


Abstract
Kaposi’s sarcoma-associated herpesvirus encodes four viral homologues to cellular interferon regulatory factors (IRFs), where the most studied is vIRF-1. Even though vIRF-1 shows sequence homology to the N-terminal DNA-binding domain (DBD) of human IRFs, a specific role for this domain in vIRF-1’s function has remained uncertain. To provide insights into the function of the vIRF-1 DBD, we have determined the crystal structure of it in complex with DNA and in its apo-form. Using a thermal stability shift assay (TSSA), we show that the vIRF-1 DBD binds DNA, whereas full-length vIRF-1 does not, suggesting a cis-acting regulatory mechanism in similarity to human IRFs. The complex structure of vIRF-1 DBD reveals interactions with the DNA backbone and the positioning of two arginines for specific recognition in the major grove. A superimposition with human IRF-3 reveals a similar positioning of the two specificity-determining arginines, and additional TSSAs indicate binding of vIRF-1 to an IRF-3 operator consensus sequence. The results from this study, therefore, provide support that vIRF-1 has evolved to bind DNA and plays a role in DNA binding in the context of transcriptional regulation and might act on some of the many operator sequences controlled by human IRF-3.
INTRODUCTION
Kaposi’s sarcoma-associated herpesvirus (KSHV), also known as human herpesvirus 8, is currently one of the seven identified human tumor viruses, which includes Epstein–Barr virus, hepatitis B virus, hepatitis C virus, human T-lymphotropic virus, Merkel cell polyomavirus and human papillomavirus (1). Since its discovery in 1994 by Chang et al. (2), it has been shown to be the causative agent of Kaposi’s sarcoma, multicentric Castleman’s disease and primary effusion lymphoma (3–5). Infection by KSHV is currently being attributed as the most common cause of adult cancers in some sub-Saharan countries, as well as being the cause of major complications within immunosuppressed individuals worldwide (6,7).
Although tumor formation induced by oncogenic viruses is a multi-step event, host immune response suppression induced by the virus is a major contributor for the development of virus-induced tumors (1,8). Well-known examples for downregulation of host responses are the targeting of human tumor suppressor genes, such as p53 and the interferon (IFN) signaling pathways (1,4,8,9). During the annotation of the KSHV genome, it was noted that about one-third of the KSHV genes shared significant sequence homology to cellular genes, and it was proposed that KSHV had acquired these through molecular piracy, which in many cases would serve in the repression of host responses (10). Among these pirated genes was a group of four proteins dubbed viral IFN regulatory factors (IRF), vIRF-1, vIRF-2, vIRF-3 and vIRF-4, because of their homology to human IFN regulatory factors (human IRFs) (11,12). The human IRFs constitute a family of transcription factors that regulate the production of IFNs during innate immune responses, as well as a large number of so called IFN-stimulated genes (13,14). The inhibition of IRFs leads to downstream disturbances of cell proliferation, differentiation, cell migration and apoptosis (13,14).
The human IRFs have been characterized in great detail, and several structures have been presented both in their apo-forms and in complexes with different DNA operators, as well as with additional transcriptional regulators (15–19). The IRF homologues from KSHV are still relatively poorly characterized, and detailed insights into their mechanisms of action remain elusive. Of the four KSHV vIRFs, vIRF-1 is by far the most studied, as can be observed by the amount of literature about this particular protein. For instance, vIRF-1 has been shown to inhibit anti-viral responses by IRF-3 and IRF-7 (20–24) and thereby avoiding the downstream pathways activated by type I IFN. On top of IFN-inhibitory activities, several studies have indicated vIRF-1 as a direct interaction partner of other important host proteins, such as p53, GRIM19, transforming growth factor-β and the pro-apoptotic BH3 protein Bim (21,22,25–29). The cumulative downstream effects of these interactions are resistance of the host cell towards cell growth arrest and apoptosis. Interestingly, the overexpression of vIRF-1 in NIH3T3 cells was able to cause tumor formation when injected into nude mice and was, hence, labeled as an oncogene (21).
vIRF-1 has two predicted domains—an N-terminal DNA-binding domain (DBD) and a C-terminal IRF interaction domain (IAD) (Figure 1a) (30). Although vIRF-1 shares an overall low sequence homology with the human IRFs (e.g. with IRF-3: 26.6% and IRF-7: 26.2%), the individual domains of the protein share higher sequence homology. The DBD of vIRF-1 shares a slightly higher sequence homology with the DBD of the human IRFs (e.g. with IRF-3: 41.5% and IRF-7: 38.3 %), whereas the IAD of vIRF-1 shares higher sequence similarity with the IAD of the human IRFs (e.g. IRF-3: 32.5% and IRF-7: 31.8%) (13,14). Although the sequence similarities with the other KSHV vIRFs are low, some vIRFs share similar functions, like type I IFN-inhibitory activities (31,32), whereas some have different functions from the others (11,20,31,32).
Figure 1.

(a) The predicted domain boundaries of vIRF-1: DBD and the IAD (30). Below are the different domain boundary truncations of our four vIRF-1 protein constructs. (b) A TSSA was performed for all the four vIRF-1 protein constructs with several dsDNA, including the PRDIII-I sequence from the human IRF operator (PRDIII-I), a two times repeat of S0 (S0_x2), two sequences from the viral operator region that contains sequences that are highly similar to S0 (v1 and v2) and a random DNA sequence that contains 25 bp (N25). The TSSA shows that only vIRF-188–196 is stabilized by the various dsDNA. (c) A dose–response experiment results of vIRF-188–196 with varying concentrations of the aforementioned dsDNA. A dose–responsive stabilization of vIRF-188–196 by the different dsDNA was observed.
It was initially speculated that vIRF-1 inhibited the IFN signaling pathways by binding to the IFN-sensitive response elements through its DBD, but this theory was abandoned when overexpressed vIRF-1 was found to be unable to bind to the operator regions that the human IRFs bind to (21–23,33). Instead, its functions as an IFN transcription repressor and a proto-oncogene have been suggested to be mediated through its IAD and the non-conserved secondary structures between the DBD and the IAD (18,20–23,25–29,34,35). Therefore, much of the studies done on vIRF-1 have been focused on the IAD and its inhibiting role. The role of the vIRF-1 DBD has remained largely undefined, and it has been suggested that as it lacks three of five conserved tryptophan residues that are part of DNA binding for the human IRFs, it might have lost its DNA-binding ability (21). True enough, studies had showed that vIRF-1 was unable to bind to the consensus DNA sequence of the human IRFs (13,22,33). However, in 2007, a study by Park et al. (36) showed that vIRF-1 indeed could bind DNA oligonucleotides in vitro, and they suggested that it acted as a transcription factor on operator regions within the viral genome.
To shed light on whether vIRF-1 has the essential molecular features to bind DNA, we followed up on the study by Park et al. (36) by performing structural and biophysical DNA-binding studies. The crystal structure of vIRF-1 DBD in complex with a double-stranded DNA was determined to a resolution of 1.5 Å, revealing the principle DNA-binding features of vIRF-1. The crystal structure also revealed two arginines located in a similar position as two sequence-specificity determining arginine residues that are unique to the IRF-3 DBD (16,19). By also solving the structure of the substrate-free vIRF-1 DBD (subsequently known as the apo vIRF-1 DBD structure) and by comparing it with the DNA bound vIRF-1 complex structure, it can be seen that minor conformational changes of the DBD occur on DNA binding. Using a thermal stability shift assay (TSSA), we demonstrated that several DNA sequences stabilize a vIRF-1 construct containing only the vIRF-1 DBD in a dose–responsive manner, indicative of specific binding in vitro. In addition, constructs containing both the DBD and the IAD domains were not stabilized, suggesting that the vIRF-1 DBD is cis-inhibited, presumably by some type of interaction of the IAD.
MATERIALS AND METHODS
Cloning and expression
The vIRF-1 protein from KSHV contains two predicted domains (30). Four gene constructs of vIRF-1 (vIRF-11−449, vIRF-188−449, vIRF-188−196 and vIRF-184−449) (Figure 1a) (GeneBank Protein ID: ABD28909.1) were subcloned and inserted into the vector pNIC28-Bsa4 (37) that contains an N-terminal hexa-histidine tag (His-tag) and a tobacco etch virus (TEV)-protease digestion site, using ligation-independent cloning (38). The gene constructs were expressed in BL21-DE3 Rosetta strain (Novagen). Cells were grown in 750 ml of terrific broth medium supplemented with 50 μg/ml of kanamycin and 34 μg/ml of chloramphenicol at 37°C to OD600 = 2 and induced with 0.5 mM IPTG at 18°C overnight using the large expression (LEX) system (Harbinger Biotech).
Osmotic shock
Natural metal chelators, like dicarboxylic acids produced from the citric acid cycle, present in the periplasmic space of the Escherichia coli cells (39), can lower the protein purification efficiency by causing nickel ion leakage from the immobilized affinity chromatography (IMAC) columns (40). As some of the constructs gave poor protein yield, we decided to add an additional step of osmotic shock after the protein expression to increase the yield of all vIRF-1 constructs.
The cells were harvested by centrifugation (4500g for 10 min at 15°C), and the cell pellet was re-suspended in a sucrose buffer (50 mM HEPES, pH 7.9, 20% sucrose and 1 mM EDTA). The E. coli cells were centrifuged (7000g for 30 min at 4°C) before re-suspension in 5 mM MgSO4 solution. The cells were pelleted by centrifugation (4500g for 20 min at 4°C), re-suspended with lysis buffer (100 mM HEPES, pH 8.0, 500 mM NaCl, 10 mM imidazole, 10% glycerol and 0.5 mM TCEP) supplemented with 0.1 mg/ml of lysozyme (Sigma-Aldrich), 1 μl/ml of protease inhibitor (Calbiochem) and 125 U/ml of Benzonase (Merck) and 1 mM MgSO4 and stored at −80°C.
Purification and concentration
All vIRF-1 constructs were purified with a two-step purification procedure, including IMAC, using a 1-ml HisTrap™ HP column (GE Healthcare), and gel filtration chromatography, using HiLoad 16/60 Superdex 75 prep grade gel filtration column (GE Healthcare) on the ÄKTAxpress system (GE Healthcare).
The frozen cell lysate was thawed and sonicated. The lysate was clarified by centrifugation (47 000 g for 25 min at 4°C), and filtration of the supernatant was carried out through a 1.2-μm syringe filter. The filtrate was loaded onto a pre-equilibrated IMAC column (20 mM HEPES, pH 7.5, 500 mM NaCl, 10 mM imidazole, 10% glycerol and 0.5 mM TCEP). The column was washed with 20 column volumes (CVs) of wash buffer 1 and 15 CVs of wash buffer 2 (20 mM HEPES, pH 7.5, 500 mM NaCl, 25 mM imidazole, 10% glycerol and 0.5 mM TCEP) before eluting with 5 CVs of elution buffer (20 mM HEPES, pH 7.5, 500 mM NaCl, 500 mM imidazole, 10% glycerol and 0.5 mM TCEP). The eluate was loaded onto a pre-equilibrated (20 mM HEPES, pH 7.5, 500 mM NaCl, 10% glycerol and 0.5 mM TCEP) gel filtration column and collected in 2-ml fractions. The fractions were analyzed on 4–12% Nu–polyacrylamide gel electrophoresis (PAGE) gels (Invitrogen), and the fractions with the highest protein purity were pooled for TEV-protease digestion. The protein concentration was determined by measuring the ultraviolet absorbance at 280 nm on a Nanodrop spectrophotometer (Thermo Scientific).
The N-terminal His-tag on the vIRF-1 constructs was removed by TEV-protease digestion. vIRF-1 constructs were incubated with TEV-protease with a molar ratio 1:40 (protease:construct) at 4°C overnight. The digestions were analyzed on a 4–12% Nu–PAGE gel to ensure complete TEV-protease digestion. The final step of IMAC was used to recover the digested protein. The recombinant proteins were collected and concentrated using a 10 kDa cut-off centrifugal driven filter concentrator for vIRF-11−449, vIRF-188−449 and vIRF-188−196 and a 5 kDa cut-off centrifugal driven filter concentrator for vIRF-184−449 (Satorius Stedium Biotech).
Oligonucleotides
De-ionized distilled water was added into the lyophilized oligonucleotides (1st Base Asia) used for the TSSA and crystallization to achieve a final stock concentration of 2 and 5 mM, respectively. Before any experiment, all the oligonucleotides were heated at 80°C for 10 min before cooling down to room temperature. Double-stranded oligonucleotides were prepared by mixing equimolar ratios of complementary oligonucleotides before cooling down to room temperature to obtain a final concentration of 1 mM.
Thermal stability shift assay
The TSSA was used to analyze the binding of both the single-stranded (ssS0) (Forward: 5′-GCGTCGAGACGC-3′) and doubled-stranded (dsS0) (Forward: 5′-GCGTCGAGACGC-3′; Reverse: 5′-GCGTCTCGC-3′) forms of the reported consensus sequence (36) (Supplementary Figure S1a) with the different protein constructs of vIRF-1 as previously described by Ericsson et al. (41). The following modifications were made: triplicates were performed for each reaction. The various protein constructs of vIRF-1 and the oligonucleotides were diluted to 0.2 mg/ml and 10 μM, respectively, with the TSSA buffer (20 mM HEPES, pH 7.5, 500 mM NaCl and 0.5 mM TCEP). Only the TSSA buffer was added to the control samples to obtain the melting temperature (Tm) of the un-complexed protein. The change in melting temperature (ΔTm) of each individual reaction was calculated with ΔTm = T0 − Tm, where T0 is the observed melting temperature of the various vIRF-1 constructs in the presence of the different oligonucleotides. To verify the stabilizing effect of ssS0, a dose–response assay was set-up by repeating the TSSA with ssS0 at concentrations of 100, 50, 25, 12.5, 6.3, 3.2, 1.6, 0.8 and 0.4 μM.
To investigate the vIRF-1 DBD’s stabilization in the presence of other biologically relevant DNA sequences, we ran a dose–response assay with an IRF-3 recognition sequence [PRDIII-I (Forward: 5′-GAGGAAAACTGAAAGGGAACTGAAA-3′; Reverse: 5′-TTTCAGTTCCCTTTCAGTTTTCCTC-3′)] (14), S0 at twice its length [S0_x2 (Forward: 5′-AGCGTCGAGACGCGCGTCGAGACGC-3′; Reverse: 5′-TGCGTCTCGACGCGCGTCTCGACGC-3′)], two oligonucleotides similar to the proposed viral operator sequences from Park et al. (36) [v1 (Forward: 5′-AGGCACGCGTCCAGGCAACCGGG-3′; Reverse: 5′-TCCCGGTTGCCTGGACGCGTGCC-3′) and v2 (Forward: 5′-ATAACTGGGAGGCGACGCTGGCG-3′; Reverse: TCGCCAGCGTCGCCTCCCAGTTA-3′)] and a random 25 bp sequence [N25 (Forward: 5′-TATACAAACCCCTTACCAATATCATA-3′; Reverse: 5′-ATATGATATTGGTAAGGGGTTTGTAT-3′)].
Crystallization and data collection
In all, 0.1 μM of vIRF-188−196 (also known as the vIRF-1 DBD) (18 mg/ml) and 0.2 μM of the ssS0 were mixed and incubated at room temperature for 10 min. The crystals of vIRF-1 DBD with DNA were obtained by adding 3 μl of the complex solution to 3 μl of the reservoir solution containing 0.1 M phosphate citrate, pH 4.2, and 40% PEG 300 in a 24-well hanging drop plate (Hampton) at room temperature.
The apo-vIRF-1 DBD crystals were obtained by adding 0.2 μl of the vIRF-188−196 protein solution (18 mg/ml) to 0.1 μl of the reservoir solution (10 mM adenosine triphosphate, 0.1 M HEPES, pH 7, 27% Jeffamine ED-2001), in a 96-wells sitting drop Intelli-plate® (Art Robbins) at 4°C.
The crystals were flash frozen in liquid nitrogen, and diffraction data for the vIRF-1 DBD with DNA were collected at National Synchrotron Radiation Research Center (NSRRC, Taiwan, ROC), Diamond Light Source (DLS, UK) and the Australian Synchrotron (AS, Australia). The data sets were integrated and scaled using HKL2000 (42).
Structure determination
The vIRF-1 DBD with DNA complex structure was solved by molecular replacement (MR), to a resolution of 1.5 Å. Of all the available structures of the DBD of the human IRFs (15–19,24,43,44), the measured diffraction data agree to the largest extent with the data computed from the structure of the DBD of human IRF-2 (45). Therefore, PDB ID: 2IRF (18) was used as a search model using PHASER (45) from the CCP4 suite (46). Structure refinement of the complex structure was carried out with the maximum likelihood refinement using REFMAC (47) and automatic model rebuilding using ARP/wARP solvent (48). Manual model building of the DNA was performed using COOT (49).
The apo-structure was solved by MR, to a resolution of 2.4 Å, with vIRF-1A from the complex structure as a search model using PHASER-MR (45) from the Phenix suite (50). Structure refinement of the apo-structure was carried out with the maximum likelihood refinement using phenix.refine from the Phenix suite (50). Manual model building was performed using COOT (49).
The models for vIRF-1 DBD were validated using the Phenix suite (50). All figures were generated with Pymol (51).
RESULTS
Thermal stability shift assay
Because of the lack of a known target sequence for vIRF-1, Park et al. (36) used a library strategy to select oligonucleotides that interacted with the purified vIRF-1. A consensus sequence (5′-GCGTCnnGACGC-3′), which is different from the consensus sequence for the human IRFs (AAnnGAAA), was reported (36). This consensus sequence, which we have chosen to call S0, was proposed to be related to transcriptional regulatory regions within the viral DNA (36). We used this consensus sequence as a tool to study the DNA-binding properties of our different vIRF-1 constructs in a TSSA (Figure 1a). Both single-stranded (ssS0) and double-stranded DNA (dsS0) forms of S0 were used (Supplementary Figure S1a). The Tm of vIRF-188−196 increased significantly by ∼4°C in the presence of both ssS0 and dsS0, whereas very small shifts could be observed for the other vIRF-1 constructs (Supplementary Figure S1b). Our TSSA results show that S0 stabilizes vIRF-188−196 in a dose–responsive manner (Supplementary Figure S1c).
To further investigate the DNA-binding properties of the vIRF-1 DBD, we also tested other and perhaps more biologically relevant DNA sequences: the human positive regulatory domain of the IFN-β enhancer (PRDIII-I) that human IRF-3 is able to bind to (16), a two times repeat of the S0 sequence (S0_x2), the proposed viral operator sequences (v1 and v2) and a random DNA sequence of 25 bp (N25) (Figure 1b). All sequences were made ∼25-bp long, to maximize potential protein–DNA interactions. Our TSSA results show that all these sequences also stabilized the vIRF-1 DBD in a dose–responsive manner (Figure 1c).
Overall structure of vIRF-1 DBD in complex with DNA
Well diffracting co-crystals of vIRF-1 DBD (vIRF-188−196) with ssS0 were obtained. The complex structure was subsequently determined with molecular replacement (45) at a resolution of 1.5 Å. The asymmetric unit of the complex structure consists of two vIRF-1 DBD monomers, labeled vIRF-1A (amino acids 90–195) and vIRF-1B (amino acids 90–195), and two DNA duplexes stacked on top of each other. Each DNA duplex consists of two ssS0 that form a distorted dsDNA with two mismatching G–A base pairs in the center (Figure 2a). The dsDNA that is formed by two ssS0 is hereafter referred to as dsS0mm (Figure 2a).
Figure 2.

The crystal structure of vIRF-1 DBD in complex with DNA. (a) The asymmetric unit of the complex structure is made up of two vIRF-1 DBDs (vIRF-188–196), namely, vIRF-1A (orange) and vIRF-1B (green), and two dsS0mm stacked on top of each other. Each dsS0mm is made up of two molecules of ssS0 (strands C and D) that form a distorted double-stranded DNA with two mismatching G–A base pairs in the center. Both vIRF-1A and vIRF-1B bind to the major grooves on the opposite sides of the DNA duplex (gray). vIRF-1A binds to the major groove of one DNA duplex, whereas vIRF-1B binds to the major groove that is created by the imperfect stacking of two DNA duplexes. The individual DNA bases that interact with vIRF-1A and vIRF-1B are colored light orange and light green, respectively. (b) Both vIRF-1A and vIRF-1B are essentially the same. The DBD is made up of three α-helices (α1–α3), four β-sheets (β1–β4) and three long loops (L1–L3). It forms the typical winged HTH motif.
Although vIRF-1A and vIRF-1B bind at different positions along the elongated DNA duplex, both protein molecules are essentially the same, superimposing with a root mean square deviation (RMSD) of 0.249 Å. The vIRF-1 DBD consists of three α-helices (α1–α3), four β-strands (β1–β4) and three long loops, termed L1–L3 (Figure 2b). It has the typical winged helix-turn-helix (HTH) motif, which is commonly found in human IRFs as well as other transcription factors (52).
The dsS0mm forms a semi-continual DNA duplex in the crystal with direct end-to-end interactions, which is frequently seen in structures with dsDNA. Although both vIRF-1A and vIRF-1B are the same protein, they bind different sequences in the crystal structure. Although vIRF-1A binds the whole face of the dsS0mm duplex (GCGTCGAGACGC) (Figures 2a and 3a–c), vIRF-1B binds two halves of the continued duplex corresponding to the sequence GAGACGCxGCGTC, where ‘x’ denotes a break and an end-to-end contact (Figures 2a and 3a–c).
Protein–DNA interactions
Both vIRF-1A and vIRF-1B make extensive and related interactions with the phosphate backbone of the DNA duplex (Figure 3a). Both protein molecules make similar phosphate interactions by the main chain amides of Ser 92 and Ile 93, the amino acids at the N-termini of helix α1 and the side chains of Arg 147 and Asn 149 (Figure 3a). The canonical DNA recognition helix α3 is well positioned for specific DNA recognition in the major grooves by both monomers (Figure 3b and c). However, the specific interactions are made differently by the two monomers. vIRF-1B makes nice hydrogen bonds with DNA bases through Arg 163 and Arg 171 that are located on the DNA recognition helix α3 (Figure 3c). The side chain of Arg 171 is in an extended conformation forming hydrogen bonds with G-3* and T-4* of the DNA chain D, whereas the side chain of Arg 163 forms hydrogen bonds with G-11 of the DNA chain C (Figure 3c). Although the corresponding Arg 163 from vIRF-1A is also hydrogen bonded to the DNA, it binds to G-6 of the distorted G–A base pair on DNA chain C (Figure 3a). Also, the side chains of Arg 163 from both vIRF-1A and vIRF-1B are extending in different directions (Figure 3d). The side chain of Arg 171 from both monomers also extends in different directions. In fact, Arg 171 from vIRF-1A does not interact with any DNA bases (Figure 3d). One other notable difference is that Arg 164 from vIRF-1A is hydrogen bonded to G-6 of the distorted G–A base pair on the DNA chain D (Figure 3b), but the corresponding residue in vIRF-1B does not bind to any DNA bases.
Figure 3.

An illustration of the interactions between the vIRF-1 DBD and the DNA duplex. (a) The DNA bases are labeled (guanine = G; adenine = A; cytosine = C; and thymine = T) and numbered according to their sequence from the 5′- to 3′-end. The DNA bases that are labeled with an asterisk are bases from the neighboring asymmetrical unit that are included in the complex structure. The pentose sugars of these bases are colored orange. The black and the red dotted lines represent the interactions between the respective amino acids with the DNA phosphate backbone and the DNA bases. The interacting amino acids from vIRF-1A and vIRF-1B are colored orange and green, respectively. The DNA nucleotides that interact with vIRF-1A and vIRF-1B are also colored orange and green, respectively. (b) Arg 163 and Arg 164 from vIRF-1A and (c) Arg 163 and Arg 171 from vIRF-1B are the only amino acids that were found to interact with the DNA bases through hydrogen bonds (blue dotted lines). (d) A superimposition of vIRF-1A and vIRF-1B is shown to illustrate the difference in the arrangement of Arg 163 and Arg 171 in both molecules.
The difference in DNA recognition by the two monomers could be interpreted as if vIRF-1B binds the DNA duplex in a semi-cognate mode, whereas vIRF-1A binds the DNA in a more non-cognate recognition mode. This is consistent with that the interactions for vIRF-1B and the DNA in the vIRF-1B are tighter on the protein. It is possible that the highly distorted A–G pair positioned in the middle of the α3 recognition helix blocks a more cognate DNA recognition for vIRF-1A. In contrast, the end-to-end DNA connection where vIRF-1B binds, it creates a wider major groove that adds extra flexibility allowing a more compact DNA interaction.
Structure of apo-vIRF-1
The crystal structure of apo-vIRF-1 was determined at a resolution of 2.4 Å with four monomers in the asymmetric unit (Table 1). The structures of the four monomers are similar, superimposing with an average RMSD of 0.302 Å (Figure 4a). As no continuous electron density could be seen for residues 148–154, which corresponds to loop L2, this part could not be modeled. This suggests a highly disordered secondary structure in this area. A comparison with the complex structure reveals similar structures with an average RMSD of 0.437 Å (Figure 4b). Differences are seen in some side chain conformations and L2. L2 can be modeled in the complex structure, and several amino acids in the loop are found to interact with the DNA phosphate backbone. Another difference between the apo and complex structure is a clearly visible disulphide bond between two cysteine (Cys 99) residues in neighboring monomers. There is no direct reason to believe that these dimeric interactions are functionally important. We instead assume that these disulphide bonds were formed as the result of crystal packing contacts (the protein purifies as a monomer on size-exclusion chromatography, and it runs as a monomer on native gels; data not shown).
Table 1.
Data collection and refinement statistics
| Criterion | PDB ID 4HLY (complex) | PDB ID 4HLX (apo) |
|---|---|---|
| X-ray source | NSRRC BL13C1 | NSRRC BL13B1 |
| Wavelength (Å) | 0.96722 | 0.96722 |
| Space group | P1 | F222 |
| Unit cell parameters | a = b = 38.5 Å, c = 48.6 Å | a = 65.8 Å b = 128.8 Å, c = 228.5 Å |
| α = 91.3°, β = 88.5°, γ = 63.1° | α = β = γ = 90° | |
| Resolution range (Å) | 50.00-1.48 (1.53-1.48)a | 23.29 - 2.38 (2.46 - 2.38)b |
| Total reflections | 1326707 | 481953 |
| Unique reflections | 41661 | 18285 |
| Multiplicity | 3.8 (3.8)a | 6.6 (6.7)b |
| Rsymc | 0.052 (0.507)a | 0.119 (0.414)b |
| I/σ(I) | 21.29 (2.30)a | 9.43 (4.29)b |
| Completeness (%) | 94.7 (92.8)a | 92.58 (99.33)b |
| Refinement statistics | ||
| Rfactord/Rfree e(%) | 17.2/21.0 | 25.1/28.6 |
| Non-hydrogen atoms | 2267 | 6492 |
| Protein residues | 212 | 397 |
| DNA bases | 24 | 0 |
| Solvent molecules | 232 | 117 |
| RMSD bonds (Å) | 0.0296 | 0.002 |
| RMSD angles (°) | 3.52 | 0.57 |
| Ramachandran quality plot | ||
| In preferred region (%) | 97.6 | 96.6 |
| In allowed regions (%) | 2.4 | 3.4 |
| Outliers (%) | 0 | 0 |
aValues within parentheses represent the highest resolution shell (1.53–1.48 Å).
bValues within parentheses represent the highest resolution shell (2.46–2.38 Å).
cRsym = 100 × ∑(|Ij − [I]|)/ ∑(|I|), where the sum is calculated over all observations of a measured reflection (Ij), and [I] is the mean intensity of all the measured observations (Ij).
dRfactor = 100 × ∑(|F0| − |Fc|)/ ∑(|F0|), where F0 and Fc are the observed and calculated structure factors, respectively.
eRfree is equivalent to Rfactor, but where 5% of the measured reflections have been excluded from refinement and set aside for cross validation.
Figure 4.
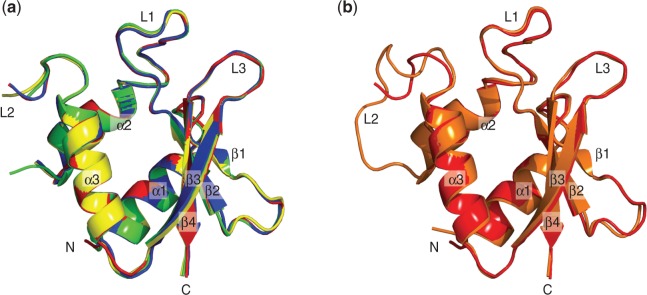
The superimpositions of the vIRF-1 DBDs. (a) The superimpositions of the four molecules of the apo-vIRF-1 DBD found in an asymmetrical unit of the crystal structure. (b) The superimposition of the DNA bound vIRF-1 (orange) and the apo vIRF-1 DBD (red). Both the DNA bound form and the apo-vIRF-1 DBD are similar, except at the L2.
DISCUSSION
KSHV has acquired genes to aid in the evasion of the host immune response. These genes often show significant homology to host genes; therefore, they are likely to have been acquired through piracy (9). Examples of KSHV proteins that have been found to be acquired through molecular piracy [the KSHV opening reading frames (ORFs) are indicated in the parentheses] include the cellular complement 2 (ORF 4), the interleukin-6 (ORF K2), the D-type cyclin (ORF 72) and the G-protein–coupled receptor (ORF 74) (9,53). vIRF-1 is also a KSHV protein that is encoded by a pirated gene (ORF K9) and has been implicated to play an important role in immune evasion by attenuating the expression of genes in the IFN response cascade (13–19,53). The current dominant view is that vIRF-1 primarily attenuates IFN signaling through the C-terminal IAD and its flanking sequences, by interacting with CBP/p300 and thereby scavenging this co-activator from the host IRFs (20–23,25–29,34,35). In contrast, the role of the DBD of vIRF-1 has remained enigmatic because of the lack of positive and conclusive evidences for DNA binding in vivo. Recently, Park et al. (36) presented results that showed that vIRF-1 could bind DNA oligonucleotides that resembled upstream regulatory regions of viral genes.
We have solved the crystal structure of the vIRF-1 DBD in complex with DNA. This constitutes the first crystal structure of a viral IRF’s DBD. The vIRF-1 DBD consists of a typical HTH-motif commonly seen in other IRFs and DNA-binding proteins. This structure provides a detailed view of the DNA interaction mode of vIRF-1, which gives strong support for the role of this protein in DNA binding. Extensive interactions are made with the DNA phosphate backbone, and two arginine residues are well positioned in the major groove for specific DNA recognition (Figure 3a). Only co-crystallization trials with the partially palindromic ssS0 gave crystals of sufficient quality for structure determination. Hence, the DNA duplex seen in the structure originates from two ssS0 molecules (dsS0mm). Therefore, the dsS0mm in the structure has two distorted base pairs in the middle of the duplex, resulting in significant distortions of the DNA at this position (Figure 2). This DNA duplex in turn forms an elongated continuous duplex by head-to-tail interactions that run through the entire crystal lattice. The complex structure also reveals that two similar vIRF-1 DBD (vIRF-1A and vIRF-1B) bind to the DNA duplex in the asymmetric unit. The vIRF-1A interacts at one face of the DNA duplex, whereas the vIRF-1B interacts at the opposite face of the DNA duplex that is created through the head-to-tail interactions of two dsS0mm in the asymmetric unit. It is likely that neither of the two DNA–protein interaction modes seen in the crystal structure correspond to a fully cognate interaction. Instead, vIRF-1A is closer to a non-cognate interaction mode, whereas vIRF-1B is closer to a cognate-binding mode. This is consistent with that only vIRF-1B makes direct DNA base interactions through two outstretched residues, Arg 163 and Arg 171, located on the recognition region of the major groove. However, the energetics of DNA binding are complex, and the direct assignment of a recognition sequence from DNA–protein structures remains challenging (54).
Extensive structural information of the DBD of human IRFs is available. IRF-1, -2, -3, -4 and -7 have been determined in DNA complexes (15–19) and IRF-3 and -7 in their respective apo-forms (22). Models for two larger transcription complexes serving as a basis for the reconstitution of a model of the enhanceosome containing IRF-3 and IRF-7 have also been determined (16,19,43). The overall structures of the DBDs of the human IRFs are similar, but significant variation is seen in the loop structures as well as in DNA specificity-determining residues of the canonical DNA recognition helix, which binds in the major groove. Structural studies of IAD from IRF-3 and -5 support a mechanism where phosphorylation of the IAD induces conformational changes of this domain, exposing residues for dimer formation (16,19,43). In the reconstituted enhanceosome structure, besides making interactions with the DNA operator sequences, IRF-3 and IRF-7 also make interactions with neighboring transcription factors (16,19,43).
Superimposition of the structure of the vIRF-1 DBD on the structure of the human IRFs DBD show a significant structural similarity in the overall fold with a RMSD ranging from 1.825 to 2.951 Å. The vIRF-1 DNA recognition helix α3 is similarly positioned in the major groove as in the human proteins. However, significant differences are seen in the loop regions involved in interactions with the DNA phosphate backbone. For example, the L2 containing Arg 147 and Asn 149 in vIRF-1 seems to make main chain interactions with the phosphate backbone, instead of lysine and arginine side chain interactions that are dominating in the human IRFs. Differences are also seen for the loop structure of L3 and its side chains. Residues 183–186 in vIRF-1 do not make any direct interactions with the DNA, whereas the corresponding loop in the human IRFs seems to make direct interactions with the DNA backbone. Another notable difference is the absence of an important and highly conserved histidine residue in L1 that has been shown to be part of sequence recognition (Figure 5a and b). In the human IRFs, this particular histidine makes a water-mediated hydrogen bond to an adenine base in the minor groove but in vIRF-1, the corresponding residue in this position is a proline, and no direct recognition is made in the minor groove (Figure 5a) (16,18,19,43). Analyzing the conserved residues in the region for recognition of the major groove, we can conclude that IRF-3 has the most extensive similarities to vIRF-1B, where arginines are also present at positions of Arg 163 and Arg 171 (Figure 5a and b). This could indicate that there might be some similarities in the recognition sequences for IRF-3 and vIRF-1. These particular and unique arginines in the recognition helix of IRF-3 have been proposed to be responsible for IRF-3’s ability to recognize both consensus and non-consensus sequences, depending on their conformation. Even though the specific interactions made by these arginines with the DNA bases differ in IRF-3 and vIRF-1, such differences have also been observed for among different IRF-3–DNA complexes (16,19,43). This suggests a similar versatility in vIRF-1 DNA recognition, as IRF-3 and vIRF-1B interacts with the DNA in a more cognate mode than vIRF-1A.
Figure 5.

Comparison of the vIRF-1 DBD with the human IRFs DBD. (a) The sequence alignment of the DBDs of vIRF-1 and the human IRF-1, -2, -3, -4 and -7. The completely conserved amino acids are highlighted in red, and the similar amino acids are red. The light blue boxes indicate the amino acids that take part in the direct and water-mediated DNA interactions. (b) The DBD of the human IRF-1 (green), IRF-2 (yellow), IRF-3 (blue), IRF-4 (cyan), IRF-7 (orange) and vIRF-1 (red) was superimposed. The amino acid residues that were found to interact with the DNA base(s) as marked in the sequence alignment are shown as sticks and are colored accordingly. Most of the protein residues that participate in the protein–DNA interactions are located on the recognition helix (α3). (c) The superimposition of IRF-3 and vIRF-1B in complex with DNA. The specificity determining arginines of both IRF-3 (blue) and vIRF-1B (red) are shown in sticks and are colored accordingly.
Prompted by our observation that vIRF-1 has two DNA specificity-recognizing arginine residues in similar positions as IRF-3, we wanted to further examine the DNA-binding ability of vIRF-1 by using a TSSA study. TSSA constitutes a biophysical technique, which has emerged as a convenient and reproducible assay for studies of protein–ligand interactions, including protein–DNA interactions (41,55). Although some proteins are not responsive to the technique, responding proteins give information on direct interactions where shift sizes and saturation curves correlate to binding affinities generated with other methods (56). For studies using purified proteins, this assay is also more convenient and direct than electrophoretic shift assays and, therefore, less likely to give erroneous results. Importantly, false positive results are in our experience rarely seen with the TSSA.
The results from our TSSA show that the DBD of vIRF-1 does bind to the IRF-3 operator region (PRDIII-I) (16), as well as the viral operator regions (v1 and v2) described by Park et al. (36) (Figure 1b). This is in contrast to conclusions from previous in vivo studies that have shown that vIRF-1 is unable to bind to the operator region that human IRFs bind to (21–23,33). By performing a dose–response experiment, with the PRDIII-I, S0_x2, v1, v2 and N25, we can observe that all of the aforementioned sequences gave dose–responsive curves. However, PRDIII-I and S0_x2 seem to stabilize vIRF-1 DBD to a greater extent (Figure 1c). Taken together, these data support the possibility that vIRF-1 may have a broad DNA recognition capacity, like IRF-3, and that it is also capable of specific cognate DNA interactions, including with regulatory elements recognized by IRF-3. The positive shifts in the TSSA for all the sequences suggest that there is basal level of binding, which could be due to non-cognate recognition of the phosphate backbone.
Interestingly, TSSA studies of several vIRF-1 constructs suggest that constructs containing the IAD do not bind DNA (Figure 1b). This implies that the full-length vIRF-1 requires activation to release the DBD from some type of cis-inhibition. Cis-inhibition has been shown for human IRFs (57,58). The observation that the full-length protein probably requires appropriate activation opens the possibility that in previous transfection and in vitro translation experiments, the DNA-binding properties of vIRF-1 may have been obscured by challenges in generating appropriate conditions for vIRF-1 activation (21–23,33). Even though the vIRF-1 lacks the characteristic C-terminal serine cluster that exists in the IAD of all the known human IRFs (44,59–61), sequence analyses of vIRF-1 indicate several potential phosphorylation sites in the N-terminal and the C-terminal region (data not shown). Phosphorylation of these residues could potentially affect the DNA binding of vIRF-1.
In addition to solving the structure of vIRF-1 DBD in complex with DNA, we also solved the structure of the DBD in its apo-form. On superimposition of the two forms, we can see that basically all structural features are conserved, with the exception of L2. We propose that on DNA binding, L2 undergoes a conformational change and becomes more ordered and adds to the overall DNA-binding capacity of vIRF-1. In the complex structure, as already mentioned, several residues in L2 do in fact interact with the phosphate backbone. Regardless of the interrupted L2 in the apo-structure, we propose that no major conformational changes occur on DNA binding in contrast to what has been observed for IRF-3 (24). Intriguingly, Arg 163 and Arg 171 seem to be highly flexible and differ in conformation among the four apo-molecules in the asymmetric unit, where none of them displays a similar outstretched conformation as can be seen in vIRF-1B. This further supports the importance and versatility of these residues in DNA recognition that have also been proposed for IRF-3 (16).
In conclusion, our structural and biophysical studies of vIRF-1 give strong additional support that the protein is in fact a DNA-binding protein that is regulated by some kind of cis-inhibition and might act directly on several different operator sequences like IRF-3. With these results in hand, we hope to shift some of the focus on vIRF-1, back to its DBD to propose more efforts into finding the core recognition sequence and shed light on the regulation of the viral genome as well as the identification of possible co-transcriptional regulators that could cooperate in DNA binding.
ACCESSION NUMBERS
4HLY and 4HLX.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figure 1.
FUNDING
Centre grant from Nanyang Technological University; Swedish Research Council (Vetenskapsrådet). Funding for open access charge: Nanyang Technological University start-up grant.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr Juergen Haas for kindly supplying the original vIRF-1 clone. Portions of this research were carried out at the National Synchrotron Radiation Research Center, a national user facility supported by the National Science Council of Taiwan, ROC. The Synchrotron Radiation Protein Crystallography Facility is supported by the National Research Program for Genomic Medicine. This research was also undertaken on the MX1 and MX2 beamlines at the Australian Synchrotron, Victoria, Australia (56).
REFERENCES
- 1.Moore PS, Chang Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat. Rev. Cancer. 2010;10:878–889. doi: 10.1038/nrc2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science. 1994;266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 3.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 1995;332:1186–1191. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- 4.Moore PS, Chang Y. Kaposi's Sarcoma Associated Herpesvirus Immunoevasion And Tumorigenesis: two sides of the same coin? Annu. Rev. Microbiol. 2003;57:609–639. doi: 10.1146/annurev.micro.57.030502.090824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bouvard V, Baan R, Straif K, Grosse Y, Secretan B, Ghissassi FE, Benbrahim-Tallaa L, Guha N, Freeman C, Galichet L, et al. A review of human carcinogens?Part B: biological agents. Lancet Oncol. 2009;10:321–322. doi: 10.1016/s1470-2045(09)70096-8. [DOI] [PubMed] [Google Scholar]
- 6.Parkin DM, Sitas F, Chirenje M, Stein L, Abratt R, Wabinga H. Part I: Cancer in Indigenous Africans—burden, distribution, and trends. Lancet Oncol. 2008;9:683–692. doi: 10.1016/S1470-2045(08)70175-X. [DOI] [PubMed] [Google Scholar]
- 7.Uldrick TS, Whitby D. Update on KSHV epidemiology, Kaposi Sarcoma pathogenesis, and treatment of Kaposi Sarcoma. Cancer Lett. 2011;305:150–162. doi: 10.1016/j.canlet.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zur Hausen H. The search for infectious causes of human cancers: where and why. Virology. 2009;392:1–10. doi: 10.1016/j.virol.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 9.Moore PS, Chang Y. Antiviral activity of tumor-suppressor pathways: clues from molecular piracy by KSHV. Trends Genet. 1998;14:144–150. doi: 10.1016/s0168-9525(98)01408-5. [DOI] [PubMed] [Google Scholar]
- 10.Russo JJ, Bohenzky RA, Chien MC, Chen J, Yan M, Maddalena D, Parry JP, Peruzzi D, Edelman IS, Chang Y, et al. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8) Proc. Natl Acad. Sci. 1996;93:14862–14867. doi: 10.1073/pnas.93.25.14862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee H-R, Kim MH, Lee J-S, Liang C, Jung JU. Viral interferon regulatory factors. J. Interferon Cytokine Res. 2009;29:621–627. doi: 10.1089/jir.2009.0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cunningham C, Barnard S, Blackbourn DJ, Davison AJ. Transcription mapping of human herpesvirus 8 genes encoding viral interferon regulatory factors. J. Gen. Virol. 2003;84:1471–1483. doi: 10.1099/vir.0.19015-0. [DOI] [PubMed] [Google Scholar]
- 13.Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF family transcription factors in immunity and oncogenesis. Annu. Rev. Immunol. 2008;26:535–584. doi: 10.1146/annurev.immunol.26.021607.090400. [DOI] [PubMed] [Google Scholar]
- 14.Taniguchi T, Ogasawara K, Takaoka A, Tanaka N. IRF family of transcription factors as regulators of host defense. Annu. Rev. Immunol. 2001;19:623–655. doi: 10.1146/annurev.immunol.19.1.623. [DOI] [PubMed] [Google Scholar]
- 15.Escalante CR, Brass AL, Pongubala JMR, Shatova E, Shen L, Singh H, Aggarwal AK. Crystal Structure of PU.1/IRF-4/DNA Ternary Complex. Mol. Cell. 2002;10:1091–1105. doi: 10.1016/s1097-2765(02)00703-7. [DOI] [PubMed] [Google Scholar]
- 16.Escalante CR, Nistal-Villan E, Shen L, Garcia-Sastre A, Aggarwal AK. Structure of IRF-3 bound to the PRDIII-I regulatory element of the human interferon-beta enhancer. Mol. Cell. 2007;26:703–716. doi: 10.1016/j.molcel.2007.04.022. [DOI] [PubMed] [Google Scholar]
- 17.Escalante CR, Yie J, Thanos D, Aggarwal AK. Structure of IRF-1 with bound DNA reveals determinants of interferon regulation. Nature. 1998;391:103–106. doi: 10.1038/34224. [DOI] [PubMed] [Google Scholar]
- 18.Fujii Y, Shimizu T, Kusumoto M, Kyogoku Y, Taniguchi T, Hakoshima T. Crystal structure of an IRF-DNA complex reveals novel DNA recognition and cooperative binding to a tandem repeat of core sequences. EMBO J. 1999;18:5028–5041. doi: 10.1093/emboj/18.18.5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Panne D, Maniatis T, Harrison SC. Crystal structure of ATF-2/c-Jun and IRF-3 bound to the interferon-b enhancer. EMBO J. 2004;23:4384–4393. doi: 10.1038/sj.emboj.7600453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li M, Lee H, Guo J, Neipel F, Fleckenstein B, Ozato K, Jung JU. Kaposi’s sarcoma-associated herpesvirus viral interferon regulatory factor. J. Virol. 1998;72:5433–5440. doi: 10.1128/jvi.72.7.5433-5440.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao S-J, Bosho C, Jayachandra S, Weiss RA, Chang Y, Moore PS. KSHV ORF K9 (vIRF) is an oncogene which inhibits the interferon signaling pathway. Oncogene. 1997;15:1979–1985. doi: 10.1038/sj.onc.1201571. [DOI] [PubMed] [Google Scholar]
- 22.Flowers CC, Flowers SP, Nabel GJ. Kaposi's sarcoma-associated herpesvirus viral interferon regulatory factor confers resistance to the antiproliferative effect of interferon-alpha. Mol. Med. 1998;4:402–412. [PMC free article] [PubMed] [Google Scholar]
- 23.Burýšek L, Yeow W-S, Lubyová B, Kellum M, Schafer SL, Huang YQ, Pitha PM. Functional analysis of human herpesvirus 8-encoded viral interferon regulatory factor 1 and its association with cellular interferon regulatory factors and p300. J. Virol. 1999;73:7334–7342. doi: 10.1128/jvi.73.9.7334-7342.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Ioannes P, Escalante CR, Aggarwal AK. Structures of apo IRF-3 and IRF-7 DNA binding domains: effect of loop L1 on DNA binding. Nucleic Acids Res. 2011;39:7300–7307. doi: 10.1093/nar/gkr325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seo T, Park J, Lee D, Hwang SG, Choe J. Viral interferon regulatory factor 1 of Kaposi's sarcoma-associated herpesvirus binds to p53 and represses p53-dependent transcription and apoptosis. J. Virol. 2001;75:6193–6198. doi: 10.1128/JVI.75.13.6193-6198.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakamura H, Li M, Zarycki J, Jung JU. Inhibition of p53 tumor suppressor by viral interferon regulatory factor. J. Virol. 2001;75:7572–7582. doi: 10.1128/JVI.75.16.7572-7582.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seo T, Park J, Choe J. Kaposi's sarcoma-associated herpesvirus viral IFN regulatory factor 1 inhibits transforming growth factor-beta signaling. Cancer Res. 2005;65:1738–1747. doi: 10.1158/0008-5472.CAN-04-2374. [DOI] [PubMed] [Google Scholar]
- 28.Choi YB, Nicholas J. Bim nuclear translocation and inactivation by viral interferon regulatory factor. PLoS Pathog. 2010;6:e1001031. doi: 10.1371/journal.ppat.1001031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seo T, Lee D, Shim YS, Angell JE, Chidambaram NV, Kalvakolanu DV, Choe J. Viral interferon regulatory factor 1 of Kaposi's sarcoma-associated herpesvirus interacts with a cell death regulator, GRIM19, and inhibits interferon/retinoic acid-induced cell death. J. Virol. 2002;76:8797–8807. doi: 10.1128/JVI.76.17.8797-8807.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunesekaran P, Ceric G, Forslund K, et al. The Pfam protein families database. Nucleic Acids Res. 2010;38:211–222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mutocheluh M, Hindle L, Aresté C, Chanas SA, Butler LM, Lowry K, Shah K, Evans DJ, Blackbourn DJ. Kaposi's sarcoma-associated herpesvirus viral interferon regulatory factor-2 inhibits type 1 interferon signalling by targeting interferon-stimulated gene factor-3. J. Gen. Virol. 2011;92:2394–2398. doi: 10.1099/vir.0.034322-0. [DOI] [PubMed] [Google Scholar]
- 32.Aresté C, Mutocheluh M, Blackbourn DJ. Identification of caspase-mediated decay of interferon regulatory factor-3, exploited by a Kaposi sarcoma-associated herpesvirus immunoregulatory protein. J. Biol. Chem. 2009;284:23272–23285. doi: 10.1074/jbc.M109.033290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zimring JC, Goodbourn S, Offermann MK. Human herpesvirus 8 encodes an interferon regulatory factor (IRF) homolog that represses IRF-1-mediated transcription. J. Virol. 1998;72:701–707. doi: 10.1128/jvi.72.1.701-707.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seo T, Lee D, Lee B, Chung JH, Choe J. Viral interferon regulatory factor 1 of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) binds to, and inhibits transactivation of, CREB-binding protein. Biochem. Biophys. Res. Commun. 2000;270:23–27. doi: 10.1006/bbrc.2000.2393. [DOI] [PubMed] [Google Scholar]
- 35.Lin R, Genin P, Mamane Y, Sgarbanti M, Battistini A, Harrington WJ, Jr, Barber GN, Hiscott J. HHV-8 encoded vIRF-1 represses the interferon antiviral response by blocking IRF-3 recruitment of the CBP/p300 coactivators. Oncogene. 2001;20:800–811. doi: 10.1038/sj.onc.1204163. [DOI] [PubMed] [Google Scholar]
- 36.Park J, Lee MS, Yoo SM, Jeong KW, Lee D, Choe J, Seo T. Identification of the DNA sequence interacting with Kaposi's sarcoma-associated herpesvirus viral interferon regulatory factor 1. J. Virol. 2007;81:12680–12684. doi: 10.1128/JVI.00556-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stols L, Gu M, Dieckman L, Raffen R, Collart FR, Donnelly MI. A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr. Purif. 2002;25:8–15. doi: 10.1006/prep.2001.1603. [DOI] [PubMed] [Google Scholar]
- 38.Savitsky P, Bray J, Cooper CD, Marsden BD, Mahajan P, Burgess-Brown NA, Gileadi O. High-throughput production of human proteins for crystallization: the SGC experience. J. Struct. Biol. 2010;172:3–13. doi: 10.1016/j.jsb.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grigoriy SC. Twenty-five years of immobilized metal ion affinity chromatography: past, present and future. J. Biochem. Biophys. Methods. 2001;49:313–334. doi: 10.1016/s0165-022x(01)00206-8. [DOI] [PubMed] [Google Scholar]
- 40.Magnusdottir A, Johansson I, Dahlgren LG, Nordlund P, Berglund H. Enabling IMAC purification of low abundance recombinant proteins from E. coli lysates. Nat. Methods. 2009;6:477–478. doi: 10.1038/nmeth0709-477. [DOI] [PubMed] [Google Scholar]
- 41.Ericsson UB, Hallberg BM, Detitta GT, Dekker N, Nordlund P. Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal. Biochem. 2006;357:289–298. doi: 10.1016/j.ab.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 42.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 43.Panne D, Maniatis T, Harrison SC. An atomic model of the interferon-beta enhanceosome. Cell. 2007;129:1111–1123. doi: 10.1016/j.cell.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takahasi K, Horiuchi M, Fujii K, Nakamura S, Noda NN, Yoneyama M, Fujita T, Inagaki F. Ser386 phosphorylation of transcription factor IRF-3 induces dimerization and association with CBP/p300 without overall conformational change. Genes Cells. 2010;15:901–910. doi: 10.1111/j.1365-2443.2010.01427.x. [DOI] [PubMed] [Google Scholar]
- 45.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Collaborative Computational Project N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 47.Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011;50:760–763. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Langer G, Cohen SX, Lamzin VS, Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 2008;3:1171–1179. doi: 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC (2011)
- 52.Luscombe NM, Austin SE, Berman HM, Thornton JM. An overview of the structures of protein-DNA complexes. Genome Biol. 2005;1:465–469. doi: 10.1186/gb-2000-1-1-reviews001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moore PS, Chang Y. Molecular virology of Kaposi's sarcoma-associated herpesvirus. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2001;356:499–516. doi: 10.1098/rstb.2000.0777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rohs R, Jin X, West SM, Joshi R, Honig B, Mann RS. Origins of specificity in protein-DNA recognition. Annu. Rev. Biochem. 2010;79:233–269. doi: 10.1146/annurev-biochem-060408-091030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dahlroth SL, Gurmu D, Schmitzberger F, Engman H, Haas J, Erlandsen H, Nordlund P. Crystal structure of the shutoff and exonuclease protein from the oncogenic Kaposi's sarcoma-associated herpesvirus. FEBS J. 2009;276:6636–6645. doi: 10.1111/j.1742-4658.2009.07374.x. [DOI] [PubMed] [Google Scholar]
- 56.Fedorov O, Marsden B, Pogacic V, Rellos P, Muller S, Bullock AN, Schwaller J, Sundstrom M, Knapp S. A systematic interaction map of validated kinase inhibitors with Ser/Thr kinases. Proc. Natl Acad. Sci. USA. 2007;104:20523–20528. doi: 10.1073/pnas.0708800104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yoneyama M, Suhara W, Fukuhara Y, Fukuda M, Nishida E, Fujita T. Direct triggering of the type I interferon system by virus infection- activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J. 1998;17:1087–1095. doi: 10.1093/emboj/17.4.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lin R, Mamane Y, Hiscott J. Multiple regulatory domains control IRF-7 activity in response to virus infection. J. Biol. Chem. 2000;275:34320–34327. doi: 10.1074/jbc.M002814200. [DOI] [PubMed] [Google Scholar]
- 59.Qin BY, Liu C, Lam SS, Srinath H, Delston R, Correia JJ, Derynck R, Lin K. Crystal structure of IRF-3 reveals mechanism of autoinhibition and virus-induced phosphoactivation. Nat. Struct. Biol. 2003;10:913–921. doi: 10.1038/nsb1002. [DOI] [PubMed] [Google Scholar]
- 60.Qin BY, Liu C, Srinath H, Lam SS, Correia JJ, Derynck R, Lin K. Crystal structure of IRF-3 in complex with CBP. Structure. 2005;13:1269–1277. doi: 10.1016/j.str.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 61.Chen W, Lam SS, Srinath H, Jiang Z, Correia JJ, Schiffer CA, Fitzgerald KA, Lin K, Royer WE., Jr Insights into interferon regulatory factor activation from the crystal structure of dimeric IRF5. Nat. Struct. Mol. Biol. 2008;15:1213–1220. doi: 10.1038/nsmb.1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.