

Abstract
Background
Genome-wide association studies (GWASs) have identified multiple genetic susceptibility loci for breast cancer. However, these loci explain only a small fraction of the heritability. Very few studies have evaluated copy number variation (CNV), another important source of human genetic variation, in relation to breast cancer risk.
Methods
We conducted a CNV GWAS in 2623 breast cancer patients and 1946 control subjects using data from Affymetrix SNP Array 6.0 (stage 1). We then replicated the most promising CNV using real-time quantitative polymerase chain reaction (qPCR) in an independent set of 4254 case patients and 4387 control subjects (stage 2). All subjects were recruited from population-based studies conducted among Chinese women in Shanghai.
Results
Of the 268 common CNVs (minor allele frequency ≥ 5%) investigated in stage 1, the strongest association was found for a common deletion in the APOBEC3 genes (P = 1.1×10−4) and was replicated in stage 2 (odds ratio =1.35, 95% confidence interval [CI] = 1.27 to 1.44; P = 9.6×10−22). Analyses of all samples from both stages using qPCR data produced odds ratios of 1.31 (95% CI = 1.21 to 1.42) for a one-copy deletion and 1.76 (95% CI = 1.57 to 1.97) for a two-copy deletion (P = 2.0×10−24).
Conclusions
We provide convincing evidence for a novel breast cancer locus at the APOBEC3 genes. This CNV is one of the strongest common genetic risk variants identified so far for breast cancer.
Breast cancer is one of the most common malignancies diagnosed among women worldwide, including those living in East Asian countries. Genetic factors play an important role in the etiology of both sporadic and familial breast cancer. Recent genome-wide association studies (GWASs) focusing on evaluating common single nucleotide polymorphisms (SNPs) have identified approximately 67 genetic susceptibility loci for breast cancer (1–14). However, the vast majority of risk variants identified to date have small effect sizes (per allele odds ratio [OR] < 1.20) and only explain a very small portion of the heritability (4).
Recent studies indicate that copy number variations (CNVs) occur frequently in the genome and are an important source of human genetic variation (15,16). It has been proposed that CNVs may explain some of the missing heritability for complex diseases after the findings from GWASs (17). CNVs may affect a wider spectrum of genomic sequences and are more likely to be causal variants compared with common SNPs (18). CNVs have been associated with several complex diseases, including HIV infection/AIDS (19), psoriasis (20), Crohn’s disease (21), and autism (22). With the exception of a common CNV in the NBPF23 gene associated with neuroblastoma risk, no other common CNVs have been convincingly identified in relation to cancer risk. Herein, we conducted a GWAS to search for common CNV markers for breast cancer risk using data from the Shanghai Breast Cancer Genetics Study.
Methods
Study Populations
Included in this project were 5792 case patients and 5830 control subjectss from the Shanghai Breast Cancer Genetics Study (Table 1). The study subjects were drawn from four population-based studies conducted in Shanghai—the Shanghai Breast Cancer Study (SBCS), Shanghai Women’s Health Study (SWHS), Shanghai Breast Cancer Survival Study (SBCSS), and the Shanghai Endometrial Cancer Study (SECS, contributed control data only). Detailed descriptions of participating studies have been published elsewhere (13). Demographic characteristics of study participants are provided in Table 1. In brief, the SBCS is a two-stage (SBCS-I and SBCS-II), population-based, case–control study. SBCS-I recruitment occurred between August 1996 and March 1998; SBCS-II recruitment occurred between April 2002 and February 2005. The SBCSS included newly diagnosed breast cancer case patients ascertained by the population-based Shanghai Cancer Registry between April 2002 and December 2006. The SECS is a population-based, case–control study of endometrial cancer conducted between January 1997 and December 2003 using a protocol similar to the SBCS; only community control subjects from the SECS were included in the present study. The SWHS is a population-based prospective cohort study of women from urban communities in Shanghai who were recruited between 1996 and 2000. The cohort has been followed by a combination of record linkage and active follow-ups. All these studies are conducted among Chinese women in Shanghai, a genetically homogenous population, using virtually identical protocols in data and sample collection. Genomic DNA for all included participants was extracted using commercial DNA purification kits. Written, informed consent was obtained from all participants before interview, and the study protocols have been approved by the institutional review boards of all institutions involved in the study.
Table 1.
Distribution of demographic characteristics and known breast cancer risk factors for case patients and control subjects included in the study*
| Category | Case patients (n = 5792) | Control subjects (n = 5830) |
|---|---|---|
| Source of study subjects, No. | ||
| SBCS | 2638 | 2707 |
| SWHS | 138 | 2261 |
| SBCSS/SECS | 3016 | 862 |
| Demographic factors | ||
| Age, y | 52.3±9.8 | 52.9±9.3 |
| Education ≥ middle school, % | 63.4 | 53.5 |
| Reproductive risk factors | ||
| Age at menarche, y | 14.4±1.7 | 14.8±1.8 |
| Postmenopausal, % | 46.9 | 49.6 |
| Age at menopause, y† | 49.1±4.4 | 48.5±4.3 |
| Age at first live birth, y‡ | 26.8±4.3 | 26.2±3.9 |
| Other risk factors | ||
| First-degree relative with breast cancer, % | 5.0 | 2.4 |
| Body mass index, kg/m2 | 23.95±3.45 | 23.69±3.38 |
| Waist-to-hip ratio | 0.83±0.06 | 0.81±0.06 |
* Unless otherwise specified, data are mean ± standard deviation. SBCS = Shanghai Breast Cancer Study; SBCSS/SECS = Shanghai Breast Cancer Survival Study/Shanghai Endometrial Cancer Study; SWHS = Shanghai Women’s Health Study.
† Among postmenopausal women.
‡ Among parous women.
Genotyping Methods and CNV Detection
Affymetrix SNP Array 6.0 Genotyping.
The genotyping protocol using the Affymetrix Genome-Wide Human SNP Array 6.0 (Affymetrix, Inc., Santa Clara, CA) has been described previously (13). We included one negative control and three positive quality control (QC) samples from Coriell Cell Repositories (http://ccr.coriell.org/) in each 96-well plate. CNVs were called based on the signal intensities of more than 1.8 million SNPs or copy number probes on the Affymetrix SNP 6.0 array. The Affymetrix Power Tools (APT-1.14.3) package was used for normalization for each 96-well plate. CNV calls were conducted using the Canary program in Birdsuite (version 1.4; http://www.broad.mit.edu/mpg/birdsuite/analysis.html) (23). This study only focused on the common CNVs previously identified in the HapMap project (24). Sample QC procedure based on SNP data has been described previously (13). We removed additional subjects who met any of the following criteria based on CNV data: 1) standard deviations (SDs) of log R ratio (LRR) greater than 0.3; 2) total number of CNV calls are greater than the 95th percentile. The three positive QC samples on each 96-well plate were used to verify the integrity of genotyping quality on each plate. CNVs with number of SNPs/probes less than six or CNVs with length less than 100 base pairs were excluded.
Real-Time Qualitative Polymerase Chain Reaction (qPCR).
Primers and probes highly specific to the target gene, APOBEC3B (assay ID: Hs04504055_cn), and the reference gene, RNase P, were purchased from Applied Biosystems (Foster City, CA). The Coriell DNA NA18635, which carried two copies of the APOBEC3B gene, was used as the calibrator. In our standard-curve assays, the slopes of the APOBEC3B and RNase P were −3.48 and −3.49, respectively, and the calculated amplification efficiencies were 93.8% and 93.4%, respectively. The ΔΔCt was calculated by the formula: (Ct reference genesample − Ct target genesample) − (Ct reference genecalibrator − Ct target genecalibrator) (25). If there were no PCR amplification for the APOBEC3B gene after 45 cycles, whereas the RNase P gene was successfully amplified, the ΔΔCt value could not be estimated, and these subjects were determined to carry a two-copy deletion of the APOBEC3B gene. When the ΔΔCt was estimated, the APOBEC3B gene was called a two-copy deletion (2X2ΔΔCt < 0.2), a one-copy deletion (0.8 < 2X2ΔΔCt < 1.2), and no deletion (2X2ΔΔCt > 1.8). If the 2X2ΔΔCt value was not within the above ranges, these subjects were repeated with triplicates scattered on another 384-well plate. All qPCR assays were performed by a single lab staff member (G. Li), and all CNV calls were conducted by two independent staff members. An equal number of breast cancer case patients and control subjects were included in each of the genotyping plates in both Affymetrix SNP Array 6.0 and qPCR assays.
Genotyping SNP rs12628403 and rs5750715 in Stage 2 Samples.
Of all SNPs analyzed using Affymetrix SNP Array 6.0, SNP rs5750715 showed the strongest linkage disequilibrium (LD) with the APOBEC3 deletion (r 2 = 0.50, the 1000 Genomes Project Asian data; and r 2 =0.44, this study). Based on the 1000 Genomes Project Asian data, the APOBEC3 deletion is in strong LD with SNP rs12628403 (r 2 =0.91). These two SNPs were genotyped in stage 2 samples using the iPLEX Sequenom MassArray platform in the Vanderbilt Molecular Epidemiology Laboratory. Included in each 96-well plate as QC samples were one negative control (water), two blinded duplicates, and two samples from the HapMap project. The mean concordance rate was 99.6% for the blind duplicates and 98.9% for HapMap samples.
Statistical Analyses
Associations between the CNV and breast cancer risk were assessed using odds ratios and 95% confidence intervals (CIs) derived from logistic regression models. Odds ratios were estimated for one-copy and two-copy deletion genotypes compared with no deletion genotype. The odds ratio was also estimated for per-copy deletion based on a log-additive model and adjusted for age and study stage (if applicable). Potential confounding by population structure was adjusted for using principal components estimated in EIGENSTRAT (26). Additional adjustment for education, menarche age, and body mass index were also performed, and the results did not change materially. Analyses stratified by menopausal status, estrogen receptor status, years of menstruation, and age group were carried out. The interaction between the CNV and breast cancer genetic risk variants identified in a previous GWAS (http://www.genome.gov/26525384) was also investigated. Multiplicative interactions between CNV and demographic variables or GWAS-identified SNPs were evaluated using the likelihood ratio test when interaction terms were added to logistic regression models. The population attributable risk PAR was estimated as follows (8):
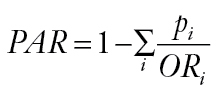 |
where p i represents the proportion of total cases in the population with the ith genotype, OR i is the odds ratio for the ith genotype. SAS 9.2 was used to conduct these analyses.
We conducted multiple additional analyses to evaluate potential batch effects in CNV calling in stage 1. We directly checked the association stratified by DNA source (blood or buccal cell) using the qPCR data. A linear discriminant function analysis, which has been shown robust to differential errors and noisy data in CNV studies (27), was conducted using the CNVtools package (version 1.48.0; http://www.bioconductor.org/packages/2.11/bioc/html/CNVtools.html) (27). The principal component analysis summarized intensity data from 24 probes on Affymetrix SNP 6.0 array was also tested in relation to breast cancer risk by using CNVTools package.
The 1000 Genomes Project 2011 October release phased data (http://www.sph.umich.edu/csg/abecasis/MACH/download/ 1000G.2012-02-14.html) were used as reference for imputation for a 4-Mb region centered on the APOBEC3 deletion in the stage 1 samples. We used the recommended two-step imputation approach and recommended parameters of 50 iterations of the Markov sampler and 200 states. First, MACH (http://www.sph.umich.edu/csg/abecasis/MACH/index.html) was used to estimate haplotypes for the GWAS data. Then, minimac (http://genome.sph.umich.edu/wiki/Minimac#Imputation_quality_evaluation) was used to impute missing genotypes for SNPs included in the 1000 Genomes Project but not in the GWAS. Mach2dat (http://genome.sph.umich.edu/wiki/Mach2dat:_Association_with_MACH_output) was used to conduct logistic association between the dosage data (imputation uncertainty taken into account) with breast cancer risk.
All statistical tests were two-sided and a P value less than.05 was considered statistically significant.
Results
In stage 1, among the 1320 CNVs previously identified in the HapMap project (24), 268 common CNVs with a minor allele frequency greater than or equal to 5% were observed. Among them, 18 were associated with breast cancer risk at P less than or equal to.05. Of them, CNP2576 (31.4% in control subjects) showed the strongest association (OR per-copy loss = 1.19, 95% CI = 1.09 to 1.31; P = 1.1×10−4) (Supplementary Table 1, available online). A histogram of the principal component summarized probe intensity showed a clear three-component CNV (Supplementary Figure 1, available online). This is very consistent with the CNV calls from Birdsuite with three genotypes, no deletion, one-copy deletion, and two-copy deletion. The linear discriminant function analyses based on the summarized probe intensity data using CNVTools also showed a statistically significant association with breast cancer risk. To verify the accuracy of CNV calls, we reanalyzed 2981 of 4569 samples included in initial stage 1 using qPCR and obtained very similar results as shown using Affymetrix SNP Array 6.0 data (Supplementary Table 1, available online). Association between the APOBEC3 gene deletion and breast cancer risk in stage 1 subjects was similar within the top five principal components adjusted for each subject (Supplementary Table 2, available online). To minimize the effect of assay method on our study results, only qPCR data were included in the final analyses (Table 2).
Table 2.
Association between the APOBEC3 gene deletion and breast cancer risk, results from the Shanghai Breast Cancer Genetics Study*
| Genotypes | No. of case patients | No. of control subjects | OR (95% CI)† | OR (95% CI)‡ |
|---|---|---|---|---|
| Per-copy deletion | 5792 | 5830 | 1.32 (1.25 to 1.40) | 1.33 (1.26 to 1.40) |
| No deletion | 2045 | 2530 | 1.00 (referent) | 1.00 (referent) |
| 1-copy deletion | 2805 | 2638 | 1.31 (1.21 to 1.42) | 1.31 (1.21 to 1.42) |
| 2-copy deletion | 942 | 662 | 1.76 (1.57 to 1.97) | 1.77 (1.58 to 1.99) |
| P trend | 2.0×10−24 | 3.0×10−24 |
* All subjects were genotyped by real-time quantitative polymerase chain reaction. CI = confidence interval; OR = odds ratio.
† Adjusted for age and study stage.
‡ Adjusted for age, study stage, education, body mass index, and age at menarche.
In stage 2 independent samples, a highly statistically significant association was again observed (OR =1.35, 95% CI = 1.27 to 1.44 per-copy loss; P = 9.6×10−22) (Supplementary Table 3, available online). Analyses of all samples from both stages using qPCR data produced odds ratios of 1.31 (95% CI = 1.21 to 1.42) for one-copy deletion and 1.76 (95% CI = 1.57 to 1.97) for two-copy deletion (P = 2.0×10−24) (Table 2), providing unequivocal evidence for an association of this deletion with breast cancer risk. This deletion was associated with a population attributable risk of 18.4% in our study population, larger than virtually any of the common genetic susceptibility variants identified to date. However, the population attributable risk may be much lower in populations of European or African ancestry that have a lower prevalence of this deletion. Additional adjustment for education, body mass index, and age at menarche did not change results appreciably (Table 2). Association of this CNV with breast cancer risk was similar when stratified by menopausal or estrogen receptor status, age group, and duration of menstruation. None of the heterogeneity tests were statistically significant (Table 3). No interaction was observed between this CNV and genetic risk variants identified in previous GWASs on the risk of breast cancer (Supplementary Table 4, available online). We also conducted stratification analyses based on DNA source (blood or buccal cell) within the whole study population or SBCS, which contributed the largest number of samples to this study, with both both blood and buccal cell samples collected. A consistent association between the CNV marker and breast cancer risk was observed regardless of the type of samples used (Supplementary Table 5, available online). Similarly, histogram plots of ΔΔCt value for all subjects included in this study indicated that there is no difference across the study origin (SBCS, SBCSS, SWHS, and SECS) and DNA sources (blood or buccal cell) (Supplementary Figure 2, available online). We also investigated the association between the CNV marker and breast cancer risk stratified by study origin. A statistically significant association was observed in all studies (Table 4).
Table 3.
Association of the APOBEC3 gene deletion and breast cancer stratified by breast cancer risk factors*
| Category | No. of case patients | No. of control subjects | OR (95% CI)† | P trend | P heterogeneity | |
|---|---|---|---|---|---|---|
| 1-copy deletion | 2-copy deletion | |||||
| Menopausal status | ||||||
| Premenopausal | 3074 | 2939 | 1.22 (1.09 to 1.36) | 1.68 (1.43 to 1.97) | 1.7×10−10 | .18 |
| Postmenopausal | 2718 | 2888 | 1.42 (1.27 to 1.59) | 1.84 (1.56 to 2.17) | 1.2×10−15 | |
| ER status | ||||||
| Positive | 3390 | 5830 | 1.29 (1.18 to 1.42) | 1.80 (1.58 to 2.06) | 1.9×10−19 | .99 |
| Negative | 1896 | 5830 | 1.33 (1.19 to 1.49) | 1.78 (1.52 to 2.09) | 1.3×10−13 | |
| Years of menstruation (by median) | ||||||
| >33 y | 2960 | 2702 | 1.36 (1.22 to 1.53) | 1.88 (1.59 to 2.22) | 1.7×10−15 | .16 |
| ≤33 y | 2827 | 3118 | 1.26 (1.12 to 1.40) | 1.62 (1.38 to 1.90) | 6.4×10−10 | |
| Age group, y | ||||||
| ≤40 | 407 | 331 | 1.11 (0.80 to 1.53) | 1.67 (1.06 to 2.63) | 4.4×10−2 | .39 |
| 41–50 | 2433 | 2265 | 1.29 (1.14 to 1.46) | 1.77 (1.48 to 2.13) | 1.1×10−10 | |
| 51–60 | 1677 | 1804 | 1.28 (1.11 to 1.48) | 1.74 (1.41 to 2.14) | 7.9×10−8 | |
| 61–70 | 883 | 1318 | 1.47 (1.22 to 1.77) | 1.61 (1.22 to 2.11) | 2.2×10−5 | |
| >70 | 392 | 112 | 1.48 (0.95 to 2.31) | 3.38 (1.52 to 7.54) | 1.8×10−3 | |
* Adjusted for age. CI = confidence interval; ER = estrogen receptor; OR = odds ratio.
† No deletion as referent. P values are two-sided. P trend are estimated through logistic regression. P heterogeneity are estimated using the likelihood ratio test
Table 4.
Association between the APOBEC3 gene deletion and breast cancer stratified by study
| No. of deletion | SBCS-I* (941 case patients/ 1,064 control subjects) | SBCS-II* (1,697 case patients/ 1,643 control subjects) | SWHS (138 case patients/ 138 control subjects) | SBCSS/SECS/SWHS† (3,016 case patients/ 2,985 control subjects) |
|---|---|---|---|---|
| No deletion | 1.00 (referent) | 1.00 (referent) | 1.00 (referent) | 1.00 (referent) |
| 1-copy deletion | 1.28 (1.06 to 1.56) | 1.18 (1.02 to 1.37) | 1.25 (0.74 to 2.11) | 1.42 (1.27 to 1.59) |
| 2-copy deletion | 1.52 (1.14 to 2.02) | 1.33 (1.07 to 1.66) | 3.30 (1.48 to 7.39) | 2.12 (1.80 to 2.49) |
| Per-copy deletion | 1.25 (1.09 to 1.43) | 1.16 (1.05 to 1.29) | 1.63 (1.14 to 2.34) | 1.44 (1.34 to 1.56) |
| P trend | 1.4×10−3 | 3.3×10−3 | 7.5×10−3 | 3.6×10−21 |
* SBCS is a two-stage (SBCS-I and SBCS-II) study. SBCS = Shanghai Breast Cancer Study; SBCSS = Shanghai Breast Cancer Survival Study; SECS = Shanghai Endometrial Cancer Study; SWHS = Shanghai Women’s Health Study.
† From SWHS, 2123 control subjects were selected to serve as control subjects for case patients in SBCSS.
Based on the 1000 Genomes Project Asian data, the APOBEC3 deletion is in strong LD with SNP rs12628403 (r 2 =0.91). A strong association was observed between this SNP with breast cancer risk (OR = 1.29, 95% CI = 1.14 to 1.45; P = 2.8×10−5) in stage 1 samples (Table 5). Such strong association was replicated in stage 2 samples (P = 7.8×10−6) (Table 4). Analyses of all samples from both stages yielded an odds ratio of 1.18 (95% CI = 1.12 to 1.25) with a P value of 2.9×10−9. SNP rs5750715 is in moderate LD with the APOBEC3 deletion (r 2 = 0.50) in 1000 Genomes Project Asian data. Again, a statistically significant association was observed between this SNP and breast cancer risk in both stages. These results provide an independent replication of our results for the CNV marker.
Table 5.
Association of single nucleotide polymorphisms (SNPs) rs5750715 and rs12628403 with breast cancer risk, results from the Shanghai Breast Cancer Genetics Study
| SNP* | Stage | No. of case patients | No. of control subjects | Per-allele OR (95% CI)† | P† |
|---|---|---|---|---|---|
| rs5750715 (T/A)‡ | Stage 1 | 1531 | 1441 | 1.16 (1.05 to 1.29) | 3.5×10−3 |
| Stage 2 | 4079 | 4211 | 1.11 (1.04 to 1.18) | 1.2×10−3 | |
| Combined | 5610 | 5652 | 1.12 (1.07 to 1.18) | 1.6×10−5 | |
| rs12628403 (A/C)§ | Stage 1 | 2918 | 2324 | 1.29 (1.14 to 1.45) | 2.8×10−5 |
| Stage 2 | 4096 | 4136 | 1.16 (1.09 to 1.23) | 7.8×10−6 | |
| Combined | 7014 | 6460 | 1.18 (1.12 to 1.25) | 2.9×10−9 |
* Reference/effect alleles based on forward strand. CI = confidence interval; OR = odds ratio.
† Adjusted for age and study stage (if applicable) using logistic test trend test. All P values are two-sided. No copy deletion was the reference group.
‡ rs5750715 in moderate linkage disequilibrium with the deletion (r 2 = 0.50).
§ SNP rs12628403 in strong linkage disequilibrium with the deletion (r 2 =0.91).
Discussion
The CNP2576, defined by 24 probes on the Affymetrix SNP 6.0 array (hg19, chromosome 22: 39363619–39375307), is located in the APOBEC3 gene cluster (Figure 1). It overlaps with a deletion that was first discovered by mapping end-sequence pairs from a human fosmid library against the human genome reference sequence (28). Recently, the 1000 Genomes Project (2011 October genotype data release) refined this APOBEC3 deletion to a 29936 base-pair fragment (hg19, 39358280–39388216).
Figure 1.

A regional plot of the –log10 P values for single nucleotide polymorphisms at flanking 500kb of the APOBEC3 deletion. The linkage disequilibrium is estimated using data from 1000 Genome Asian population (2011 October data release). Also shown are the SNP Build 37 coordinates in kilobases (kb) and genes in the region (below) based on the February 2009 UCSC genome browser assembly (http://genome.ucsc.edu/).
The APOBEC3 gene family encodes cytosine deaminases that have been implicated in innate cellular immunity against retroviruses (29). The APOBEC3 genes, as well as related cytosine deaminases, including activation-induced deaminase, have been shown to deaminate 5-methylcytosine and 5-hydroxymethylcytosine, with base excision repair of the resulting mismatch providing a mechanism for active DNA demethylation (29). Furthermore, the APOBEC3 genes may play a role in carcinogenesis by triggering DNA mutation (30). Activation-induced deaminase–mediated DNA double strand breaks have been linked to the generation of chromosomal translocations frequently observed in prostate cancer (31). Very recently, two studies highlighted the APOBEC genes’ mutagenesis function in cancer, including breast cancer. Nik-Zainal et al. (32) sequenced the complete genomes of 21 primary breast cancers and matched normal DNAs from the same individuals. A remarkable phenomenon of localized hypermutation, termed “kataegis,” and multiple mutation signatures were observed. The APOBEC family is proposed to play a role in this kataegis and/or in the mutational process. Also, Roberts et al. (33) reported that mutations in C- or G-coordinated clusters in human cancer often fell into motifs of APOBEC gene family, again indicating that APOBEC plays an important role in carcinogenesis.
The APOBEC3 deletion is located between the fifth exon of APOBEC3A and the eighth exon of APOBEC3B, resulting in complete elimination of the APOBEC3B gene-coding region. The resultant fusion gene has a protein sequence identical to APOBEC3A, but has a 3′ untranslated region of the APOBEC3B gene (34). The expression level of this fusion gene may differ from the undeleted APOBEC3A because of different stability of its RNA or different transcription levels (34). This deletion has been associated with decreased expression of the APOBEC3B gene in lymphoblastoid cell lines (35) and breast cancer cells (36).
The APOBEC3 gene families are expressed in most types of cells and tissues, including mammary epithelial cells. They are overexpressed in multiple cancer cell lines and cancer tissues, including breast cancer (37). Moreover, expression of the APOBEC3 genes is regulated by estrogen (38), a hormone that plays a central role in the etiology of breast cancer. Somatic APOBEC3 gene deletion has also been observed in breast and oral cancer tumor tissue (36). An earlier small case−control study of 50 case patients and 50 control subjects in Japan reported an elevated, although non-statistically significant, risk of breast cancer associated with homozygous deletion of this region (OR = 3.91, 95% CI = 0.77 to 19.83) (36). This deletion has been suggested to be associated with increased risk of other diseases or conditions, including HIV-1 infection and its progression to AIDS (39) and autism (40).
There are some limitations in this study. The study subjects were from multiple studies. However, all subjects were recruited in Shanghai, China, with almost identical recruitment instrument and protocol. In addition, the DNA was isolated from blood samples in some subjects and from buccal cells in other participants. However, the association between the APOBEC3 deletion and breast cancer risk was not affected by the study origin and DNA source. This deletion is common in East Asians but rare in European and African ancestry populations, with frequency of 6% and 0.9%, respectively (41). The statistical power is very limited to investigate the association of this deletion with breast cancer risk in these two populations. Research in the region identified in this study may be fruitful to discover risk variants for breast cancer in non-Asian populations.
In summary, in this large study conducted among Chinese women, we provide convincing evidence for an association with a common CNV located in the APOBEC3 gene cluster. CNVs are understudied compared with SNPs. This CNV is one of the strongest common genetic risk variants identified so far for breast cancer (GWAS catalog, http://www.genome.gov/26525384).
Funding
This work was supported in part by grants from the US National Institutes of Health grants (R01CA137013 to JL; R01CA148667, R01CA124558, R01CA064277, and R37CA070867 to WZ; R01CA118229 and R01CA092585 to X-OS; R01CA122756 to QC); Ingram Professorship and Research Reward funds (to WZ); and Department of Defense (DOD) Idea Awards (BC011118 to X-OS; BC050791 to QC). Sample preparation and genotyping assays were conducted at the Survey and Biospecimen Shared Resources and Vanderbilt Microarray Shared Resource, which are supported in part by the Vanderbilt-Ingram Cancer Center (P30 CA68485).
Supplementary Material
The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agents. The authors wish to thank the study participants and research staff for their contributions and commitment to this project; Regina Courtney for DNA preparation; Wanqing Wen, Bingshan Li, Jing He, and Dexter Duncan for data processing and analyses; and Bethanie Rammer for assistance in the preparation of this manuscript.
References
- 1. Ahmed S, Thomas G, Ghoussaini M, et al. Newly discovered breast cancer susceptibility loci on 3p24 and 17q23.2. Nat Genet. 2009; 41(5):585–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cai Q, Long J, Lu W, et al. Genome-wide association study identifies breast cancer risk variant at 10q21.2: results from the Asia Breast Cancer Consortium. Hum Mol Genet. 2011; 20(24):4991–4999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Easton DF, Pooley KA, Dunning AM, et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007; 447(7148):1087–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fletcher O, Houlston RS. Architecture of inherited susceptibility to common cancer. Nat Rev Cancer. 2010; 10(5):353–361 [DOI] [PubMed] [Google Scholar]
- 5. Gold B, Kirchhoff T, Stefanov S, et al. Genome-wide association study provides evidence for a breast cancer risk locus at 6q22.33. Proc Natl Acad Sci U S A. 2008; 105(11):4340–4345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hunter DJ, Kraft P, Jacobs KB, et al. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet. 2007; 39(7):870–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Long J, Cai QY, Sung H, et al. Genome-wide association study in East Asians identifies novel susceptibility loci for breast cancer. PLoS Genet. 2012; 8(2):e1002532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Long J, Cai Q, Shu XO, et al. Identification of a functional genetic variant at 16q12.1 for breast cancer risk: results from the Asia Breast Cancer Consortium. PLoS Genet. 2010; 6(6):e1001002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stacey SN, Manolescu A, Sulem P, et al. Common variants on chromosomes 2q35 and 16q12 confer susceptibility to estrogen receptor-positive breast cancer. Nat Genet. 2007; 39(7):865–869 [DOI] [PubMed] [Google Scholar]
- 10. Stacey SN, Manolescu A, Sulem P, et al. Common variants on chromosome 5p12 confer susceptibility to estrogen receptor-positive breast cancer. Nat Genet. 2008; 40(6):703–706 [DOI] [PubMed] [Google Scholar]
- 11. Thomas G, Jacobs KB, Kraft P, Yeager M, Wacholder S, Cox DG, et al. A multistage genome-wide association study in breast cancer identifies two new risk alleles at 1p11.2 and 14q24.1 (RAD51L1). Nat Genet. 2009; 41(5):579–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Turnbull C, Ahmed S, Morrison J, et al. Genome-wide association study identifies five new breast cancer susceptibility loci. Nat Genet. 2010; 42(6):504–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zheng W, Long J, Gao YT, et al. Genome-wide association study identifies a new breast cancer susceptibility locus at 6q25.1. Nat Genet. 2009; 41(3):324–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Michcailidou K, Hall P, Gonzalez-Neira A, et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat Genet. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sebat J, Lakshmi B, Troge J, et al. Large-scale copy number polymorphism in the human genome. Science. 2004; 305(5683):525–528 [DOI] [PubMed] [Google Scholar]
- 16. Iafrate AJ, Feuk L, Rivera MN, et al. Detection of large-scale variation in the human genome. Nat Genet. 2004; 36(9):949–951 [DOI] [PubMed] [Google Scholar]
- 17. Eichler EE, Flint J, Gibson G, et al. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet. 2010; 11(6):446–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Willer CJ, Speliotes EK, Loos RJ, et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. 2009; 41(1):25–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gonzalez E, Kulkarni H, Bolivar H, et al. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science. 2005; 307(5714):1434–1440 [DOI] [PubMed] [Google Scholar]
- 20. de Cid R, Riveira-Munoz E, Zeeuwen PL, et al. Deletion of the late cornified envelope LCE3B and LCE3C genes as a susceptibility factor for psoriasis. Nat Genet. 2009; 41(2):211–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McCarroll SA, Huett A, Kuballa P, et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s disease. Nat Genet. 2008; 40(9):1107–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Weiss LA, Shen Y, Korn JM, et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008; 358(7):667–675 [DOI] [PubMed] [Google Scholar]
- 23. Korn JM, Kuruvilla FG, McCarroll SA, et al. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat Genet. 2008; 40(10):1253–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McCarroll SA, Kuruvilla FG, Korn JM, et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet. 2008; 40(10):1166–1174 [DOI] [PubMed] [Google Scholar]
- 25. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25(4):402–408 [DOI] [PubMed] [Google Scholar]
- 26. Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006; 38(8):904–909 [DOI] [PubMed] [Google Scholar]
- 27. Barnes C, Plagnol V, Fitzgerald T, et al. A robust statistical method for case-control association testing with copy number variation. Nat Genet. 2008; 40(10):1245–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tuzun E, Sharp AJ, Bailey JA, et al. Fine-scale structural variation of the human genome. Nat Genet. 2005; 37(7):727–732 [DOI] [PubMed] [Google Scholar]
- 29. Conticello SG. The AID/APOBEC family of nucleic acid mutators. Genome Biol. 2008; 9(6):229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Suspene R, Aynaud MM, Guetard D, et al. Somatic hypermutation of human mitochondrial and nuclear DNA by APOBEC3 cytidine deaminases, a pathway for DNA catabolism. Proc Natl Acad Sci U S A. 2011; 108(12):4858–4863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lin C, Yang L, Tanasa B, et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell. 2009; 139(6):1069–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nik-Zainal S, Alexandrov LB, Wedge DC, et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012; 149(5):979–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Roberts SA, Sterling J, Thompson C, et al. Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol Cell. 2012; 46(4):424–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Abe H, Ochi H, Maekawa T, Hatakeyama T, Tsuge M, Kitamura S, et al. Effects of structural variations of APOBEC3A and APOBEC3B genes in chronic hepatitis B virus infection. Hepatol Res. 2009; 39(12):1159–1168 [DOI] [PubMed] [Google Scholar]
- 35. Schlattl A, Anders S, Waszak SM, Huber W, Korbel JO. Relating CNVs to transcriptome data at fine resolution: assessment of the effect of variant size, type, and overlap with functional regions. Genome Res. 2011;21(12):2004–2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Komatsu A, Nagasaki K, Fujimori M, Amano J, Miki Y. Identification of novel deletion polymorphisms in breast cancer. Int J Oncol. 2008; 33(2):261–270 [PubMed] [Google Scholar]
- 37. Lemaire F, Millon R, Young J, et al. Differential expression profiling of head and neck squamous cell carcinoma (HNSCC). Br J Cancer. 2003; 89(10):1940–1949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pauklin S, Sernandez IV, Bachmann G, Ramiro AR, Petersen-Mahrt SK. Estrogen directly activates AID transcription and function. J Exp Med. 2009; 206(1):99–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. An P, Johnson R, Phair J, et al. APOBEC3B deletion and risk of HIV-1 acquisition. J Infect Dis. 2009; 200(7):1054–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Celestino-Soper PB, Shaw CA, Sanders SJ, et al. Use of array CGH to detect exonic copy number variants throughout the genome in autism families detects a novel deletion in TMLHE. Hum Mol Genet. 2011; 20(22):4360–4370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kidd JM, Newman TL, Tuzun E, Kaul R, Eichler EE. Population stratification of a common APOBEC gene deletion polymorphism. PLoS Genet. 2007; 20;3(4):e63 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.