

Abstract
Background
The objective of this study was to prepare a novel fentanyl wafer formulation by a freeze-drying method, and to evaluate its in vitro and in vivo release characteristics, including its bioavailability via the sublingual route.
Methods
The wafer formulation was prepared by freeze-drying an aqueous dispersion of fentanyl containing sodium carboxymethylcellulose and amylogum as matrix formers. Uniformity of weight, friability, and dissolution testing of the fentanyl wafer was achieved using standard methods, and the residual moisture content was measured. The fentanyl wafer was also examined using scanning electron microscopy and x-ray diffraction. The absolute bioavailability of the fentanyl wafer was evaluated in 11 opioid-naïve adult female patients using a randomized crossover design.
Results
In vitro release showed that almost 90% of the fentanyl dissolved in one minute. In vivo, the first detectable plasma fentanyl concentration was observed after 3.5 minutes and the peak plasma concentration between 61.5 and 67 minutes. The median absolute bioavailability was 53.0%.
Conclusion
These results indicate that this wafer has potential as an alternative sublingual fentanyl formulation.
Keywords: absolute bioavailability, fentanyl wafer, in vitro dissolution, in vivo study, pharmacokinetics, sublingual
Video abstract
Introduction
Fentanyl is an opioid analgesic suitable for adult and pediatric patients. It is commonly used in doses of 1–2 μg per kg per dose in children, or 50–100 μg in adults, every 2–3 hours as required.1 Among the current products on the market, fentanyl is the only opioid formulated for transmucosal administration. The lollipop or lozenge formulation (Actiq®, Abbott Laboratories Inc, Abbott Park, IL, USA), known as oral transmucosal fentanyl citrate, was designed to allow rapid absorption of fentanyl through the mucosa as a result of its lipophilicity and appropriate molecular weight.23 However, this dosage form stimulates saliva production and swallowing, such that some fentanyl is swallowed and metabolized due to a significant first-pass effect. Typical opioid dose-related side effects occur.4
An effervescent buccal fentanyl tablet (Fentora®, Cephalon Inc, Frazer, PA, USA) is based on a proprietary OraVescent® drug delivery system (Cima Labs Inc, Brooklyn Park, MN, USA) that changes the local pH, the purpose of which is to enhance the absorption of fentanyl through the buccal mucosa. Pharmacokinetic data from effervescent buccal fentanyl tablet studies show that fentanyl is rapidly absorbed, with a time to maximum concentration of 35–45 minutes and an average onset of analgesia at approximately 15 minutes. The side effects are also typical of opioid analgesics.5 Another formulation, developed as a buccal-soluble fentanyl film, shows an absolute bioavailability of 71%.6 Fentanyl is clearly an effective analgesic when administered via the oral cavity.
Interest in noninvasive but fast-acting formulations for opioid administration has generated investigation of the sublingual route.7,8 Compared with the transmucosal and buccal routes, the sublingual route may be even more attractive for fentanyl delivery.9–11 Clinical studies have shown that sublingual fentanyl formulations give rise to detectable plasma concentrations (Cfirst) 8–11 minutes after administration, but there are no data on its bioavailability.10 We observed that a sublingual fentanyl wafer had an absolute bioavailability of 78.9% in healthy volunteers.11 In addition, fentanyl is particularly suitable for sublingual administration because it lacks the bitter taste of some other opioids.12
This study describes the development and evaluation of the physicochemical properties of a sublingual fentanyl wafer, including a pilot investigation of its bioavailability and pharmacokinetic profile in surgical patients.
Materials and methods
Materials
Fentanyl powder was purchased from Professional Compounding Centers of America (Houston, TX, USA). Intravenous fentanyl was purchased from Mayne Pharma Ltd (Melbourne, Australia) as Fentanyl DBL 500 μg in 10 mL. Amylogum® CLS was obtained from Avebe Food (Veendam, The Netherlands). D-Mannitol, D-lactose, and glycine were supplied by Sigma-Aldrich (St Louis, MO, USA). Sodium carboxymethylcellulose was purchased from BDH (Poole, UK). Avicel® PH 101 and polyethylene glycol 2000 were obtained from Fluka (Steinheim, Germany). All other chemicals were purchased commercially as analytical grade reagents.
Preparation of fentanyl wafer
The fentanyl (0.12% as the citrate salt) wafer was prepared by freeze-drying an aqueous solution using carboxymethylcellulose (1.62% w/w) and Amylogum (13.16% w/w) as matrix forming agents, lactose (30.87% w/w), mannitol (46.48% w/w), and microcrystalline cellulose (Avicel PH101, 1.55% w/w) as filling agents, glycine (3.1% w/w) as an anticollapsing agent, and polyethylene glycol 2000 (3.1% w/w) as a lubricant. The formulae for the ingredients were all dry weight. Amylogum and carboxymethylcellulose were first dissolved in MilliQ water (Millipore, Bedford, MA, USA) at 50°C, and when the solution had cooled to room temperature, polyethylene glycol 2000, glycine, Avicel PH101, lactose, and mannitol were added individually, stirring mechanically to obtain a homogenous mixture. Fentanyl (as the citrate salt) was subsequently dissolved in the mixture by stirring (freely soluble; the solubility of the citrate salt in water is 1 in 40 mL).
The resulting mixture was transferred by pipette and accurately weighed into preformed foil blister pack wells, resulting in a fentanyl dose of 50 μg per wafer, before freezing at −30°C for approximately 24 hours. After freezing, the sample was freeze-dried (Dynavac Engineering Pty Ltd, Bayswater, Australia) for 24 hours. The prepared sample was subsequently stored in a desiccator over silica gel at a room temperature of 22°C ± 1°C.
Characterization of wafers
Uniformity of weight
The uniformity of the weight of the fentanyl wafer was tested in accordance with the British Pharmacopoeia 2009 test.13 That is, 20 wafers were individually weighed, and the average weight and relative standard deviation was calculated.
Friability
The strength of the fentanyl wafer, ie, its ability to be reduced from a solid matrix into smaller pieces or powder, was measured. The test13 was also conducted according to the method (ie, friability of uncoated tablets), using a friability tester (Erweka GmbH, Heusenstamm, Germany). A sample of 20 fentanyl wafers was weighed accurately and placed in the apparatus. A rotation time of 4 minutes at 25 rpm was used. Whole fentanyl wafers were removed and reweighed, and the percentage weight loss was calculated.
Moisture analysis
The moisture content of the fentanyl wafer after freeze-drying was analyzed using a 870 Karl Fisher Titrino Plus device (Metrohm Ag, Filderstadt, Germany). A fentanyl wafer was weighed and powdered, then dispensed into the cell containing Hydranal® methanol (Sigma-Aldrich) and titrated using Hydranal Composite A. The test results are presented as the mean ± standard deviation (n = 4).
Scanning electron microscopy
Surface morphology and a cross-section of samples from randomly selected wafers were observed by scanning electron microscopy (Zeiss, EVO 40 XVP, Oxford Instruments, Abingdon, UK). Cross-sectional samples were prepared by cutting a thin slice of the wafer using a scalpel. Samples were then coated with carbon prior to examination. The accelerating voltage was 10 kV.
Powder x-ray diffraction
Powder x-ray diffraction experiments were performed using a D8 Advance instrument (Bruker, Karlsrube, Germany) with a LynEye detector. The radiation used was nickel-filtered CuKα, which was generated using an acceleration voltage of 40 kV and a cathode current of 40 mA. The samples were scanned over a 2-theta range of 7.5° to 70°, with a counting time of one second per 0.02°.
In vitro dissolution studies
Dissolution tests were carried out using BP apparatus I. The temperature of the medium was 37°C ± 0.5°C. A fentanyl wafer was used to determine the amount of drug release from the formulation. Dissolution rates for the fentanyl wafer were determined in 200 mL of phosphate-buffered solution (25 mM, pH 6.8) with a basket rotation speed of 75 rpm. At given intervals (ie, 0.5, 1, 5, and 10 minutes), 1.0 mL samples were replaced with an equal volume of fresh medium. The drug released was measured by high-pressure liquid chromatography using a C18 column (150 × 4.6 mm, 5 μm, Alltech Associates Inc, Deerfield IL, USA), a mobile phase of methanol to 0.04 M phosphoric acid (50:50, v/v, pH 3.0) and a flow rate of 1.2 mL per minute. The monitoring wavelength was at 210 nm.9 The calibration curve for concentrations in the range of 5–100 μg/mL (seven-point calibration) was linear. The coefficient of variation for the high-pressure liquid chromatography assay was 0.57% at a fentanyl concentration of 0.5 μg/mL (n = 8) and 0.54% at a fentanyl concentration of 4 μg/mL (n = 8).
In vivo study
Study subjects and design
After receiving The Women and Newborn Health Service of King Edward Memorial Hospital for Women ethics committee approval, the trial was registered with the Australian Therapeutic Goods Administration under the Clinical Trial Notification scheme and with the Australian and New Zealand Clinical Trials Registry (12609000743268). Twenty-one adult female opioid-naïve patients requiring major gynecological surgery gave their written informed consent to participate in the study. All patients were free of sublingual/buccal ulceration or disease, weighed 50–100 kg, and consented to general anesthesia including intravenous morphine as the sole opioid followed by patient-controlled intravenous morphine.
Sampling and data collection
Each patient was assigned to receive the equivalent of 50 μg of fentanyl by both the sublingual and intravenous routes, with the order of administration randomized according to a Latin crossover square design and allocated using sealed opaque envelopes.
Patients given a fentanyl wafer were requested to avoid swallowing for at least 10 minutes to minimize loss of fentanyl via the oral route. A dedicated intravenous cannula was placed in the forearm for subsequent venous blood sampling. Blood samples (5 mL) were collected into sterile polypropylene tubes at 0, 2, 5, 10, 15, 30, 60, 120, and 180 minutes after fentanyl administration. Self-administration of further doses of morphine was permitted at any time for the duration of the trial to ensure adequate pain relief.
After collection, the blood samples were immediately centrifuged at 4°C and 2000–2500 g for 15 minutes. The plasma was then extracted and placed into polypropylene tubes for storage at −70°C until analysis.
Pharmacokinetic analysis
Sample extracts were analyzed on an API 4000 LC-MS/MS analyzer (Applied Biosystems, Foster City, CA, USA), preceded by a Prominence HPLC system (Shimadzu, Tokyo, Japan) with d5-fentanyl as the internal standard. This assay has a limit of detection of 0.01 ng/mL. Precision was determined by duplicate analyses of plasma containing 0.03, 2.00, and 8.00 ng/mL fentanyl. The results were precise to within ±3.01%, ±1.76%, and ±2.16% of the mean measured concentrations of 0.03, 2.14, and 8.47 ng/mL, respectively, and accurate to within 6.67%, 7.00%, and 5.87% of the nominal concentrations of 0.03, 2.00, and 8.00 ng/mL, respectively. At each concentration, the number of replicates was six.
The following parameters were determined by model-independent pharmacokinetic analyses of the plasma fentanyl concentration-time profiles, using WinNonlin pharmacokinetic program version 4.1 (Pharsight Corporation, Mountain View, CA, USA). The pharmacokinetic parameters, including maximum plasma concentration (Cmax), Cfirst, time taken to reach maximum plasma concentration tmax, and time to Cfirst (tfirst) were read directly from the plasma fentanyl concentration-time curves from each subject.
The terminal elimination rate constant (kel) was determined as the slope of the regression line of best fit to the approximately log-linear terminal elimination phase. All fitting was performed with unity weighting of the data. The terminal elimination half-life (t1/2) was obtained from kel and equaled ln 2/kel. The area under the curve from 0 to 3 hours (AUC0–180 ) and AUC0–t values were obtained using the trapezoidal rule. Extrapolation to AUC0–∞ was calculated from AUC0–t + Ct/kel.
The absolute bioavailability (F) of fentanyl was calculated using the following equation:
Clinical evaluation of wafers
In vivo dissolution time of the wafer was assessed by frequent inspection of the sublingual area. Patient-rated effectiveness (four-point scale: excellent, good fair, poor), acceptability (four-point Likert scale: unacceptable to very acceptable), and satisfaction (numerical 0–10, verbal rating scale) were assessed.
Statistical analysis
Continuous demographic data were summarized using the median and interquartile range (IQR), and categorical data were summarized using frequency distributions. SAS version 9.2 (SAS Institute Inc, Cary, NC, USA) statistical software was used for the data analysis. All tests were two-sided and P values < 0.05 were considered to be statistically significant.
Results and discussion
Characterization of fentanyl wafers
The wafers prepared showed a weight variation of 65.2 ± 0.89 mg, and the fentanyl content was 96.3% ± 1.05%. The friability test gave a weight loss for 20 wafers of 8.65%. Although this weight loss does not comply with British Pharmacopoeia 2009 standard13 for compressed tablets, there is no such standard for wafers in either the British or US pharmacopoeias.14–16 This implies that the wafers would not withstand robust handling, but in practice they did not crumble when handled by patients. The diameter and thickness of the wafers were approximately 10 mm and 4 mm, respectively. The moisture content was 3.51% ± 0.10%, indicating that water had been removed efficiently by the freeze-drying process. Stability studies showed that the fentanyl wafers remained stable for more than 2 years when stored at room temperature (22°C ± 1°C) in a desiccator.
Scanning electron microscopy
Two cross-sectional scanning electron microscopic images are shown in Figure 1. Both the blank and fentanyl wafers formed highly porous inner structures, supporting the rapid disintegration time and dissolution profiles. In addition, there were no noticeable morphological differences between the blank and fentanyl-containing wafers, indicating that the fentanyl was homogenously dispersed and did not change the matrix structure.
Figure 1.

Scanning electron micrographs of a cross-section of a (A) blank wafer and (B) fentanyl wafer.
Powder x-ray diffraction
The physical state of the materials in the wafer was evident in the x-ray diffraction spectra. Spectra for the fentanyl, blank matrix, and fentanyl wafer are shown in Figure 2. It was observed that the powder patterns of all wafers prepared were dominated by intense scattering peaks located at approximately 2-theta of 9.58°, 19.68°, and 20.05°, indicating the crystalline nature of the excipient, Avicel. Absence of the characteristic peaks for fentanyl pointed to amorphization of fentanyl during the freeze-drying process. This finding was also supported by the data generated from the scanning electron microscopic analysis. Indeed the excipients used in the formulations, including glycine, lactose, mannitol and microcrystalline cellulose, were crystalline in nature, but amorphization appeared to have occurred during freeze-drying.
Figure 2.

Powder x-ray diffraction spectra (A) blank wafer, (B) fentanyl citrate and (C) fentanyl wafer.
In vitro dissolution studies
The dissolution profiles are shown in Figure 3. The cumulative percentage of fentanyl dissolved from four wafers was approximately 90% within one minute. Total release occurred within five minutes. This result indicates that the fentanyl wafer was a solid dispersion of fentanyl within a porous matrix that permits rapid fluid disintegration and release of the contents. The fentanyl wafer formulation complied with the British Pharmacopoeia standard for orodispersible tablets of disintegration within 3 minutes. An excellent correlation has been reported between in vitro and oral disintegration times in volunteers taking rapidly disintegrating tablets.17
Figure 3.
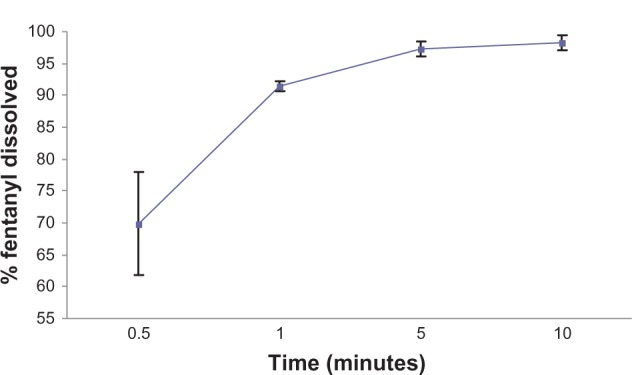
Dissolution profiles for the fentanyl wafer formulation in phosphate-buffered solution at pH 6.8 (average with error bars) at 37°C (n = 4).
In vivo studies
Twenty-one patients were randomized, 11 to the sublingual followed by intravenous sequence and 10 to the intravenous followed by sublingual sequence. Of these 21 patients, 11 received both sublingual and intravenous fentanyl; six patients were only given intravenous fentanyl (sublingual fentanyl was subsequently not required because of no further pain) and two patients received only sublingual fentanyl (intravenous fentanyl was not subsequently required). Two patients did not complete the sublingual and/or the intravenous administration arm of the study and were eliminated from all analyses. The demographic characteristics of the patients are reported in Table 1.
Table 1.
Demographic characteristics of the study patients
| Number# | 11 |
| Mean age in years (range) | 45 (21–75) |
| Mean weight in kg (range) | 69 (50–100) |
| Study dose of fentanyl (μg) | 50 |
| Gynecological abdominal surgery | 76% |
| Vaginal surgery | 24% |
Notes:
Two patients did not complete the sublingual and/or the intravenous administration arm of the study. Also not included were six patients receiving only intravenous fentanyl and two patients only sublingual fentanyl.
On commencing the study, it was noted that the wafer did not disintegrate/dissolve in the first five patients until approximately 12 minutes, as a result of dry mouth immediately after general anesthesia. Subsequently, the study patients were given a mouth rinse using 3 mL of water before placing the wafer sublingually, and this reduced the mean disintegration time to 6.6 minutes. Accordingly, the pharmacokinetic results were later divided into two subgroups, ie, a dry mouth group and a wet mouth group.
Pharmacokinetic data
Plasma fentanyl concentration versus time curves for the intravenous and sublingual routes are shown in Figure 4. The median (±IQR) AUCinf data for intravenous and sublingual administration are shown in Table 2. With respect to bioavailability and tmax, the pharmacokinetic data for the sublingual 50 μg fentanyl wafer were generally similar to those previously reported for a sublingual fentanyl 100 μg tablet and wafer.10,11 The Cmax (0.11 ng/mL) of the 50 μg fentanyl wafer was approximately half that reported for a double-strength sublingual fentanyl tablet and wafer, namely 0.24 ng/mL and 0.22 ng/mL, respectively. Compared with previous data, the tmax of the 50 μg fentanyl wafer tended to be longer (120 and 61 minutes for the dry and wet mouth groups, respectively) compared with 39.7 ± 17.4 minutes for the 100 μg sublingual fentanyl tablet and 54.6 ± 0.73 minutes for the 100 μg wafer. The median (IQR) bioavailability of the sublingual wafer was 53.0% (IQR 51.4%) for the wet mouth group (See Table 3 for all bioavailability results), which is comparable with the 65% bioavailability of an effervescent buccal fentanyl tablet18 and the 47% bioavailability of oral transmucosal fentanyl citrate.19 The buccal mucosa is thicker (500–800 μm) than the sublingual mucosa (100–200 μm),20 which may explain the more prolonged absorption phase and elimination half-life reported for oral transmucosal fentanyl citrate.
Figure 4.

Median (±interquartile range) plasma concentration (ng/mL) over time profiles for sublingual fentanyl wafer and intravenous fentanyl (given as an intravenous push over one minute).
Note: Inset figure shows profiles of the wafer administered to the dry mouth and wet mouth groups.
Table 2.
Median values (IQR) of plasma pharmacokinetic parameters for fentanyl
| Pharmacokinetic parameter | Sublingual fentanyl wafer (n = 11)
|
Intravenous fentanyl (n = 12) | |
|---|---|---|---|
| Dry mouth (n = 5) | Wet mouth (n = 6) | ||
| AUCinf (ng/mL/min), median | 16.7 (15.2) | 20.4 (19.8) | 38.5 (32.4) |
| AUCt (ng/mL/min), median | NA | 15.2 (14.5) | 27.7 (20.8) |
| tfirst (min), median | 30 (15) | 3.5 (8.8) | NA |
| Cmax (ng/mL), median | 0.11 (0.08) | 0.12 (0.11) | 0.87 (0.69) |
| tmax (min), median | 67 (60) | 61.5 (60) | 2 (2) |
| t1/2 (h), median | NA | 4.9 (3.4) | 1.7 (1.5) |
| Bioavailability (%) using AUCinf | NA | 53.0 | NA |
Abbreviations: AUC, area under the concentration-time curve; NA, not applicable; IQR, interquartile range.
Table 3.
Median values (IQR) of the absolute bioavailability of fentanyl 50 μg wafer
| Dry mouth | Wet mouth |
|---|---|
| Absolute bioavailability (%) of fentanyl 50 μg wafer | |
| 46.7 | 60.6 |
| N/A | 53.2 |
| 54.5 | 52.8 |
| N/A | 51.0 |
| N/A | 50.5 |
| N/A | 54.5 |
| Median (IQR) = 53.0 (51.4) | |
Abbreviations: IQR, interquartile range; N/A, not available (inability to obtain three final points for calculating AUCt).
The lower bioavailability found in this study compared with that from a study in human volunteers11 probably related to the different durations of these studies. The human volunteer study had a duration of 24 hours, whereas this study was for 3 hours. This may have affected the AUCinf values, leading to the lower bioavailability.
Cfirst after sublingual administration was observed at 30 (IQR 15) minutes in the dry mouth group and 3.5 (IQR 8.8) minutes in the wet mouth group (Table 2). This was expected because a dry mouth markedly delayed wafer dissolution, preventing fentanyl being available for sublingual absorption.
The tfirst of 3.5 minutes in the wet mouth group was similar to that in a 100 μg fentanyl wafer group (2–10 minutes)11 and faster than that from a sublingual tablet (8–11 minutes).10 This reflects the high permeability of fentanyl in the rich blood flow of the sublingual mucosa and avoidance of the hepatic “first-pass” effect.21,22 The t1/2 was considerably shorter after intravenous administration, being 1.7 (IQR 1.5) hours compared with 4.9 (IQR 3.4) hours in the wet mouth group. This may have been influenced by the relatively short sampling time.
Clinical results
The median time taken for the sublingual wafer to dissolve was 5 (range 1–12) minutes. The median satisfaction score was 9 (range 2–10), with 80% reporting that it dissolved completely and 87% reporting that there was no bitter or unpleasant taste. Effectiveness was rated as excellent by 31%, good by 54%, fair by 8%, and poor by 8%, compared with reports of excellent by 56% and good by 44% after intravenous fentanyl administration. Sixty-two percent found the wafer to be very acceptable, 33% found it acceptable, and 5% found it reasonably acceptable. Among nine patients who were able to compare the sublingual and intravenous routes, 33% (three of nine) expressed a preference for the sublingual wafer, 44% (four of nine) a preference for the intravenous injection, and 22% (two out of nine) had no preference.
There are several limitations to the design of this study, which reflect its pilot nature. These included the limited clinical information that could be obtained from a pharmacokinetic study with a small sample size within a heterogeneous surgical population, and the clinical difficulty in obtaining data for both intravenous and sublingual administration from all participants. It is noteworthy that dry mouth is a critical factor in drug delivery by the sublingual route if a rapid response is desired. This finding should be noted for other drugs, such as glyceryl trinitrate, where a rapid response is important.
Conclusion
The fentanyl wafer studied was a solid dispersion of amorphized fentanyl in a porous matrix. The fast in vitro release pattern correlated well with in vivo absorption profiles. Sublingual administration of the fentanyl wafer resulted in rapidly detectable plasma fentanyl concentrations (at a mean of 3.5 minutes) after administration to patients with a moist mouth. The absolute bioavailability of the fentanyl wafer was approximately 53%. The high interindividual variability of tfirst was due to slow dissolution of the wafer in patients with a dry mouth postoperatively. Advice to rinse the mouth with water prior to insertion may be advisable using other sublingual products for which a rapid onset of action is desired.
Acknowledgments
The authors would like to thank iX BioPharma Pte Ltd, Singapore, for financing this project, and the Department of Physics, Curtin University, for scanning electron microscopy and x-ray analysis. Moreover, we wish to thank the study coordinators, Desiree Cavill and Tracy Bingham, anesthetic research fellows Aneeta Sinha, David Law, and Rupert Ledger, and the recovery room and ward nursing staff.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.AHFS Drug Information . Fentanyl Citrate Monograph. Vol. 98. Bethesda, MD: American Society of Health-System Pharmacist; 1996. pp. 1466–1470. [Google Scholar]
- 2.Mock DL, Streisand JB, Hague B, et al. Transmucosal narcotic delivery: an evaluation of fentanyl (lollipop) premedication in man. Anesth Analg. 1986;65:S102. [Google Scholar]
- 3.Lee M, Kern SE, Kisicki JC, Egan D. A pharmacokinetic study to compare two simultaneous 400 μg doses with a single 800 μg dose of oral transmucosal fentanyl citrate. J Pain Symptom Manage. 2003;26:743–747. doi: 10.1016/s0885-3924(03)00241-0. [DOI] [PubMed] [Google Scholar]
- 4.Zhang H, Zhang J, Streisand J. Oral mucosal drug delivery clinical pharmacokinetics and therapeutic applications. Clin Pharmacokinet. 2002;41:661–680. doi: 10.2165/00003088-200241090-00003. [DOI] [PubMed] [Google Scholar]
- 5.Lichtor JL, Sevarino FB, Joshi GP, Busch MA, Nordbrock E, Ginsberg B. The relative potency of oral transmucosal fentanyl citrate compared with intravenous morphine in the treatment of moderate to severe postoperative pain. Anesth Analg. 1999;89:732–738. doi: 10.1097/00000539-199909000-00038. [DOI] [PubMed] [Google Scholar]
- 6.Vasisht N, Gever LN, Tagarro I, Finn AL. Single-dose pharmacokinetics of fentanyl buccal soluble film. Pain Med. 2010;11:1017–1023. doi: 10.1111/j.1526-4637.2010.00875.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reisfield GM, Wilson GR. Rational use of sublingual opioids in palliative medicine. J Palliat Med. 2007;10:465–475. doi: 10.1089/jpm.2006.0150. [DOI] [PubMed] [Google Scholar]
- 8.Überall MA, Müller-Schwefe GH. Sublingual fentanyl orally disintegrating tablet in daily practice: efficacy, safety and tolerability in patients with breakthrough cancer pain. Curr Med Res Opin. 2011;27:1385–1394. doi: 10.1185/03007995.2011.583231. [DOI] [PubMed] [Google Scholar]
- 9.Bredenberg S, Duberg M, Lennernäs B, et al. In vitro and pharmacokinetic evaluation of a new sublingual tablet system for rapid oromucosal absorption using fentanyl citrate as the active substance. Eur J Pharm Sci. 2003;20:327–334. doi: 10.1016/j.ejps.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 10.Lennernas B, Hedner T, Holmberg M, Bredenberg S, Nystrom C, Lennernas H. Pharmacokinetics and tolerability of different doses of fentanyl following sublingual administration of a rapidly dissolving tablet to cancer patients: a new approach to treatment of incident pain. Br J Clin Pharmacol. 2004;59:249–253. doi: 10.1111/j.1365-2125.2004.02264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lim CBS, Schug SA, Sunderland VB, Paech JM, Liu Y. A phase I pharmacokinetic and bioavailability study of a sublingual fentanyl wafer in healthy volunteers. Anesth Analg. 2012;115:554–559. doi: 10.1213/ANE.0b013e3182575cbf. [DOI] [PubMed] [Google Scholar]
- 12.Gardner-Nix J. Oral transmucosal fentanyl and sufentanil for incident pain. J Pain Symptom Manage. 2001;22:627–630. doi: 10.1016/s0885-3924(01)00321-9. [DOI] [PubMed] [Google Scholar]
- 13.British Pharmacopoeia Commission . British Pharmacopoeia 2009. London, UK: The Stationery Office; 2008. [Google Scholar]
- 14.Garsuch V, Breitkreutz J. Novel analytical methods for the characterization of oral wafers. Eur J Pharm Biopharm. 2009;73:195–201. doi: 10.1016/j.ejpb.2009.05.010. [DOI] [PubMed] [Google Scholar]
- 15.Boateng JS, Matthews KH, Auffret AD, Humphrey MJ, Stevens HNE, Eccleston GM. In vitro drug release studies of polymeric freeze-dried wafers and solvent-cast films using paracetamol as model soluble drug. Int J Pharm. 2009;378:66–72. doi: 10.1016/j.ijpharm.2009.05.038. [DOI] [PubMed] [Google Scholar]
- 16.Boateng JS, Auffret AD, Matthews KH, Humphrey MJ, Stevens HNE, Eccleston GM. Characterisation of freeze-dried wafers and solvent evaporated films as potential drug delivery system to mucosal surfaces. Int J Pharm. 2010;389:24–31. doi: 10.1016/j.ijpharm.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 17.Abdelbary G, Eouani C, Prinderre P, Joachim J, Reynier J, Piccerelle P. Determination of the in vitro disintegration profile of rapidly disintegrating tablets and correlation with oral disintegration. Int J Pharm. 2005;292:29–41. doi: 10.1016/j.ijpharm.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 18.Darwish M, Kirby M, Robertson P, Tracewell W, Jiang JG. Absolute and relative bioavailability of fentanyl buccal tablet and oral transmucosal fentanyl citrate. J Clin Pharmacol. 2007;47:343–350. doi: 10.1177/0091270006297749. [DOI] [PubMed] [Google Scholar]
- 19.Aronoff G, Brennan M, Pritchard D, Ginsberg B. Evidence-based oral transmucosal fentanyl citrate (OTFC) dosing guide lines. Pain Med. 2005;6:305–314. doi: 10.1111/j.1526-4637.2005.00045.x. [DOI] [PubMed] [Google Scholar]
- 20.Shojaei A. Buccal mucosa as a route for systemic drug delivery. J Pharm Sci. 1998;1:15–30. [PubMed] [Google Scholar]
- 21.Ghosh TK, Pfister WR. Drug Delivery to the Oral Cavity. Boca Raton, FL: CRC Press; 2005. [Google Scholar]
- 22.Mercadante S, Radbruck L, Davies A, et al. A comparison of intranasal fentanyl spray with oral transmucosal fentanyl citrate for the treatment of breakthrough cancer pain: an open-label, randomized, crossover trial. Curr Med Res Opin. 2009;25:2805–2815. doi: 10.1185/03007990903336135. [DOI] [PubMed] [Google Scholar]