

Abstract
Conventional PET methods to estimate [11C]raclopride binding potential (BP ND) assume that endogenous dopamine concentration does not change during the scan time. However, this assumption is purposely violated in studies using pharmacological or behavioral stimuli to invoke acute dopamine release. When the assumption of steady-state dopamine is violated, conventional analysis methods may produce biased or even unusable estimates of BP ND. To illustrate this problem, we examined the effect of scan duration on ΔBP ND estimated by three common analysis methods (simplified reference tissue model, Logan graphical reference method, and equilibrium analysis) applied to simulated and experimental single-scan activation studies. The activation – dopamine release – in both the simulated and experimental studies was brief. Simulations showed ΔBP ND to be highly dependent on the window of data used to determine BP ND in the activation state. A similar pattern was seen in the data from human smoking studies. No such pattern of ΔBP ND dependence on the window of data used was apparent in simulations where dopamine was held constant. The dependence of ΔBP ND on the duration of data analyzed illustrates the inability of conventional methods to reliably quantify short-lived increases in endogenous dopamine.
Keywords: Binding potential (BPND), transient dopamine release, model selection, scan duration, smoking, time-invariant
Introduction
Traditional analysis of dynamic [11C]raclopride positron emission tomography (PET) studies includes estimating [11C]raclopride binding potential (BP ND) – a static parameter that represents the potential for specific binding of [11C] raclopride to dopamine (DA) D2/D3 receptors in the brain. This is usually accomplished by fitting the dynamic data with compartmental or graphical (linearized) models, such as the simplified reference tissue model (SRTM) [1] or the Logan graphical method [2,3]. There are no time-varying parameters in any of the conventional models. Thus, a basic assumption of these models is that endogenous dopamine (DA) concentration is at steady-state and does not change during the course of the scan.
However, experiments are often conducted for which the assumption of steady-state DA concentration is purposely violated in order to measure changes in synaptic DA due to a pharmacological or behavioral challenge. In these studies, BP ND is measured at rest (no challenge) and after a challenge. An increase in endogenous DA in the challenge condition competes with the radiotracer for binding at the receptor and results in lower BP ND values relative to the rest condition [4]. In this “competition model”, decreases in BP ND are used as an index of DA release following a wide variety of pharmacological stimuli, including amphetamine [5-7] and nicotine [8-15].
A decrease in BP ND has been shown to be a robust and reliable index of amphetamine-induced DA release. Amphetamine has been shown to produce large increases in DA concentration [16-18] that peak over 600% above baseline [17]. It has also been shown to provoke long-lasting displacement of [11C]raclopride (potentially due, in part, to receptor internalization [4,19]) and greater than 20% decrease in BP ND [5]. Conventional analysis methods, such as SRTM, Logan graphical method (“Logan plot”), and equilibrium methods, have been able to reproducibly estimate sustained decreases in BP ND for varying doses of amphetamine administered under a variety of experimental conditions in both humans and animals.
In contrast, it is unclear whether nicotine-induced DA release can be detected by decreases in [11C]raclopride BP ND. Based on a study of human subjects smoking 1 cigarette during a break in a scan with bolus-plus-constant-infusion (B/I) of [11C]raclopride, Brody et al. reported 30% decrease in BP ND after smoking in the left ventral caudate [13]. In later studies with larger cohorts and a more advanced scanner, but the same protocol, Brody et al. reported only 8% decrease in [11C]raclopride BP ND [9-12]. Scott et al. reported a similar decrease in BP ND (~10%) between regular and denicotinized cigarettes smoked while in the scanner [15]. However, only 1% decrease was reported by Montgomery et al. in [11C]raclopride BP ND in response to 2mg nicotine nasal spray administered during scanning [20], and a study by Barrett et al. requiring subjects to smoke up to 6 cigarettes during the scan measured highly variable changes in [11C]raclopride BP ND (range of +57% and -67% change in BP ND) in the ventral striatum and caudate [8].
We believe that the between-study and within-study variability in nicotine-induced decrease in BP ND reported in the literature may be due to violations of the assumption that endogenous DA concentration remains at steady-state during the challenge condition. Unlike amphetamine, which is a direct releaser of DA, nicotine only indirectly causes transient increases in DA concentration and transient displacement of [11C]raclopride. Microdialysis studies have shown that nicotine, in doses comparable to what a human would self-administer by smoking, produces a brief 125% increase in DA which returns to baseline within 30 min [21]. Nicotine challenges in PET studies may also cause transient decreases in [11C]raclopride signal which return to baseline levels just as quickly. Because of amphetamine’s long-lasting effects on DA release and tracer displacement, [11C]raclopride reaches a new equilibrium state after challenge and BP ND can be estimated by conventional models without appreciably violating the assumption that endogenous DA concentration is not changing during the scan. For nicotine challenges, DA concentration and tracer binding are continuously changing and [11C]raclopride does not reach a new equilibrium state. In this scenario, specific binding of [11C]raclopride will not be constant during the challenge scan and any attempt to estimate BP ND with conventional methods will produce an estimate which is a weighted-average of the specific tracer binding as it changes over the course of the challenge scan [22]. This could result in a reduced measure of nicotine-induced decrease in BP ND, which, coupled with a small effect size and inter-subject variability, could easily obscure detection of a significant decrease in BP ND due to DA release from nicotine.
To investigate if inappropriate application of conventional modeling approaches could obscure detection of nicotine-induced DA release, we examined the effect of scan duration on decrease in BP ND estimated by SRTM, Logan plot, and equilibrium analysis. We applied these analyses to simulated single-scan activation studies and human smoking studies. An observed dependence of ΔBP ND on the duration of data analyzed suggests that conventional methods are not well-suited to reliable quantification of short-lived increases in endogenous DA.
Materials and methods
Simulations
Noiseless and noisy striatal [11C]raclopride time activity curves (TACs) with varying amounts of DA release were simulated using an enhanced tracer kinetic model (Figure 1) [23]. This model includes state variables for free and bound dopamine and it includes rate constants for the tracer as well as binding and dissociation of DA to and from the receptor. TACs based on a B/I of tracer were simulated for a scan duration of 120 min consisting of 1-min frames.
Figure 1.
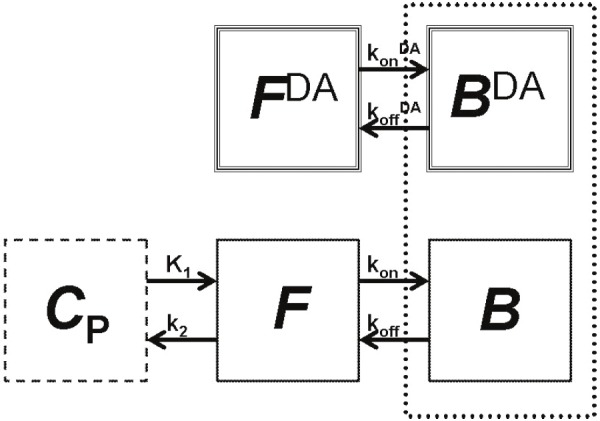
The enhanced tracer kinetic model accounts for variations in tracer concentration as well as endogenous dopamine (DA) [23,40]. Free (F DA) and bound (B DA) endogenous DA are represented by the compartments at top, where k on DA and k off DA are the association and dissociation rate constants for DA at the receptor. The standard two-tissue compartment model at bottom includes compartments for the tracer free in tissue (F) and specifically bound (B) to the receptor of interest. Rate constants describe the movement of tracer between plasma (C p) and tissue (K 1, k 2) and binding to the receptor (k on and k off). The competition between the tracer and endogenous DA for a limited number of receptor sites is represented by the dotted-line box around B DA and B compartments.
For all simulations, striatal parameters were set as K 1 = 0.0918 mL/min/g, k 2 = 0.4484 min-1, k on= 0.0282 mL/(pmol · min), k off = 0.1363 min-1, B max = 44 pmol/mL, and F v = 0.04 mL/mL [24]. DA binding parameters (association and dissociation rate) were set as k on DA = 0.25 mL/(pmol · min) and k off DA = 25 min-1 [23]. A noiseless cerebellum TAC was also simulated using the same model by setting k on and k off to 0. All simulations were implemented in MATLAB (R2006a, MathWorks, Inc., Natick, MA) using modeling functions provided by a library of COMKAT [25].
Dopamine release
Free DA (F DA) was modeled as a gamma variate plus a constant baseline with the start of DA release, t d, set at 40 min post tracer injection (Equation 1):
Baseline DA level (DA basal) was set to 100 pmol/mL [26]. Free DA curves with a fixed peak height but increasing area-under-the-curve (AUC) were created by varying α, β, and γ, parameters which specify the shape and magnitude of a gamma variate. Five different DA signals were created: [α, β, γ] = [2, 2, 800], [2, 1, 200], [2, 0.5, 50], [2, 0.25, 12.5], [2, 0.15, 4.5] (Figure 2). All DA signals were designed to represent plausible cases of transient DA release (defined as returning to baseline during the course of the simulated scan). Total DA released was measured as the area under F DA(t) - DA basal from t d to the end of the scan (e.g., 40 min to 120 min post injection) and ranged from 190-2665 (pmol·min)/mL. Noiseless and noisy Rest TACs with no DA perturbation were simulated by setting γ to 0.
Figure 2.
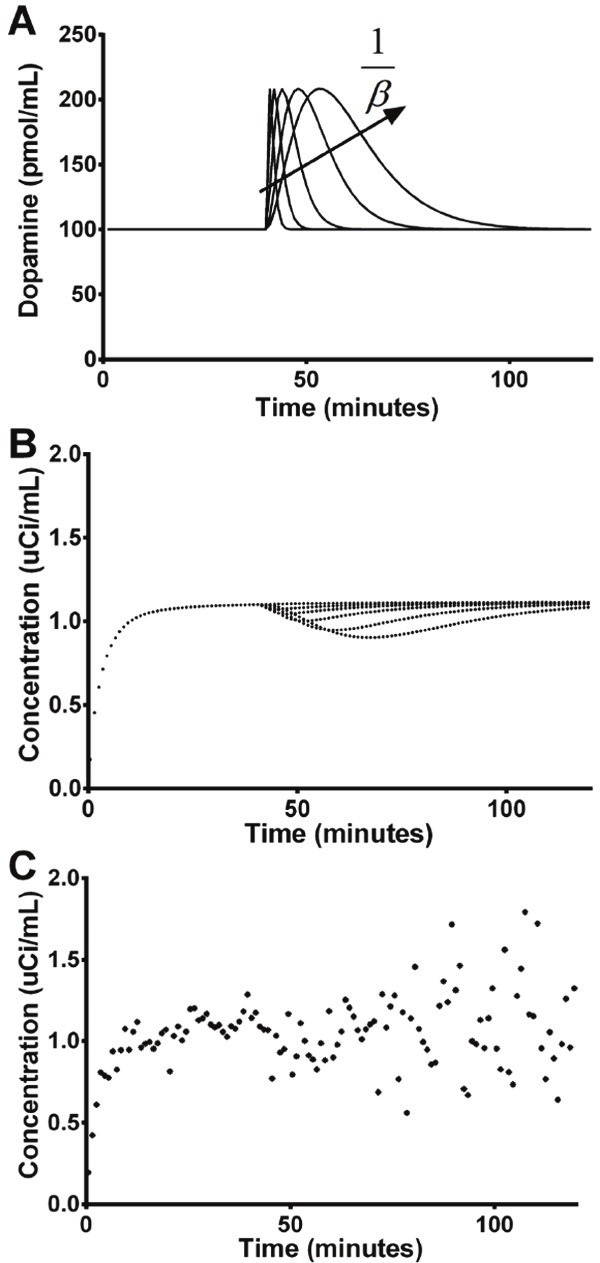
Dopamine (DA) signals with increasing area-under-the-curve (AUC) but same peak height (A) were used to simulate noiseless (B) and noisy (C) striatal time activity curves (TACs). All DA signals produced visually noticeable decreases in the noiseless simulated TACs. These decreases were less visually apparent for transient DA signals in the noisy simulated data (for ease of visualization, only one noisy curve is shown here). In (A), DA signals increase in width as β decreases.
Noise
Monte-Carlo simulations were used to produce 100 noisy PET TACs for each DA signal. Additive Gaussian noise proportional to the imaged PET activity was added to the noiseless, simulated curves [27,28] (Equation 2):
where ROI(t) is the noiseless, decay-corrected TAC, G(0,1) is a pseudo-random number from a Gaussian distribution with 0 mean and standard deviation of 1, and SD(t) is the standard deviation of the noise (Equation 3):
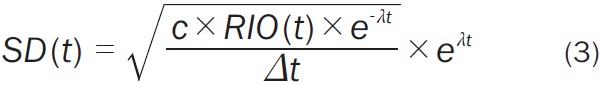 |
In this noise model, c is the proportionality constant which determines the noise level, λ is the decay constant for C-11, and Δt is the simulated frame length. The term e -λt applies decay to the decay-corrected ROI(t) while the term eλt decay-corrects the standard deviation. The constant c was set to 0.002 to produce a noise level similar to that seen in regional-level TACs extracted from the ventral striatum in [11C] raclopride studies completed on the High Resolution Research Tomograph (HRRT; Siemens/CTI, Knoxville, TN, USA).
Human experiments
Subjects
Healthy smokers (n = 4, 2M/2F) were recruited as subjects for a study investigating the effect of cigarette smoking and the temporal pattern of DA release. Subjects were screened to determine medical history and personal and familial psychiatric and smoking history. They were excluded from the study if they met criteria for any Structured Clinical Interview for DSM-IV Axis 1 Disorders except Nicotine Dependence. Severity of nicotine dependence was assessed by the Fagerström Test for Nicotine Dependence (FTND). Only subjects with an FTND score > 3 were included in this study. Smoking status was confirmed by breath CO levels > 12 ppm and urine cotinine levels > 150ng/mL at intake. Smokers smoked 15 ± 6 cig/day for 13 ± 15 yr. Written informed consent was obtained from all subjects before the study but only after complete explanation of the study procedures. All study procedures were approved by the Yale University Human Investigations Committee (HIC).
Experimental design
Prior to PET imaging, each subject received a 3T structural MRI using a 3D fast spoiled grass (FSPGR) MR pulse acquisition sequence with an IR prep of 300 ms (TE = 3.3 ms, flip angle = 17 degrees; slice thickness = 1.2 mm). Subjects received up to two [11C]raclopride scans (rest and smoking) on separate days. Only the data from the smoking scan were used in these analyses. [11C]raclopride was synthesized as reported previously [29]. Coincident with a B/I of [11C]raclopride (685 ± 55 MBq, mean ± standard deviation), list-mode data were acquired for 120 min on the HRRT. A transmission scan was conducted immediately prior to the dynamic scan for attenuation correction. The Vicra optical tracking system (Vicra, NDI Systems, Waterloo, Canada) was used to measure and record subject head movement during each scan. The Vicra records subject head motion in quarternions at a rate of 20Hz from a rigid tool with reflective spheres attached to each subject’s head via a swim cap.
Smoking challenge
During the smoking scan, 3 of 4 subjects were asked to smoke 2 consecutive cigarettes, beginning 45 min after the initial bolus of [11C]raclopride. One female subject was asked to smoke two cigarettes ~30 min apart. The first cigarette was at 53 min; the second was cigarette at 87 min after time-of-injection (TOI).
All subjects were allowed to smoke their own brand of cigarette. Immediately prior to the start of smoking, a study investigator informed the subject that they were about to start smoking. The investigator handed the subject a cigarette, which the investigator lit with a long-reach lighter once the subject had placed the cigarette in his/her mouth. Subjects were instructed to smoke at their typical pace while remaining as still as possible. A small basin was provided at the subject’s chest as an ashtray. An investigator monitored the smoking session, and when subjects finished smoking each cigarette, they dropped it into the basin which the study investigator removed immediately to extinguish the cigarette. Each subject was asked if he/she felt they could smoke a second cigarette (either immediately following the first cigarette or 30 min later). All subjects accepted and smoked a second cigarette. A freestanding air exhaust and filtration system (Movex Inc, Northampton, PA) that had been previously approved by the Yale Environmental Health and Safety Office was used to capture and filter second-hand smoke.
Data processing
Dynamic scan data were reconstructed iteratively with all corrections, including motion using the information collected by the Vicra, by the MOLAR algorithm [30] with the following frame timing: 40 x 3 min. Frame-by-frame motion correction was also performed by registering each frame to an early summed (0 – 9 min) image using a six-parameter mutual information algorithm (FLIRT, FSL 3.2, Analysis Group, FMRIB, Oxford, UK). A summed (0 – 9 min) motion-corrected image was then registered to each subject’s MR, which was warped to an AAL MR template. Anatomically-based striatal regions of interest (ROIs) were defined on the template in the left and right dorsal caudate, left and right dorsal putamen, and left and right ventral striatum [31] (based on Martinez et al. and Mawlawi et al. [6,32]). TACs were extracted from these regions as well as from an AAL-defined cerebellum (reference region).
Estimating BPND
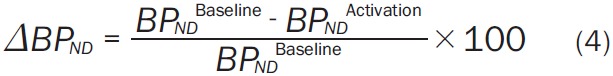
BP ND was estimated for each simulated TAC and all experimental data via SRTM [1], the Logan graphical method using a reference tissue as input (“Logan plot”) [2], and equilibrium analysis (EQ) [33,34]. We chose these methods because they have been used previously to estimate [11C]raclopride BP ND in studies seeking to measure smoking- or nicotine-induced DA release. The main outcome measure considered in these studies, ΔBP ND, was calculated as the percent difference between BP ND Baseline and BP ND Activation (Equation 4):
 |
Because we focused on a single-scan study design, BP ND Baseline was estimated by fitting 0-35 min of data (simulated or experimental) with SRTM and Logan plot (0 min represents time of injection). Previous studies have indicated that cues for reward may contribute to DA release [35]. Subjects in human experiments were aware that they would be smoking 45 min after the start of the scan, and study investigators entered the scanning suite up to 5 min before the start of the smoking session. To ensure that the potential cue of study investigators entering the scanning suite did not induce DA release (i.e., expectation prior to smoking) which could bias the BP ND Baseline value, only 35 min of data were used to calculate BP ND Baseline. For EQ, BP ND Baseline was taken as the average BP ND from 30-40 min. To test the effect of activation scan duration on ΔBP ND, BP ND Activation was estimated from increasingly wider data windows by fitting 0 - m minutes of simulated and experimental data with SRTM and Logan plot, where m = [40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 105, 115, 120]. For EQ, BP ND Activation was taken as the average BP ND from 40 - n minutes, where n was [45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 105, 110, 115, 120].
Simplified reference tissue model (SRTM)
SRTM [1] assumes that tracer activity in both the ROI and reference region can be described by a 1-tissue compartment model. The three model parameters, R1, k2’, and k2a, were estimated by fitting SRTM to TACs using the following operational equation (Equation 5):
where CT(t) is the activity of the tracer in the tissue, C Ref(t) is the activity of the tracer in the reference region, R 1 = K 1/K 1’, the ratio of K 1 in the target tissue to K 1’ in the reference tissue, k 2’ is the efflux rate from the reference region, and k 2a is the apparent efflux rate of tracer from the target region. The cerebellum was used as the reference region and cerebellar TACs were presmoothed by fitting to a sum of exponentials. Parameters were estimated using unweighted least squares. Nonlinear parameter estimation was performed using a Marquardt-Levenberg algorithm [36] and custom software for IDL (8.0 ITT Visual Information Solutions, Boulder, CO). BP ND was calculated as (Equation 6):
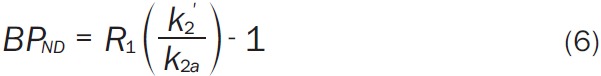 |
Logan graphical method using a reference tissue as input
The Logan graphical method using a reference tissue as input relies on the integration of tracer kinetic equations to yield a simple, linear equation whose slope equals the distribution volume ratio (DVR) of the tracer [2] (Equation 7):
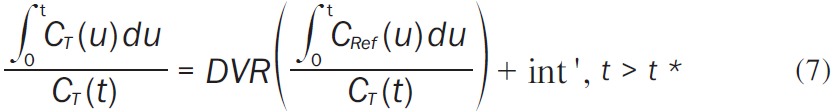 |
The Logan graphical technique proposes that after some time t*, int’ is constant and the plot of ∫ 0 tCT(u)du/CT(t) vs. ∫0tCRef(u)du/CT(t) becomes linear with slope DVR. In all Logan graphical method analyses, t* was fixed at 15 min and the cerebellum was used as the reference region. BP ND was then calculated as DVR-1.
Equilibrium analysis (EQ)
When tracer concentration is at equilibrium, BP ND is the ratio of the difference between target tissue concentration CT,EQ to reference tissue concentration CRef,EQ, relative to the reference tissue concentration (Equation 8). BP ND is calculated as an average over multiple time frames:
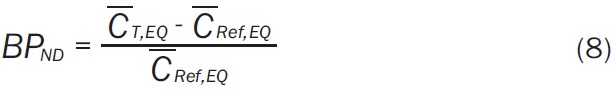 |
where the superscript bar indicates average over some range of time frames. Equilibrium was achieved by 30 min in both simulated and experimental data.
Results
Simulations
Effect of dopamine signal on simulated time activity curves
Inclusion of a time-varying DA signal (Figure 2A) produced decreases in the simulated TACs dependent on the magnitude of the DA signal input (Figure 2B). Degree of deflection in TAC values following onset of DA release was significantly correlated (R2=0.96, p<0.005) with total DA concentration. Consistent with the transient nature of the DA inputs, activity level in many of the noiseless simulated TACs returned to baseline during the course of the scan (Figure 2B). In noisy simulations (Figure 2C), this effect was not easily visible with the naked eye.
Effect of transient dopamine signal on SRTM fitting
Fits of SRTM to simulated TACs containing DA release were poor (Figure 3A). Fits to simulated TACs containing DA release had greater residual sum of squares than fits to rest TACs for activation scan window durations of 40 min or greater (the time of DA release start in simulations). After initiation of DA release, the shape of each fit became dependent on the duration of data used (Figure 3A). As the data window was widened to include more of the transient dip in the TAC, the fitted curves were pulled down relative to the simulated data. Fitted curves went back up again as the data window was widened even more to include later data points unaffected by the transient DA release. The same effect was seen when fitting SRTM to experimental data, though a deflection in experimental TACs at time of smoking was not easily visible (Figure 3B).
Figure 3.

Example of SRTM fits of varying data window length to representative simulated (A) and experimental (B) data which contains dopamine DA release. DA release in the simulated curve is represented by a gamma variate function (Equation 1) with [α, β, γ]=[2, 0.5, 50]. Solid lines are fits. The inset in A is a close-up of the boxed section of the graph. To aid visualization in the inset, the last time point included in the fit to the data for each data window is indicated by a vertical line. The shape of each fit (and thus the BP ND) is dependent on the amount of data used. Logan plots of the full 120-min simulated (C) and experimental (D) curves in A and B. In the simulated data, the slope of the Logan plot during DA release is changing (indicated by arrow), but the slopes before and after DA release are the same. This same effect is not visible in the Logan plot of experimental data.
Effect of transient dopamine signal on Logan plot
Fits to the linear portion of the Logan plots were also dependent on the duration of data used. The fits varied with data window width and the size of the DA-induced changes in TACs. Subtle deviations from linearity, beginning at the onset of DA increase, were observed in the transformed data in Logan plot space for noiseless data (Figure 3C). However, the slopes of the transformed data before and after the time of DA release were the same. Deviations from linearity were not observed in the Logan plots of experimental data (Figure 3D).
Effect of scan duration on ΔBPND
ΔBP ND varied based on the duration of data used to determine BP ND in the activation state for both noiseless (Figure 4) and noisy (Figure 5) simulations. This effect was seen for all DA signals tested. In simulations, a distinct pattern was seen: ΔBP ND increased as activation window duration increased, until reaching a maximum at some window width (generally 20-40 min after onset of DA release), after which ΔBP ND decreased with even wider activation window. The activation window width for which ΔBP ND was maximal varied by DA signal shape. Maximum ΔBP ND was found at longer activation windows for greater total DA release (Table 1). Note: greater total DA release was associated with later DA peak in our simulations. For a given DA signal, ΔBP ND values were highest when estimated by SRTM and lowest when estimated by the Logan graphical approach. For smaller DA responses (e.g., β=1), this resulted in negative ΔBP ND values for some activation window widths, an effect which was exacerbated by noise (Figure 5).
Figure 4.
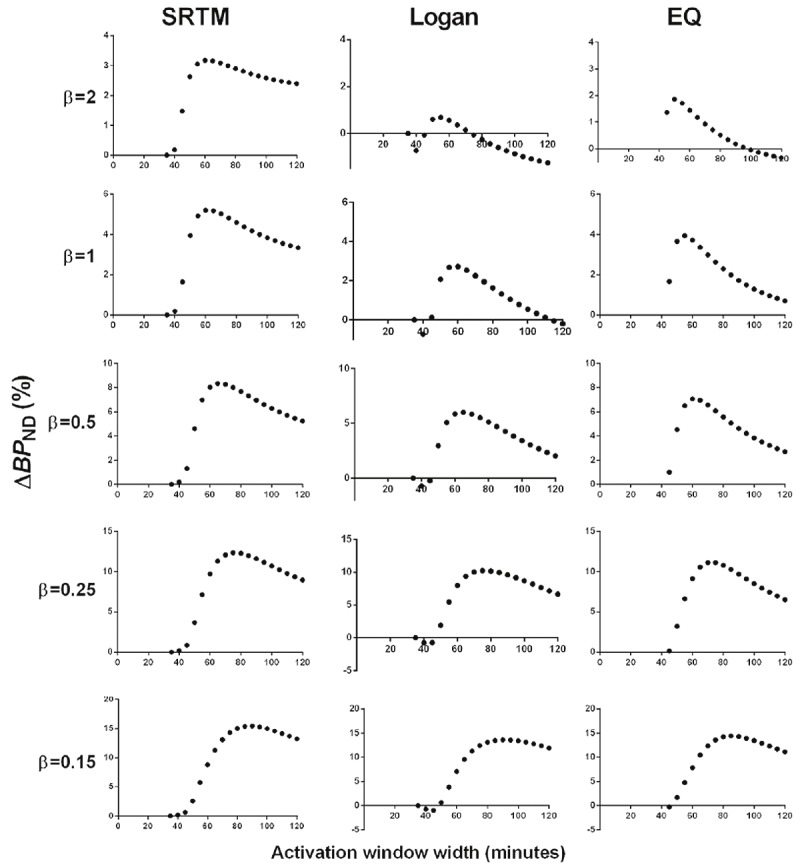
Effect of scan duration on ΔBP ND estimated by SRTM (left column), Logan (middle column), and EQ (right column) for all noiseless simulated TACs. Each row represents the results from a noiseless simulated TAC containing the DA signal indicated by the β value. For all graphs, the dependent axis is ΔBP ND and the independent axis is activation window width in min.
Figure 5.
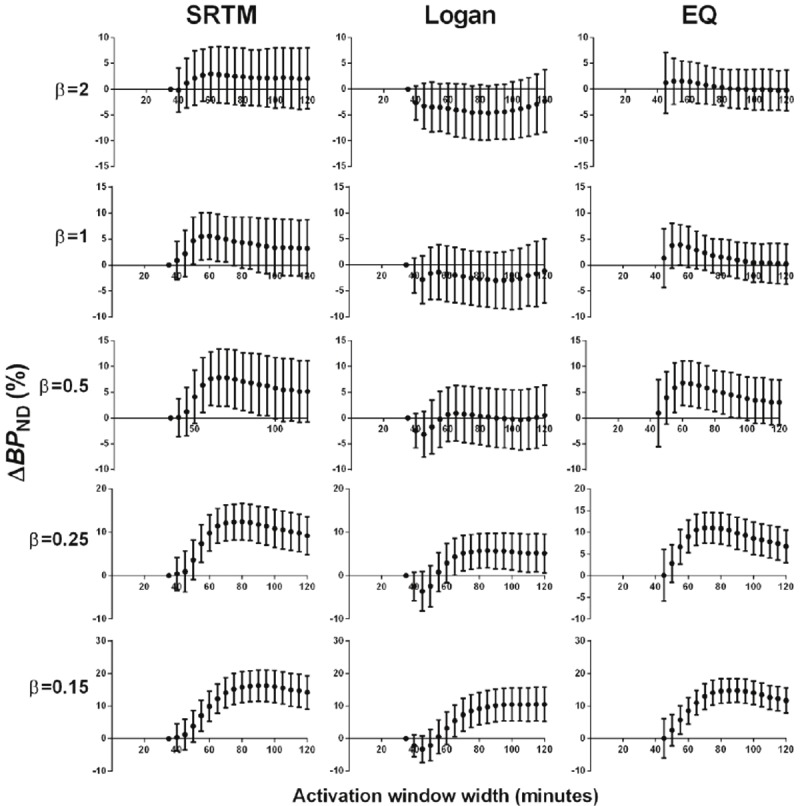
Effect of scan duration on ΔBP ND estimated by SRTM (left column), Logan (middle column), and EQ (right column) for all noisy simulated TACs. Points are mean values and error bars are SD of 100 noisy curves. Each row represents the results from noisy simulated TACs containing the DA signal indicated by the β value. For all graphs, the dependent axis is ΔBP ND and the independent axis is activation window width in min.
Table 1.
Maximum ΔBP ND values from noiseless simulation data
| Maximum ΔBP ND (%) | Activation window width for maximum ΔBP ND (min) | ||||||
|---|---|---|---|---|---|---|---|
|
|
|||||||
| DA signal | SRTM | Logan | EQ | SRTM | Logan | EQ | DA peak time |
| β=2 | 3% | 1% | 2% | 60 | 55 | 40-50 | 41 |
| β=1 | 5% | 3% | 4% | 60 | 60 | 40-55 | 42 |
| β=0.5 | 8% | 6% | 7% | 65 | 65 | 40-60 | 44 |
| β=0.25 | 12% | 10% | 11% | 75 | 75 | 40-75 | 48 |
| β=0.15 | 15% | 14% | 14% | 90 | 90 | 40-85 | 53 |
Rest data
All analyses were also performed on noiseless and noisy simulated Rest data (containing no DA release). ΔBP ND from Rest scan data (ΔBP ND Rest) did not vary noticeably by activation scan duration in any method tested (SRTM, Logan, or EQ) (Figure 6). In noisy simulations, there were biases in ΔBP ND Rest; for an activation window of 120 min ΔBP ND was 0.8% ± 6.1% for SRTM, -4.1% ± 7.0% for the Logan graphical approach, and -1.0% ± 3.7% for EQ.
Figure 6.
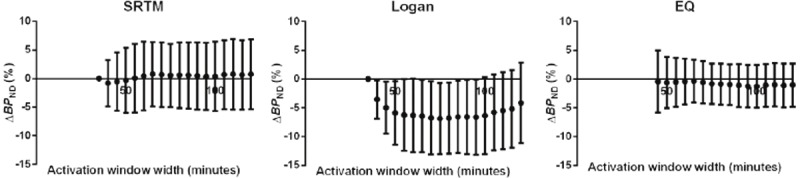
Effect of scan duration on ΔBP ND estimated by SRTM (left column), Logan (middle column), and EQ (right column) for Rest data. Points are mean values and error bars are SD of 100 noisy curves. ΔBP ND Rest values did not vary with activation scan window duration.
Human experiments
ΔBP ND was observed to vary with activation scan duration in experimental data from 4 human subjects (Figure 7). A similar pattern to that seen from analysis of simulated data can be seen clearly in the left ventral striatum in the 2 male subjects and left dorsal caudate in the 2 female subjects (Figure 8). Dependence of ΔBP ND on activation scan duration was not seen in other regions examined in these 4 subjects.
Figure 7.
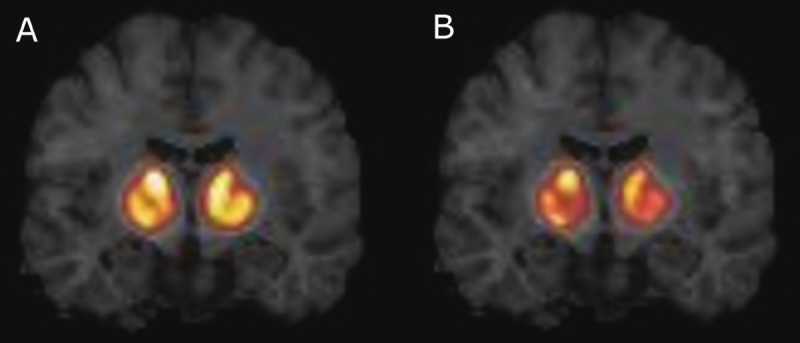
Binding potential (BP ND) images from a male subject who smoked two cigarettes at 45 min after time of injection. A. BP ND estimated by SRTM from 120 min of data. B. BP ND estimated by SRTM from 70 min of data. Both images show the same coronal slice through the striatum, and images are on the same intensity scale. Throughout the striatum, BP ND values estimated from only 70 min of smoking scan data are lower than those estimated from the full 120 min of data. In ΔBP ND calculations, this will result in greater ΔBP ND for 70 min of smoking scan data than 120 min of smoking scan data.
Figure 8.
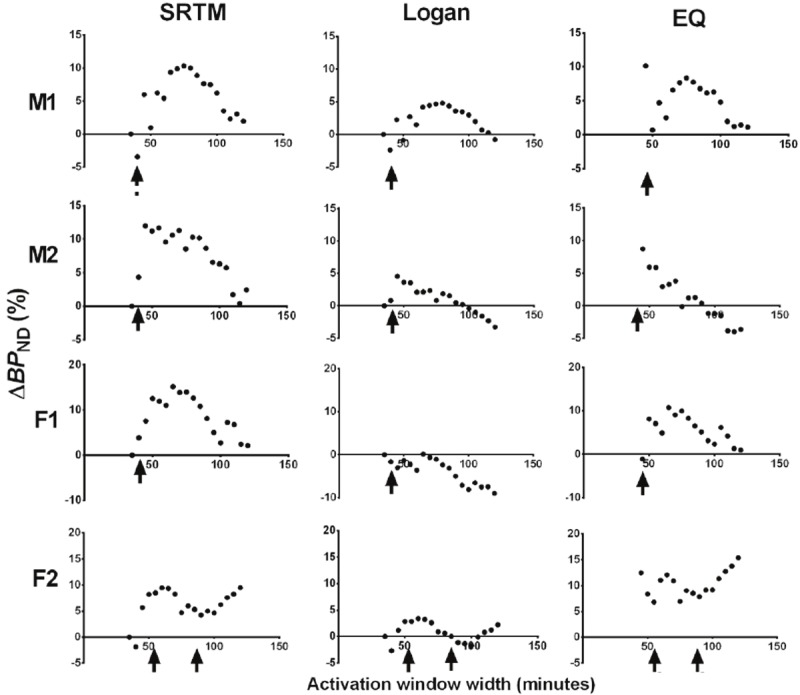
Effect of scan duration on ΔBP ND estimated by SRTM (left column), Logan (middle column), and EQ (right column) for experimental data. Arrows indicate times at which cigarettes were smoked. ΔBP ND values estimated from human smoking studies were dependent on the activation scan window duration. The pattern seen in simulations was identified in the left ventral striatum (top two rows) of two male subjects (M1 and M2) and in the left dorsal caudate (bottom two rows) of two female subjects (F1 and F2). Note: Unlike subjects M1, M2, and F1, who smoked 2 cigarettes at 45 min after time of injection (TOI), subject F2 smoked two cigarettes ~30 min apart, at 53 min and 87 min after TOI.
Discussion
This study examined the effect of the duration of the activation scan on ΔBP ND estimated with SRTM, Logan plot, and EQ applied to PET data containing effects of transient DA release. Through analysis of simulations, we showed that the magnitude of ΔBP ND is dependent on the amount of activation scan data used in estimation of BP ND Activation. We found a similar effect in human data from a study of smoking-induced DA release. We believe that together, these findings illustrate the limitation of applying standard analysis methods to data acquired while the endogenous DA signal was varying rapidly during the scan.
The methods used in this study assume that BP ND is a static parameter. For a method to be fruitful and reliable, varying the length of the scan data fitted should not change the BP ND value estimated. As we show in our analysis of simulated Rest data (Figure 6), ΔBP ND Rest didn’t vary with activation scan duration. A slightly biased ΔBP ND Rest was found by all methods and was greatest with the Logan plot. This suggests that more than 35 min of data may be necessary for these methods to produce unbiased BP ND estimates. In the case of the Logan plot, a longer t* for shorter scan durations (e.g. 35 min) may reduce this bias. Noise has also been shown to introduce a negative bias in BP ND estimated by the Logan plot [37].
Unlike simulated Rest data, ΔBP ND varied with the activation scan duration for simulated data containing transient DA release. ΔBP ND tended to decrease as more data after onset of DA release was included in the analysis (i.e. the data window was widened). Because the DA release in our simulations was transient and the effect of DA release on the tracer signal was brief, inclusion of data beyond the period of DA release appears to have washed out the effect of the DA release on ΔBP ND. This suggests that at typical scan durations (90 min-120 min) used in [11C]raclopride studies, ΔBP ND measured by these common methods may have low sensitivity to transient DA release.
Endres and Carson [22] showed that when neurotransmitter release is time-varying, distribution volume (and hence binding potential) ΔBP ND is a time-weighted average of ΔBP ND(t) weighted by the free tracer concentration [22]. Our simulations are a demonstration of this principle. We also show that if DA release is transient and total DA release is modest, it may not be detectable via ΔBP ND depending on the duration of activation scan data analyzed. This is due not only to the kinetics of endogenous DA but also those of the tracer. Yoder et al. [38] showed that ΔBP ND is dependent on the relative timing and kinetics of both the tracer and the endogenous ligand. In our simulation studies, onset of DA release was 40 min after time of injection. Had DA release occurred earlier during the activation scan, ΔBP ND would be even smaller for longer (90, 120 min) scan durations because there would be more PET data which had returned to baseline levels after DA release included in the estimation of BP ND Activation. This would further decrease the likelihood of detection of transient DA release by ΔBP ND for typical [11C]raclopride scan durations of 90 or 120 min.
Our simulations suggest that the discrepancy in ΔBP ND between findings in two separate smoking studies by Brody et al. [11,13] may be due solely to differences in the amount of data used to estimate BP ND Activation. In the studies by Brody et al., subjects were scanned for 50 min, taken off the camera for 10 min with the tracer infusion still running and allowed to smoke 1 cigarette outside, then returned to the camera and scanned for an additional 30 min. EQ was used to estimate BP ND. In an early study [13], BP ND Activation was estimated from the average of 10 min of data collected after each subject smoked a cigarette (60-70 min post-injection), and a 30% decrease in BP ND due to smoking was reported in the left ventral caudate. In later studies [9-12], 30 min of data (60-90 min post-injection) were used to estimate BP ND Activation, and only an 8% decrease in BP ND was found in the left ventral caudate. Brody et al. did not offer a persuasive argument to explain this large discrepancy between studies. However, the drop in effect size with increased activation scan window is consistent with our simulation findings which show that for β=1, an activation scan window of 40-60 min gives ΔBP ND = 4% but an activation scan window of 40-80 min gives ΔBP ND = 2% (Figure 4). The actual ΔBP ND from the Brody et al studies depends on how big the DA effect was and its temporal kinetics, as well as other factors such as how the regions were drawn, but our simulations predict that there would be a discrepancy in ΔBP ND reported using 20 min vs. 40 min of activation scan data precisely in the direction of the observed discrepancy in the Brody et al. studies.
The dependence of ΔBP ND on activation scan duration demonstrated by simulations was also observed in analysis of our human data. This pattern seen in simulations was seen only in the left ventral striatum of the two male subjects studies and only in the left dorsal caudate of the two female subjects. Based on our simulations, this suggests the presence of transient DA release in these regions. The regional difference in DA release in males and females seen here may suggest a gender-effect in DA response to smoking, one which we are currently pursuing.
The interpretation of these results is limited to the framework in which these studies were completed. We tested SRTM, the Logan graphical method, and EQ on B/I data from a single-scan activation study design. While application of EQ to this study design is common, SRTM and the Logan graphical method are typically applied to data from dual-scan (aka “paired-bolus”) activation studies. Nonetheless, we believe that application of the methods as done here demonstrates a general danger of applying conventional analysis methods to [11C]raclopride data which contain transient DA release. When the data violate the assumption that DA concentration is at steady-state, BP ND is no longer a static parameter and the measured ΔBP ND becomes a time-weighted average of the instantaneous ΔBP ND(t). Practically speaking, this may increase inter-subject variability in ΔBP ND since the kinetics of DA release may vary across subjects. This variability will, in turn, reduce effect size.
This would seem to suggest that shorter scan durations may improve the detection of transient DA release via ΔBP ND. However, because ΔBP ND depends on the timing and kinetics of DA relative to that of the tracer, interpretation of ΔBP ND as an index of the amount of DA released is not straightforward. Instead of optimizing conventional methods to detect a significant ΔBP ND, it may be better to estimate DA release using advanced models, which take into account the time-variation in DA signal. The linear extension of the simplified reference region model (LSRRM) [39] and the neurotransmitter PET (ntPET) suite of methods [40-43] both allow endogenous neurotransmitter to vary during the course of the scan. These methods may be better suited for estimating transient DA release, such as smoking-induced DA release, from [11C]raclopride PET data.
Conclusions
The dependence of ΔBP ND, as estimated by SRTM, Logan plot, and EQ method, on the duration of activation data analyzed illustrates the inability of conventional methods based on time-invariant parameters to reliably quantify short-lived increases in endogenous DA. Ultimately, more complicated models which take into account time-variation in DA are needed to properly measure the effect of stimulus-induced DA release when the DA response is brief.
Acknowledgments
We’d like to thank Drs. Karmen Yoder and Dianne Lee for their assistance with defining our striatal template. We thank Erin McGovern and Sabrina Helmbrecht for subject recruitment and screening. We also acknowledge the chemistry staff, nuclear medicine technologists, nurses, and research staff of the Yale PET Center without whom this work would not be possible.
Supported by ORWH and NIDA (P50DA033945, K02 DA031750, R21DA032791).
Conflict of interest statement
The authors have no conflicts of interest to declare.
References
- 1.Lammertsma AA, Hume SP. Simplified reference tissue model for PET receptor studies. Neuroimage. 1996;4:153–158. doi: 10.1006/nimg.1996.0066. [DOI] [PubMed] [Google Scholar]
- 2.Logan J, Fowler JS, Volkow ND, Wang GJ, Ding YS, Alexoff DL. Distribution volume ratios without blood sampling from graphical analysis of PET data. J Cereb Blood Flow Metab. 1996;16:834–840. doi: 10.1097/00004647-199609000-00008. [DOI] [PubMed] [Google Scholar]
- 3.Logan J, Fowler JS, Volkow ND, Wolf AP, Dewey SL, Schlyer DJ, MacGregor RR, Hitzemann R, Bendriem B, Gatley SJ, et al. Graphical analysis of reversible radioligand binding from time-activity measurements applied to [N-11C-methyl] -(-)-cocaine PET studies in human subjects. J Cereb Blood Flow Metab. 1990;10:740–747. doi: 10.1038/jcbfm.1990.127. [DOI] [PubMed] [Google Scholar]
- 4.Laruelle M. Imaging synaptic neurotransmission with in vivo binding competition techniques: a critical review. J Cereb Blood Flow Metab. 2000;20:423–451. doi: 10.1097/00004647-200003000-00001. [DOI] [PubMed] [Google Scholar]
- 5.Drevets WC, Price JC, Kupfer DJ, Kinahan PE, Lopresti B, Holt D, Mathis C. PET measures of amphetamine-induced dopamine release in ventral versus dorsal striatum. Neuropsychopharmacology. 1999;21:694–709. doi: 10.1016/S0893-133X(99)00079-2. [DOI] [PubMed] [Google Scholar]
- 6.Martinez D, Slifstein M, Broft A, Mawlawi O, Hwang DR, Huang Y, Cooper T, Kegeles L, Zarahn E, Abi-Dargham A, Haber SN, Laruelle M. Imaging human mesolimbic dopamine transmission with positron emission tomography. Part II: amphetamine-induced dopamine release in the functional subdivisions of the striatum. J Cereb Blood Flow Metab. 2003;23:285–300. doi: 10.1097/01.WCB.0000048520.34839.1A. [DOI] [PubMed] [Google Scholar]
- 7.Munro CA, McCaul ME, Oswald LM, Wong DF, Zhou Y, Brasic J, Kuwabara H, Kumar A, Alexander M, Ye W, Wand GS. Striatal dopamine release and family history of alcoholism. Alcohol Clin Exp Res. 2006;30:1143–1151. doi: 10.1111/j.1530-0277.2006.00130.x. [DOI] [PubMed] [Google Scholar]
- 8.Barrett SP, Boileau I, Okker J, Pihl RO, Dagher A. The hedonic response to cigarette smoking is proportional to dopamine release in the human striatum as measured by positron emission tomography and [11C] raclopride. Synapse. 2004;54:65–71. doi: 10.1002/syn.20066. [DOI] [PubMed] [Google Scholar]
- 9.Brody AL, London ED, Olmstead RE, Allen-Martinez Z, Shulenberger S, Costello MR, Abrams AL, Scheibal D, Farahi J, Shoptaw S, Mandelkern MA. Smoking-induced change in intrasynaptic dopamine concentration: effect of treatment for Tobacco Dependence. Psychiatry Res. 2010;183:218–224. doi: 10.1016/j.pscychresns.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brody AL, Mandelkern MA, Olmstead RE, Allen- Martinez Z, Scheibal D, Abrams AL, Costello MR, Farahi J, Saxena S, Monterosso J, London ED. Ventral striatal dopamine release in response to smoking a regular vs a denicotinized cigarette. Neuropsychopharmacology. 2009;34:282–289. doi: 10.1038/npp.2008.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brody AL, Mandelkern MA, Olmstead RE, Scheibal D, Hahn E, Shiraga S, Zamora-Paja E, Farahi J, Saxena S, London ED, McCracken JT. Gene variants of brain dopamine pathways and smoking-induced dopamine release in the ventral caudate/nucleus accumbens. Arch Gen Psychiatry. 2006;63:808–816. doi: 10.1001/archpsyc.63.7.808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brody AL, Olmstead RE, Abrams AL, Costello MR, Khan A, Kozman D, Saxena S, Farahi J, London ED, Mandelkern MA. Effect of a history of major depressive disorder on smoking-induced dopamine release. Biol Psychiatry. 2009;66:898–901. doi: 10.1016/j.biopsych.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brody AL, Olmstead RE, London ED, Farahi J, Meyer JH, Grossman P, Lee GS, Huang J, Hahn EL, Mandelkern MA. Smoking-induced ventral striatum dopamine release. Am J Psychiatry. 2004;161:1211–1218. doi: 10.1176/appi.ajp.161.7.1211. [DOI] [PubMed] [Google Scholar]
- 14.Montgomery AJ, Lingford-Hughes AR, Egerton A, Nutt DJ, Grasby PM. The effect of nicotine on striatal dopamine release in man: A [(11)C] raclopride PET study. Synapse. 2007;61:637–645. doi: 10.1002/syn.20419. [DOI] [PubMed] [Google Scholar]
- 15.Scott DJ, Domino EF, Heitzeg MM, Koeppe RA, Ni L, Guthrie S, Zubieta JK. Smoking modulation of mu-opioid and dopamine D2 receptor-mediated neurotransmission in humans. Neuropsychopharmacology. 2007;32:450–457. doi: 10.1038/sj.npp.1301238. [DOI] [PubMed] [Google Scholar]
- 16.Adams BW, Bradberry CW, Moghaddam B. NMDA antagonist effects on striatal dopamine release: microdialysis studies in awake monkeys. Synapse. 2002;43:12–18. doi: 10.1002/syn.1114. [DOI] [PubMed] [Google Scholar]
- 17.Endres CJ, Kolachana BS, Saunders RC, Su T, Weinberger D, Breier A, Eckelman WC, Carson RE. Kinetic modeling of [11C] raclopride: combined PET-microdialysis studies. J Cereb Blood Flow Metab. 1997;17:932–942. doi: 10.1097/00004647-199709000-00002. [DOI] [PubMed] [Google Scholar]
- 18.Laruelle M, Iyer RN, al-Tikriti MS, Zea-Ponce Y, Malison R, Zoghbi SS, Baldwin RM, Kung HF, Charney DS, Hoffer PB, Innis RB, Bradberry CW. Microdialysis and SPECT measurements of amphetamine-induced dopamine release in nonhuman primates. Synapse. 1997;25:1–14. doi: 10.1002/(SICI)1098-2396(199701)25:1<1::AID-SYN1>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 19.Houston GC, Hume SP, Hirani E, Goggi JL, Grasby PM. Temporal characterisation of amphetamine-induced dopamine release assessed with [11C] raclopride in anaesthetised rodents. Synapse. 2004;51:206–212. doi: 10.1002/syn.10296. [DOI] [PubMed] [Google Scholar]
- 20.Montgomery AJ, Lingford-Hughes AR, Egerton A, Nutt DJ, Grasby PM. The effect of nicotine on striatal dopamine release in man: A [11C] raclopride PET study. Synapse. 2007;61:637–645. doi: 10.1002/syn.20419. [DOI] [PubMed] [Google Scholar]
- 21.Pontieri FE, Tanda G, Orzi F, Di Chiara G. Effects of nicotine on the nucleus accumbens and similarity to those of addictive drugs. Nature. 1996;382:255–257. doi: 10.1038/382255a0. [DOI] [PubMed] [Google Scholar]
- 22.Endres CJ, Carson RE. Assessment of dynamic neurotransmitter changes with bolus or infusion delivery of neuroreceptor ligands. J Cereb Blood Flow Metab. 1998;18:1196–1210. doi: 10.1097/00004647-199811000-00006. [DOI] [PubMed] [Google Scholar]
- 23.Morris ED, Fisher RE, Alpert NM, Rauch SL, Fischman AJ. In vivo imaging of neuromodulation using positron emission tomography: Optimal ligand characteristics and task length for detection of activation. Hum Brain Mapp. 1995;3:35–55. [Google Scholar]
- 24.Pappata S, Dehaene S, Poline JB, Gregoire MC, Jobert A, Delforge J, Frouin V, Bottlaender M, Dolle F, Di Giamberardino L, Syrota A. In vivo detection of striatal dopamine release during reward: a PET study with [(11)C] raclopride and a single dynamic scan approach. Neuroimage. 2002;16:1015–1027. doi: 10.1006/nimg.2002.1121. [DOI] [PubMed] [Google Scholar]
- 25.Muzic RF Jr, Cornelius S. COMKAT: compartment model kinetic analysis tool. J Nucl Med. 2001;42:636–645. [PubMed] [Google Scholar]
- 26.Fisher RE, Morris ED, Alpert NM, Fischman AJ. In vivo imaging of neuromodulatory synaptic transmission using PET: A review of relevant neurophysiology. Hum Brain Mapp. 1995;3:24–34. [Google Scholar]
- 27.Oikonen V. Noise model for PET time-radioactivity curves. 2012 2003-01-19. http://www.turkupetcentre.net/reports/tpcmod0008.pdf. [Google Scholar]
- 28.Varga J, Szabo Z. Modified regression model for the Logan plot. J Cereb Blood Flow Metab. 2002;22:240–244. doi: 10.1097/00004647-200202000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Langer O, Någren K, Dolle F, Lundkvist C, Sandell J, Swahn CG, Vaufrey F, Crouzel C, Maziere B, Halldin C. Precursor synthesis and radiolabelling of the dopamine D2 receptor ligand [11C] raclopride from [11C] methyl triflate. J Labelled Compd Rad. 1999;42:1183–1193. [Google Scholar]
- 30.Carson RE, Barker WC, Jeih-San L, Johnson CA. Design of a motion-compensation OSEM list-mode algorithm for resolution-recovery reconstruction for the HRRT. IEEE Nucl Sci Sym Conf Rec. 2003;5:3281–3285. [Google Scholar]
- 31.Yoder KK, Albrecht DS, Kareken DA, Federici LM, Perry KM, Patton EA, Zheng QH, Mock BH, O’Connor S, Herring CM. Test-retest variability of [11C] raclopride-binding potential in nontreatment-seeking alcoholics. Synapse. 2011;65:553–561. doi: 10.1002/syn.20874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mawlawi O, Martinez D, Slifstein M, Broft A, Chatterjee R, Hwang DR, Huang Y, Simpson N, Ngo K, Van Heertum R, Laruelle M. Imaging human mesolimbic dopamine transmission with positron emission tomography: I. Accuracy and precision of D(2) receptor parameter measurements in ventral striatum. J Cereb Blood Flow Metab. 2001;21:1034–1057. doi: 10.1097/00004647-200109000-00002. [DOI] [PubMed] [Google Scholar]
- 33.Frey KA, Ehrenkaufer RL, Beaucage S, Agranoff BW. Quantitative in vivo receptor binding. I. Theory and application to the muscarinic cholinergic receptor. J Neurosci. 1985;5:421–428. doi: 10.1523/JNEUROSCI.05-02-00421.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lassen NA. Neuroreceptor quantitation in vivo by the steady-state principle using constant infusion or bolus injection of radioactive tracers. J Cereb Blood Flow Metab. 1992;12:709–716. doi: 10.1038/jcbfm.1992.101. [DOI] [PubMed] [Google Scholar]
- 35.Volkow ND, Wang GJ, Fowler JS, Tomasi D, Telang F. Addiction: beyond dopamine reward circuitry. Proc Natl Acad Sci U S A. 2011;108:15037–15042. doi: 10.1073/pnas.1010654108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marquardt DW. An Algorithm for Least-Squares Estimation of Nonlinear Parameters. J Soc Ind and Appl Math. 1963;11:431–441. [Google Scholar]
- 37.Slifstein M, Laruelle M. Effects of statistical noise on graphic analysis of PET neuroreceptor studies. J Nucl Med. 2000;41:2083–2088. [PubMed] [Google Scholar]
- 38.Yoder KK, Wang C, Morris ED. Change in binding potential as a quantitative index of neurotransmitter release is highly sensitive to relative timing and kinetics of the tracer and the endogenous ligand. J Nucl Med. 2004;45:903–911. [PubMed] [Google Scholar]
- 39.Alpert NM, Badgaiyan RD, Livni E, Fischman AJ. A novel method for noninvasive detection of neuromodulatory changes in specific neurotransmitter systems. Neuroimage. 2003;19:1049–1060. doi: 10.1016/s1053-8119(03)00186-1. [DOI] [PubMed] [Google Scholar]
- 40.Morris ED, Yoder KK, Wang C, Normandin MD, Zheng QH, Mock B, Muzic RF Jr, Froehlich JC. ntPET: a new application of PET imaging for characterizing the kinetics of endogenous neurotransmitter release. Mol Imaging. 2005;4:473–489. doi: 10.2310/7290.2005.05130. [DOI] [PubMed] [Google Scholar]
- 41.Normandin MD, Schiffer WK, Morris ED. A linear model for estimation of neurotransmitter response profiles from dynamic PET data. Neuroimage. 2012;59:2689–2699. doi: 10.1016/j.neuroimage.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Constantinescu CC, Bouman C, Morris ED. Nonparametric Extraction of Transient Changes in Neurotransmitter Concentration From Dynamic PET Data. IEEE Trans Med Imaging. 2007;26:359–373. doi: 10.1109/TMI.2006.891501. [DOI] [PubMed] [Google Scholar]
- 43.Morris ED, Constantinescu CC, Sullivan JM, Normandin MD, Christopher LA. Noninvasive visualization of human dopamine dynamics from PET images. Neuroimage. 2010;51:135–144. doi: 10.1016/j.neuroimage.2009.12.082. [DOI] [PMC free article] [PubMed] [Google Scholar]