


Abstract
Transcription activator-like effectors (TALEs) have revolutionized the field of genome engineering. We present here a systematic assessment of TALE DNA recognition, using quantitative electrophoretic mobility shift assays and reporter gene activation assays. Within TALE proteins, tandem 34-amino acid repeats recognize one base pair each and direct sequence-specific DNA binding through repeat variable di-residues (RVDs). We found that RVD choice can affect affinity by four orders of magnitude, with the relative RVD contribution in the order NG > HD ∼ NN ≫ NI > NK. The NN repeat preferred the base G over A, whereas the NK repeat bound G with 103-fold lower affinity. We compared AvrBs3, a naturally occurring TALE that recognizes its target using some atypical RVD-base combinations, with a designed TALE that precisely matches ‘standard’ RVDs with the target bases. This comparison revealed unexpected differences in sensitivity to substitutions of the invariant 5′-T. Another surprising observation was that base mismatches at the 5′ end of the target site had more disruptive effects on affinity than those at the 3′ end, particularly in designed TALEs. These results provide evidence that TALE–DNA recognition exhibits a hitherto un-described polarity effect, in which the N-terminal repeats contribute more to affinity than C-terminal ones.
INTRODUCTION
Transcription activator-like effectors (TALEs) are sequence-specific DNA-binding proteins that the bacterial pathogen Xanthomonas injects into plant cells. Inside the plant cell, they bind to and activate specific host promoters (1). Their promoter specificity is conferred by a series of tandem protein repeats, typically 34 amino acids in length. Unlike any previously described DNA-binding domain, each repeat recognizes a single DNA base pair. Amino acids at positions 12 and 13, known as repeat variable di-residues (RVDs), determine the base preferences of a repeat. Deciphering the correspondence between RVD composition and target DNA bases created the ‘TALE DNA binding code’, making TALEs the first DNA-binding protein class for which robust and comprehensive rules of DNA recognition are known (2,3). Sequence-specific DNA binding is achieved by simple assembly of individual repeats with desired base specificities.
Recent crystallographic work revealed the structural basis for TALE–DNA recognition (4,5). Each repeat consists of two alpha helices connected by a three-residue loop that contains the RVDs (the ‘RVD loop’). Sequential repeats interact to form a solenoid that binds to one DNA strand, with the TALE N-terminal to C-terminal direction aligned with the DNA 5′ to 3′ direction. Position 13 contacts the target base in the major groove through hydrogen bonds or van der Waals interactions, while position 12 stabilizes the RVD loop structure. Thus, repeat sequence preferences are essentially determined by a single amino acid–base interaction.
Because of their modularity, easy programmability and reliability, TALE proteins have become the preferred DNA-binding domain to create artificial transcription factors (ATFs) and nucleases (TALENs), and have rapidly transformed the field of genome engineering (6–10). These properties also provide a special opportunity for synthetic biology, in which quantitative and predictable interactions of modular transcription factor and promoter parts are required to engineer gene regulatory circuits (11). However, few direct quantitative assessments of TALE–DNA affinities and specificities have been reported and, as of yet, there is no predictive framework in place. Extant data largely consist of cell-based transcription factor reporter assays in which the readout can be complicated by numerous factors in addition to TALE DNA-binding affinity. To understand and predict TALE repeat affinities, specificities and functionalities, quantitative binding data are required for members of this important new class of DNA-binding domain.
We present here the first systematic quantitative assessment of TALE DNA affinity and recognition. We combined quantitative gel shift and transcription reporter assays to explore the relative affinities of individual RVDs in vitro and their relation to activity in vivo. We also examined the specificity of two G-recognition RVDs, NN and NK, and the distribution of binding affinity over the length of the repeat region. Our data provide physical explanations and quantification of previously reported trends and suggest some complexities in this seemingly simple mode of DNA recognition. In particular, we demonstrate that the N-terminal TALE repeats interact more strongly with DNA than the C-terminal repeats, suggesting a polarity effect of TALE binding.
MATERIALS AND METHODS
Designed TALE construction
Designed TALE (dTALE) repeat arrays were modularly assembled using the Golden Gate cloning reagents described in (12), with slight modifications to the procedures. The 17.5-repeat arrays were assembled in two steps of cut-ligation reactions. The first reaction assembled two five-repeat arrays and one seven-repeat array. Each cut-ligation reaction used 75 ng of appropriate plasmids with BsaI-HF (New England Biolabs) and T4 ligase (New England Biolabs) that were incubated at 37°C for 5 h. On sequence verification, the three segments were assembled in a second cut-ligation reaction using a vector containing the last half-repeat to form a complete 17.5-repeat array (5 + 5 + 7 + 0.5 = 17.5). Final 17.5-repeat arrays were cloned by StuI/AatII digestion into pPreTALE111-42 and pPreTALE94-42, which contained truncated N- and C-termini of the naturally occurring TALE PthXo1 in pAH103 (5), generated by polymerase chain reaction (PCR) using primers listed in Supplementary Table S1. XhoI and AgeI sites incorporated at the termini allowed subcloning of entire dTALEs into expression vectors.
In this work, the RVD-containing repeat region is taken as starting at the beginning of the ‘0 repeat’ (residue 255 for AvrBs3, LTDGQ … ) and ending at the end of the complete ‘0.5 repeat’ (residue 897, … SRPDP). Thus, the 111-42 truncation refers to a variant that retains 111 N-terminal and 42 C-terminal residues from the full-length TALE, appended to the RVD repeat region (Supplementary Figure S1A).
Protein preparations
AvrBs3254-180 and dTALEs were cloned using BamHI/AgeI and XhoI/AgeI, respectively, into pMAL-TEV, a prokaryotic expression plasmid derived from pMAL-c5x (New England Biolabs) that contained a site for the Tobacco Etch Virus (TEV) protease. TALE reading frames were bounded by an N-terminal maltose-binding protein (MBP) tag, a TEV protease cleavage site and a His6 C-terminal His-tag (Supplementary Figure S1B). Tandem affinity purification allowed isolation of homogeneous full-length MBP-TALE–His6 fusion proteins (Supplementary Figure S2B). BL21 cells (Novagen) were transformed and grown overnight on Luria Broth agar containing 100 µg/ml carbenicillin. Single colonies were inoculated into 25 ml of Luria Broth containing 100 µg/ml carbenicillin, and grown with vigorous shaking at 37°C. At an OD600 of 0.4, incubation was continued at 30°C to an OD600 of 0.6–0.8. Isopropyl-β-D-thiogalactopyranoside (IPTG) (0.1 mM final) was added and the cultures were shaken at 30°C for 3–4 h. Cells were pelleted (10 min, 2000g) and stored at −80°C. Purification was carried out at 4°C, and all buffers contained 2 mM sodium azide. Cells were resuspended in 40 ml lysis/wash buffer (500 mM NaCl, 5 mM imidazole, 20 mM Tris-Cl, pH 7.9) and lysed using a microfluidizer (Microfluidics Corp., Model M100-Y). The resulting lysate, including washes (100 ml total), was clarified by centrifugation (40 min, 15 000g), and the supernatant was passed through a 2-ml column bed of Ni-IDA resin (2–3 ml/min, Novagen). The column was washed with 100 ml of lysis/wash buffer, 100 ml of high-salt wash buffer (2 M NaCl, 5 mM imidazole, 20 mM Tris-Cl, pH 7.9) to completely remove bound nucleic acids and another 100 ml of lysis/wash buffer. The MBP–TALE fusion proteins were eluted in five 2-ml fractions using His elution buffer (500 mM NaCl, 500 mM imidazole, 20 mM Tris-Cl, pH 7.9). Fractions containing more than 0.1 OD280 were passed through a 1-ml Luer lock syringe column containing 0.75 ml of amylose resin (New England Biolabs) (∼0.3 ml/min). The columns were then washed with 20 ml of TALE storage buffer (480 mM KCl, 1.6 mM ethylenediaminetetraacetic acid (EDTA), 2 mM dithiothreitol (DTT), 12 mM Tris-Cl, pH 7.5). The highly purified fusion protein was eluted in 0.5-ml aliquots with TALE storage buffer containing 10 mM maltose. The most concentrated fractions (1 to 4 OD280) were dialyzed against 2 × 300 ml of TALE storage buffer, quantified by ultraviolet absorbance and flash-frozen at −80°C in 50 -μL aliquots. The zinc finger DNA-binding domain of Zif268 (13) was subcloned into pMAL-TEV and purified as described for dTALEs, except buffers contained 100 µM ZnCl2. The molar extinction coefficients at 280 nm were 81820, 81820, 92820 and 69330 for the MBP-TALE111-42, MBP-TALE94-42, MBP-AvrBs3254-180 and MBP-Zif268 proteins, respectively (EXPASY). Typical final concentrations were 10–20 µM, with an overall yield of 1–5 mg protein (4–20 mg/l media). These proteins maintained binding activity for at least 1 week at 4°C. For cases in which the MBP tag was removed, quantitative cleavage was achieved by incubation of 10–20 µM dTALE with TEV proteinase (20 µg/ml final, a gift from Chris Fraser, UC Davis) and 5 mM DTT overnight at 4°C.
Electrophoretic mobility shift assay
Biotin-labeled DNA targets were generated by PCR amplification using a 5′ biotinylated forward primer of 69-mer oligonucleotides containing 19-base pair TALE target sites or the Zif268 site (Supplementary Table S2). PCR reactions contained unlabeled reverse primer in a 4:1 ratio over the biotinylated primer. Amplified targets were column purified (Qiagen). Binding reactions were mixed on ice and placed in the dark for 1 h at room temperature (22°C) in 1× TALE electrophoretic mobility shift assay (EMSA) buffer (12 mM Tris-Cl, pH 7.5, 60 mM KCl, 2 mM DTT, 0.05% NP-40, 50 ng/µL double-stranded poly (deoxyinosine-deoxycytosine)n (dIdC), 0.1 mg/ml bovine serum albumin (BSA), 5% glycerol, 5 mM MgCl2, 0.2 mM EDTA). As indicated, zinc finger binding reactions were performed in 1 x TALE EMSA buffer supplemented with 100 µM ZnCl2, or in Zinc Buffer A (ZBA: 100 mM Tris, 90 mM KCl, 1 mM MgCl2, and 90 µM ZnCl2, pH 7.5) with 5% glycerol, 0.1 mg/ml BSA, 0.05% NP-40. All binding reactions contained 25–55 pM target DNA, and purified proteins with a concentration of 0.1 – 2500 nM. After the room-temperature binding reaction, samples were placed at 4°C for 30 min. For all experiments, besides the ‘polarity’ assays, gel electrophoresis was performed on a 1.3% agarose gel using Amresco Biotechnology Grade Agarose I in 0.5× tris-borate-EDTA (TBE) buffer (Bio-Rad). Gels were pre-run at 105 V in 0.5× TBE buffer at 4°C for 30 min before loading. Binding reactions were loaded onto the gel while the current was on, and run for 20–30 min. Using a wet-transfer apparatus (Bio-Rad), the DNA was blotted onto a Biodyne B nylon membrane (Pierce) for 20 min at 100 V at 4°C. The DNA was cross-linked to the membrane with an ultraviolet cross-linker (Stratagene) for 4 min. The biotinylated DNA was visualized using the LightShift Chemiluminescent EMSA Kit (Pierce) according to the manufacturer’s protocol. Equilibrium binding constants (apparent KD) were calculated from protein titration experiments. Gel images on X-ray film (Denville Scientific) were scanned and then quantitated using ImageJ. All reported EMSA measurements were averages of at least three experiments performed with independent protein dilutions. For the ‘polarity’ gel shifts, the protocol was the same, except that tris-borate (TB) buffer was substituted for TBE buffer (in the gel and running buffer) at identical concentrations. Representative EMSA data are shown in Supplementary Figure S3.
ATF Assay
dTALEs were cloned using XhoI and AgeI into the phosphoglycerate kinase (PGK) promoter-driven mammalian expression vector pPGK-VP64 (14), which appended an N-terminal HA epitope tag and nuclear localization sequence, and a C-terminal VP64 transcriptional activation domain (15) (Supplementary Figure S1C). Target sites for the dTALEs were cloned between NotI and XhoI sites upstream of the SV40 promoter in pGL3-control plasmids (Promega), using primers listed in Supplementary Table S4. In 24-well plates, HEK293T cells at 80% confluency in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal calf serum, 1 U/ml of penicillin and 1 µg/ml of streptomycin were co-transfected with 100 ng of dTALE ATF expression plasmid, 25 ng of modified pGL3-control firefly luciferase reporter plasmid containing a dTALE target site and 25 ng of pRL-TK-Renilla Luciferase plasmid (as a transfection control, Promega), using Lipofectamine 2000 (Invitrogen). Cells were harvested 48 h post-transfection by removing media, washing with 500 µL of 1× Dulbecco's Phosphate-Buffered Saline (DPBS) and then followed by lysis in 100 µL of 1× passive lysis buffer (Promega) with 1× complete protease inhibitors (Roche). Clarified cell lysates (20 µL) were used to determine luciferase activity using DualGlo reagents (40 µL, Promega) in a Veritas microplate luminometer (Turner Biosystems). All experiments were performed in duplicate and repeated on two different days.
Binding site specificity assay using massively parallel sequencing (Bind-n-Seq)
Bind-n-Seq was performed essentially as described (13) using a full-length MBP–AvrBs3 fusion protein (AvrBs3254-267) as bait. AvrBs3254-267 was purified from induced cells using amylose affinity resin (New England Biolabs) according to manufacturer’s instructions. Bar-coded 93-mer double-stranded oligonucleotide targets containing Illumina primer binding sites and a 21-nt random region were incubated with 450 nM AvrBs3254-267 in 1× TALE EMSA buffer. Bound complexes were precipitated using amylose resin and enriched by six wash steps in the corresponding salt buffer. Eluted DNA was sequenced on an Illumina sequencer. Sequencing reads were filtered and sorted using custom Perl scripts found in the MERMADE package, an updated version of the Bind-n-Seq data analysis pipeline. MERMADE is freely available with user documentation at http://korflab.ucdavis.edu/Datasets/BindNSeq. Briefly, high-quality reads (composed only of A, C, T or G, with a valid constant region [‘AA’] and unique random region) were retained and split into separate files based on their unique 3-nt barcode (MERMADE scripts: sequence_converter.pl, debarcode.pl). For motif analysis, recovered sequences were analyzed relative to a file of unenriched background 21-mer sequences using a sliding window of 6-12 bp (MERMADE scripts: kmer_counter.pl, kmer_selector.pl). Sequences showing ≥2-fold enrichment relative to background were then analyzed by MERMADE using an iterative motif searching approach (MERMADE scripts: mermade.pl, motif_expander.pl). The graphical representation of the sequence motif was rendered using WebLogo.
RESULTS
Generating a scaffold
Natural TALE proteins consist of a series of DNA-binding repeats flanked by ∼250-residue N-terminal and C-terminal extensions that direct transcriptional regulation and protein localization (Figure 1A). To develop a biochemically tractable scaffold suitable for DNA affinity measurements, we truncated the N- and C-termini of a TALE composed of an AvrBs3 StuI/AatII fragment central core bounded by PthXol flanking sequences (Supplementary Figure S1A). Using Jpred secondary-structure predictions (17) to indicate ordered boundaries in the flanking sequences, we designed two N-terminal truncations containing 111 or 94 residues upstream of the 0 repeat and one C-terminal truncation containing 42 residues downstream of the terminal repeat, AvrBs3111-42 and AvrBs394-42, respectively (Figure 1A and Supplementary Figure S1A). Significantly, digestion of an MBP–AvrBs3 fusion protein with Factor Xa, a site-specific protease that also cleaves after Arg residues in unstructured regions, yielded a ∼77-kD fragment with full DNA-binding activity (Supplementary Figure S2A). Edman sequencing indicated that Factor Xa cleaved 114 residues N-terminal to the 0 repeat, whereas fragment size and Factor Xa arginine specificity suggested that the C-terminal flank was cleaved 37 or 39 residues after the terminal repeat. As a reference, we also produced a nearly full-length BamHI/AgeI fragment of the natural AvrBs3 (18), which contained 254 - and 180-residue native N- and C-terminal extensions, respectively (AvrBs3254-180). The proteins were expressed as fusions with an N-terminal MBP affinity tag and a TEV protease-cleavable linker, as well as a C-terminal His6 affinity tag. A two-column affinity purification scheme yielded milligram quantities of homogeneous, full-length, soluble MBP–His6 fusion proteins (Supplementary Figure S2B).
Figure 1.

Affinity and transcriptional activation data for several AvrBs3 variants. (A) Schematic of a TALE polypeptide showing the 18 RVD-containing repeats with N- and C-terminal flanking regions. The ‘0 repeat’ is shown in white. The numbers indicate the lengths of the N- and C-terminal extensions outside the repeat region used in the different constructs described in this work. A comprehensive survey of N- and C-terminal boundaries used in previous TALE studies is given in Supplementary Figure S1. (B) RVD amino acid composition of AvrBs3 (first row), along with the sequence of a natural DNA target, Bs3 (third row), and the consensus AvrBs3 site, UPA (2) (fourth row). The RVD composition of the dAvrBs3 variant, which contains only the standard NI, HD and NG RVDs and no mismatches to the Bs3 box target site, is also shown (second row). AvrBs3 RVDs that are ‘non-standard’ or mismatched to Bs3, and the corresponding RVDs in dAvrBs3, are underlined. The UPA site bases that differ from Bs3 are also underlined. (C) EMSA and ATF activation data were obtained as described in Materials and Methods. Target site sequences and RVD compositions are listed in Supplementary Tables S2 and S3. The affinity of Zif268 was measured in TALE 1× binding buffer. Zif268 affinity measured in a standard zinc-finger binding buffer (16) was more typical, 11 ± 4 nM.
EMSAs (Supplementary Figure S3) were performed with a DNA target, Bs3, which contained the 19-bp ‘Bs3 box’ bound by AvrBs3 (3) (Figure 1B, Supplementary Table S2). The presence of the MBP tag did not affect binding affinity, as its removal by TEV protease cleavage had no significant effect on apparent dissociation constant (KD) values (Figure 1C). AvrBs3254-180 and AvrBs3111-42 had nearly identical KD values for the Bs3 box site, 3-4 nM. In contrast, AvrBs394-42 bound Bs3 poorly, with a KD of 220 nM. To compare the functionality of the two scaffolds in cells, we developed an ATF reporter assay, in which a TALE–VP64 activation domain fusion protein drove expression of a luciferase reporter gene through an SV40 promoter with an upstream TALE target site. In agreement with the affinity data, the 111-42 framework showed a nearly 4-fold activation over background, whereas the 94-42 framework did not activate, with the promoter containing the Bs3 box target site (Figure 1C). Interestingly, AvrBs3254-180 produced ∼3-fold more gene activation than AvrBs3111-42. This result was unexpected because in vitro AvrBs3111-42 bound as well as AvrBs3254-180. Xanthomonas-delivered AvrBs3 has been shown to activate multiple host plant promoter sequences. Alignment of these sequences resulted in the identification of an AvrBs3 consensus target sequence known as the UPA box (2) (Figure 1B, Supplementary Table S3). We inserted the UPA box in place of the Bs3 box and repeated the ATF assay (Figure 1C). Interestingly, all three proteins performed better on the UPA box as compared with the Bs3 box-containing promoter. However, unlike with the Bs3 box, the 254-180 and 111-42 frameworks produced a similar 13-fold activation. Again, the 94-42 framework yielded 3 - to 4-fold lower activation, indicating that >94 N-terminal flanking residues are required for high-affinity binding comparable with AvrBs3254-180. The 111-42 framework was used for all subsequent experiments.
Using dTALEs to interrogate RVD relative affinities
To compare the binding affinities of the five RVDs that have been widely used to program dTALE specificities (HD, NI, NG, NN and NK), we used a ‘host-guest’ design in which 10 ‘guest’ positions containing the RVDs to be tested were interspersed with eight constant ‘host’ RVDs in a largely alternating pattern. The base 5′ to the target site was kept constant as T. Host contexts I – III (Table 1) sampled all base-step and base-triple combinations to examine potential context effects. Importantly, this setup avoided the structural peculiarities of homopolymeric runs in the target DNA sequences. Because the host repeats remained constant in each of the three contexts, we reasoned that their contribution to binding could be accounted for, allowing us to make direct comparisons based on only the guest repeat identity.
Table 1.
RVD composition, DNA target sites and affinity and transcriptional activation data for a series of 15 dTALEs
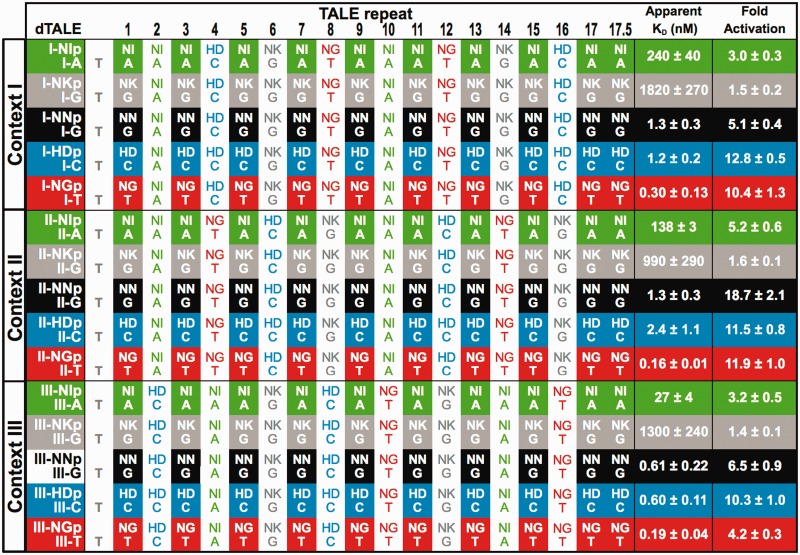 |
Three host contexts, I, II and III, with 10 guest RVDs indicated (shaded). The RVD types are color-coded (NI, green; NK, gray; NN, black; HD, blue; and NG, red). Host RVD compositions are the same but differently ordered in the three contexts. The DNA target sequences match the corresponding RVD pattern given above them, including the invariant 5′-T base that is present in all targets. EMSA dissociation constants (KD values) and ATF fold activation measurements were made as described in ‘Materials and Methods’ section.
Fifteen dTALEs were constructed and matched with corresponding DNA targets (Table 1). dTALE I-NIp refers to a protein with host context I and NI RVD-containing repeats at the guest positions, whereas the cognate DNA target is referred to as I-A. dTALEs containing G-recognizing NN and NK guests were compared against identical G-containing target sites (e.g. I-G, II-G and III-G). The proteins were expressed in E. coli and purified to homogeneity (Supplementary Figure S2B). EMSA revealed that their apparent KDs spanned four orders of magnitude, from 160 pM to 1.8 µM (Figure 2A, Table 1). Several trends became immediately apparent. The repeat type was the largest factor in affinity differences. The dTALEs with NG, HD and NN guest RVDs bound their targets with high affinity (160 pM – 2.4 nM). The strongest affinities were for the three NG guest proteins, whereas III-NNp and III-HDp also had picomolar affinity. The NK guest dTALEs bound least well in all three contexts. Although consistently better than NK, the NI guest dTALEs also bound poorly but with more variation. I-NIp had a KD of 240 nM, but III-NIp bound with a KD of 27 nM. This 9-fold difference in affinity, despite the two proteins sharing the same overall distribution of repeat types, clearly demonstrates the potential for significant contextual effects. Excluding the NK-containing proteins, context III was the most favorable setting for NI, NN and HD proteins, with a 3 - to 9-fold advantage over contexts I and II. Taken in whole, the gel shift data suggested that the relative affinities of individual repeats can be ordered as NG > HD ∼ NN ≫ NI > NK.
Figure 2.

Affinity and transcriptional activation data of 15 dTALEs. Comparison of (A) EMSA affinity constants for 15 dTALEs for their cognate DNA targets (KA = 1/KD, vertical axis, logarithmic scale), and (B) fold activation in an ATF assay. Guest RVD types (horizontal axis) and host contexts (I, white; II, gray; III, black bars) are indicated. Data were taken from Table 1. (C) Comparison of NN RVD interaction with G and A. EMSA affinities (left panel) and ATF fold activation (right panel) are shown for NN RVD TALE proteins with corresponding G and A DNA targets. Numerical data are given in Supplementary Table S4.
In the ATF assay, reporter gene activation by the dTALE series ranged from 1.4 - to 19-fold (Figure 2B, Table 1). All three HD guest proteins were strong activators, with levels at least 10-fold over background. As predicted from the binding data, the three NK guest proteins were the poorest activators. The correlation of affinity to activation was not linear. A simple log-log model produced a reasonable fit of the data (R2 = 0.68, P = 0.0002, Supplementary Figure S4), but several dTALEs displayed considerable deviation. For example, the tight-binding III-NGp produced relatively low activation (4-fold) compared with the two other NG guest proteins (>10-fold activation levels). Conversely, moderate-binding II-NNp showed the highest activation of the entire set (19-fold), higher than the other NN guests. There was an apparent demarcation in activation between 2.4 nM and 27 nM affinities (P = 2 × 10−12, based on a comparison of 60 individual non-averaged fold activation values in the two categories using a two-tailed heteroscedastic Student t-test). The six dTALEs with apparent KD ≥ 27 nM had an average fold activation of 2.7 fold, whereas those with KD ≤ 2.4 nM had an average fold activation of 10.2. Considering fold activation alone, the effectiveness of the five repeat types studied here would be HD ≥ NG ≥ NN ≫ NI > NK.
The NN RVD prefers G over A
According to correlations of natural TALE RVDs and their target sites (2,3), the NN repeat has a similar preference for G and A, and there are cases where this has been shown in dTALEs (2,19). However, other reports (8,20) have shown instances where NN displays an apparent preference for G. We compared NN repeat binding to G and A by measuring I-NNp, II-NNp and III-NNp affinities to the corresponding hosts containing G or A guests (Figure 2C and Supplementary Table S4). When the NN guest repeats were paired with A rather than G in contexts I and II, binding was reduced 49 - and 41-fold, respectively. In context III, the reduction was less severe, but was still >17-fold. The same trend was apparent in the ATF assay, with an 8-fold reduction in activation in context II, and reduction to background levels in contexts I and III. These data indicate that NN RVDs prefer binding G over A, although on a per-repeat basis, the binding energy differences are relatively small (see Discussion).
Natural and designed versions of AvrBs3 differ in their requirement for a 5′T
Most naturally occurring TALEs bind to DNA sequences beginning with a T (2,3), but dTALEs have been reported to recognize targets that have bases other than T in the 5′ position (8,21) and in one case, the 5′-T requirement was dependent on the N-terminal extension length (21). One major difference between natural TALEs and dTALEs produced by available assembly kits is that the artificial proteins (22,23) are predominantly constructed using just the HD, NG, NI, NN and NK RVDs. In contrast, most natural TALEs contain one or more ‘non-standard’ RVDs, such as NS, N* (residue 13 deleted), HG and others (1,2). In addition, ‘mismatches’ between RVDs and their consensus bases are rather typical. In fact, there has not been a single documented case of a naturally occurring TALE and a plant target promoter with a perfect code-predicted match. For example, AvrBs3, when bound to its Bs3 box target, has two HD-A mismatches and an NG-C mismatch. Additionally, AvrBs3 contains three ‘non-standard’ NS RVDs.
To assess the influence of the non-standard and mismatched RVDs in AvrBs3, we used the TALE code to create dAvrBs3111-42, composed of standard RVDs that match perfectly to the Bs3 box (Figure 1B). Both dAvrBs3111-42 and AvrBs3111-42 performed similarly in EMSA and ATF reporter assays (Figure 1C), suggesting that the non-standard RVDs did not confer an obvious binding advantage to AvrBs3111-42. Surprisingly, the two proteins displayed markedly different behavior against target sites substituted at the 5′-T (Figure 3 and Supplementary Table S5). Substitution with A, C or G reduced AvrBs3111-42 binding affinity by 13 - to 20-fold (Figure 3A), and reduced ATF reporter activity to background levels (Figure 3B). Thus, for AvrBs3111-42, a 5′-T is essential. In contrast, 5′ A, C or G reduced affinity for dAvrBs3111-42 by only 2 - to 3-fold, and activated gene expression only slightly less than with a 5′-T. These data suggest that the non-standard and/or mismatched repeats in naturally occurring proteins, which are generally not included in engineered dTALEs, may play an important role in binding specificity.
Figure 3.

Natural and designed TALEs show differential dependence on 5′-T. The 5′ base of the Bs3 box target site affects the (A) affinity (KA = 1/KD, linear scale) and (B) fold activation of the natural AvrBs3111-42 (black bars) more dramatically than for a designed dAvrBs3111-42 (white bars) (see Figure 1B for RVD compositions).
TALE proteins display a binding polarity, favoring the target sequence 5′ end
The N- to C-terminal directionality of the TALE–DNA interaction gives rise to the question of whether DNA affinity and specificity are evenly distributed over the length of the repeats, or concentrated in particular regions. We used the Bind-n-Seq assay (13) to probe the sequence binding preferences of AvrBs3254-267. Bind-n-Seq is an in vitro target site selection assay that presents proteins with a 21-bp randomized DNA library, and bound oligonucleotides are analyzed by high-throughput sequencing. DNAs bound to AvrBs3254-267 were enriched for bases on the 5′ end of the consensus UPA target (Figure 4A, top). In contrast, no enrichment was seen for the specified bases at the 3′ end. To ensure the apparent enrichment was not an artifact of the motif-finding method, a sliding window was used to identify 6-mer segments of the Bs3 target site in the sequencing reads from the AvrBs3254-267-selected library. Again, the library was enriched with only 6-mers from the 5′ end of the binding site but not the 3′ end (Figure 4A, bottom). These data suggested that N-terminal repeats might contribute more to the overall affinity than C-terminal repeats.
Figure 4.

Polarity effects of truncating substitutions at the 5′ and 3′ ends of the target site. (A) The most frequent binding motif produced by the Bind-n-Seq target site selection assay for AvrBs3254-267 is aligned below the expected UPA target site. The graph below the motif shows the enrichment of 6-mers corresponding to the 5′ end of the target site, but no enrichment of 3′ 6-mers, in the protein-bound DNA pool. (B) EMSA target sets used to test truncations (underlined) of the Bs3 box binding site are shown using IUPAC nomenclature. Target sets for the III-HDp and III-NGp are provided in Supplementary Table S2. (C, D, F and H) EMSA data are expressed as a percentage of affinity retained, compared with the 19-bp substrate, when the indicated protein binds the corresponding site with the indicated number (horizontal axis) of either 5′ (blue) or 3′ (yellow) truncated bases. (E, G and I) ATF reporter assay data are expressed as fold activation when the indicated protein binds the indicated truncated site. The activation level using the wild-type target site is shown (dashed line). Numerical data are provided in Supplementary Table S6.
To explore this bias quantitatively, we tested dTALE binding on a series of target sites shortened on either the 5′ or 3′ ends by substitutions of three, six and nine terminal bases (Figure 4B). To avoid biases inherent in making specific replacements, a random mixture of the three non-target bases replaced the target bases at each substituted position. For example, if a binding site position was T, the modified target site would contain an evenly distributed mixture of A, G or C. In our naming convention, target variant ‘3m3’ has three bases on the 3′ end randomized, whereas ‘5m6’ has six bases randomized on the 5′ end. An invariant 5′-T was maintained, reasoning that changing this base might unfairly bias the outcome because of its known importance. We first examined two high-affinity proteins, III-HDp and III-NGp (Figure 4C and D and Supplementary Table S6). Substitutions at the 5′ end of the target site reduced affinity considerably more than the 3′ substitutions. In the most dramatic case of III-NGp, substitutions of three or six 3′ bases had essentially no effect, whereas substitutions of the first three or six bases after the 5′-T reduced binding 15-fold and 370-fold, respectively. Put another way, mismatches of the first three, six and nine bases reduced affinity 15 -, 180 - and 150-fold more, respectively, than equivalent mismatches at the 3′ end. Importantly, III-NGp still bound tightly to a site with nine 3′ mismatches (KD = 2.5 nM). III-HDp showed a similar trend.
Interestingly, the polarity effect for AvrBs3111-42 was much smaller than for III-HDp or III-NGp. Truncating substitutions at either the 3′ and 5′ ends caused strong reductions in affinity (Figure 4F), indicating that binding affinity and/or specificity were more equally distributed across the repeats. Nonetheless, the 5′-end mismatches had greater effect (1.3 - to 3.1-fold). In contrast, as with the 5′-T preferences, dAvrBs3111-42 differed from AvrBs3111-42 in displaying marked polarity effects (Figure 4H). Mismatches at the 5′ end reduced binding 26-fold more than at the 3′ end, but substitutions beyond the first 3′ and 5′ triplets had little additional effect.
To determine if binding polarity is corroborated by in vivo activation, we tested III-NGp, AvrBs3111-42, and dAvrBs3111-42 against mutated target sites in the ATF reporter assay. Unlike the EMSA experiments, the activation assay required a specific target sequence for each protein. The base least represented in the TALE binding code for the particular RVD was used (Supplementary Table S3). For AvrBs3111-42 and dAvrBs3111-42, no activation was observed for any of the truncated sites (Figures 4E, G and I). This was not surprising, because the in vitro dissociation constants were higher than 46 nM and, as described previously, KD values ≥27 nM generally correlated to weak or no reporter gene activation. In contrast, III-NGp showed little reduction or increased activation levels for the three- and six-base truncations on the 3′ end, but similar truncations on the 5′ end dramatically reduced activity (Figure 4E).
DISCUSSION
The discovery of the TALE DNA-binding code was one of the most exciting recent developments in the field of engineered DNA-binding proteins. The modular nature of TALE specificity, the accessibility of materials and protocols for assembly and their comparatively robust programmability are significant advantages over other technologies such as zinc fingers or meganucleases (24). TALE technology was rapidly incorporated into designed site-specific nucleases, transcription factors and recombinases (6–10). Although the targeting activities are generally reliable, efficiencies vary widely and are near background in 10–15% of the cases (23). In one case, a single base change in the TALE-target pair can elicit 5-fold changes in activity (25). Plausible reasons include low protein expression, inefficient dTALE folding, target DNA modifications, chromatin structure variations and the affinity of the dTALE for its target (23). However, cell-based measurements make it difficult to disentangle the intrinsic properties of dTALE-DNA interactions from the multitude of other influences. Here, we directly quantified dTALE DNA affinity with purified proteins and well-defined target substrates. In parallel, we assessed dTALE activity in vivo using an ATF reporter assay.
A central conclusion of our work is that repeat composition significantly influences affinity. The NG RVD contributed most strongly. NN and HD repeats were also strong binders, whereas NI and NK RVDs contributed much less to affinity than the other three repeat types. Overall, based on the averaged affinities in the three host contexts, the relative contributions are NG (1) > NN (0.18) ∼ HD (0.16) ≫ NI (0.0016) > NK (0.00016). On a per-repeat basis, the free-energy differences vary from 0.4 to 2.2 kJ/mol, relative to the NG repeat. These values, in conjunction with the affinities, suggest that the average binding contribution for a single repeat is also small, 1 – 4 kJ/mol. However, the modest correlation between dTALE DNA affinity and ATF activity supports the idea that cell-based assays are complex and likely dependent on several factors, of which affinity is only one. For example, the synthetic DNA targets and plasmids used in the activation assay may contain cryptic target sites or sequences that influence binding and/or promoter activity. Nonetheless, our quantitative studies should provide a good baseline for building a better model of transcriptional responses.
Our results indicating that the NG RVD confers the highest affinity are in disagreement with conclusions by Streubel et al. (20), who suggested that NG-containing repeats are ‘weak’. A possible explanation for this discrepancy could be the use of uninterrupted runs of six identical repeats in their assays. Transcriptional activation was reduced concomitant with increasing numbers of adjacent NG and NI repeats, and for most other RVDs, a run of six had the lowest activations [Supplementary Data from reference (20)]. When just two repeats were used, the relative dTALE efficacies correlated more closely with our DNA-binding data, NG being the strongest.
Our dTALEs containing NN guest RVDs had a marked preference for G over A guest bases. The TALE DNA-binding code suggests that NN RVDs are paired just as often with A as G (2,3). Cell-based assays suggest that NN RVDs tolerate A bases but prefer G (19,20), and Systematic Evolution of Ligands by Exponential Enrichment (SELEX) results (8) further support this G preference, although A is preferred in some contexts. However, the energetic difference for individual repeats appears to be small. As 10 guest repeats in the three host contexts, NN RVD dTALEs discriminated G from A substrates by 17- to 49-fold, but the average discrimination per repeat is on the order of one kilojoule per mole, a factor of 2-3 in equilibrium constant. Further, NN RVD dTALEs bound A substrates more tightly than the corresponding ‘A-specific’ NI RVD dTALEs, 2.6- to 3.8-fold, suggesting that ‘off-target’ A recognition by NN RVDs is a significant concern.
Another clear result was the poor performance of the NK repeat. The NK guest proteins displayed the lowest affinities and activation levels of all the dTALEs. Our data provide a biochemical rationale for the low activities of NK-rich dTALEs in cell-based studies (19,20,26,27), strongly indicating that NN is a better choice than NK for G recognition. Recently, the novel NH RVD has shown promise for superior G specificity (19,20), although quantitative affinity measurements have yet to be reported.
The weak affinity of NI repeats for A agrees with the activation data of others (20,25), although the quantity and density of NI RVDs did not always correlate with activation levels (25). In at least one context, NI apparently encoded G specificity (8). Thus, it seems that the contribution of NI repeats may vary more by context than other RVDs. Given its higher affinity, the NN RVD might be a better choice for A recognition in cases where G discrimination is not critical.
What are the structural rationales for the different repeat affinity contributions? In the crystal structures (4,5,28), only the second RVD residue (position 13) contacts the recognition base in the major groove. Tight binding specified by NN and HD RVDs can be rationalized by the direct H-bonds from Asn13 and Asp13 to major groove base atoms. The NN preference for G over A might be due to subtle differences between the Asn-amide-purine-N7 H-bonds. Perhaps these differences could be modulated by context, leading to the occasional NN preference for A over G (8).
The high affinity of NG repeats for T reveals something unexpected about specificity. Crystal structures (4,5,28) depict the thymine 5-methyl contacting Gly13 alpha carbon through a van der Waals interaction, whereas the non-glycine residues of other RVDs would be expected to clash with this base. The RVD comparisons here suggest that the NG RVD not only provides a void for the 5-methyl, but the T 5-methyl-Gly alpha carbon interaction actually enhances binding over NN and HD repeats. Although this van der Waals interaction may be more favorable than the NN or HD hydrogen bonds, another idea is that other TALE repeat residues flanking the RVD provide the binding energy rather than the RVD itself. A prime candidate is adjacent Gln 17, the only repeat residue that directly contacts the phosphate backbone. In this light, the phosphate contact mediates DNA affinity, modulated by the RVD loop. Cognate interactions would fix and orient the loop position favorably for the Gln-phosphate hydrogen bond, whereas non-cognate interactions exert a disruptive effect.
A significant novel insight of this study is the clear affinity bias of TALEs for the 5′-end of the target sequence. The dTALEs dAvrBs3111-42, III-HDp and III-NGp bound targets with blocks of 3′ substitutions 10 - to 180-fold better than targets with analogous 5′ substitutions, sometimes retaining sufficient affinity for potent activity. For example, III-NGp bound a target with just 10 matching 5′-end repeats with a KD of 2.5 nM (Supplementary Table S6). As such relatively short 10-mer sites might occur thousands of times in a eukaryotic genome, this polarized promiscuity indicates large potential for off-target events. This result suggests that the 5′ end of the dTALE target sequences should be selected for their uniqueness in particular.
In contrast to dTALEs, the differences between 5′ and 3′ target modifications in the natural TALE AvrBs3111-42 were small, ∼1- to 3-fold (Figure 4F, Supplementary Table S6). Furthermore, unlike the dTALEs described here, AvrBs3 variants with substituted or additional repeats at the C-terminus showed high specificity for the cognate 3′ target modifications in cell-based reporter assays (7). Why is this? One reason may be the asymmetric base composition of the target site. The 3′ side, rich in T and C, might be expected to contribute more to affinity than the A- and T-rich 5′ side, with the greater relative effect of 3′-side substitutions offsetting the polarity effect. However, this explanation does not rationalize the polarized behavior of dAvrBs3111-42, which is designed to recognize the same target. Even more curiously, AvrBs3111-42 was much more sensitive to substitutions of the invariant 5′-T than dAvrBs3111-42 (13 - to 20-fold vs 2 - to 3-fold, respectively), but dAvrBs3111-42 was much more sensitive to the 5m3 modification than AvrBs3111-42 (345-fold vs 29-fold, respectively, Supplementary Table S6). These results may suggest that natural features like the non-standard RVDs and non-canonical RVD-base alignments play a yet-to-be-understood role in distributing affinity contributions more evenly over the target site (see later in the text).
The polarization of the TALE–DNA interaction was not apparent in earlier SELEX studies (8). Perhaps, the SELEX procedure was optimized to probe specificity rather than relative affinities of different positions, which would require collecting sequence data at every SELEX cycle (29). In contrast, the single enrichment cycle used in Bind-n-Seq is ideal for revealing relative affinity contributions of different RVD positions.
DNA binding by dTALEs was sensitive to the N-terminal and C-terminal scaffold boundaries. The 111-42 framework is stable enough to allow production of soluble dTALEs in milligram amounts and provided binding and activation behavior comparable with AvrBs3254-180. Using the dHAX3/DNA co-crystal structure (4) as a guide, TALE–DNA contacts involve at least 27 C-terminal residues. However, another AvrBs3 deletion variant retaining only 20 C-terminal residues exhibited full DNA-binding activity (data not shown), and a TALEN construct containing just two C-terminal residues was fully functional (30). Thus, the C-terminus seems tolerant to deletions.
The N-terminal extension seems more critical. The PthXol-DNA (3UGM) crystal structure depicts one additional repeat-like module, the ‘-1 repeat’, contacting the conserved 5′-T, but no well-ordered structures or DNA contacts are observed N-terminal to that (5). Nonetheless, a 111-residue N-terminal extension was required for full DNA-binding and ATF activity, whereas 94 residues reduced DNA binding by 50-fold. Miller et al. (8) used ‘optimized’ TALENs with a 102-residue extension, suggesting that just eight extra residues confer optimal DNA binding. What is the function of the N-terminal extension? Secondary structure predictions and proteolysis by Factor Xa, which cleaves arginine residues in unstructured peptides, suggest that this region is a boundary between ordered and disordered segments. Comprising potentially one or two additional 34-residue repeats, the N-terminus may be an organizing center for DNA binding. One idea is that this region, including the -1 repeat, forms a folding unit that contacts the 5′-T (5). This productive DNA-binding interaction then initiates nucleation of the repeat superhelical filament that wraps around the target. Recent work by Gao et al. (31) provides support for this idea by demonstrating that the TALE N-terminal region 148-288 autonomously binds DNA non-specifically.
Our finding that the 5′-repeat interactions contribute more to DNA affinity is consistent with the model of an N-terminal organizing center. If the N-terminus serves as an anchor, then the diminishing contribution of the more distant repeats could result from registration mismatch between the repeat and DNA helices. Variations in helical pitch and geometries of adjacent-repeat and DNA base-step transitions would compound with increasing distance from the 5′-T, de-phasing the protein–DNA interaction and degrading the quality of the contacts. Indeed, in the PthXol-DNA structure, the last three RVD repeats do not contact the DNA, even though the DNA sites are available for binding.
Extending this idea further, the local geometry of particular DNA sequences might be more or less compatible with the TALE superhelix geometry, leading to an additional level of TALE DNA discrimination through indirect readout. Runs of the same nucleotides, in particular polydA/polydT (32,33), have characteristic helical parameters and deformability that differ from those for ‘average’ B-DNA. Registration mismatches could explain the dramatic reductions in activation with increasing run lengths described previously (20). We attempted to minimize homopolymeric runs in our host-guest system out of concern about this possibility. The insensitivity of III-NGp binding to the 3m3 target truncation may be a manifestation of this phenomenon, as the last four target residues are T. This idea finds structural support in the dHAX3/DNA co-crystal structure (3VPT), which contains a T3 run. While, in the first NG-T interaction, the T 5-Methyl and Gly13 C-alpha contact each other closely, 3.7 Å, either of the subsequent NG repeats take on unusual RVD loop conformations that increase the 5-methyl-C-alpha separation by 2 Å, out of van der Waals contact distance. Perhaps these RVD conformational variations serve to re-phase the downstream contacts but sacrifice affinity to do so. Utilization of non-standard repeats like NS and N*, and non-canonical ‘mismatched’ RVD-base combinations may carry out the same function, by loosening local structural constraints to realign the TAL and DNA helices. Taken in this light, it is perhaps not surprising that dAvrBs3111-42 and AvrBs3111-42 differ in their sensitivity to substitutions at the 5′ and 3′ ends. It may be that a number of apparent dTALE specificity anomalies may be rooted in the registration differences between TALEs and DNA.
The overall implication is that for some proportion of dTALEs, we do not yet have rules that reliably predict their interaction with their targets. However, from surveys of TALE efficacy, it should be possible to deduce rules for the more complex behavior. The discrepancies between DNA-binding affinity and transcriptional activation suggest that affinity is a necessary, but not sufficient, property for highly active synthetic dTALEs. High specificity for the target is another desirable trait. It may be that, as with zinc fingers (34), some dTALEs with very high affinities may prove to be problematic. Consider that III-NGp, which binds with low nanomolar affinity to targets containing only the first 10 of 19 bases, could occupy thousands of perfect 10-base-pair matches expected in a mammalian genome. Such an example underscores the potential for significant off-target activity by some dTALEs.
Overall, these first systematic biochemical measurements of TALE DNA affinities reveal the potential for both high affinity and high variability, as well as complexities underlying the TALE DNA-binding code. These measurements will also provide calibration for bioengineering of regulatory networks that are constructed using the TALE–DNA platform.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR online: Supplementary Tables 1–6 and Supplementary Figures 1–4.
FUNDING
National Institutes of Health (NIH) [GM097073 to D.J.S. and E.P.B.]. Funding for open access charge: NIH [GM097073].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We kindly thank Adam Bogdanove, Iowa State University, for the PthXo1 clone in pAH103, and the Chris Fraser Lab, MCB, UC Davis, for providing purified TEV protease. We thank Jose Zarasoga for assistance in protein production.
REFERENCES
- 1.Boch J, Bonas U. Xanthomonas AvrBs3 Family-Type III Effectors: Discovery and Function. Annu. Rev. Phytopathol. 2010;48:419–436. doi: 10.1146/annurev-phyto-080508-081936. [DOI] [PubMed] [Google Scholar]
- 2.Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, Lahaye T, Nickstadt A, Bonas U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326:1509–1512. doi: 10.1126/science.1178811. [DOI] [PubMed] [Google Scholar]
- 3.Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science. 2009;326:1501. doi: 10.1126/science.1178817. [DOI] [PubMed] [Google Scholar]
- 4.Deng D, Yan C, Pan X, Mahfouz M, Wang J, Zhu JK, Shi Y, Yan N. Structural basis for sequence-specific recognition of DNA by TAL effectors. Science. 2012;335:720–723. doi: 10.1126/science.1215670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mak AN, Bradley P, Cernadas RA, Bogdanove AJ, Stoddard BL. The crystal structure of TAL effector PthXo1 bound to its DNA target. Science. 2012;335:716–719. doi: 10.1126/science.1216211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, Bogdanove AJ, Voytas DF. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics. 2010;186:757–761. doi: 10.1534/genetics.110.120717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morbitzer R, Romer P, Boch J, Lahaye T. Regulation of selected genome loci using de novo-engineered transcription activator-like effector (TALE)-type transcription factors. Proc. Natl Acad. Sci. USA. 2010;107:21617–21622. doi: 10.1073/pnas.1013133107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, Meng X, Paschon DE, Leung E, Hinkley SJ, et al. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011;29:143–148. doi: 10.1038/nbt.1755. [DOI] [PubMed] [Google Scholar]
- 9.Mercer AC, Gaj T, Fuller RP, Barbas CF. Chimeric TALE recombinases with programmable DNA sequence specificity. Nucleic Acids Res. 2012;40:11163–11172. doi: 10.1093/nar/gks875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perez-Pinera P, Ousterout DG, Gersbach CA. Advances in targeted genome editing. Curr. Opin. Chem. Biol. 2012;16:268–277. doi: 10.1016/j.cbpa.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garg A, Lohmueller JJ, Silver PA, Armel TZ. Engineering synthetic TAL effectors with orthogonal target sites. Nucleic Acids Res. 2012;40:7584–7595. doi: 10.1093/nar/gks404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morbitzer R, Elsaesser J, Hausner J, Lahaye T. Assembly of custom TALE-type DNA binding domains by modular cloning. Nucleic Acids Res. 2011;39:5790–5799. doi: 10.1093/nar/gkr151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zykovich A, Korf I, Segal DJ. Bind-n-Seq: high-throughput analysis of in vitro protein-DNA interactions using massively parallel sequencing. Nucleic Acids Res. 2009;37:e151. doi: 10.1093/nar/gkp802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Szczepek M, Brondani V, Buchel J, Serrano L, Segal DJ, Cathomen T. Structure-based redesign of the dimerization interface reduces the toxicity of zinc-finger nucleases. Nat. Biotechnol. 2007;25:786–793. doi: 10.1038/nbt1317. [DOI] [PubMed] [Google Scholar]
- 15.Beerli RR, Segal DJ, Dreier B, Barbas CF. Toward controlling gene expression at will: specific regulation of the erbB-2/HER-2 promoter by using polydactyl zinc finger proteins constructed from modular building blocks. Proc. Natl Acad. Sci. USA. 1998;95:14628–14633. doi: 10.1073/pnas.95.25.14628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim MS, Stybayeva G, Lee JY, Revzin A, Segal DJ. A zinc finger protein array for the visual detection of specific DNA sequences for diagnostic applications. Nucleic acids research. 2011;39:e29. doi: 10.1093/nar/gkq1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cuff JA, Clamp ME, Siddiqui AS, Finlay M, Barton GJ. JPred: a consensus secondary structure prediction server. Bioinformatics. 1998;14:892–893. doi: 10.1093/bioinformatics/14.10.892. [DOI] [PubMed] [Google Scholar]
- 18.Schornack S, Meyer A, Romer P, Jordan T, Lahaye T. Gene-for-gene-mediated recognition of nuclear-targeted AvrBs3-like bacterial effector proteins. J. Plant Physiol. 2006;163:256–272. doi: 10.1016/j.jplph.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 19.Cong L, Zhou R, Kuo YC, Cunniff M, Zhang F. Comprehensive interrogation of natural TALE DNA-binding modules and transcriptional repressor domains. Nat. Commun. 2012;3:968. doi: 10.1038/ncomms1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Streubel J, Blucher C, Landgraf A, Boch J. TAL effector RVD specificities and efficiencies. Nat. Biotechnol. 2012;30:593–595. doi: 10.1038/nbt.2304. [DOI] [PubMed] [Google Scholar]
- 21.Sun N, Liang J, Abil Z, Zhao H. Optimized TAL effector nucleases (TALENs) for use in treatment of sickle cell disease. Mol. Biosyst. 2012;8:1255–1263. doi: 10.1039/c2mb05461b. [DOI] [PubMed] [Google Scholar]
- 22.Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, Baller JA, Somia NV, Bogdanove AJ, Voytas DF. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011;39:e82. doi: 10.1093/nar/gkr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reyon D, Tsai SQ, Khayter C, Foden JA, Sander JD, Joung JK. FLASH assembly of TALENs for high-throughput genome editing. Nat. Biotechnol. 2012;30: 460–465. doi: 10.1038/nbt.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Silva G, Poirot L, Galetto R, Smith J, Montoya G, Duchateau P, Paques F. Meganucleases and other tools for targeted genome engineering: perspectives and challenges for gene therapy. Curr. Gene Ther. 2011;11:11–27. doi: 10.2174/156652311794520111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang F, Cong L, Lodato S, Kosuri S, Church GM, Arlotta P. Efficient construction of sequence-specific TAL effectors for modulating mammalian transcription. Nat. Biotechnol. 2011;29:149–153. doi: 10.1038/nbt.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang P, Xiao A, Zhou M, Zhu Z, Lin S, Zhang B. Heritable gene targeting in zebrafish using customized TALENs. Nat. Biotechnol. 2011;29:699–700. doi: 10.1038/nbt.1939. [DOI] [PubMed] [Google Scholar]
- 27.Christian ML, Demorest ZL, Starker CG, Osborn MJ, Nyquist MD, Zhang Y, Carlson DF, Bradley P, Bogdanove AJ, Voytas DF. Targeting G with TAL Effectors: A Comparison of Activities of TALENs Constructed with NN and NK Repeat Variable Di-Residues. PloS One. 2012;7:e45383. doi: 10.1371/journal.pone.0045383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deng D, Yin P, Yan C, Pan X, Gong X, Qi S, Xie T, Mahfouz M, Zhu JK, Yan N, et al. Recognition of methylated DNA by TAL effectors. Cell Res. 2012;22:1502–1504. doi: 10.1038/cr.2012.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roulet E, Busso S, Camargo AA, Simpson AJ, Mermod N, Bucher P. High-throughput SELEX SAGE method for quantitative modeling of transcription-factor binding sites. Nat. Biotechnol. 2002;20:831–835. doi: 10.1038/nbt718. [DOI] [PubMed] [Google Scholar]
- 30.Mussolino C, Morbitzer R, Lutge F, Dannemann N, Lahaye T, Cathomen T. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res. 2011;39:9283–9293. doi: 10.1093/nar/gkr597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao H, Wu X, Chai J, Han Z. Crystal structure of a TALE protein reveals an extended N-terminal DNA binding region. Cell Res. 2012;22:1716–1720. doi: 10.1038/cr.2012.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nelson HC, Finch JT, Luisi BF, Klug A. The structure of an oligo(dA).oligo(dT) tract and its biological implications. Nature. 1987;330:221–226. doi: 10.1038/330221a0. [DOI] [PubMed] [Google Scholar]
- 33.Peck LJ, Wang JC. Sequence dependence of the helical repeat of DNA in solution. Nature. 1981;292:375–378. doi: 10.1038/292375a0. [DOI] [PubMed] [Google Scholar]
- 34.Pattanayak V, Ramirez CL, Joung JK, Liu DR. Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat. Methods. 2011;8:765–770. doi: 10.1038/nmeth.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.