial thought was to install the pyrrole through classical Vilsmeier-type chemistry (POCl3/pyrrole);9 however, this failed to provide the desired 22. We surveyed a number of known strategies to convert the lactam carbonyl into a suitable electrophile, followed by treatment with pyrrole under a variety of reaction conditions, but none proved successful. The lactam was converted into the corresponding triflate 23 through treatment with Tf2O or PhNTf2, followed by a Suzuki coupling with various forms of N-protected, 2-pyrrole boronic acid. Unfortunately, all attempts with multiple Pd(0) and Ni(0) sources, bases, and solvents afforded either no product or only trace amounts of 22 (Scheme 6).
Scheme 6.
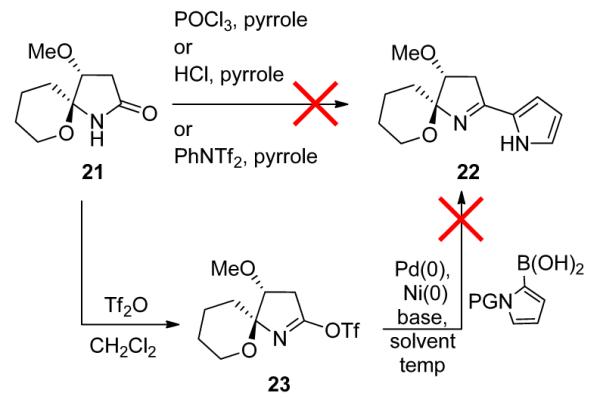
Attempts to Install the Pyrrole Moiety at C5.
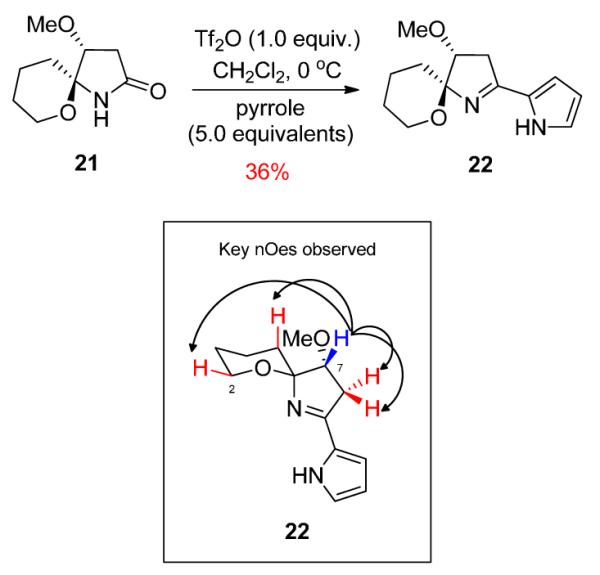
A deeper perusal of the literature led us to consider the chemistry of triflic anhydride/amide adducts, and the opportunity to potentially intercept the in situ generated triflate with the pyrrole nucleophile in a single pot reaction.10,11 It has been demonstrated that treatment of an indolin-2-one with Tf2O, to generate the iminium triflate salt, followed immediately by the additon of a functionalized indole affords the bis-indole product.11 With this lone precedent, we treated 21 with 2.0 equivalents of Tf2O, to generate the iminium triflate salt, followed by the addition of 5.0 equivalents of pyrrole in CH2Cl2 at 0 °C. Unfortunately, these conditions afforded only a trace (<5%) of the desired 22. Evaluation and refinement of the reaction conditions identified that employing 1.0 equivalent of Tf2O, to generate the iminium triflate salt, followed by the addition of 5.0 equivalents of pyrrole in CH2Cl2 at 0 °C did provide the desired model system 22 of marineosin A, 1, in 36% yield (Scheme 7).19 The stereochemistry was further confirmed at this stage by nOe studies on 22. Irradiation of H-7 supported the 6R,7S stereochemical assignment of 22; no equilibration to the syn O-1,O-7 isomer had occurred after installation of the pyrrole, even after a period of two weeks in CDCl3.4,9 Identical nOe data was seen in model system 22. Although the configuration of the spirocenter in model 22 is opposite to marineosins A, we envision that a syn O-1,O-7 isomer can be obtained by increasing the steric demands of the pyran ring through stereoselective functionalization of a carbon fragment similar to Grignard 13. Repetition of this sequence, starting from the (S)-hydroxy succinic acid, afforded the model system 24 reminiscent of marineosin B in ~9% overall yield. Once again, literature preceent and extensive nOe data confimred the sterochemcial assignment.
Scheme 7.
Late Stage Installation of the Pyrrole and Completion of the Model Systems of 1.
As both 1 and 2 displayed inhibition of human colon carcinoma (HCT-116 IC50s of 0.5 μM and 46 μM, resectively), and due to the fact that many related, bi- and tricyclic prodigiosin natural products have potent cytotoxicity,13,14 we evaluated 22 and 24 in our HCT-116 cytotoxicity assay in order to ascertain if the model systems represented a minimum pharmacophore for 1 and 2, respectively. Interestingly, both model systems were inactive in this assay, suggesting the larger construct, and/or stereochemical conformation, of 1 and 2 are important for the observed biological activity, thus warranting completion of the total synthesis of 1 and 2.
In summary, we have developed chemistry to enable late stage introduction of the pyrrole moiety at C5 in marineosin A (1) and B (2) via a novel application of the nucleophilic displacement of an iminium triflate salt by pyrrole. Moreover, we have performed an enantioselective synthesis of two spiroaminal model systems reminiscent of 1 and 2 starting from chiral pool molecules. Overall yields for both 22 and 24 averaged ~9% from commercial tetrahydrofuran. This synthetic approach is currently being applied to the total synthesis of 1, and results will be presented in due course.
Acknowledgments
This work was supported, in part, by the Department of Pharmacology (Vanderbilt University) and William K. Warren, Jr.. Funding for the NMR instrumentation was provided in part by a grant from NIH (S10 RR019022). The authors thank Brenda Crews (Marnett lab) for performing the HCT-116 viability/toxicity assays.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Boonlarppradab C, Kauffman CA, Jensen PR, Fenical W. Org. Lett. 2008;10:5505–5508. doi: 10.1021/ol8020644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fuerstner A. Angew. Chem. Int. Ed. 2003;42:3582–3603. doi: 10.1002/anie.200300582. [DOI] [PubMed] [Google Scholar]
- 3.Aldrich LN, Dawson EW, Lindsley CW. Org. Lett. 2010;12:1048–1051. doi: 10.1021/ol100034p. [DOI] [PubMed] [Google Scholar]
- 4.Cai X-C, Wu X, Snider BB. Org. Lett. 2010;12:1600–1603. doi: 10.1021/ol100333d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zheng J-F, Chen W, Huang S-Y, Ye J-L, Huang P-Q. Beilstein J. Org. Chem. 2007;3:1–6. doi: 10.1186/1860-5397-3-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grieco PA, Larsen SD. J. Org. Chem. 1986;51:3553–3555. [Google Scholar]
- 7.Naylor A, Judd DB, Scopes DIC, Hayes AG, Birch PJ. J. Med. Chem. 1994;37:2138–2144. doi: 10.1021/jm00040a004. [DOI] [PubMed] [Google Scholar]
- 8.Zheng J-L, Liu H, Zhang Y-F, Zhao W, Tong J-S, Ruan Y-P, Huang P-Q. Tetrahedron Asymm. 2011;22:257–263. [Google Scholar]
- 9.Rapoport H, Castagnoli N., Jr. J. Am. Chem. Soc. 1962;84:2178–2181. [Google Scholar]
- 10.Sforza S, Dossena A, Corradini R, Virgili E, Marchelli R. Tetrahedron Lett. 1998;39:711–714. [Google Scholar]
- 11.Baraznenok IL, Nenajdenko VG, Balenkova ES. Tetrahderon. 2000;56:3077–3119. [Google Scholar]
- 12.Black D.St. C., Ivory AJ, Kumar N. Tetrahedron. 1996;52:4697–4705. [Google Scholar]
- 13.Aldrich LN, Stoops SL, Crews BC, Marnett LJ, Lindsley CW. Bioorg. Med. Chem. Lett. 2010;20:5207–5211. doi: 10.1016/j.bmcl.2010.06.154. [DOI] [PubMed] [Google Scholar]
- 14.Melvin MS, Calcutt MW, Noftle RE, Manderville RA. Chem. Res. Toxicol. 2002;15:742–748. doi: 10.1021/tx025508p. [DOI] [PubMed] [Google Scholar]
- 15.A flame-dried flask was charged with magnesium powder (447 mg, 18.4 mmol) and placed under an inert argon atmosphere. The magnesium was suspended in anhydrous THF (21 mL). To this mixture was added pyran 12 (1.4 mL, 7.5 mmol). After warming to 50 °C, pyran 12 (2.0 mL, 10.7 mmol) was added dropwise. The reaction mixture was heated periodically until it sustained reflux. A separate flame-dried flask was charged with ether 17 (1.5 g, 6.0 mmol) and THF (30 mL). After cooling to −20 °C, the solution of Grignard 13 was added dropwise via syringe. The reaction mixture was kept between −10 °C and −15 °C. After 2.5 h, water (5 mL) was added and the reaction was allowed to reach rt. The product was extracted with diethyl ether (3 × 20 mL). The combined organics were washed with brine, dried over Na2SO4, and concentrated. The residue was purified on silica gel (30:70 EtOAc:hexanes) to provide 6.61 g (80%) of tertiary alcohol 18. 1H NMR (CDCl3, 400 MHz) δ 1.48-1.59 (m, 5H), 1.61-1.75 (m, 2H), 1.77-1.90 (m, 1H), 2.12-2.24 (m, 2H), 2.57 (d, J = 17.9 Hz, 1H), 2.71 (dddd, J = 1.9, 7.2, 17.8 Hz, 1H), 3.27 (s, 3H), 3.29-3.34 (m, 1H), 3.45-3.50 (m, 1H), 3.63-3.71 (m, 1H), 3.77 (s, 3H), 3.80-3.85 (m, 1H), 4.49-4.52 (m, 1H), 4.55-4.66 (m, 3H), 4.85 (q, J = 6.8 Hz, 1H), 6.81 (d, J = 8.6 Hz, 2H), 7.13 (d, J = 8.6 Hz, 2H). 13C NMR (CDCl3, 100 MHz) δ 19.6, 19.8, 23.4, 23.5, 25.4 (2C), 30.1, 30.2, 30.7, 30.8, 36.0 (2C), 42.9 (2C), 55.1, 55.2, 62.3, 62.6, 66.5, 66.7, 72.0 (2C), 98.9, 99.0, 107.0, 107.1, 113.9 (2C), 127.9, 128.3, 128.4, 139.2, 139.3, 158.8 (2C), 173.0, 173.1. HRMS (TOF ES+): C22H33NO6Na [M+Na]+ calcd 430.2206, found 430.2210. [α]22D= −15.0 (c 0.6, CHCl3).
- 16.To a stirred solution of tertiary alcohol 18 (390 mg, 1.0 mmol) in CH2Cl2 (6 mL) at 0 °C was added p-toluenesulfonic acid monohydrate (41 mg, 0.2 mmol). After 30 minutes the solvent was removed. The residue was purified on silica gel (30:70 EtOAc:hexanes) to provide 244 mg (80%) of spiroaminal 20. 1H NMR (CDCl3, 400 MHz) δ 1.39-1.51 (m, 4H), 1.63-1.70 (m, 1H), 1.85-1.92 (m, 1H), 2.47 (d, J = 17.4 Hz, 1H), 2.65 (dd, J = 5.5, 17.4 Hz, 1H), 3.32 (s, 3H), 3.61 (m, 1H), 3.73 (s, 3H), 3.84 (dd, J = 2.4, 11.2 Hz, 1H), 3.95 (d, J = 5.5 Hz, 1H), 4.11 (d, J = 16.0 Hz, 1H), 4.70 (d, J = 16.0 Hz, 1H), 6.78 (d, J = 8.7 Hz, 2H), 7.14 (d, J = 8.7 Hz, 2H). 13C NMR (CDCl3, 100 MHz) δ 20.0, 24.6, 27.9, 34.3, 41.8, 55.0, 56.6, 64.7, 74.9, 94.9, 113.5, 127.9, 130.5, 158.2, 174.5. HRMS (TOF, ES+): C17H24NO4 [M+H]+ calcd 306.1705, found 306.1702. [α]22D = −50.6 (c 1, CHCl3).
- 17.To a stirred solution of spiroaminal 20 (258 mg, 0.85 mmol) in acetonitrile (27 mL) and water (3.5 mL) was added ceric ammonium nitrate (1.4 g, 2.5 mmol). After 1.5 h, a second portion of ceric ammonium nitrate (467 mg, 0.8 mmol) was added. After 1 h, the acetonitrile was removed under reduced pressure. The product was extracted with CH2Cl2 (4 × 15 mL). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated. The residue was purified on silica gel (30:70 EtOAc/hexanes) to provide 105 mg (67%) of amide 21. 1H NMR (CDCl3, 400 MHz) δ 1.54-1.63 (m, 2H), 1.63-1.73 (m, 1H), 1.73-1.83 (m, 3H), 2.29 (dd, J = 1.7, 17.2 Hz, 1H), 2.70 (dd, J = 5.6, 17.2 Hz, 1H), 3.34 (s, 3H), 3.66-3.72 (m, 3H), 8.64 (bs, 1H). 13C NMR (CDCl3, 100 MHz) δ 19.4, 25.2, 29.3, 35.7, 57.3, 62.7, 82.4, 91.8, 177.3. HRMS (TOF, ES+): C9H16NO3[M+H]+ calcd 186.1130, found 186.1131. [α]22D = −97.1 (c 1.1, CHCl3).
- 18.A flame-dried flask was charged with amide 21 (64 mg, 0.3 mmol) and placed under an inert argon atmosphere. After cooling to 0° C, CH2Cl2 (4 mL) and trifluoromethanesulfonic anhydride (58.2 μL, 0.3 mmol) were added. After 2 minutes, pyrrole (119.8 μL, 1.7 mmol) was added. After 10 minutes, the reaction was quenched with saturated NaHCO3 and allowed to reach rt. The product was extracted with CH2Cl2 (3 × 5 mL). The combined organics were dried over Na2SO4 and concentrated. The residue was purified on silica gel (30:70 EtOAc/hexanes) to provide 29 mg (36%) of pyrrole 22. 1H NMR (CDCl3, 400 MHz) δ 1.54-1.78 (m, 5H), 1.86-1.99 (m, 2H), 2.78 (dd, J = 5.3, 16.5 Hz, 1H), 3.21 (dd, J = 6.8, 16.5 Hz, 1H), 3.44 (s, 3H), 3.72 (d, J = 10.8 Hz, 1H), 3.84 (t, J = 6.2 Hz, 1H), 4.15 (dt, J = 2.8, 11.1 Hz, 1H), 6.21 (t, J = 3.2 Hz, 1H), 6.57 (d, J = 3.5 Hz, 1H), 6.90 (s, 1H). 13C NMR (CDCl3, 100 MHz) δ 19.6, 25.8, 29.2, 38.7, 58.2, 64.2, 85.3, 102.8, 110.7, 112.8, 121.1, 126.3, 164.6. HRMS (TOF, ES+): C13H19N2O2 [M+H]+ calcd 235.1447, found 235.1447. [α]22D = −65.2 (c 1.6, CHCl3).