

CONSPECTUS
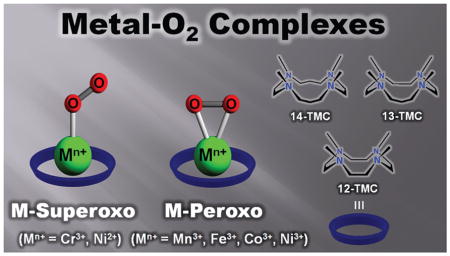
Metalloenzymes activate dioxygen to carry out a variety of biological reactions, including the biotransformation of naturally occurring molecules, oxidative metabolism of xenobiotics, and oxidative phosphorylation. The dioxygen activation at the catalytic sites of the enzymes occurs through several steps, such as the binding of O2 at a reduced metal center, the generation of metal–superoxo and –peroxo species, and the O–O bond cleavage of metal–hydroperoxo complexes to form high-valent metal-oxo oxidants. Because these mononuclear metal–dioxygen (M–O2) adducts are implicated as key intermediates in dioxygen activation reactions catalyzed by metalloenzymes, studies of the structural and spectroscopic properties and reactivities of synthetic biomimetic analogues of these species have aided our understanding of their biological chemistry. One particularly versatile class of biomimetic coordination complexes for studying dioxygen activation by metal complexes is M–O2 complexes bearing the macrocyclic N-tetramethylated cyclam (TMC) ligand.
This Account describes the synthesis, structural and spectroscopic characterization, and reactivity studies of M–O2 complexes bearing tetraazamacrocyclic n-TMC ligands, where M = Cr, Mn, Fe, Co, and Ni and n = 12, 13, and 14, based on recent results from our laboratory. We have used various spectroscopic techniques, including resonance Raman and X-ray absorption spectroscopy, and density functional theory (DFT) calculations to characterize several novel metal–O2 complexes. Notably, X-ray crystal structures had shown that these complexes are end-on metal-superoxo and side-on metal-peroxo species. The metal ions and the ring size of the macrocyclic TMC ligands control the geometric and electronic structures of the metal–O2 complexes, resulting in the end-on metal–superoxo versus side-on metal–peroxo structures. Reactivity studies performed with the isolated metal-superoxo complexes reveal that they can conduct electrophilic reactions such as oxygen atom transfer and C–H bond activation of organic substrates. The metal–peroxo complexes are active oxidants in nucleophilic reactions, such as aldehyde deformylation. We also demonstrate a complete intermolecular O2-transfer from metal(III)–peroxo complexes to a Mn(II) complex. The results presented in this Account show the significance of metal ions and supporting ligands in tuning the geometric and electronic structures and reactivities of the metal–O2 intermediates that are relevant in biology and in biomimetic reactions.
1. Introduction
Metalloenzymes activate dioxygen to carry out a variety of biological reactions, including biotransformation of naturally occurring molecules, oxidative metabolism of xenobiotics, and oxidative phosphorylation.1 The dioxygen activation at the active sites of the enzymes occurs through several steps, such as the binding of O2 at a reduced metal center, the generation of metal–superoxo and –peroxo species, and the O–O bond cleavage of metal–hydroperoxo complexes to form high-valent metal-oxo oxidants.1 In heme and nonhemeiron and copper enzymes, mononuclear metal–dioxygen adducts (M–O2), such as end-on and side-on metal-O2 species (Scheme 1), are generated in the O2 activation and play important roles as key intermediates in the oxidation of organic substrates.1–4 Such metal–O2 intermediates have been investigated extensively in biomimetic studies to understand the structural and chemical properties of the intermediates that are short-lived and thus difficult to study in enzymatic reactions.5–11 For example, the binding modes of the O2 unit and the bond lengths of O–O and M–O bonds of synthetic metal–superoxo and –peroxo complexes were elucidated by X-ray crystallographic analysis. Spectroscopic characterization of the intermediates, such as O–O and M-O stretching frequencies determined in resonance Raman (rRaman) measurements,12 supported the assignment of the binding modes of the O2 unit in M–O2 complexes. The reactivities of these M–O2 complexes have been extensively investigated in electrophilic and nucleophilic oxidation reactions. 13,14 For instance, Fe(III)–O2 complexes with heme and nonheme ligands have been synthesized as chemical models of cytochrome P450 aromatases and Rieske dioxygenases, respectively, and have shown reactivities in nucleophilic reactions such as aldehyde deformylation.4,14 As biomimetic compounds of copper-containing enzymes, peptidylglycine-α-hydroxylating monooxygenase (PHM) and dopamine-β-monooxygenase (DβM), mononuclear Cu–O2 complexes have been synthesized and characterized and their reactivities have been investigated in electrophilic reactions.7,8,10,15–17 Fe(III)–superoxo species have been proposed as active oxidants in C–H bond activation of substrates by nonheme iron enzymes, such as isopenicillin N-synthase and myo-inositol oxygenase.2 Biomimetic studies provided indirect evidence that mononuclear nonheme Fe(III)–superoxo complexes are capable of activating C–H bonds of organic substrates, although such Fe(III)–superoxo species have yet to be isolated and characterized spectroscopically.18
SCHEME 1.
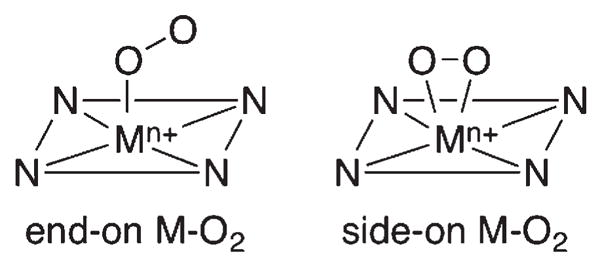
End-on and Side-on Metal–O2 Complexes
In addition to the iron and copper enzymes, Mn–O2 complexes have been invoked as reactive intermediates in manganese-containing enzymes, such as in the oxygen evolving complex of photosystem II, manganese superoxide dismutase, and catalase.19 In biomimetic studies, the first crystal structure of a side-on Mn(III)–peroxo complex, [Mn(TPP)(O2)]− (TPP = meso-tetraphenylporphyrin), was reported by Valentine and co-workers,20 followed by the crystal structure of a nonheme side-on Mn(III)–peroxo complex by Kitajima and co-workers.21 The chemistry of other M–O2 complexes (M = Ti, V, Cr, Co, Ni, and the second and third row transition metals) has been well documented. 13,22,23 One representative example is the Co–O2 complexes bearing salen, porphyrin, and tetraazamacrocyclic ligands as models of dioxygen-carrying proteins, such as hemoglobin and myoglobin, and as active oxidants in the oxidation of organic substrates.22,24,25
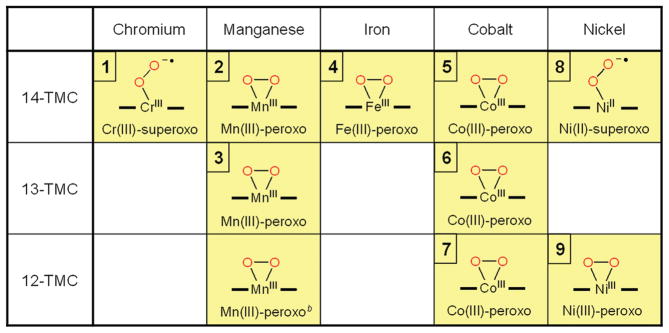
It has been demonstrated recently that N-tetramethylated cyclams (TMC),26 such as n-TMC (n = 14 for 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane, 13 for 1,4,7,10-tetramethyl-1,4,7,10-tetraazacyclotridecane, and 12 for 1,4,7,10-tetramethyl-1,4,7,10-tetraazacyclodecane; see Scheme 2 for ligand structures), are versatile ligands in the biomimetic chemistry of dioxygen activation by metal complexes. A variety of metal–TMC complexes of oxo, superoxo, and peroxo ligands have been synthesized and characterized spectroscopically and/or structurally.27–40 In particular, the first high-resolution crystal structures of Fe(IV)–oxo and Fe(III)–peroxo complexes with the 14-TMC ligand, such as [FeIV(14-TMC)(O)]2+ and [FeIII(14-TMC)-(O2)]+,31,38 were successfully obtained in nonheme iron models. In these studies, it has been shown that the ring size of the TMC ligands is an important factor that controls the geometric and electronic structures of the M–O2 adducts. Such a ring size effect has been highlighted by two Ni–O2 complexes bearing 14-TMC and 12-TMC ligands;34–36 [Ni(14-TMC)(O2)]+ is an end-on Ni(II)-superoxo species,34 whereas [Ni(12-TMC)(O2)]+ is a side-on Ni(III)-peroxo species.35 Thus, significant progress has been achieved in understanding the chemical and physical properties of the biologically important metal–oxygen intermediates, such as high-valent metal–oxo and metal–O2 complexes, by synthesizing their biomimetic compounds using the versatile tetraazamacrocyclic ligands, n-TMC. In this Account, we describe our recent results on the synthesis, structural and spectroscopic characterization, and reactivity studies of the M–O2 complexes bearing n-TMC ligands (M = Cr, Mn, Fe, Co, Ni; n = 12, 13, 14) (Table 1). The effects of the metal ions and macrocyclic ring size on the structures and reactivities of the biologically important M-O2 intermediates are discussed as well.
SCHEME 2.
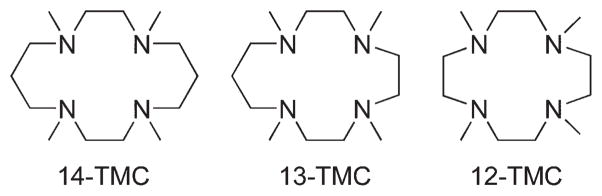
TMC Ligands Used in the Synthesis of M–O2 Complexes
TABLE 1.
Structures of Metal–O2 Complexes Bearing Macrocyclic n-TMC Ligandsa
|
There are no high-resolution crystal structures available for [CoIII(14-TMC)(O2)]+ (5) and [NiII(14-TMC)(O2)]+ (8).
The details of the high-resolution crystal structure of [MnIII(12-TMC)(O2)]+ will be reported elsewhere.
2. Synthesis and Characterization
Metal–O2 complexes were synthesized by reacting metal(II) complexes, [M(n-TMC)]2+ (M = Cr, Mn, Fe, Co, Ni; n = 12, 13, 14), with O2 or H2O2 in the presence of base (e.g., triethylamine (TEA) or tetramethylammonium hydroxide (TMAH)), as summarized in Scheme 3. First, bubbling of O2 to the solution of [CrII(14-TMC)(Cl)]+ in CH3CN afforded the color change from blue to violet (Scheme 3, route A).27 The violet intermediate, [CrIII(14-TMC)(O2)(Cl)]+ (1), was characterized with various spectroscopic methods (see Table 2, entry 1 for spectroscopic data). The superoxo character of the O2 ligand was unambiguously assigned by rRaman spectroscopic and X-ray crystallographic analyses. In rRaman measurements, 1 prepared with 16O2 exhibits an isotopically sensitive band at 1170 cm−1, which shifts to 1104 cm−1 in samples of 1 prepared with 18O2 (Table 2). The single crystal structure of 1 revealed the mononuclear end-on Cr–superoxo complex in a distorted octahedral geometry (Figure 1A). The O–O bond length (1.231 Å) of 1 is shorter than that of the side-on Cr(III)–superoxo complex (1.327 Å).41 The relatively short O–O bond distance is consistent with the high frequency of the O–O stretching vibration observed in the rRaman measurements. All N-methyl groups of the 14-TMC ligand are oriented anti to the superoxo ligand, while the syn site is occupied by a Cl ligand.
SCHEME 3.
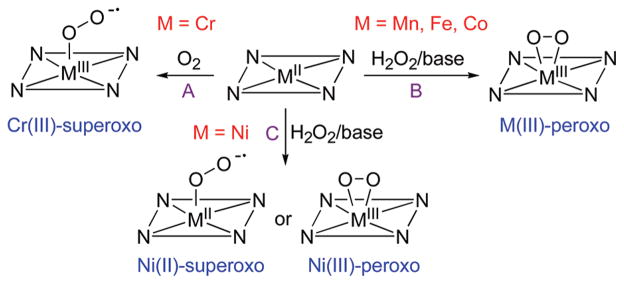
Reactions Showing the Synthesis of Metal–Superoxo and –Peroxo Complexes
TABLE 2.
UV–Vis, ESI-MS, rRaman, EPR, Spin State, and X-ray Data of M–O2 Complexes
| entry | M–O2 complex | UV–vis λ [nm] (ε [M−1 cm−1]) | ESI-MS m/z 16O2 (m/z 18O2) | rRaman [cm−1] v(16O–16O) (v(18O–18O)) | EPR | spin state | bond lengths M–O/O–O [Å] | ref |
|---|---|---|---|---|---|---|---|---|
| 1 | [CrIII(14-TMC)(O2)(Cl)]+ (1) | 331 (3800), 391 (290), 469 (150), 549 (240), 643 (130), 675 (140) | 375.0 (379.0) | 1170 (1104) | silent | S = 1 | 1.876(4)/1.231(6) | 27 |
| 2 | [MnIII(14-TMC)(O2)]+ (2) | 453 (490), 630 (120) | 343.1 (347.1) | silent | S = 2 | 1.884(2)/1.403(4) | 28 | |
| 3 | [MnIII(13-TMC)(O2)]+ (3) | 288 (2400), 452 (390), 615 (190) | 329.1 (333.1) | silent | S = 2 | 1.859/1.410(4) | 29 | |
| 4 | [FeIII(14-TMC)(O2)]+ (4) | 750 (600) | 344.1 (348.1) | 825 (781) | g = 8.8, 5.9, 4.3 | S = 5/2 | 1.910/1.463(6) | 31 |
| 5 | [CoIII(14-TMC)(O2)]+ (5) | 436 (250), 575 (100), 801 (90) | 347.1 (351.1) | silent | S = 1 | 32 | ||
| 6 | [CoIII(13-TMC)(O2)]+ (6) | 348 (620), 562 (210), ~500 (170), ~710 (100) | 333.1 (337.1) | 902 (846) | silent | S = 0 | 1.855/1.438(4) | 33 |
| 7 | [CoIII(12-TMC)(O2)]+ (7) | 350 (450), 560 (180), ~500 (150), ~710 (90) | 319.1 (323.1) | 902 (845) | silent | S = 0 | 1.866/1.4389(17) | 33 |
| 8 | [NiII(14-TMC)(O2)]+ (8) | 336 (610), 414 (130), 690 (50) | 346.1 (350.0) | 1131 (1067) | g = 2.29, 2.21, 2.09 | S = 1/2 | 34 | |
| 9 | [NiIII(12-TMC)(O2)]+ (9) | 345 (300), 400 (250), 646 (100), 899 (70) | 318.0 (322.0) | 1002 (945) | g = 2.22, 2.17, 2.06 | S = 1/2 | 1.889/1.386(4) | 35 |
FIGURE 1.

Crystal structures of M-O2 complexes: (A) [CrIII(14-TMC)(O2)(Cl)]+ (1); (B) [MnIII(14-TMC)(O2)]+ (2); (C) [MnIII(13-TMC)(O2)]+ (3); (D) [FeIII(14-TMC)-(O2)]+ (4); (E) [CoIII(13-TMC)(O2)]+ (6); (F) [CoIII(12-TMC)(O2)]+ (7); (G) [NiIII(12-TMC)(O2)]+ (9).
Different from the Cr–O2 complex, other metal(II) complexes, such as Mn(II), Fe(II), Co(II), and Ni(II), did not react with O2 to form M-O2 complexes. Alternatively, metal–O2 complexes were generated by reacting metal(II) complexes with H2O2 in the presence of base (Scheme 3, routes B and C). First, [MnIII(14-TMC)(O2)]+ (2) was synthesized by reacting [MnII(14-TMC)]2+ with H2O2 in the presence of TEA in CH3CN (Scheme 3, route B; Table 2, entry 2 for spectroscopic data).28 The ESI-MS of the green intermediate 2 exhibits a prominent ion peak at a mass-to-charge ratio (m/z) of 343.1, whose mass and isotope distribution pattern correspond to [Mn(14-TMC)(O2)]+. When 2 was synthesized using isotopically labeled H2 18O2, a mass peak corresponding to [Mn(14-TMC)-(18O2)]+ appeared at m/z 347.1. The four mass unit increase upon the substitution of 16O with 18O indicates the binding of an O2 unit in 2. The X-ray crystal structure of 2 revealed the mononuclear side-on Mn(III)–peroxo complex in a distorted octahedral geometry arising from the triangular MnO2 moiety (Figure 1B). The O–O bond length of 1.403(4) Å is typical for a peroxo ligand bound to a transition metal ion, but is slightly shorter than those found in [Mn(TPP)(O2)]− (1.43 Å)20 and [Mn(TpPr)(3,5-Pr2pzH)(O2)] (1.421 Å).21 All N-methyl groups of the 14-TMC ligand are oriented syn to the peroxo ligand residing in the N-methyl pocket. Similarly, [MnIII(13-TMC)(O2)]+ (3) was synthesized by reacting [MnII(13-TMC)]2+ with H2O2 in the presence of TEA in CH3CN (Scheme 3, route B; Table 2, entry 3 for spectroscopic data).29 The X-ray crystal structure of the green intermediate 3 revealed a monomeric six-coordinate MnIII cation with a side-on peroxo ligand and the O–O bond length of 1.403(4) Å (Figure 1C).
The Fe(III)–peroxo complex, [FeIII(14-TMC)(O2)]+ (4), was prepared by reacting [FeII(14-TMC)]2+ with H2O2 in the presence of TEA in CF3CH2OH (Scheme 3, route B).30,31 The intermediate persisted for several hours at 0 °C, and the greater thermal stability of 4 in alcoholic solvents allowed us to isolate crystals.31 The isolated intermediate 4 was fully characterized with various spectroscopic techniques (Table 2, entry 4), including UV–vis, ESI-MS, EPR, rRaman, and X-ray absorption spectroscopy (XAS). The rRaman spectrum of 4 shows two peaks that shift upon 18O-substitution. The peak at 487 cm−1 shifts to 468 cm−1 with a 16,18Δ value of 19cm−1 (16,18Δ(calcd) =21 cm−1) and is assigned as an Fe–O stretch. The other isotopically sensitive peak in the rRaman spectrum appears at 825 cm−1, which shifts to 781 cm−1 in the 18O-labeled 4; this feature is assigned as the O–O stretch with the 16,18Δ value of 44 cm−1 (16,18Δ (calcd) = 47 cm−1). These values are comparable to the Fe–O and O–O stretches in spectroscopically characterized high-spin side-on Fe(III)–peroxo complexes in heme and non-heme iron systems.31,42 The X-ray crystal structure of 4 revealed the mononuclear side-on Fe(III)–peroxo complex in a distorted octahedral geometry (Figure 1D). The FeO2 geometry is similar to the crystallographically characterized structure of naphthalene dioxygenase (NDO), where dioxygen binds in a side-on fashion at the mononuclear iron center (1.75 Å resolution, rO–O ca. 1.45 Å).4 The structurally determined O–O bond length of 1.463(6) Å is indicative of peroxo character of the O2 group,12 as assigned by rRaman experiments (vide supra). All four N-methyl groups of the 14-TMC ligand are oriented syn to the peroxo ligand. In the case of an Fe(IV)–oxo complex bearing the same 14-TMC ligand, the four N-methyl groups of [FeIV(14-TMC)(O)(CH3CN)]2+ are anti to the oxo ligand,38 whereas those in a Sc3+-bound [FeIV(14-TMC)(O)]2+ are syn to the oxo ligand.39 In addition, a good correlation was observed when the O–O bond length and O–O stretching frequency of 4 was fitted in the plot of O–O stretching frequency versus O–O bond length for side-on metal–O2 complexes (Figure 2).12
FIGURE 2.

Plot of O–O stretching frequency (cm−1) versus O–O bond distance (Å) for side-on metal–O2 complexes. Circles represent experimental data points, and squares represent theoretical ones.12 The solid line represents a least-squares linear fit of the experimental and theoretical data. Data points for [FeIII(14-TMC)(O2)]+ (4) (red right triangle), [CoIII(13-TMC)(O2)]+ (6) (magenta up triangle), [CoIII(12-TMC)- (O2)]+ (7) (blue down triangle), and [NiIII(12-TMC)(O2)]+ (9) (green tilted square) are included in the diagram.
The reaction of [CoII(14-TMC)(CH3CN)]2+ with H2O2 in the presence of TEA in CH3CN afforded a gradual color change from pale purple to green (Scheme 3, route B).33 The green intermediate, [CoIII(14-TMC)(O2)]+ (5), was assigned by ESI-MS and EPR experiments (Table 2, entry 5), and the structural information was obtained by XAS and density functional theory (DFT) calculations (vide infra). The Co(III)–peroxo complexes bearing 13-TMC and 12-TMC were synthesized as follows: Addition of H2O2 to [CoII(13-TMC)(CH3CN)]2+ and [CoII(12-TMC)(CH3CN)]2+ in the presence of TEA in CH3CN afforded purple intermediates [CoIII(13-TMC)(O2)]+ (6) and [CoIII(12-TMC)(O2)]+ (7), respectively.33 The spectroscopic (Table 2, entries 6 and 7) and structural (Figure 1E and F) characterization revealed that the peroxo ligand is bound to the cobalt ion in a side-on fashion. The structural properties of the CoO2 core in 6 and 7 are nearly identical; the X-ray structures of 6 and 7 revealed a mononuclear side-on Co(III)–peroxo complex in a distorted octahedral geometry arising from the triangular CoO2 moiety, and the O–O bond distances of 6 and 7 were 1.438(6) and 1.4389(17) Å, respectively (Table 2). All four N-methyl groups of the 13-TMC and 12-TMC ligands are oriented syn to the peroxo ligand in both complexes. The O–O bond stretching frequency of 6 and 7 was determined to be 902 cm−1 by rRaman measurements, and we have observed a good correlation between the O–O stretching frequencies and the O–O bond lengths in 6 and 7 (Figure 2).12
Different from the metal(III)–peroxo complex formation discussed above, a Ni(II)–superoxo complex, [NiII(14-TMC)-(O2)]+ (8), was formed in the reaction of [NiII(14-TMC)(CH3CN)]2+ and H2O2 in the presence of base (TEA or TMAH) in CH3CN (Scheme 3, route C).34 The formation of 8 was also observed when a low valent [NiI(14-TMC)]+ complex was reacted with O2.34 The formation of 8 was confirmed by various spectroscopic techniques (Table 2, entry 8). The rRaman data show that 8 exhibits an isotopically sensitive band at 1131 cm−1, which shifts to 1067 cm−1 in samples of 8 prepared with 18O-labeled H2 18O2. The O–O stretching frequency of 8 (1131 cm−1) is comparable to that of 1 (1170 cm−1), indicating the superoxo character of the O2 unit in 8. The structural information was obtained by XAS and DFT calculations (vide infra). Interestingly, a Ni(III)–peroxo complex, [NiIII(12-TMC)(O2)]+ (9), was obtained by changing the supporting ligand from 14-TMC to 12-TMC.35 The reaction of [NiII(12-TMC)(CH3CN)]2+ with H2O2 in the presence of TEA in CH3CN produces the green intermediate, 9, which exhibits characteristic absorption bands that are distinct from those of 8 (Scheme 3, route C; Table 2, entry 9 for spectroscopic data). The rRaman spectra of 9 show the peroxo character of the O2 group; 9 exhibits an isotopically sensitive band at 1002 cm−1, which shifts to 945 cm−1 upon substitution of 16O with 18O. The observed O–O stretching frequency of 9 (1002 cm−1) is significantly lower than that of 8 (1131 cm−1). The X-ray crystal structure of 9 revealed the mononuclear side-on Ni(III)–peroxo complex in a distorted octahedral geometry (Figure 1G).35 The O–O bond length of 9 (1.386(4) Å) is longer than those of Ni(II)–superoxo complexes, such as 8 (1.301 Å, obtained from DFT calculations)34 and NiO2 with a β-diketiminato ligand (1.347 Å).43 Further, the Ni–O average bond distance of 9 (1.889 Å) is shorter than that of 8 (1.984 Å, obtained from DFT calculations),34 supporting a Ni3+ oxidation state, which was also shown by XAS experiments and supported by DFT calculations. All four N-methyl groups of the 12-TMC ligand are oriented syn to the peroxo ligand in 9. There is a correlation between the observed O–O bond length and the O–O stretching frequency in 9, as observed in other side-on M–O2 complexes (Figure 2).12
2.1. Metal Ion Effect
We have shown above that the nature of O2 binding (e.g., side-on vs end-on and superoxo vs peroxo), the O–O and M-O bond distances in the M–O2 unit, and the orientation of methyl groups in TMC ligands are markedly influenced by the central metal ions of M–O2 complexes (Figure 1; Tables 1 and 2). For example, in the series of M–O2 complexes bearing 14-TMC ligand, the O2 ligands of the Cr(III)–superoxo and Ni(II)–superoxo complexes are bound to the metal ions in an end-on fashion, whereas those of the Mn(III)–peroxo, Fe(III)–peroxo, and Co(III)–peroxo complexes are coordinated to the metal ions in a side-on fashion (see the structures in the first row in Table 1). Thus, there is a trend that metal ions with a relatively high oxidation state prefer to form a side-on metal–peroxo complex, probably due to the stronger donor interaction by the peroxo ligand (e.g., Mn(III)–O2, Fe(III)–O2, and Co(III)–O2), whereas an end-on metal–superoxo complex is a preferred structure for metal ions with a relatively low oxidation state (e.g., Ni(II)–O2) (see Scheme 4).
SCHEME 4.
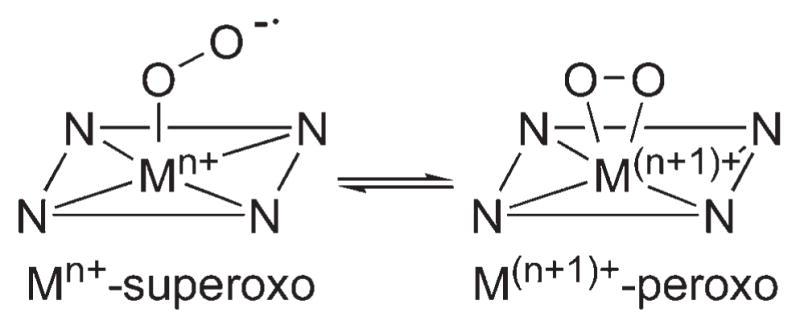
Preferred Geometry of M–O2 Complex Depending on the Oxidation State of Metal Ion
Second, the M-O and O–O bond distances are inversely correlated (R2 = 0.794), although there is one exception with 4 (Figure 3). Such a correlation has been observed in other mononuclear side-on metal–O2 complexes, and the correlation was interpreted with the orbital overlap between in-plane O2 π* orbital and metal dxy orbital.12 Thus, the decrease of the O–O bond distances is correlated with the increase of the M–O bond distances, resulting from the decrease of metal–oxygen covalency. Co(III)–peroxo (7) and Ni(III)–peroxo (9) complexes bearing 12-TMC ligand represent another good example for the correlation between the M–O and O–O bond distances. The average Co–O bond distance of 7 (1.866 Å) is shorter than the average Ni–O bond distance of 9 (1.889 Å), whereas the O–O bond distance of 7 (1.4389(17) Å) is longer than that of 9 (1.386(4) Å).33,35 A similar trend in M–O bond distances has been observed in Mn(III)–peroxo (2) and Fe(III)–peroxo (4) complexes with 14-TMC. The Mn–O bond distance of 2 (1.884(2) Å) is shorter than the average Fe–O bond distance of 4 (1.910 Å),28,31 which is comparable to those of previously reported Mn–O(H) and Fe–O(H) complexes.44 However, the O–O bond distance of 4 (1.463(6) Å) is significantly longer than that of 2 (1.403(4) Å).28,31 The extraordinary O–O bond elongation in 4, which is supported by the rRaman data (vide supra), makes it a dramatic exception to the correlation otherwise demonstrated in Figure 3.
FIGURE 3.

Plot of M-O versus O–O bond distances (Å) of M–O2 complexes. The dashed line represents a least-squares linear fit (y = −0.52x + 2.61, R2 = 0.794) of the experimental data except 4, although the data point of 4 is included in the diagram.
Third, all four N-methyl groups of TMC ligands in metal–peroxo complexes point to the same side of the peroxo moiety irrespective of metal ions and the ring size of TMC ligands (see Figure 1).28,29,31,33,35 In contrast, all N-methyl groups of the 14-TMC ligand in the Cr–superoxo complex, [CrIII(14-TMC)(O2)(Cl)]+ (1), are oriented anti to the superoxo ligand.27 Thus, the orientation of the N-methyl groups of TMC ligands in M–O2 complexes may be determined by the binding mode of the O2 ligand (e.g., syn to the peroxo ligand vs anti to the superoxo ligand), although there is only one crystal structure of a M–superoxo species binding a chloride ligand trans to the O2 group (i.e., [CrIII(14-TMC)(O2)(Cl)]+).27
2.2. Ring Size Effect
The macrocyclic ligands, n-TMC (n = 12, 13, 14), give several transition metal–O2 complexes in different oxidation states, suggesting that the ring size of the macrocycles is an important factor that controls the nature of M–O2 geometric and electronic structures (e.g., Mm+ superoxo vs M(m+1)+–peroxo in Scheme 4).34,35 To understand the ring size effect, XAS studies and DFT calculations were performed on [Co(n-TMC)(O2)]+ and [Ni(n-TMC)(O2)]+ complexes, where n = 12 or 14.36 In the case of [Ni(n-TMC)(O2)]+ (e.g., 8 and 9), Ni K-edge energies combined with extended X-ray absorption fine structure (EXAFS) data revealed that the smaller 12-TMC macrocycle forms a Ni(III)–peroxo complex, while the bigger 14-TMC ligand forms a Ni(II)–superoxo species. In 9, the smaller ring size allows O2 to bind in a side-on fashion and stabilizes the high oxidation state of Ni3+. In the case of [Co(n-TMC)(O2)]+, 7 bearing the 12-TMC ligand forms a side-on bound Co(III)–peroxo complex similar to 9 in the nickel system. However, while 8 forms a Ni(II)–superoxo complex, 5 forms a Co(III)–peroxo complex despite having the 14-TMC ligand. This is confirmed by Co K-edge XAS data, which show similar pre-edge and edge energy positions for 5 and 7.36 This discrepancy is investigated by DFT calculations, which show that there is a significantly higher thermodynamic driving force for the oxidation of Co(II) to Co(III) compared to Ni(II) to Ni(III).36 Thus, the results discussed above suggest that there is an interplay of several factors in determining the final stable geometric and electronic structures of [M(n-TMC)(O2)]m+ species, such as the macrocyclic ring size effect and the inherent thermodynamic stability of the metal ion in different oxidation states.
3. Reactivity
3.1. Electrophilic and Nucleophilic Reactions
Reactivities of the synthetic metal–O2 complexes have been investigated in electrophilic and nucleophilic reactions in order to understand the chemical properties and mechanisms of the oxidation of organic substrates by metal–superoxo and –peroxo species in enzymatic reactions. The substrates used in electrophilic reactions were PPh3 and alkylaromatics with weak C–H bond dissociation energies (BDEs) for oxygen atom transfer and hydrogen atom (H-atom) abstraction reactions, respectively, whereas the nucleophilic character of the metal–O2 complexes was examined in aldehyde deformylation reactions (Scheme 5). In electrophilic reactions, the nickel(II)–superoxo species, [Ni(14-TMC)(O2)]+ (8), oxidized PPh3 to produce OPPh3 in quantitative yield (Scheme 5A), and the source of oxygen in the product was confirmed using 18O-labeled [Ni(14-TMC)(18O2)]+.34,45 The electrophilic character of metal–superoxo species was further investigated in H-atom abstraction reactions by the chromium(III)–superoxo complex, [Cr(14-TMC)(O2)(Cl)]+ (1), using substrates with weak C–H bond dissociation energies (BDEs), such as xanthene, dihydroanthracene, and cyclohexadiene.27,40 In the latter reactions, the order of reaction rates (k2) was xanthene > DHA > CHD, and a linear correlation was observed between the log k2 values and the C–H BDE of the substrates. In addition, a kinetic isotope effect (KIE) value of 50 was obtained in the oxidation of DHA by 1. Such a large KIE value, with the good correlation between k2 and BDE of the substrates, implicates that the H-atom abstraction is the rate-determining step for the C–H bond activation by the Cr-superoxo complex.
SCHEME 5.
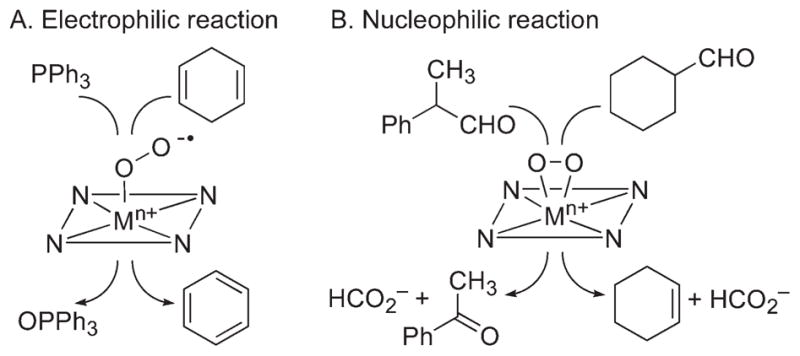
Reactivities of End-on Metal–Superoxo and Side-on Metal–Peroxo Complexes
Although metal–peroxo complexes are not reactive in electrophilic reactions, they are capable of conducting nucleophilic reactions, such as the deformylation of aldehydes. For example, the reaction of [Co(14-TMC)(O2)]+ (5) with 2-phenylpropionaldehyde (2-PPA) afforded acetophenone as the major product (Scheme 5B).32 Similar results were obtained in the reaction of cyclohexane carboxaldehyde (CCA), and product analysis of the resulting solution revealed the formation of cyclohexene (Scheme 5B).32 The reactivity of 5 was further investigated using benzaldehydes with a series of electron-donating and -withdrawing substituents at the para position of the phenyl group (para-Y–Ph–CHO; Y = OMe, Me, F, H, Cl). A positive ρ+ value of 1.8 in the Hammett plot was obtained, which is consistent with the nucleophilic character of 5 in the oxidation of aldehydes. The reactivity of metal–peroxo complexes was further investigated using primary (1°-CHO), secondary (2°-CHO), and tertiary (3°-CHO) aldehydes, and the observed reactivity order of 1°-CHO > 2°-CHO > 3°-CHO supports the nucleophilic character of the metal–peroxo species.31,35 Other metal–peroxo complexes, [MIII(n-TMC)(O2)]+, showed reactivities in the aldehyde deformylation reactions with positive ρ+ values in the reactions of para-X-substituted benzaldehydes and the reactivity order of 1°-CHO > 2°-CHO > 3°-CHO.28–33,35 The aldehyde deformylation reactions by metal–peroxo complexes are initiated by attack of the carbonyl group by the M-O2 moiety (Scheme 6).14
SCHEME 6.
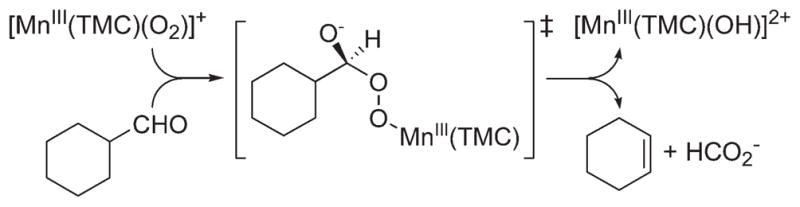
Proposed Mechanism for the Deformylation of CCA by [MnIII(14-TMC)(O2)]+ (2)28
A remarkable axial ligand effect on the reactivity of metal–peroxo complexes in heme and nonheme systems was observed in oxidative nucleophilic reactions.14,29 For example, [Mn(13-TMC)(O2)(X)] bearing different axial ligands (3-X; X = N3−, CF3CO2−, NCS−, CN−) was studied in the oxidation of CCA and observed the reactivity order of 3-N3 > 3-CF3CO2 > 3-NCS > 3-CN > 3.29 These results indicate that an increase in the nucleophilicity of the Mn(III)–peroxo unit upon binding of anionic axial ligands makes 3-X more electron-rich. The electronic effect was confirmed by plotting the potential of 3-X against the reaction rates of CCA oxidation by 3-X, showing that the Mn(III)–peroxo complexes with more electron-donating axial ligands are more reactive in oxidative nucleophilic reactions (Figure 4).29 In addition, the geometric effect on the binding of the anionic ligand trans to the peroxo ligand may facilitate the conversion of the side-on peroxo ligand to an end-on peroxo ligand, which probably becomes more nucleophilic.29 That is, upon binding of an axial ligand, the axial ligand pulls down the metal ion into the TMC ring plane (Scheme 7, A), as proposed in heme models.14 As a consequence, the peroxo group experiences more repulsive interactions from atoms in the TMC ring, resulting in the shift of the equilibrium toward the end-on conformation (Scheme 7, B). DFT calculations on the side-on and end-on Mn(III)–peroxo species illustrate that the peroxo group in the end-on structure has much more anionic character than that in the side-on structure where the electron densities at the oxygen atoms in the side-on and end-on peroxo ligands are −0.24 and −0.63, respectively. 29 Therefore, the oxygen atom in the end-on structure is predicted to be more reactive than the side-on bound structure as a nucleophile.
FIGURE 4.
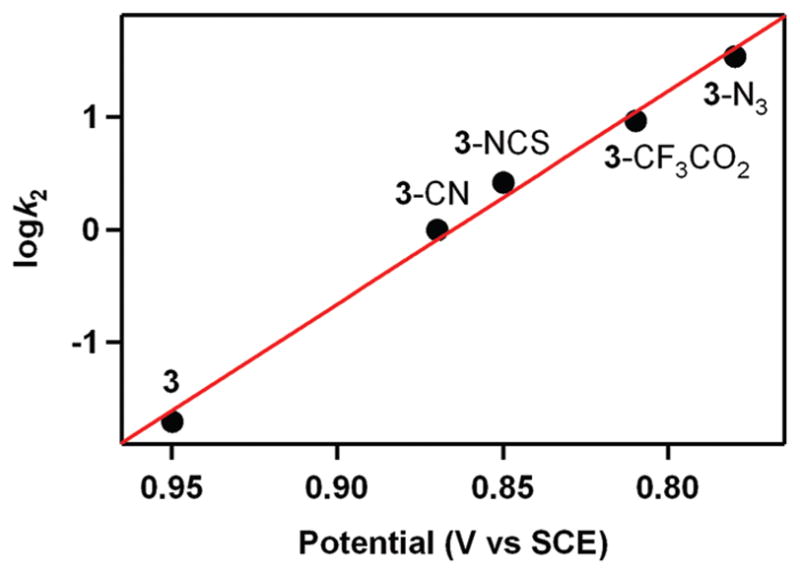
Plot of log k2 of [Mn(13-TMC)(O2)(X)]+ (3-X; X = N3−, CF3CO2−, NCS−, CN−) at 0 °C against Ep,a values of 3-X.29
SCHEME 7.
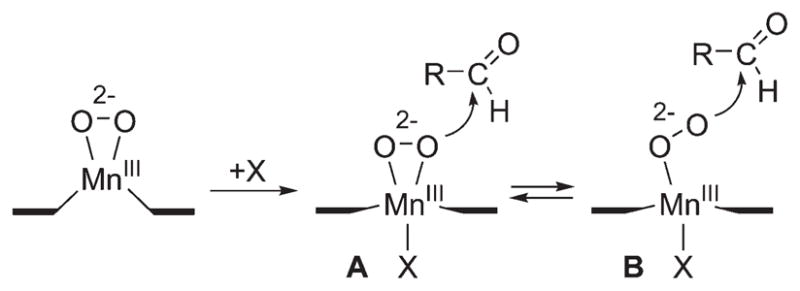
Axial Ligand Effect of Metal–Peroxo Complexes in Nucleophilic Reactions29
3.2. O2-Transfer Reactions
It has been shown that an intermolecular O2-tranfer occurs from a Ni(III)–peroxo complex, [Ni(12-TMC)(O2)]+ (9), to a Mn(II) complex, giving the corresponding Ni(II) complex and Mn(III)–peroxo complex, 2 (Scheme 8).35 Addition of [Mn(14-TMC)]2+ to a solution of 9 resulted in the disappearance of the characteristic bands of 9 with the concomitant growth of an absorption band (λmax = 453 nm) corresponding to 2 (Figure 5). It was suggested that the O2-transfer reaction proceeds via a bimolecular mechanism in which the formation of an undetected [(12-TMC) Ni–O2-Mn(14-TMC)]3+ intermediate is the rate-determining step (Scheme 8).35
SCHEME 8.
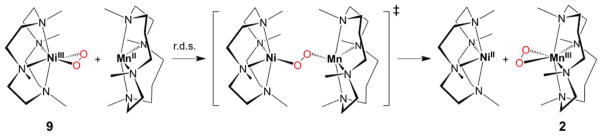
Reaction Scheme Showing an Intermolecular O2-Transfer between Metal Complexes35
FIGURE 5.
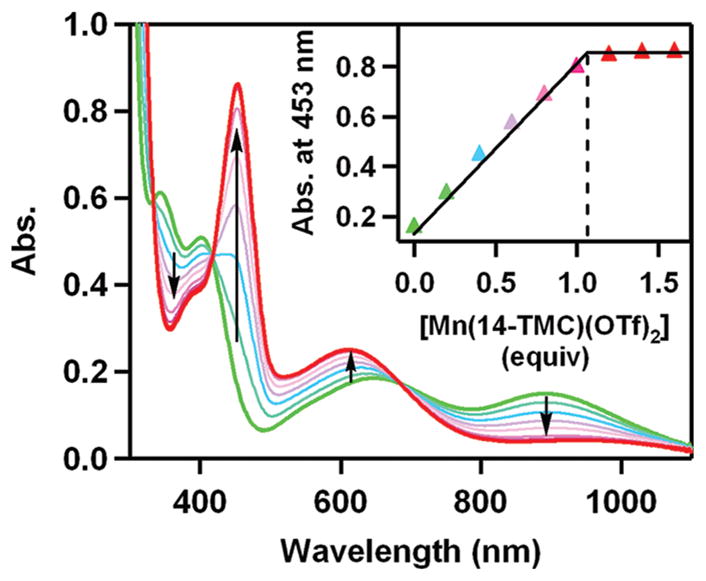
UV–vis spectral changes showing the formation of [Mn(14-TMC)(O2)]+ (2) (red) and the disappearance of [Ni(12-TMC)(O2)]+ (9) (green) by addition of [Mn(14-TMC)]2+ to a solution of 9. Inset shows the spectroscopic titration at 453 nm for the formation of 2 as a function of the equivalents of [Mn(14-TMC)]2+ added to a solution of 9.35
In order to obtain further insight into the O2-transfer reaction, isolated [Co(13-TMC)(O2)]+ (6) and [Co(12-TMC)(O2)]+ (7) complexes were used in the intermolecular O2-transfer reactions.33 The cobalt–peroxo complexes transferred their O2 unit to [Mn(14-TMC)]2+, giving the corresponding Co(II) and Mn(III)–peroxo complexes. Interestingly, the O2-tranfer from [Co(12-TMC)(O2)]+ (7) to [Mn(14-TMC)]2+ is ~1100 times slower than that from[Ni(12-TMC)(O2)]+ (9) to [Mn(14-TMC)]2+ (k2 = 1.9 × 10−4M−1 s−1 for 7 and 0.2M−1 s−1 for 9), indicating that the Ni(III)–peroxo complex is much more reactive than the Co(III)–peroxo analogue toward the attack of the Mn(II) ion. These results were elucidated by the difference of the enthalpy values of 65kJmol−1 for 7 and 49 kJmol−1 for 9, and the difference of ΔΔG between 7 and9 is ~13 kJ mol−1. In addition, as the ring size of macrocyclic ligands in the Co(III)–peroxo complexes increases from 12-TMC to 13-TMC, the O2-transfer reaction is facilitated, and the reaction rate is increased ~600 times (k2 = 1.9 × 10−4 M−1 s−1 for 7 and 1.2 × 10−1M−1 s−1 for 6). This phenomenon in the complete O2-transfer between metal complexes bearing TMC ligands is different from the behavior of other systems; in the reactions of metal (M)–O2 complexes (M = Cu and Ni) and a second metal complex, M or M′, formation of homo- or heterodinuclear complexes containing [M2(μ-O)2]n+, [M2(O2)]n+, or [MM′(μ-O)2]n+ cores is observed.37,46
4. Concluding Remarks
Metal–O2 intermediates are short-lived and highly reactive in many cases, thus making it difficult to study their chemical and physical properties in the catalytic cycle of dioxygen activation by metalloenzymes. Further, it is especially challenging to obtain crystal structures of the thermally unstable and reactive metal–O2 intermediates. In this Account, we have described our recent results on the synthesis of a number of metal–O2 complexes bearing tetraazamacrocyclic TMC ligands and their spectroscopic characterization and crystal structures, by emphasizing the significant role of the macrocyclic TMC ligands in controlling the redox potentials and spin states of metal ions as well as the stability of the bound O2 ligands. We have also demonstrated the importance of metal ions and the ring size of the supporting ligands on the geometric and electronic structures of the metal–O2 complexes. Reactivities of the isolated metal–O2 complexes are discussed in electrophilic and nucleophilic reactions as well as in intermolecular O2-transfer reactions. This Account, with the previous review on mononuclear nonheme Fe(IV)–oxo complexes,47 has shown that the field of nonheme metal–oxygen intermediates (e.g., M–O, M–O2, and M–O2H) has been developed greatly in the past decade in bioinorganic chemistry through intense synthetic, spectroscopic, and theoretical efforts; however, there is still much to be explored in trapping, characterizing, and understanding the key intermediates involved in dioxygen activation chemistry by metalloenzymes and their biomimetic compounds.
Acknowledgments
Financial support from the NRF/MEST of Korea through CRI, GRL (2010-00353), and WCU (R31-2008-000-10010-0) programs and the 2011 KRICT OASIS Project is greatly acknowledged. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393) and the National Center for Research Resources (P41RR001209).
Biographies
Jaeheung Cho received his B.S. and M.S. in Chemistry from Pukyong National University, Korea, working under the guidance of Professor Ju Chang Kim. In 2002, he joined the group of Professor Masatatsu Suzuki at Kanazawa University, Japan, and received his Ph.D. degree in 2005. Following postdoctoral research with Professor Charles G. Riordan at University of Delaware, he moved to Ewha Womans University, Korea, as a Full-Time Lecturer.
Ritimukta Sarangi received her B.S. (Honors) degree in Chemistry from Saint Xaviers College, Kolkata, India, M.Sc. degree in Chemistry from Indian Institute of Technology, Kanpur, India and Ph.D. in Inorganic Chemistry from Stanford University under the guidance of Professors Edward I. Solomon and Keith O. Hodgson. Since 2007, she has been at the Stanford Synchrotron Radiation Light source as a Staff Scientist in the Structural Molecular Biology division.
Wonwoo Nam received his B.S. degree in Chemistry from California State University, Los Angeles and his Ph.D. degree in Inorganic Chemistry from UCLA under the direction of Professor Joan S. Valentine in 1990. After one year postdoctoral experience at UCLA, he became an Assistant Professor at Hong Ik University. He moved to Ewha Womans University in 1994, where he is currently a Distinguished Professor of Ewha Womans University.
Footnotes
The authors declare no competing financial interest.
References
- 1.Nam W. Dioxygen activation by metalloenzymes and models. Acc Chem Res. 2007;40:465. and review articles in the special issue. [Google Scholar]
- 2.Bollinger JM, Jr, Krebs C. Enzymatic C-H activation by metal-superoxo intermediates. Curr Opin Chem Biol. 2007;11:151–158. doi: 10.1016/j.cbpa.2007.02.037. [DOI] [PubMed] [Google Scholar]
- 3.Kovaleva EG, Lipscomb JD. Crystal structures of Fe2+ dioxygenase superoxo, alkylperoxo, and bound product intermediates. Science. 2007;316:453–457. doi: 10.1126/science.1134697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karlsson A, Parales JV, Parales RE, Gibson DT, Eklund H, Ramaswamy S. Crystal structure of naphthalene dioxygenase: Side-on binding of dioxygen to iron. Science. 2003;299:1039–1042. doi: 10.1126/science.1078020. [DOI] [PubMed] [Google Scholar]
- 5.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee SK, Lehnert N, Neese F, Skulan AJ, Yang YS, Zhou J. Geometric and electronic structure/function correlations in non-heme iron enzymes. Chem Rev. 2000;100:235–349. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 6.Costas M, Mehn MP, Jensen MP, Que L., Jr Dioxygen activation at mononuclear nonheme iron active sites: Enzymes models intermediates. Chem Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 7.Mirica LM, Ottenwaelder X, Stack TDP. Structure and spectroscopy of copper-dioxygen complexes. Chem Rev. 2004;104:1013–1045. doi: 10.1021/cr020632z. [DOI] [PubMed] [Google Scholar]
- 8.Cramer CJ, Tolman WB. Mononuclear Cu–O2 complexes: Geometries, spectroscopic properties, electronic structures, and reactivity. Acc Chem Res. 2007;40:601–608. doi: 10.1021/ar700008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suzuki M. Ligand effects on dioxygen activation by copper and nickel complexes: Reactivity and intermediates. Acc Chem Res. 2007;40:609–617. doi: 10.1021/ar600048g. [DOI] [PubMed] [Google Scholar]
- 10.Himes RA, Karlin KD. Copper–dioxygen complex mediated C–H bond oxygenation: Relevance for particulate methane monooxygenase (pMMO) Curr Opin Chem Biol. 2009;13:119–131. doi: 10.1016/j.cbpa.2009.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yao S, Driess M. Lessons from isolable nickel(I) precursor complexes for small molecule activation. Acc Chem Res. 2012;44:276–287. doi: 10.1021/ar200156r. [DOI] [PubMed] [Google Scholar]
- 12.Cramer CJ, Tolman WB, Theopold KH, Rheingold AL. Variable character of O–O and M–O bonding in side-on (η2) 1:1 metal complexes of O2. Proc Natl Acad Sci US A. 2003;100:3635–3640. doi: 10.1073/pnas.0535926100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bakac A. Kinetic and mechanistic studies of the reactions of transition metal-activated oxygen with inorganic substrates. Coord Chem Rev. 2006;250:2046–2058. [Google Scholar]
- 14.Wertz DL, Valentine JS. Nucleophilicity of iron-peroxo porphyrin complexes. Struct Bonding (Berlin) 2000;97:37–60. [Google Scholar]
- 15.Peterson RL, Himes RA, Kotani H, Suenobu T, Tian L, Siegler MA, Solomon EI, Fukuzumi S, Karlin KD. Cupric superoxo-mediated intermolecular C-H activation chemistry. J Am Chem Soc. 2011;133:1702–1705. doi: 10.1021/ja110466q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kunishita A, Kubo M, Sugimoto H, Ogura T, Sato K, Takui T, Itoh S. Mononuclear copper(II)-superoxo complexes that mimic the structure and reactivity of the active center of PHM and Dβ. M J Am Chem Soc. 2009;131:2788–2789. doi: 10.1021/ja809464e. [DOI] [PubMed] [Google Scholar]
- 17.Maiti D, Lee DH, Gaoutchenova K, Würtele C, Holthausen MC, Narducci Sarjeant AA, Sundermeyer J, Schindler S, Karlin KD. Reactions of a copper(II) superoxo complex lead to C-H and O-H substrate oxygenation: Modeling copper-monooxygenase C-H hydroxylation. Angew Chem, Int Ed. 2008;47:82–85. doi: 10.1002/anie.200704389. [DOI] [PubMed] [Google Scholar]
- 18.Lee YM, Hong S, Morimoto Y, Shin W, Fukuzumi S, Nam W. Dioxygen activation by a nonheme iron(II) complex: Formation of an iron(IV)-oxo complex via C-H activation by a putative iron(III)-superoxo species. J Am Chem Soc. 2010;132:10668–10670. doi: 10.1021/ja103903c. [DOI] [PubMed] [Google Scholar]
- 19.Wu AJ, Penner-Hahn JE, Pecoraro VL. Structural, spectroscopic, and reactivity models for the manganese catalases. Chem Rev. 2004;104:903–938. doi: 10.1021/cr020627v. [DOI] [PubMed] [Google Scholar]
- 20.VanAtta RB, Strouse CE, Hanson LK, Valentine JS. Peroxo- (tetraphenylporphinato)manganese(III) and chloro(tetraphenylporphinato)manganese(II) anions. Syntheses, crystal structures, and electronic structures. J Am Chem Soc. 1987;109:1425–1434. [Google Scholar]
- 21.Kitajima N, Komatsuzaki H, Hikichi S, Osawa M, Moro-Oka Y. A monomeric side-on peroxo manganese(III) complex: Mn(O2)(3,5-iPr2pzH)(HB(3,5-iPr2pz)3. J Am Chem Soc. 1994;116:11596–11597. [Google Scholar]
- 22.Hikichi S, Akita M, Moro-oka Y. New aspects of the cobalt-dioxygen complex chemistry opened by hydrotris(pyrazoly)borate ligands (TpR): Unique properties of TpRCo-dioxygen complexes. Coord Chem Rev. 2000;198:61–87. [Google Scholar]
- 23.Dickman MH, Pope MT. Peroxo and superoxo complexes of chromium, molybdenum, and tungsten. Chem Rev. 1994;94:569–584. [Google Scholar]
- 24.Tiné MR. Cobalt complexes in aqueous solutions as dioxygen carriers. Coord Chem Rev. 2012;256:316–327. [Google Scholar]
- 25.Bailey CL, Drago RS. Utilization of O2 for the specific oxidation of organic substrates with cobalt(II) catalysts. Coord Chem Rev. 1987;79:321–332. [Google Scholar]
- 26.Barefield EK. Coordination chemistry of N-tetraalkylated cyclam ligands-A status report. Coord Chem Rev. 2010;254:1607–1627. [Google Scholar]
- 27.Cho J, Woo J, Nam W. An “end-on” chromium(III)-superoxo complex: Crystallographic and spectroscopic characterization and reactivity in C-H bond activation of hydrocarbons. J Am Chem Soc. 2010;132:5958–5959. doi: 10.1021/ja1015926. [DOI] [PubMed] [Google Scholar]
- 28.Seo MS, Kim JY, Annaraj J, Kim Y, Lee YM, Kim SJ, Kim J, Nam W. [Mn(tmc)(O2)]+: A side-on peroxido manganese(III) complex bearing a non-heme ligand. Angew Chem Int Ed. 2007;46:377–380. doi: 10.1002/anie.200603414. [DOI] [PubMed] [Google Scholar]
- 29.Annaraj J, Cho J, Lee YM, Kim SY, Latifi R, de Visser SP, Nam W. Structural characterization and remarkable axial ligand effect on the nucleophilic reactivity of a nonheme manganese(III)–peroxo complex. Angew Chem, Int Ed. 2009;48:4150–4153. doi: 10.1002/anie.200900118. [DOI] [PubMed] [Google Scholar]
- 30.Annaraj J, Suh Y, Seo MS, Kim SO, Nam W. Mononuclear nonheme ferric-peroxo complex in aldehyde deformylation. Chem Commun. 2005:4529–4531. doi: 10.1039/b505562h. [DOI] [PubMed] [Google Scholar]
- 31.Cho J, Jeon S, Wilson SA, Liu LV, Kang EA, Braymer JJ, Lim MH, Hedman B, Hodgson KO, Valentine JS, Solomon EI, Nam W. Structure and reactivity of a mononuclear non-haem iron(III)-peroxo complex. Nature. 2011;478:502–505. doi: 10.1038/nature10535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jo Y, Annaraj J, Seo MS, Lee YM, Kim SY, Cho J, Nam W. Reactivity of a cobalt(III)-peroxo complex in oxidative nucleophilic reactions. J Inorg Biochem. 2008;102:2155–2159. doi: 10.1016/j.jinorgbio.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 33.Cho J, Sarangi R, Kang HY, Lee JY, Kubo M, Ogura T, Solomon EI, Nam W. Synthesis, structural and spectroscopic characterization, and reactivities of mononuclear cobalt(III)-peroxo complexes. J Am Chem Soc. 2010;132:16977–16986. doi: 10.1021/ja107177m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kieber-Emmons MT, Annaraj J, Seo MS, Van Heuvelen KM, Tosha T, Kitagawa T, Brunold TC, Nam W, Riordan CG. Identification of an “end-on” nickel-superoxo adduct, [Ni(tmc)(O2)]+ J Am Chem Soc. 2006;128:14230–14231. doi: 10.1021/ja0644879. [DOI] [PubMed] [Google Scholar]
- 35.Cho J, Sarangi R, Annaraj J, Kim SY, Kubo M, Ogura T, Solomon EI, Nam W. Geometric and electronic structure and reactivity of a mononuclear “side-on” nickel(III)–peroxo complex. Nat Chem. 2009;1:568–572. doi: 10.1038/nchem.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sarangi R, Cho J, Nam W, Solomon EI. XAS and DFT investigation of mononuclear cobalt(III) peroxo complexes: Electronic control of the geometric structure in CoO2 versus NiO2 systems. Inorg Chem. 2011;50:614–620. doi: 10.1021/ic101730r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kieber-Emmons MT, Riordan CG. Dioxygen activation at monovalent nickel. Acc Chem Res. 2007;40:618–625. doi: 10.1021/ar700043n. [DOI] [PubMed] [Google Scholar]
- 38.Rohde JU, In JH, Lim MH, Brennessel WW, Bukowski MR, Stubna A, Münck E, Nam W, Que L., Jr Crystallographic and spectroscopic characterization of a nonheme Fe(IV)=O complex. Science. 2003;229:1037–1039. doi: 10.1126/science.299.5609.1037. [DOI] [PubMed] [Google Scholar]
- 39.Fukuzumi S, Morimoto Y, Kotani H, Naumov P, Lee YM, Nam W. Crystal structure of a metal ion-bound oxoiron(IV) complex and implications for biological electron transfer. Nat Chem. 2010;2:756–759. doi: 10.1038/nchem.731. [DOI] [PubMed] [Google Scholar]
- 40.Cho J, Woo J, Han JE, Kubo M, Ogura T, Nam W. Chromium(V)-oxo and chromium(III)-superoxo complexes bearing a macrocyclic TMC ligand in hydrogen atom abstraction reactions. Chem Sci. 2011;2:2057–2062. [Google Scholar]
- 41.Qin K, Incarvito CD, Rheingold AL, Theopold KH. A structurally characterized chromium(III) superoxide complex features “side-on” bonding. Angew Chem, Int Ed. 2002;41:2333–2335. doi: 10.1002/1521-3773(20020703)41:13<2333::AID-ANIE2333>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 42.Li F, Meier KK, Cranswick MA, Chakrabarti M, Van Heuvelen KM, Münck E, Que L., Jr Characterization of a high-spin non-heme FeIII-OOH intermediate its quantitative conversion to an FeIV=O complex. J Am Chem Soc. 2011;133:7256–7259. doi: 10.1021/ja111742z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yao S, Bill E, Milsmann C, Wieghardt K, Driess M. A “side-on” superoxonickel complex [LNi(O2)] with a square-planar tetracoordinate nickel(II) center and its conversion into [LNi(μ-OH)2NiL. Angew Chem, Int Ed. 2008;47:7110–7113. doi: 10.1002/anie.200802234. [DOI] [PubMed] [Google Scholar]
- 44.MacBeth CE, Gupta R, Mitchell-Koch KR, Young VG, Jr, Lushington GH, Thompson WH, Hendrich MP, Borovik AS. Utilization of hydrogen bonds to stabilize M-O(H) units: Synthesis and properties of monomeric iron manganese complexes with terminal oxo and hydroxo ligands. J Am Chem Soc. 2004;126:2556–2567. doi: 10.1021/ja0305151. [DOI] [PubMed] [Google Scholar]
- 45.Latifi R, Tahsini L, Kumar D, Sastry GN, Nam W, de Visser SP. Oxidative properties of a nonheme Ni(II)(O2) complex: Reactivity patterns for C-H activation, aromatic hydroxylation and heteroatom oxidation. Chem Commun. 2011;47:10674–10676. doi: 10.1039/c1cc13993b. [DOI] [PubMed] [Google Scholar]
- 46.Aboelella NW, York JT, Reynolds AM, Fujita K, Kinsinger CR, Cramer CJ, Riordan CG, Tolman WB. Mixedmetal bis(μ-oxo) complexes with [CuM(μ-O)2]n+ (M=Ni(III) or Pd(II)) cores. Chem Commun. 2004:1716–1717. doi: 10.1039/b404640d. [DOI] [PubMed] [Google Scholar]
- 47.Nam W. High-valent iron(IV)-oxo complexes of heme and non-heme ligands in oxygenation reactions. Acc Chem Res. 2007;40:522–531. doi: 10.1021/ar700027f. [DOI] [PubMed] [Google Scholar]