


Abstract
We present Masai, a read mapper representing the state-of-the-art in terms of speed and accuracy. Our tool is an order of magnitude faster than RazerS 3 and mrFAST, 2–4 times faster and more accurate than Bowtie 2 and BWA. The novelties of our read mapper are filtration with approximate seeds and a method for multiple backtracking. Approximate seeds, compared with exact seeds, increase filtration specificity while preserving sensitivity. Multiple backtracking amortizes the cost of searching a large set of seeds by taking advantage of the repetitiveness of next-generation sequencing data. Combined together, these two methods significantly speed up approximate search on genomic data sets. Masai is implemented in C++ using the SeqAn library. The source code is distributed under the BSD license and binaries for Linux, Mac OS X and Windows can be freely downloaded from http://www.seqan.de/projects/masai.
INTRODUCTION
Next-generation sequencing allows to produce billions of base pairs within days in the form of reads of length 100 bp and more. It is an invaluable technology for a multitude of applications in biomedicine, e.g. detection of SNPs and large genomic variations, targeted or de novo genome or transcriptome assembly, isoform prediction and quantification, identification of transcription factor binding sites or methylation patterns. In many of these applications, mapping sequenced reads to their potential origin in a reference genome is the first fundamental step preceding downstream analyses.
Because of sequencing errors and genomic variations, not all reads occur exactly in a reference genome. Therefore, approximate occurrences must be considered, and algorithms for approximate string matching tolerating mismatches and indels must be applied to solve the problem. Furthermore, because of homologous and low complexity regions, not all reads occur uniquely in a reference genome. Therefore, in some applications, e.g. CNVs calling, all approximate occurrences that could be potential origins must be considered.
Previous work
All current read mappers can be broadly classified as best-mappers or all-mappers. Tools in the first class aim at finding the best mapping location for a read according to a scoring scheme eventually taking base quality values into account, whereas those in the second class aim at enumerating a comprehensive set of locations.
Most prominent best-mappers are based on backtracking algorithms for approximate string matching (1). Substrings of the reference genome within an absolute number of errors from a read are recursively enumerated using a suffix or prefix tree of the reference genome. As the time complexity of backtracking grows exponentially with the absolute number of errors considered, this method alone is impractical when mapping reads whole reads with moderate error rates. Hence, popular best-mappers (2–4) apply heuristics to reduce and prioritize enumeration and are optimized to return one or a few best mapping locations.
Conversely, most prominent all-mappers are based on filtering algorithms for approximate string matching (1). Seeds are sampled from given reads and used as anchors to quickly determine, with the help of an index, locations of the reference genome candidate to contain approximate occurrences. Each candidate location is subsequently verified with an online method (5). Increasing the error rate in filtering algorithms leads to a decrease of the seed length, which in turn deteriorates filtration efficiency. Current all-mappers (6–9) are usually slower than best-mappers, but conversely they are able to report all asked mapping locations in reasonable time.
Our contribution
We present Masai, a read mapper that combines for the first time filtering with backtracking. Our filtering approach is based on non-heuristic and full-sensitive filtration strategies using exact and approximate seeds, which are searched in the reference genome via backtracking. Approximate seeds, compared with exact seeds, increase filtration specificity while preserving sensitivity. Moreover, we introduce a multiple backtracking method, which speeds up filtration by searching all seeds simultaneously with the help of an additional index. Combined together, these methods yield a flexible and efficient filter that significantly speeds up approximate search on genomic data sets.
Masai targets all-mapping, but eventually it can be used as a best-mapper achieving even better runtimes. We extensively compared Masai with popular read mappers on simulated and real data sets. Compared with considered all-mappers, Masai is an order of magnitude faster and has comparable sensitivity. In addition, Masai is more accurate than considered best-mappers and 2–4 times faster than Bowtie 2 (2) and BWA (3). Masai is implemented in C++ using the SeqAn library and distributed under the BSD license. It can be downloaded from http://www.seqan.de/projects/masai.
MATERIALS AND METHODS
To map reads to a reference genome, we proceed as follows.
We first construct a conceptual suffix tree of the reference genome, store it on disk and reuse it for each read mapping job. Any data structure equivalent to the generalized suffix tree in terms of allowing a top-down traversal can be used to this intent. We implemented a generic algorithm using the suffix array (10), the enhanced suffix array (Esa) (11) and the FM-index (12).
At mapping time, we choose a filtration strategy according to the reference genome and the specified error rate. Our filtration strategies are based on (13), make use of exact and approximate non-overlapping seeds and are guaranteed to be full-sensitive by the pigeonhole principle. In Figure 1, we show an example providing two alternative filtration strategies.
Figure 1.
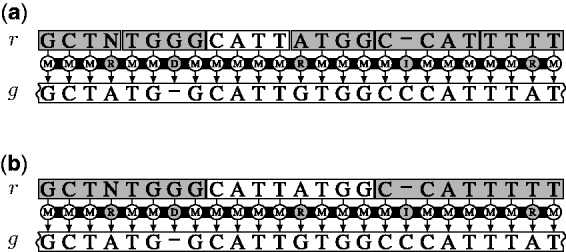
Filtration strategies. A read r occurs in the reference genome g within edit distance 5. (a) If we partition r in six seeds, at least one seed (in white) occurs exactly in g. (b) Alternatively, if we partition r in three seeds, at least one seed (in white) occurs within edit distance 1 in g.
Therefore, we partition all reads and their reverse complements in non-overlapping seeds and subsequently arrange all seeds in a conceptual radix tree. The time spent to construct the radix tree is easily justified, as the tree allows us to perform multiple backtracking. We indeed apply our multiple backtracking algorithm to the radix tree, to search simultaneously all seeds in the suffix tree of the reference genome.
Finally, we perform seed extension on each seed reported by the multiple backtracking algorithm. We extend both ends of each seed using a banded version of Myers bit-vector algorithm (14) presented in (6).
In the following of this section, we give a detailed explanation of each mapping step.
Seeds
We now consider formally the read mapping problem. Given a reference genome g, a set of reads 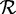 and an absolute number of errors k consisting of indels and mismatches, for each read
and an absolute number of errors k consisting of indels and mismatches, for each read 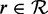 , find all mapping locations where r approximately occurs in g within k errors.
, find all mapping locations where r approximately occurs in g within k errors.
Exact seeds
A simple solution to the problem is provided by a filtering algorithm proposed in (15), which reduces an approximate search into smaller exact searches. Each read r is partitioned into k + 1 non-overlapping seeds, which are searched in g with the help of an index. As each edit operation can affect at most one seed, for the pigeonhole principle, each approximate occurrence of r in g contains an exact occurrence of some seed. However, the converse is not true; consequently, we must verify whether any candidate location induced by an occurrence of some seed corresponds to an approximate occurrence of r in g.
Filtration specificity in terms of candidate locations to verify is strongly correlated to seed length. As we want to maximize the length of the shortest seed, we let the minimum seed length be 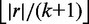 . If we want to improve filtration specificity by increasing seed length, we resort to approximate seeds.
. If we want to improve filtration specificity by increasing seed length, we resort to approximate seeds.
Approximate seeds
A more involved filtering algorithm proposed in (13) reduces an approximate search into smaller approximate searches. We partition r into 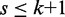 non-overlapping seeds. According to the pigeonhole principle, each approximate occurrence of r in g then contains an approximate occurrence of some seed within distance
non-overlapping seeds. According to the pigeonhole principle, each approximate occurrence of r in g then contains an approximate occurrence of some seed within distance 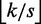 .
.
Moreover, we search 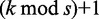 seeds within distance
seeds within distance 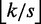 and the remaining seeds within distance
and the remaining seeds within distance 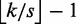 . To prove full sensitivity, it suffices to see that, if none of the seeds occurs within its assigned distance, the total distance must be at least
. To prove full sensitivity, it suffices to see that, if none of the seeds occurs within its assigned distance, the total distance must be at least 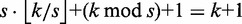 . Hence, all approximate occurrences of r in g within distance k will be found.
. Hence, all approximate occurrences of r in g within distance k will be found.
Seeds are searched approximately by backtracking on a suffix tree. We will introduce two efficient multiple backtracking algorithms to search exactly or approximately a set of seeds.
Filtration strategies
With approximate seeds, we are free to choose the number of seeds s, which in turn enforces the minimum seed length l to be 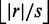 . Or vice versa, we fix l, which enforces s to be
. Or vice versa, we fix l, which enforces s to be 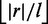 . The resulting filter is flexible, indeed by increasing l filtration becomes more specific at the expense of a higher filtration time.
. The resulting filter is flexible, indeed by increasing l filtration becomes more specific at the expense of a higher filtration time.
The optimal seed length l depends on the reference genome as well as on read length and the absolute number of errors. When mapping current next-generation sequencing data sets on short-to-medium length genomes, e.g. bacterial genomes, exact seeds are still more efficient than approximate seeds. Conversely on larger genomes, e.g. mammalian genomes, approximate seeds outperform exact seeds by an order of magnitude. Filtration results are provided in the Supplementary Table S6.
Indices
We make use of two fundamental data structures, radix and suffix trees. Here, we present these indices and give most important implementation details.
Radix tree
The radix tree (16) is a lexicographically ordered tree data structure representing a set of strings. It can be built in time and space linear in the total length of the strings. Assume w.l.o.g. that each string in the set is padded with a distinct terminator symbol, not being part of the string alphabet. The radix tree has one node designated as the root and one leaf per string in the set. Every internal node has more than one child, and edges are labeled with non-empty strings. Each path from the root to an internal node spells a different substring. Consequently, prefixes common to distinct strings are compressed.
Suffix tree
The suffix tree (17) of a string is the radix tree of all the suffixes of the string itself. It can be built in time and space linear in the length of the string (18).
The suffix tree is used for exact search. A pattern is found by starting in the root node and following the path spelling the pattern. If such path is found, each leaf below the last traversed node points to a distinct occurrence of the pattern in the text.
Approximate search is performed on the suffix tree by means of backtracking (19,20). A top-down traversal of the suffix tree spells incrementally all substrings present in the text. The distance between the pattern and the spelled text is incrementally computed while traversing a branch of the suffix tree. If the pattern approximately matches the spelled text, each leaf below the last traversed node points to a distinct approximate occurrence of the pattern in the text. Conversely, if the remaining suffix of the pattern cannot lead to any approximate occurrence, the branch is pruned, and the traversal proceeds on the next branch.
Implementation
We implemented a generic suffix tree top-down traversal on the suffix array (10), the Esa (11) and the FM-index (12). The Esa preserves the asymptotic performances of the suffix tree and consumes, as implemented in SeqAn, 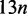 bytes for a sequence of length n. Nevertheless, the suffix array, despite consuming
bytes for a sequence of length n. Nevertheless, the suffix array, despite consuming 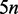 bytes and being theoretically slower, always showed better performance than the Esa (Supplementary Table S7). The FM-index implicitly represents a prefix trie in only
bytes and being theoretically slower, always showed better performance than the Esa (Supplementary Table S7). The FM-index implicitly represents a prefix trie in only 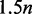 bytes and provides constant time node traversal while being slower than the other two data structures.
bytes and provides constant time node traversal while being slower than the other two data structures.
We prefer the suffix array because it provides a good compromise between speed and memory consumption, but nevertheless leaves the possibility to choose among the aforementioned data structures. We construct the (generalized) suffix array using an adaptation of the DC7 algorithm (21) to multiple sequences. For construction of the Esa or FM-index, we additionally use the algorithms proposed in (11,22) and (23). Similarly, to all read mappers relying on an index of the reference genome, we build the index of the reference genome only once, store it on disk and reuse it for each mapping job.
We implemented a lazy radix tree based on the wotd-algorithm (24), as a radix tree is a partial suffix tree only containing certain suffixes. The radix tree is constructed in linear time by subsequent radix sort steps. However, when performing multiple backtracking with exact seeds, the radix tree construction time dominates the overall filtration time. Therefore, in this case, we resort to the q-gram index to emulate the radix tree. We build the q-gram index efficiently and in linear time by bucket sort. Below depth q, the properties of the radix tree are lost; however, multiple backtracking is still applicable.
Multiple backtracking
We now introduce a method for multiple off-line approximate string matching to search simultaneously a set of patterns in a text. We start by introducing an algorithm for multiple off-line exact string matching and later extend it to approximate string matching.
For simplicity of exposition, we describe the algorithms working on tries, although they are easily extendable to work on trees. Hence, in the following, we assume the text sequence and the set of patterns to be pre-processed respectively, using a suffix trie G and a radix trie S. Given a node x, we denote with label (x) the label of the edge entering into x, with 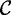 (x) the set of children of x which are internal nodes, with
(x) the set of children of x which are internal nodes, with 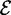 (x) the set of children of x which are leaves, and with
(x) the set of children of x which are leaves, and with 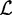 (x) the set of leaves of the subtree rooted in x. Note that entering edges of internal nodes are labeled with alphabet symbols, while entering edges of leaves are labeled with terminator symbols.
(x) the set of leaves of the subtree rooted in x. Note that entering edges of internal nodes are labeled with alphabet symbols, while entering edges of leaves are labeled with terminator symbols.
Exact search
Algorithm 1 takes as input two nodes g, s, respectively, of G, S and reports all pairs of leaves 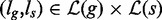 such that the path from s to ls spells a prefix of the path from g to lg.
such that the path from s to ls spells a prefix of the path from g to lg.
Consequently, by applying Algorithm 1 on the root nodes of G, S, we obtain all pairs of leaves 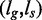 such that the pattern pointed by ls occurs in the text at the position pointed by lg.
such that the pattern pointed by ls occurs in the text at the position pointed by lg.
Algorithm 1 Multiple exact search.
1: procedure Search (g, s)
2: report
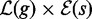
3: for all
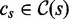 do
do
4: if
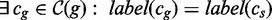 then
then
5: Search
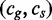
6: end if
7: end procedure
Approximate search
Algorithm 2 takes an additional input argument k, which denotes the maximum number of mismatches left and computes the union of all paths within k mismatches in the subtrees rooted in g, s. It reports all pairs of leaves 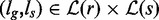 such that the path from s to ls spells a prefix of the path from g to lg with at most k mismatches.
such that the path from s to ls spells a prefix of the path from g to lg with at most k mismatches.
Therefore, by applying Algorithm 2 on the root nodes of G, S, we obtain all pairs of leaves 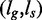 such that the pattern pointed by ls occurs within k mismatches in the text at the position pointed by lg.
such that the pattern pointed by ls occurs within k mismatches in the text at the position pointed by lg.
Algorithm 2 Multiple approximate search.
1: procedure Search (g, s, k)
2: if k = 0 then
3: Search (g, s)
4: else
5: report
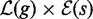
6: for all
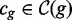 do
do
7: for all
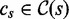 do
do
8: if
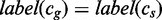 then
then
9: Search
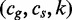
10: else
11: Search
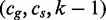
12: end if
13: end if
14: end procedure
For k = 0, lines 5–12 of Algorithm 2 are equivalent to Algorithm 1. However, Algorithm 1 is preferred to Algorithm 2 because it traverses only edges spelling common strings instead of all pairs of edges, and it is thus more efficient. Figure 2 depicts a run of Algorithm 2.
Figure 2.
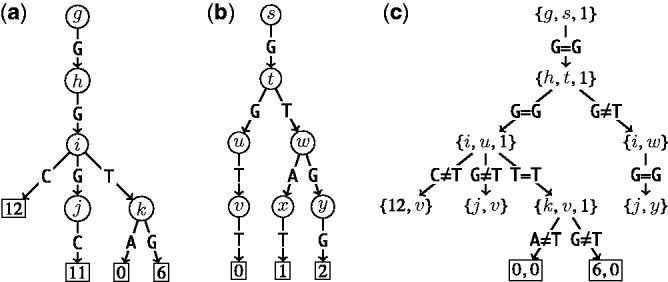
Multiple backtracking. (a) A part of the suffix trie representing the text GGTAACGGTGCGGGC (Supplementary Figure S1). Numbers on the leaves are suffix positions in the text, whereas letters on the inner nodes are arbitrary and serve to distinguish nodes from each other. (b) The trie representing the set of patterns {GGTT, GTAT, GTGG}, respectively numbered {0, 1, 2}. Labels on the leaves show pattern numbers, whereas labels on the inner nodes are again arbitrary identifiers. (c) Recursive calls performed by Algorithm 2 called with arguments {g, s, 1}. Edges represent comparisons performed by Algorithm 2 at line 10 or by Algorithm 1 at line 6, nodes with curly brackets represent recursive calls, rectangular leaves represent approximate matches reported. In this example, pattern numbered 0 (GGTT) matches the text twice, at positions 0 and 6, within 1 mismatch. For simplicity, we omitted terminator symbols in the picture.
Algorithm 2 only considers mismatches, but it can be extended to allow indels, e.g. similarly to (13). In Masai, Algorithm 2 is implemented only for mismatches, consequently full sensitivity is not attained when using approximate seeds and considering mapping locations with indels. However, in the ‘Results’ section, we show that such implementation detail sacrifices <1% sensitivity.
Seed extension
We use a banded version of Myers bit-vector algorithm (14) already presented in (6). Myers’ algorithm is an efficient dynamic programming (DP) alignment algorithm (25) for edit distance. Instead of computing DP cells one after another, it encodes the whole DP column in two bit vectors and computes the adjacent column in a constant number of 12 logical and 3 arithmetical operations. We implemented a bit-parallel version that computes only a diagonal band of the DP matrix and is faster and more specific than the original algorithm by Myers. More details can be found in the Supplementary Section S2.
However, differently from (6), instead of performing a semi-global alignment to verify a parallelogram surrounding the seed, we perform a global alignment on both ends of a seed. Given a seed occurring with e errors, we first perform seed extension on the left side within an error threshold of 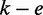 errors. Only if the seed extension on the left side succeeds, we perform a seed extension on the right side within the remaining error threshold. Moreover, we first compute the longest common prefix on each side of the seed and let the global alignment algorithm start from the first mismatching positions. We observed that this approach is up to two times faster than (6).
errors. Only if the seed extension on the left side succeeds, we perform a seed extension on the right side within the remaining error threshold. Moreover, we first compute the longest common prefix on each side of the seed and let the global alignment algorithm start from the first mismatching positions. We observed that this approach is up to two times faster than (6).
RESULTS
We thoroughly compared Masai with the best-mappers Bowtie 2, BWA and Soap 2 as well as with the all-mappers RazerS 3, Hobbes, mrFAST and SHRiMP 2. We remark that Bowtie 2, BWA, Soap 2 and SHRiMP 2 rely on scoring schemes taking into account base quality values, whereas Masai, RazerS 3, Hobbes and mrFAST use edit distance. When relevant, read mappers that accept an absolute number of errors (Masai, mrFAST, Hobbes and Soap 2) or an error rate (RazerS 3) were configured accordingly. We used default parameters, except where stated otherwise (Supplementary Section S3).
We performed runtime experiments on real data. All read sets are given by their SRA/ENA ID. As references, we used whole genomes of Escherichia coli (NCBI NC_000913.2), Caenorhabditis elegans (WormBase WS195), Drosophila melanogaster (FlyBase release 5.42) and Homo sapiens (GRCh37.p2). The mapping times were measured on a cluster of nodes with 72 GB RAM and 2 Intel Xeon X5650 processors running Linux 3.2.0. For running time comparison, we ran the tools using a single thread and used local disks for I/O.
Rabema benchmark
We first used the Rabema benchmark (26) (v1.1) for a thorough evaluation and comparison of read mapping sensitivity. Similarly to (2), we used the read simulator Mason (27) with default profile settings to simulate from each whole genome 100 k reads of length 100 bp having sequencing errors distributed like in a typical Illumina run (Supplementary Section S4).
The benchmark contains the categories all, all-best, any-best, precision and recall. In the categories all, all-best and any-best, a read mapper had to find all, all of the best or any of the best edit distance locations for each read. The categories precision and recall required a read mapper to find the original location of each read, which is a measure independent of the used scoring model, e.g. edit distance or quality based. A read is mapped correctly if the mapper reported its original location, and it is mapped uniquely if the mapper reported only one location. Rabema defines recall to be the fraction of reads that were correctly mapped and precision to be the fraction of uniquely mapped reads that were mapped correctly.
The benchmark was performed for an error rate of 5%, which corresponds to edit distance 5 for reads of length 100 bp. Therefore, we built a Rabema gold standard for each data set by running RazerS 3 in full-sensitive mode up to edit distance 5. We further classified mapping locations in each category by their edit distance.
For a more fair and thorough comparison, we also configured BWA and Bowtie 2 as all-mappers (Soap 2 could not be configured accordingly). To this extent, we parametrized them to be highly sensitive and output all found mapping locations. As BWA and Bowtie 2 were not designed to be used as all-mappers, they spent much more time than proper all-mappers, i.e. up to 3 h in a run compared with several minutes. The aim here is to investigate read mapping sensitivity, and therefore we do not report running times.
Results for H. sapiens are shown in Table 1. Additional results for E. coli, C. elegans and D. Melanogaster are shown in the Supplementary Tables S3, S4, S5.
Table 1.
Rabema benchmark results
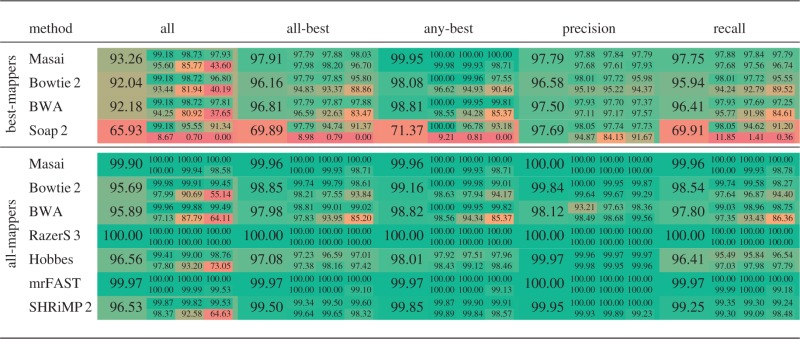 |
Rabema scores in percentage (average fraction of edit distance locations reported per read). Large numbers show total scores in each Rabema category, and small numbers show the category scores separately for reads with  errors.
errors.
Best-mappers
Masai showed the best recall values, not loosing >3.3% recall on edit distance 5. Conversely, recall values of BWA and Bowtie 2 dropped significantly with increasing edit distance up to loosing 15.4 and 11.5%, respectively, on edit distance 5. As expected, Soap 2 turned out to be inadequate for mapping reads of length 100 bp at this error rates.
Precision values have less variance than recall values. Masai showed the best precision values with 97.8%, followed by Soap 2 with 97.7%, and BWA with 97.5%. Interestingly, Bowtie 2 showed the worst precision values, loosing up to 5.6% on edit distance 5.
All-mappers
As expected, RazerS 3 showed full sensitivity and mrFAST lost only a minimal percentage of mapping locations. Overall, Masai did not loose >0.1% of all mapping locations. In particular, Masai was full sensitive for low-error locations, and it lost only a small percentage of high-error locations, i.e. its loss was limited to 0.1 and 1.4% of mapping locations at edit distance 4 and 5.
On the other side, BWA and Bowtie 2 missed 35 and 45% of all mapping locations at edit distance 5, and their recall values as all-mappers did not substantially increase. Likewise, SHRiMP 2 could not enumerate all mapping locations, although its recall values were good. Again Hobbes had the worst performance.
We remark that Masai is not full sensitive whenever approximate seeds are used, e.g. on H. sapiens. Indeed, Masai lost 0.1% overall sensitivity in respect to RazerS 3. Conversely, full sensitivity is attained whenever exact seeds are used, e.g. on E. coli, C. elegans and D. Melanogaster (Supplementary Tables S3, S4, S5). In general, RazerS 3 should be used when full sensitivity is required, i.e. for read mapper benchmarking. However, our results show that Masai can replace RazerS 3 or mrFAST as an all-mapper in practical setups.
Variant detection
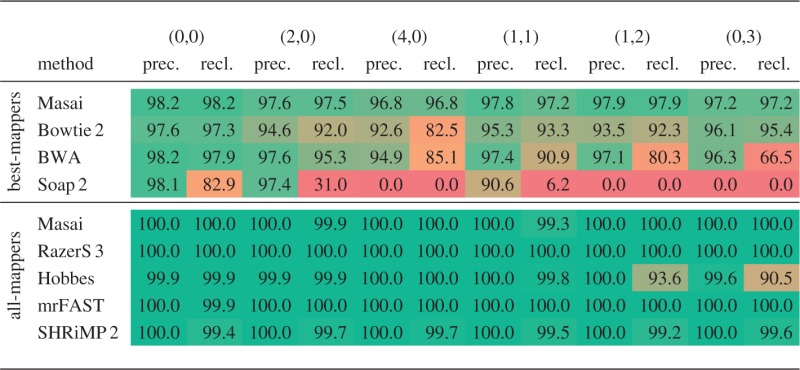
The second experiment analyses the applicability of Masai and other read mappers in genomic variation pipelines. Similarly to (9), we simulated from the whole human genome 5 million reads of length 100 bp containing sequencing errors, SNPs and indels such that each read had an edit distance of at most 5 to its genomic origin. To distribute sequencing errors according to a typical Illumina run, we used the read simulator Mason. The reads were grouped according to the numbers of contained SNPs and indels, where the class 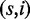 consists of all reads with s SNPs and i indels. We mapped the reads with each tool and measured its sensitivity in each class.
consists of all reads with s SNPs and i indels. We mapped the reads with each tool and measured its sensitivity in each class.
We say that a read is mapped correctly if a mapping location has been reported within 10 bp of its genomic origin. It is considered to map uniquely if only one location was reported by the mapper. For each class, we define recall to be the fraction of reads that were correctly mapped and precision to be the fraction of uniquely mapped reads that were mapped correctly. Table 2 shows the results for each read mapper and class.
Table 2.
Variant detection results
 |
We show the percentages of found origins (recall) and fraction of unique reads mapped to their origin (precision) classed by reads with s SNPs and i indels 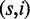 .
.
Best-mappers
Among best-mappers, Masai showed the highest precision and recall in all classes. In particular, Masai did not loose >3.2% recall in class (4,0), whether Bowtie 2 and BWA lost 17.5 and 14.9%, respectively, and Soap 2 was not able to map any read.
Interestingly, we observed that recall values of Bowtie 2, BWA and Soap 2 were negatively correlated with the amount of genomic variation. For instance, in the Rabema benchmark, Bowtie 2 lost 7.2 and 11.5%, respectively, of mapping locations at distance 4 and 5, but in this experiment, it lost 17.5% recall in class (4,0). We noticed a similar trend for BWA and Soap 2. These tools rely on quality values to guess the best mapping location for a read and tend to prefer alignments, which can be explained by sequencing errors instead of true genomic variations. The low performance of Soap 2 is also owing to its limitation to at most 2 mismatches and no support for indels.
All-mappers
Looking at all-mappers results, Masai showed 100% precision and recall in all classes, except for classes (2,0) and (1,1) where it lost only 0.1 and 0.7% recall. Masai is therefore roughly comparable with the full-sensitive read mappers RazerS 3 and mrFAST. SHRiMP 2 showed 100% precision in all classes but lost between 0.3 and 0.8% recall in each class. Hobbes had the lowest performance among all-mappers. It appears to have problems with indels; indeed, it lost 9.5% recall in class (0,3).
Performance on real data
In the last experiment, we focused on comparing read mappers performance on real data. To this end, we mapped the first 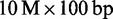 reads from an Illumina lane of E. coli (ERR022075, Genome Analyzer IIx), D. melanogaster (SRR497711, HiSeq 2000), C. elegans (SRR065390, Genome Analyzer II) and H. sapiens (ERR012100, Genome Analyzer II). To measure scalability, we also mapped the full
reads from an Illumina lane of E. coli (ERR022075, Genome Analyzer IIx), D. melanogaster (SRR497711, HiSeq 2000), C. elegans (SRR065390, Genome Analyzer II) and H. sapiens (ERR012100, Genome Analyzer II). To measure scalability, we also mapped the full 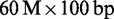 (ERR012100) and
(ERR012100) and 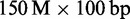 (ERR161544, HiSeq 2000) H. sapiens data sets. Whenever possible, we asked mappers to map reads within edit distance 5. We measured running times, peak memory consumptions, mapped reads and Rabema any-best scores.
(ERR161544, HiSeq 2000) H. sapiens data sets. Whenever possible, we asked mappers to map reads within edit distance 5. We measured running times, peak memory consumptions, mapped reads and Rabema any-best scores.
We could not measure precision and recall values, as real reads have unknown origins. Therefore, for the evaluation, we adopted the commonly used measure of percentage of mapped reads, i.e. the fraction of reads for which the read mapper reports a mapping location. However, as some mappers report mapping locations without constraints on the number of errors, we also included Rabema any-best scores. The Rabema any-best benchmark assigns a point for a read if the mapper reports at least one mapping location with the optimal (minimum) number of errors. Final Rabema any-best scores are normalized by the number of reads.
In this evaluation, we are interested on the capability of the mapper to retrieve the location of a single read without the help of read pairs, which can of course disambiguate mapping locations of the partner.
Results for C. elegans and H. sapiens are shown in Table 3. Additional results for E. coli, D. melanogaster and two large (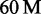 and
and 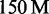 reads) H. sapiens data sets are shown in the Supplementary Table S1.
reads) H. sapiens data sets are shown in the Supplementary Table S1.
Table 3.
Runtime results
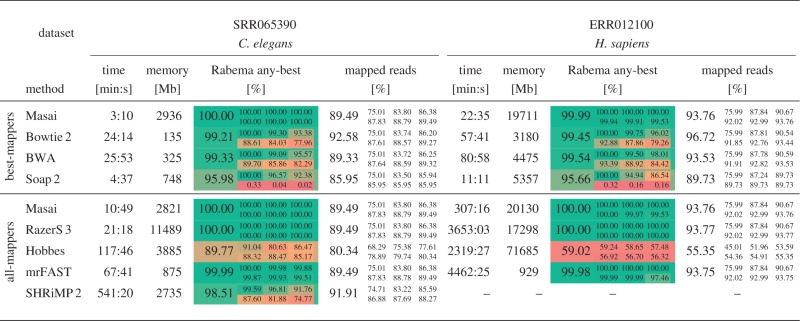 |
Results of mapping 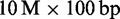 Illumina reads. Mapped reads: In large, we show the percentage of mapped reads and in small the cumulative percentage of reads that were mapped with
Illumina reads. Mapped reads: In large, we show the percentage of mapped reads and in small the cumulative percentage of reads that were mapped with 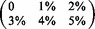 errors. Rabema any-best: In large, we show the percentage of reads mapped with the minimal number of errors (up to 5%) and in small the percentage of reads that were mapped with
errors. Rabema any-best: In large, we show the percentage of reads mapped with the minimal number of errors (up to 5%) and in small the percentage of reads that were mapped with 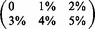 errors. Remarks: SHRiMP 2 was not able to map the H. sapiens data set within 4 days. Hobbes constantly crashed and was not able to map completely neither the C. Elegans nor the H. sapiens data set.
errors. Remarks: SHRiMP 2 was not able to map the H. sapiens data set within 4 days. Hobbes constantly crashed and was not able to map completely neither the C. Elegans nor the H. sapiens data set.
Best-mappers
On the C. elegans data set, Masai was 7.7 times faster than Bowtie 2, 8.2 times faster than BWA and 1.5 times faster than Soap 2. On the H. sapiens data set, Masai was 2.6 times faster than Bowtie 2, 3.6 times faster than BWA but 2.1 times slower than Soap 2. On one hand, Soap 2 was not able to map a consistent fraction of reads because of its limitation to two mismatches. On the other hand, Bowtie 2 reported more mapped reads than Masai but, taking any-best scores into account, it reported less mapping locations than Masai. In fact, Bowtie 2 uses a scoring scheme based on quality values and does not impose a maximal error rate threshold. On the C. elegans and H. sapiens data sets, Bowtie 2 missed 22.0 and 20.7% of reads, respectively, mappable at edit distance 5.
All-mappers
On the C. elegans data set, Masai was 2.0 times faster than RazerS 3, 10.9 times faster than Hobbes, 6.3 times faster than mrFAST and 50.1 times faster than SHRiMP 2. Hobbes constantly crashed and mapped less reads than all other mappers in this category. Likewise for Bowtie 2, also SHRiMP 2 does not impose a maximal error rate threshold and reported more mapped reads than Masai. However, its Rabema any-best score was inferior to Masai. This could be owing to the use of a different scoring scheme where two mismatches cost less than opening a gap. Anyway, this hypothesis does not explain why SHRiMP 2 did not report some mapping locations at distance 0.
On the H. sapiens data set, Masai was 11.9 times faster than RazerS 3, 14.6 times faster than mrFAST and 7.6 times faster than Hobbes. The current implementation of Hobbes often crashed and mapped only half of the reads. SHRiMP 2 was not able to map the H. sapiens data set within 4 days.
Memory requirements
In all, 20 GB of main memory are required to map a block of 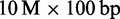 reads on H. sapiens using a suffix array index. The reference genome consumes 3 GB, its suffix array index 15 GB and
reads on H. sapiens using a suffix array index. The reference genome consumes 3 GB, its suffix array index 15 GB and 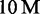 reads along with the radix tree ∼2 GB. Alternatively, the FM-index lowers the memory consumption of H. sapiens index to 4 GB. Therefore, 9 GB of main memory are sufficient to map
reads along with the radix tree ∼2 GB. Alternatively, the FM-index lowers the memory consumption of H. sapiens index to 4 GB. Therefore, 9 GB of main memory are sufficient to map 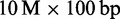 reads (Supplementary Table S7). Suffix array indices of C. elegans, D. melanogaster and E. coli are not problematic in size, as they consume only 479 MB, 575 MB and 23 MB.
reads (Supplementary Table S7). Suffix array indices of C. elegans, D. melanogaster and E. coli are not problematic in size, as they consume only 479 MB, 575 MB and 23 MB.
On low memory machines and clusters, larger read set can be always processed in blocks of appropriate size. Thus, the standard memory requirement for H. sapiens is 20 GB of main memory. Nonetheless, we mapped two huge data sets at once, merely to measure scalability. On the full ERR012100 data set (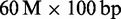 reads), Masai required 31 GB of main memory, whereas on the ERR161544 H. sapiens data set (
reads), Masai required 31 GB of main memory, whereas on the ERR161544 H. sapiens data set (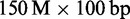 reads), Masai required 52 GB of main memory (Supplementary Table S2).
reads), Masai required 52 GB of main memory (Supplementary Table S2).
DISCUSSION
We showed that, on one hand, Masai is faster and more accurate than the best-mappers Bowtie 2 and BWA, whereas on the other hand, Masai is slightly slower but substantially more accurate than Soap 2. Masai’s accuracy becomes considerable in presence of genomic variation; therefore, we strongly advise to use Masai in small and large genomic variation pipelines.
At the same time, we showed that Masai is significantly faster than any other all-mapper while being almost full sensitive. Consequently, Masai brings all-mapping within feasible times, although with a higher memory footprint.
We finally remark that performance on short reference genomes (Supplementary Table S1) is relevant for metagenomics. On E. coli, Masai outperforms Bowtie 2 and BWA by an order of magnitude. Therefore, we strongly advise to use Masai also in metagenomic pipelines.
Masai is implemented in C++ using the SeqAn library. The source code is distributed under the BSD license, and binaries for Linux, Mac OS X and Windows can be freely downloaded from http://www.seqan.de/projects/masai.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–7 and Supplementary Figures 1–3.
FUNDING
International Max Planck Research School for Computational Biology and Scientific Computing (to E.S.); the Federal Ministry of Education and Research [16V0080 to D.W.]. Funding for open access charge: International Max Planck Research School for Computational Biology and Scientific Computing.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Manuel Holtgrewe for his joint work on experimental evaluations. They are grateful to Jochen Singer for providing them with a generic FM-index implementation.
REFERENCES
- 1.Navarro G, Baeza-Yates RA, Sutinen E, Tarhio J. Indexing methods for approximate string matching. IEEE Data Eng. Bull. 2001;24:19–27. [Google Scholar]
- 2.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li R, Yu C, Li Y, Lam T-W, Yiu S-M, Kristiansen K, Wang J. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25:1966–1967. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- 5.Navarro G. A guided tour to approximate string matching. ACM Comput. Surv. 2001;33:31–88. [Google Scholar]
- 6.Weese D, Holtgrewe M, Reinert K. RazerS 3: faster, fully sensitive read mapping. Bioinformatics. 2012;28:2592–2599. doi: 10.1093/bioinformatics/bts505. [DOI] [PubMed] [Google Scholar]
- 7.Ahmadi A, Behm A, Honnalli N, Li C, Weng L, Xie X. Hobbes: optimized gram-based methods for efficient read alignment. Nucleic Acids Res. 2012;40:e41. doi: 10.1093/nar/gkr1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alkan C, Kidd JM, Marques-Bonet T, Aksay G, Antonacci F, Hormozdiari F, Kitzman JO, Baker C, Malig M, Mutlu O, et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat. Genet. 2009;41:1061–1067. doi: 10.1038/ng.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.David M, Dzamba M, Lister D, Ilie L, Brudno M. SHRiMP2: sensitive yet practical SHort Read Mapping. Bioinformatics. 2011;27:1011–1012. doi: 10.1093/bioinformatics/btr046. [DOI] [PubMed] [Google Scholar]
- 10.Manber U, Myers G. SODA. 1990. Suffix arrays: a new method for on-line string searches; pp. 319–327. [Google Scholar]
- 11.Abouelhoda M, Kurtz S, Ohlebusch E. Replacing suffix trees with enhanced suffix arrays. J. Discrete Algorithms. 2004;2(1):53–86. [Google Scholar]
- 12.Ferragina P, Manzini G. Proceedings of the Twelfth Annual ACM-SIAM Symposium on Discrete Algorithms, Washington, D.C., USA. Philadelphia, PA, USA: Society for Industrial and Applied Mathematics; 2001. An experimental study of an opportunistic index; pp. 269–278. [Google Scholar]
- 13.Navarro G, Baeza-Yates R. A hybrid indexing method for approximate string matching. J. Discrete Algorithms. 2000;1:205–239. [Google Scholar]
- 14.Myers G. A fast bit-vector algorithm for approximate string matching based on dynamic programming. J. ACM. 1999;46:395–415. [Google Scholar]
- 15.Baeza-Yates RA, Navarro G. Faster approximate string matching. Algorithmica. 1999;23:127–158. [Google Scholar]
- 16.Morrison DR. PATRICIA-Practical algorithm to retrieve information coded in alphanumeric. J. ACM. 1968;15:514–534. [Google Scholar]
- 17.Weiner P. SWAT (FOCS) IEEE. Los Alamitos, CA, USA: IEEE Computer Society; 1973. Linear pattern matching algorithms; pp. 1–11. [Google Scholar]
- 18.Ukkonen E. On-line construction of suffix trees. Algorithmica. 1995;14:249–260. [Google Scholar]
- 19.Ukkonen E. Approximate string-matching over suffix trees. In: Apostolico A, Crochemore M, Galil Z, Manber U, editors. Lecture Notes in Computer Science. Vol. 684. Ukkonen, Esko: Springer Berlin Heidelberg; 1993. pp. 228–242. [Google Scholar]
- 20.Baeza-Yates RA, Gonnet GH. SPIRE/CRIWG IEEE. IEEE Computer Society Press; 1999. A fast algorithm on average for all-against-all sequence matching; pp. 16–23. [Google Scholar]
- 21.Dementiev R, Kärkkäinen J, Mehnert J, Sanders P. Better external memory suffix array construction. J. Exp. Algorithmics. 2008;12 3.4:1–3.4:24. [Google Scholar]
- 22.Kasai T, Lee G, Arimura H, Arikawa S, Park K. CPM. London, UK: Springer-Verlag; 2001. Linear-time longest-common-prefix computation in suffix arrays and its applications; pp. 181–192. [Google Scholar]
- 23.Grossi R, Gupta A, Vitter JS. Proceedings of the 14th annual ACM-SIAM symposium on Discrete algorithms. 2003. High-order entropy-compressed text indexes. Society for Industrial and Applied Mathematics SODA ’03, Philadelphia, PA pp. 841–850. [Google Scholar]
- 24.Giegerich R, Kurtz S, Stoye J. Efficient implementation of lazy suffix trees. Softw. Pract. Exper. 2003;33:1035–1049. [Google Scholar]
- 25.Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970;48:443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- 26.Holtgrewe M, Emde A-K, Weese D, Reinert K. A novel and well-defined benchmarking method for second generation read mapping. BMC Bioinformatics. 2011;12:210. doi: 10.1186/1471-2105-12-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holtgrewe M. 2010. Mason—a read simulator for second generation sequencing data. Technical report TR-B-10-06. Institut für Mathematik und Informatik, Freie Universität Berlin. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.