

Abstract
In Sjögren’s Syndrome (SS), inherent glandular defects, autoimmunity, and mononuclear cell infiltration within the salivary glands cause reduced salivation leading to xerostomia. Excessive production of type I interferons (IFN), triggered by environmental and genetic factors, is considered pathogenic in this disorder. However, whether type I IFN production is causative or an outcome of the disease process is not known. To address this question, we introduced a deficiency of interferon alpha receptor 1 (Ifnar1) into B6.Aec1Aec2 mice, which are known to have the genetic loci necessary for developing a SS-like disorder. This new mouse strain, B6.Aec1Aec2Ifnar1-/-, lacking type I IFN-mediated signaling, was characterized for pilocarpine-induced salivation, the presence of serum autoantibodies, sialoadenitis, and dacryoadenitis. Compared with the B6.Aec1Aec2Ifnar1+/+ (wild-type) mice, the B6.Aec1Aec2Ifnar1-/- (knockout) mice had significantly lower mononuclear cell infiltration in the salivary and lacrimal glands. The knockout mice were completely protected from salivary gland dysfunction. Surprisingly, they had a robust autoantibody response comparable with that of the wild-type mice. These findings demonstrate that, in the absence of type I IFN-mediated signaling, systemic autoantibody responses can be dissociated from glandular pathology. Our study suggests that, in genetically susceptible individuals, the type I IFN pathway can instigate certain features of SS.
Keywords: xerostomia, sialoadenitis, autoimmunity, salivary gland, mouse, interferons
Introduction
Xerostomia or dry mouth is one of the characteristic features of Sjögren’s Syndrome (SS) and affects several aspects of oral health (Carr et al., 2012). The lack of sufficient saliva often leads to an increased incidence of dental caries and periodontal disease, necessitating frequent interventions by a dental professional. Moreover, dry mouth causes difficulty in speaking and the chewing and swallowing of food and leads to a decreased quality of life.
Salivary gland dysfunction in SS has been attributed to a systemic as well as a localized autoimmune response within the salivary glands (Kang et al., 2011). While the systemic autoimmunity in SS is highlighted by the detection of autoantibodies in sera, the presence of inflammatory cells within the salivary glands is indicative of a localized immune response in the disease. Although it has been difficult to pinpoint the precise triggers responsible for initiating SS, viral infections causing excessive production of type I interferons (IFNs) have long been the suspects (Vakaloglou and Mavragani, 2011). Type I IFNs consist of several IFN proteins that bind and signal through a common interferon alpha receptor (IFNAR) (Trinchieri, 2010). Engagement of IFNAR eventually leads to the activation of a multitude of genes, known as the IFN-responsive genes, which affect several aspects of cellular metabolism. Increased expression levels of IFN-responsive genes in SS patients have led to the suggestion that type I IFNs play an important role in the pathogenesis of this disease (Hjelmervik et al., 2005; Emamian et al., 2009). Paradoxically, reduced levels of circulating IFN-α in SS patients have been used as a rationale for clinical trials with IFN-α. However, these trials have reported variable effects on the disease (Shirota et al., 2008). Thus, whether type I IFN response is causative for SS is difficult to determine in patients.
Previous work from our laboratory has demonstrated that activation of the type I IFN pathway through the Toll-like receptor 3 (TLR3) agonist poly(I:C) rapidly leads to reversible salivary gland dysfunction in mice (Deshmukh et al., 2009). This dysfunction did not occur in mice lacking IFNAR (Nandula et al., 2013). In addition, poly(I:C) treatment accelerated and increased the severity of sialoadenitis in mice genetically susceptible to the development of SS-like disease (Nandula et al., 2011). While these studies demonstrate the role played by TLR3 activation in the induction of salivary gland dysfunction and disease, we were interested in investigating whether abrogation of type I IFN signaling can prevent the development of SS-like disease. To address this question, in this study, we developed a novel genetic mouse model that lacks the IFNAR (thereby abrogating all signaling through type I IFNs), but that has all the genes necessary for the development of SS-like disorder.
Currently, there are several mouse models for SS described in the literature, and each model has its advantages and disadvantages for investigating this complicated disorder (Delaleu et al., 2011). Since the IFNAR deficiency was on the C57BL/6 (B6) background (Yasuda et al., 2007), we used the B6.Aec1Aec2 mouse for our study. The B6.Aec1Aec2 spontaneous mouse model for SS-like disorder recapitulates certain features of the human disease (Cha et al., 2002). It carries 2 genetic regions, Idd5 and Idd3, from the non-obese diabetic (NOD) mouse. The advantages of using this model are that it is spontaneous and characterized by the presence of circulating autoantibodies, lymphocytic foci within the salivary glands, and a progressive loss of glandular function. However, unlike the human disorder, there is no female-to-male preponderance of disease. In this study, we generated a novel strain, the B6.Aec1Aec2Ifnar1-/- knockout (KO) mouse, and investigated the development of SS-like disorder in this mouse.
Materials & Methods
Animals
All mice used in this study were bred and maintained under specific pathogen-free (SPF) conditions in the University of Virginia vivarium. All experiments were approved by the Institutional Animal Care and Use Committee, and all protocols followed National Institutes of Health guidelines for humane practices. The B6.Aec1Aec2 mice (Cha et al., 2002) were crossed with the B6.Ifnar1-/- mice (Yasuda et al., 2007) to generate F1 mice. The F1 mice were intercrossed and backcrossed to parental strains to generate the B6.Aec1Aec2Ifnar1-/- mouse. All progeny were genotyped for the Aec1 and Aec2 loci using a panel of microsatellite markers as described previously (Nguyen et al., 2006). The Ifnar1 deficiency was monitored by a gene-specific polymerase chain-reaction (PCR) and flow cytometry. Pilocarpine-induced salivation was measured as described previously (Deshmukh et al., 2009). All experiments were performed in female mice.
Gene Expression Analysis
Expression of IFN-responsive genes in submandibular glands was determined by quantitative PCR with Taqman probes as described previously (Deshmukh et al., 2009). A pooled RNA sample from submandibular glands of age-matched B6 mice was used as a calibrator to calculate relative gene expression by the 2-ΔΔCt method (Livak and Schmittgen, 2001).
Antibody Analysis
Immunoglobulin levels in sera were measured by ELISA as described earlier (Bagavant et al., 2002). Serum autoantibodies to a mouse submandibular gland cell line SCA-9-15 (ATCC, Manassas, VA, USA) extract were analyzed by Western blotting as described previously, with few modifications (Deshmukh et al., 1999). Goat anti-mouse IgG coupled to IRDye800 (LI-COR Biotechnology, Lincoln, NE, USA) was used as secondary antibody. Bound antibody was visualized with the ODYSSEY CLx infrared imaging system following manufacturer’s directions, and intensity of staining was quantitated with Image Studio 2.1 software (LI-COR Biotechnology). Anti-nuclear antibodies (ANA) in serum were detected by indirect immunofluorescence assay with HeLa cells as previously described (Deshmukh et al., 1999).
Stereology
Submandibular salivary gland and lacrimal gland pieces were fixed in 10% buffered formalin and embedded in paraffin, and sections were stained by hematoxylin and eosin. Quantitative analyses of glandular inflammation were done with Stereoinvestigtor software v3.0 (MBF Bioscience, Williston, VT, USA) (Glaser and Glaser, 2000). Briefly, a contour was traced around each piece of gland, and a sampling grid of 400 µm x 400 µm was randomly placed over the contour. Using the area fraction fractionator probe, we obtained a systematic random sample by positioning a counting frame of 300 µm x 250 µm containing markers at 10-micron intervals. Each marker was interrogated for the presence or absence of inflammatory cells, and the total gland area and inflammatory focus area were estimated. Salivary gland areas of 10.4 ± 0.5 x 106 sq microns and lacrimal gland areas of 3.3 ± 0.4 x 106 sq microns were studied for each mouse. Disease scores were calculated as (inflammatory area/total gland area) x100 for each mouse.
Flow Cytometry
Analysis of IFNAR expression on peripheral blood cells was carried out by flow cytometry according to standard protocols. Experimental details are provided in the Appendix.
Results
Expression of IFN-responsive Genes is Suppressed in B6.Aec1Aec2Ifnar1-/- Mice
The Ifnar1 mutation in C57BL/6 mice was moved to the B6.Aec1Aec2Ifnar1+/+ mice (WT) to generate B6.Aec1Aec2I fnar1-/- mice (KO). Appendix Fig. 1 shows the PCR products used to characterize the mouse genotype. To confirm the phenotype, we analyzed expression of IFNAR on peripheral blood mononuclear cells by flow cytometry (Fig. 1, left panel). IFNAR expression was not detected on cells obtained from the KO mice.
Figure 1.
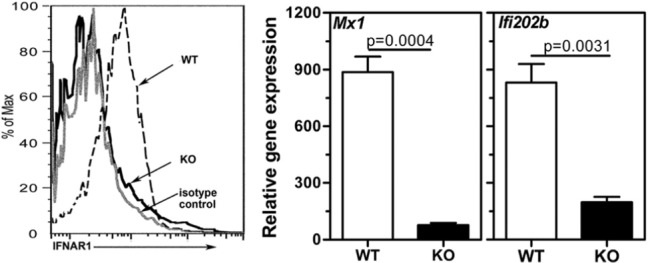
Lack of IFNAR in B6Aec1Aec2 mice suppresses the expression of type I IFN-responsive genes Mx1 and Ifi202b. (Left panel) Flow cytometry analysis showing a representative histogram of IFNAR staining on PBMCs obtained from WT (dotted line) and KO (solid line) mice. The staining by isotype control is shown by a gray line. (Right panel) Gene expression levels of Mx1 and Ifi202b in the submandibular glands obtained from WT (n = 4) and KO (n = 3) mice at 8 wks of age were determined by real-time PCR with Taqman primers. The data are represented as mean ± SEM relative gene expression over a pooled RNA sample from an age-matched C57BL/6 mouse, which was used as the calibrator. Gapdh was used to normalize the expression. Statistical significance was determined by the Mann-Whitney test. Similar results were obtained in an additional experiment.
The WT mice showed spontaneous up-regulation of type I IFN-responsive genes in their salivary glands (Nguyen et al., 2009). Therefore, expression of the IFN-responsive genes Mx1 and Ifi202b was analyzed in submandibular glands of WT and KO mice at 8 wks of age. The expression of both genes was significantly suppressed in the KO mice (Fig. 1, right panel).
KO Mice are Protected from Salivary Gland Dysfunction
Spontaneous loss of salivary gland function is one of the major characteristics of the SS-like disorder in the B6.Aec1Aec2 mouse. To determine the effect of IFNAR deficiency on this phenotype, we measured pilocarpine-induced saliva at different time-points in the WT and KO mice (Fig. 2). At an early age (11-13 wks), the difference in mean saliva volume between the WT and KO mice was not statistically significant. At later ages, there was a progressive drop in saliva volumes in the WT mice. In contrast, the mean saliva volumes in the KO mice, monitored up to 22 wks, did not decrease. At 15 to 18 wks and 19 to 22 wks, differences in mean saliva volumes between WT and KO mice were highly significant (p < .0001). Analysis of these data clearly demonstrates that genetic ablation of type I IFN signaling in the B6.Aec1Aec2 mice protects them from salivary gland dysfunction, as indexed by a ratio of salivary volume to body weight.
Figure 2.
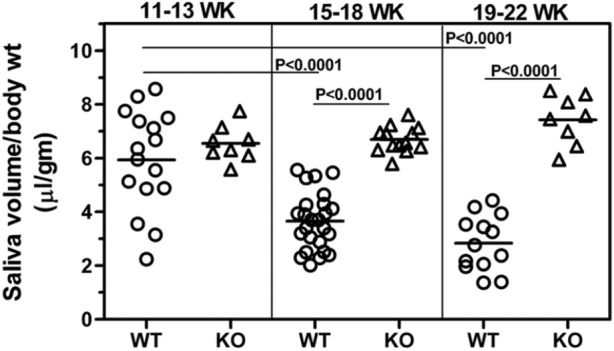
Saliva production decreases over time in WT mice but not in KO mice that lack the type I IFN signaling pathway. Pilocarpine-induced saliva production was measured at the different ages indicated in the Fig. Data are represented as the ratio of saliva volume (µL) to body weight (gm). Student’s t test was used to determine statistical significance. Saliva volumes (mean ± SEM) in WT mice at 15 to 18 wks (3.65 ± 0.21) and 19 to 22 wks (2.83 ±0.28) were significantly lower compared with those at 11 to 13 wks (5.93 ± 0.46) of age, indicating a progressive loss of function. In contrast, in KO mice, the mean saliva volumes between early and late ages were not different. Although WT and KO mice had comparable saliva production at 11 to 13 wks of age, the differences between the 2 groups were highly significant at the 15 to 18-week (p < .0001) and 19- to 22-week (p < .0001) time-points.
IFNAR Deficiency Does Not Affect Systemic Autoimmune Response
The IFNAR deficiency did not dramatically alter the total serum IgG and IgM levels (Appendix Fig. 2). Although in the WT mice at 7 to 10 wks of age, the mean IgM levels were significantly higher (p = .0101), at later time-points the differences between the 2 groups were not statistically significant. The mean IgG levels between WT and KO mice, at both early and late ages, were not significantly different.
To determine whether the protection from salivary gland dysfunction in KO mice was tied to suppression of spontaneous autoimmune responses, we analyzed the presence of ANA in sera obtained from WT and KO mice at different time-points. There was no difference in the incidence or intensity of ANA staining between the WT and KO mice at all time-points tested (Fig. 3, left panel, and Appendix Fig. 3, showing a representative ANA staining). A similar trend was observed when sera were tested for reactivity to a mouse submandibular gland cell line SCA-9-15 (data not shown). Serum reactivity to salivary gland antigens was also analyzed in Western blots (Fig. 3, right panel). Autoantibodies reactive with multiple proteins were detected in the sera of both WT and KO mice, and the intensities of staining were not different between the groups. Analysis of these data indicates that IFNAR deficiency in the B6Aec1Aec2 mice did not permanently affect total immunoglobulin levels nor did it affect autoantibody reactivity to multiple cellular antigens. Overall, these findings are consistent with the data showing no differences in T-, B-, and NK cell populations between WT and KO mice (Appendix Fig. 4).
Figure 3.
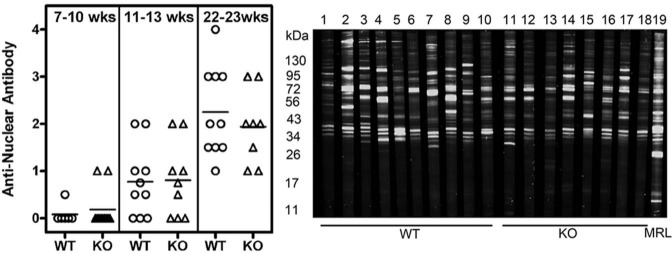
Both WT and KO mice generated a robust autoantibody response. Sera obtained from WT and KO mice at different time-points were analyzed for the presence of autoantibodies. (Left panel) Intensity of ANA staining by indirect immunofluorescence. Representative ANA pictures are shown in Appendix Fig. 3. The incidence and severity of ANA staining were not different between the WT and KO mice. (Right panel) A representative image of a Western blot showing reactivity to proteins from a mouse submandibular gland cell line. All sera were used at 1:200 dilutions, and goat anti-mouse IgG antibody coupled to IRDye 800CW was used to detect bound antibodies. Lanes 1 to 10 are sera from individual WT mice, lanes 11 to 18 are sera from individual KO mice, and lane 19 has pooled sera from lupus-prone MRL lpr/lpr mice. Similar results were obtained in an additional experiment.
IFNAR Deficiency Affects Inflammatory Cell Infiltration
H&E-stained sections of submandibular glands obtained from WT and KO mice were studied for the presence of inflammatory cell infiltrates at 20 to 22 wks, and representative histopathology is shown in Figs. 4A-4D. Quantitative analyses showed larger areas of focal inflammation in WT mice (8.49 ± 1.7 x 104 sq microns) compared with KO mice (3.8 ± 1.8 x 104 sq microns). The inflammatory area fraction of the total glandular tissue studied was calculated for each mouse and is represented as severity of sialoadenitis (Fig. 4, lower panel). The mean inflammatory area fraction was significantly lower (p = .0022) in the KO mice (0.37 ± 0.2) compared with that in age-matched WT mice (0.75 ± 0.13). Lacrimal gland inflammation (Figs. 4E-4H) was also reduced in KO mice (1.7 ± 1.2 x 104 sq microns) compared with that in WT mice (10.0 ± 2.9 x 104 sq microns). Similarly, the lacrimal inflammatory area fraction was significantly lower (p = .0003) in the KO mice (0.56 ± 0.4) compared with that in age-matched WT mice (3.43 ± 1.08).
Figure 4.

The IFNAR deficiency suppresses mononuclear cell infiltration within the submandibular and lacrimal glands. Submandibular (A-D) and lacrimal glands (E-H) obtained from WT (n = 20) and KO (n = 18) mice were evaluated for mononuclear cell infiltration in H&E-stained sections. The inflammation was quantitated by an individual blinded to experimental details. A, B, E, and F show representative low-magnification images, and C, D, G, and H are images at higher magnification. Severity of sialoadenitis and dacryoadenitis (lower panels) represents the fraction of area covered by inflammatory infiltrates in salivary and lacrimal glands, respectively, for each mouse. Statistical significance was calculated by the Mann-Whitney test. The mean severity scores of sialoadenitis and dacryoadenitis in the WT mice were significantly higher than those in the KO mice. The arrows indicate the foci of mononuclear cell infiltration. Scale bar = 50 microns.
Discussion
Type I IFNs are pluripotent cytokines that play a crucial role in host defense against viral infections (Isaacs and Lindenmann, 1957). However, they have also been implicated in the pathogenesis of autoimmune disorders such as lupus and SS (Trinchieri, 2010). Although the up-regulation of IFN-responsive genes in SS patients suggests a possible role for type I IFNs in the pathogenesis of this disease, whether they are involved in the initiation or perpetuation of SS is not known. This study, in a spontaneous mouse model for SS-like disorder, clearly demonstrates that lack of type I IFN signaling prevents the development of disease. Mice deficient for Ifnar1, but harboring other genes necessary for the development of SS-like disorder, were protected from both mononuclear cell infiltration within the lacrimal and salivary glands and the loss of saliva production. Interestingly, the KO mice had mounted a robust systemic autoimmune response, thereby dissociating the systemic humoral autoimmune response from salivary gland dysfunction.
In our model, the mice were protected from salivary gland dysfunction, yet they had a robust systemic autoimmune response. This suggests that systemic autoimmune responses need not be under the control of the type I IFN pathway. The effect of IFNAR deficiency has been studied in other models of systemic autoimmunity, particularly in lupus (Santiago-Raber et al., 2003; Agrawal et al., 2009). Although some lupus mouse models showed that the lack of Ifnar1 was responsible for decreasing autoantibody levels, others, like the FcgRIIB-/-Yaa Ifnar1-/- mice, showed that type I IFN signaling did not influence the levels of ANA and anti-Sm autoantibodies (Richez et al., 2010). Similar to our finding, the total immunoglobulin levels were not perturbed in these mice. These observations have a considerable bearing on our understanding of the pathogenic mechanisms operative in SS. The type I IFN pathway has been implicated for initiating and amplifying a systemic autoimmune response, by exerting effects on antigen-presenting cells and by influencing B-cell activation (Voulgarelis and Tzioufas, 2010). However, while depletion of B-cells with Rituximab therapy increased the expression of type I IFN-responsive genes in rheumatoid arthritis patients, in SS patients, it had no effect on the IFN signature (Vosslamber et al., 2011; St. Clair et al., 2013). Considering these observations together with our results from the KO mice, we suggest that the influence of the type I IFN pathway on autoantibody production might be dependent on the disease type and interaction with other genetic factors.
In B6.Aec1Aec2 mice, IFNAR deficiency prevents inflammatory cell infiltration in the salivary glands. Several mechanisms might be operative in exerting this protective effect. Type I IFNs are known to influence the production of chemokines (Lang et al., 2006). Our previous work has shown that activation of the type I IFN pathway leads to up-regulated gene expression for several chemokines within the submandibular glands (Nandula et al., 2011). This causes accelerated mononuclear cell infiltration within the salivary glands. A recent study suggests that 2 chemokines, CXCL10 and CXCL12, expressed in the salivary glands of B6.Aec1Aec2 mice and SS patients are responsible for the progression of disease (Horvath et al., 2012). Thus, lack of type I IFN signaling in the KO mice might decrease the localized production of such chemokines and thereby reduce inflammatory cell infiltration within the salivary glands. Alternatively or in addition, the expression patterns of different chemokine receptors on inflammatory cells can be altered in the absence of type I IFN signaling, thereby affecting their migratory properties. A systematic comparison of changes in chemokine and chemokine receptor expression between the KO and WT mice will yield targets for interfering with disease progression in SS. Indeed, administration of a CXCL10 antagonist to MRL lpr/lpr mice resulted in decreased salivary gland inflammation (Hasegawa et al., 2006).
In summary, our study demonstrates that type I IFN is responsible for initiating features of SS-like disorder in B6.Aec1Aec2 mice. This study supports the thesis that, in genetically susceptible individuals, up-regulation of type I IFN production can lead to the development of SS.
Supplementary Material
Footnotes
Saleh Mohammad provided excellent technical assistance for working with the mice. This investigation was supported in part by the U.S. Public Health Service, from the NIDCR (Research Grants DE022977 and DE019883), and from the NIAID (Grant AI079621) from the National Institutes of Health, Bethesda, MD 20892, USA.
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
A supplemental appendix to this article is published electronically only at http://jdr.sagepub.com/supplemental.
References
- Agrawal H, Jacob N, Carreras E, Bajana S, Putterman C, Turner S, et al. (2009). Deficiency of type I IFN receptor in lupus-prone New Zealand mixed 2328 mice decreases dendritic cell numbers and activation and protects from disease. J Immunol 183:6021-6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagavant H, Thompson C, Ohno K, Setiady Y, Tung KS. (2002). Differential effect of neonatal thymectomy on systemic and organ-specific autoimmune disease. Int Immunol 14:1397-1406. [DOI] [PubMed] [Google Scholar]
- Carr AJ, Ng WF, Figueiredo F, Macleod RI, Greenwood M, Staines K. (2012). Sjögren’s syndrome— an update for dental practitioners. Br Dent J 213:353-357. [DOI] [PubMed] [Google Scholar]
- Cha S, Nagashima H, Brown VB, Peck AB, Humphreys-Beher MG. (2002). Two NOD Idd associated intervals contribute synergistically to the development of autoimmune exocrinopathy (Sjögren’s syndrome) on a healthy murine background. Arthritis Rheum 46:1390-1398. [DOI] [PubMed] [Google Scholar]
- Delaleu N, Nguyen CQ, Peck AB, Jonsson R. (2011). Sjögren’s syndrome: studying the disease in mice. Arthritis Res Ther 13:217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh US, Lewis JE, Gaskin F, Kannapell CC, Waters ST, Lou YH, et al. (1999). Immune responses to Ro60 and its peptides in mice. I. The nature of the immunogen and endogenous autoantigen determine the specificities of the induced autoantibodies. J Exp Med 189:531-540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh US, Nandula SR, Thimmalapura PR, Scindia YM, Bagavant H. (2009). Activation of innate immune responses through Toll-like receptor 3 causes a rapid loss of salivary gland function. J Oral Pathol Med 38:42-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emamian ES, Leon JM, Lessard CJ, Grandits M, Baechler EC, Gaffney PM, et al. (2009). Peripheral blood gene expression profiling in Sjögren’s syndrome. Genes Immun 10:285-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser JR, Glaser EM. (2000). Stereology, morphometry and mapping: the whole is greater than the sum of the parts. J Chem Neuroanat 20:115-126. [DOI] [PubMed] [Google Scholar]
- Hasegawa H, Inoue A, Kohno M, Muraoka M, Miyazaki T, Terada M, et al. (2006). Antagonist of interferon-inducible protein 10/CXCL10 ameliorates the progression of autoimmune sialadenitis in MRL/lpr mice. Arthritis Rheum 54:1174-1183. [DOI] [PubMed] [Google Scholar]
- Hjelmervik TO, Petersen K, Jonassen I, Jonsson R, Bolstad AI. (2005). Gene expression profiling of minor salivary glands clearly distinguishes primary Sjögren’s syndrome patients from healthy control subjects. Arthritis Rheum 52:1534-1544. [DOI] [PubMed] [Google Scholar]
- Horvath S, Nazmul-Hossain AN, Pollard RP, Kroese FG, Vissink A, Kallenberg CG, et al. (2012). Systems analysis of primary Sjögren’s syndrome pathogenesis in salivary glands identifies shared pathways in human and a mouse model. Arthritis Res Ther 14:R238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacs A, Lindenmann J. (1957). Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci 147:258-267. [PubMed] [Google Scholar]
- Kang KY, Kim HO, Kwok SK, Ju JH, Park KS, Sun DI, et al. (2011). Impact of interleukin-21 in the pathogenesis of primary Sjögren’s syndrome: increased serum levels of interleukin-21 and its expression in the labial salivary glands. Arthritis Res Ther 13:R179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang KS, Georgiev P, Recher M, Navarini AA, Bergthaler A, Heikenwalder M, et al. (2006). Immunoprivileged status of the liver is controlled by Toll-like receptor 3 signaling. J Clin Invest 116:2456-2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402-408. [DOI] [PubMed] [Google Scholar]
- Nandula SR, Scindia YM, Dey P, Bagavant H, Deshmukh US. (2011). Activation of innate immunity accelerates sialoadenitis in a mouse model for Sjögren’s syndrome-like disease. Oral Dis 17:801-807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandula SR, Dey P, Corbin KL, Nunemaker CS, Bagavant H, Deshmukh US. (2013). Salivary gland hypofunction induced by activation of innate immunity is dependent on type I interferon signaling. J Oral Pathol Med 42:66-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen C, Singson E, Kim JY, Cornelius JG, Attia R, Doyle ME, et al. (2006). Sjögren’s syndrome-like disease of C57BL/6.NOD-Aec1 Aec2 mice: gender differences in keratoconjunctivitis sicca defined by a cross-over in the chromosome 3 Aec1 locus. Scand J Immunol 64:295-307. [DOI] [PubMed] [Google Scholar]
- Nguyen CQ, Sharma A, Lee BH, She JX, McIndoe RA, Peck AB. (2009). Differential gene expression in the salivary gland during development and onset of xerostomia in Sjögren’s syndrome-like disease of the C57BL/6.NOD-Aec1Aec2 mouse. Arthritis Res Ther 11:R56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richez C, Yasuda K, Bonegio RG, Watkins AA, Aprahamian T, Busto P, et al. (2010). IFN regulatory factor 5 is required for disease development in the FcgammaRIIB-/-Yaa and FcgammaRIIB-/- mouse models of systemic lupus erythematosus. J Immunol 184:796-806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago-Raber ML, Baccala R, Haraldsson KM, Choubey D, Stewart TA, Kono DH, et al. (2003). Type-I interferon receptor deficiency reduces lupus-like disease in NZB mice. J Exp Med 197:777-788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirota Y, Illei GG, Nikolov NP. (2008). Biologic treatments for systemic rheumatic diseases. Oral Dis 14:206-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Clair EW, Levesque MC, Luning Prak ET, Vivino FB, Alappatt CJ, Spychala ME, et al. (2013). Rituximab therapy for primary Sjögren’s syndrome: an open-label clinical trial and mechanistic analysis. Arthritis Rheum [Epub ahead of prnt 1/17/2013] (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinchieri G. (2010). Type I interferon: friend or foe? J Exp Med 207:2053-2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakaloglou KM, Mavragani CP. (2011). Activation of the type I interferon pathway in primary Sjögren’s syndrome: an update. Curr Opin Rheumatol 23:459-464. [DOI] [PubMed] [Google Scholar]
- Vosslamber S, Raterman HG, van der Pouw Kraan TC, Schreurs MW, von Blomberg BM, Nurmohamed MT, et al. (2011). Pharmacological induction of interferon type I activity following treatment with rituximab determines clinical response in rheumatoid arthritis. Ann Rheum Dis 70:1153-1159. [DOI] [PubMed] [Google Scholar]
- Voulgarelis M, Tzioufas AG. (2010). Pathogenetic mechanisms in the initiation and perpetuation of Sjögren’s syndrome. Nat Rev Rheumatol 6:529-537. [DOI] [PubMed] [Google Scholar]
- Yasuda K, Richez C, Maciaszek JW, Agrawal N, Akira S, Marshak-Rothstein A, et al. (2007). Murine dendritic cell type I IFN production induced by human IgG-RNA immune complexes is IFN regulatory factor (IRF)5 and IRF7 dependent and is required for IL-6 production. J Immunol 178:6876-6885. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.