

Abstract
Autophagy is a conserved homeostatic process in which cytoplasmic contents are degraded and recycled. Two ubiquitin-like conjugation pathways are required for the generation of autophagosomes, and ATG5 is necessary for both of these processes. Studies of mice deficient in ATG5 reveal a key role for autophagy in T lymphocyte function, as well as in B cell development and B-1a B cell maintenance. However, the role of autophagy genes in B cell function and antibody production has not been described. Using mice in which Atg5 is conditionally deleted in B lymphocytes, we showed here that this autophagy gene is essential for plasma cell homeostasis. In the absence of B cell ATG5 expression, antibody responses were significantly diminished during antigen-specific immunization, parasitic infection and mucosal inflammation. Atg5-deficient B cells maintained the ability to produce immunoglobulin and undergo class-switch recombination, yet had impaired SDC1 expression, significantly decreased antibody secretion in response to toll-like receptor ligands, and an inability to upregulate plasma cell transcription factors. These results build upon previous data demonstrating a role for ATG5 in early B cell development, illustrating its importance in late B cell activation and subsequent plasma cell differentiation.
Keywords: B lymphocytes, ATG5, plasma cell differentiation, antibody secretion, immunity
Introduction
Recent genome-wide association studies (GWAS) have implicated autophagy genes in human disease and ensuing investigations have uncovered the importance of this process in many aspects of immunity.1-3 Autophagy is an evolutionarily conserved process important for recycling organelles and removing unwanted cytoplasmic cargo.4 Two ubiquitin-like conjugation pathways are required for autophagosome initiation and formation, and autophagy-related gene (Atg)5 is essential in both systems.4 In brief, following activation by the E1-like enzyme ATG7 and transfer to the E2-like enzyme ATG10, ATG12 becomes conjugated to ATG5. ATG16L1, which is assembled with the ATG12–ATG5 conjugate, is able to homotetramerize and the ATG12–ATG5-ATG16L1 multimers are recruited to the nascent autophagosomal membrane. This complex serves as an E3 ligase and mediates the lipidation of ATG8/LC3 with phosphatidylethanolamine. ATG7 and ATG3 function as the E1- and E2-like enzymes in the second conjugation system. Individual homozygous deletion of several of these autophagy proteins, including ATG5,5 ATG7,6 ATG87 and ATG16L1 (Virgin HW and Xavier RJ labs, unpublished data), results in lethality in mice, highlighting the essential function of this homeostatic process.
Previous studies have demonstrated that autophagy is important at the developmental transition from pro-B to pre-B lymphocytes, as well as in the peritoneal natural antibody-producing B-1a B cell compartment.8 B lymphocytes develop in the bone marrow (BM) and migrate to secondary lymphoid organs including spleen, lymph nodes and Peyer’s patches (PP), where they secrete immunoglobulins (Ig) in response to cognate antigens. Two subsets of mature B cells, designated B-1 and B-2, exist in the periphery and are distinguished from one another by cell surface marker expression and function and may arise from distinct precursors. The majority of B-1 B cells reside in the peritoneal cavity where they produce systemic natural IgM, although some B-1 B cells reside in the gut-associated lymphoid tissues (GALT) where they produce IgA, an Ig particularly important in intestinal homeostasis.9,10 B-2 cells largely participate in classical T cell-dependent IgM and IgG responses in peripheral lymphoid organs but are also able to migrate to the intestinal lamina propria and produce IgA.9,11,12 Antibody responses derived from both mature B cell subsets have been shown to regulate murine immune responses to intestinal parasitic infections and inflammation.9-15
B cells can be activated to become antibody-secreting plasma cells (PCs) in both T cell-independent (TI) and T cell-dependent (TD) fashions, contingent upon the nature of the antigen. TI antigens, such as toll-like receptor (TLR) ligands, activate B cells to generate short-lived Ig-secreting PCs.16,17 During TD immune responses, B cells undergo B cell receptor (BCR) affinity maturation and class-switch recombination (CSR) to produce isotype-specific, long-lived PCs and memory B cells. B cells that are activated by either TI or TD antigens upregulate the PC marker SDC1/CD138 and terminally differentiate into Ig-secreting PCs. Upregulation of Prdm1/Blimp1 and Xbp1, as well as downregulation of Pax5, is necessary for B cell differentiation into Ig-secreting PCs, and members of this transcriptional program have been implicated in tumorigenic, neurological and inflammatory diseases.18-24 XBP1 is necessary for increased protein synthesis during PC differentiation through its enhancement of secretory machinery; in addition, XBP1 has been shown to mediate the crosstalk between autophagy and the unfolded protein response (UPR).19,24,25 However, whether the PC transcriptional regulator XBP1 intersects with autophagy to regulate B cell function remains unknown.
Following B cell activation, internalized BCR has been shown to traffic to the autophagosome where it recruits TLR9-containing endosomes to enhance B cell signaling.26 TLR9 ligands are known to induce antibody responses, and we therefore hypothesized that autophagy may regulate XBP1-driven B cell differentiation and subsequent antibody secretion. Moreover, a variety of secretory cell types require autophagy for appropriate function, emphasizing the importance of this cellular process in secretion.27-31 Using mice conditionally deleted for Atg5 in the B cell compartment (CD19-cre × Atg5flox/flox), we demonstrated that functional autophagy is required for B cell activation and PC terminal differentiation. Antigen-specific IgM and IgG responses were impaired in mice lacking B cell ATG5 and these mice were more susceptible to Heligmosomoides polygyrus infection and intestinal inflammation. Thus we propose autophagy is not only important during B cell development but is also essential for efficient antibody secretion in health and disease.
Results
ATG5 is required for normal B cell distribution in the peritoneum and GALT-associated tissues
In order to study the role of autophagy in B cell function, we generated CD19-cre × Atg5flox/flox mice in which Atg5 is conditionally deleted in CD19-expressing cells; we hereafter refer to this mouse as the conditional knockout (CKO).8 We used Atg5flox/flox, CD19-cre and Atg5+/+ mice as controls; all three genotypes rendered similar results and are cumulatively represented as wild-type (WT). B lymphocyte numbers were first enumerated by flow cytometric analysis in central and peripheral immune compartments (Fig. 1). As previously reported, WT and CKO mice had similar numbers of total B cells residing in the spleen and BM; however, significantly fewer B-1a B cells were present in the peritoneal cavity of CKO mice.8 In addition, we observed significantly fewer B cells in GALT including PP and lamina propria (LP) in CKO mice (Fig. 1). Thus our data confirmed that ATG5 is required for normal B cell distribution and peritoneal B-1a B cell homeostasis and suggest that ATG5 is necessary for normal B cell population in GALT compartments.
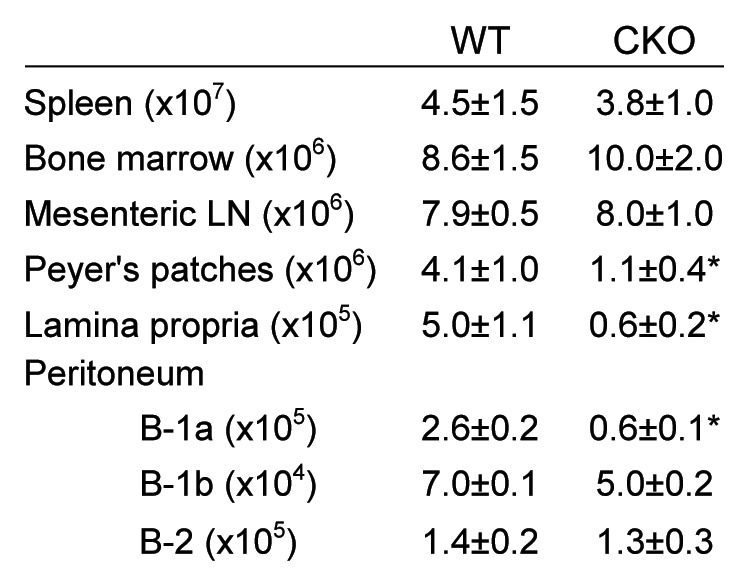
Figure 1. B lymphocyte cell numbers in immune tissues. B220+CD19+ B lymphocytes were quantified via flow cytometry. Peritoneal B cell subsets were defined as follows: B-1a (B220low, IgDlow, IgM+ and CD5+), B-1b (B220low, IgDlow, IgM+ and CD5-) and B-2 (B220high, IgD+, IgM+ and CD5-). Significance was determined using Student’s t test. *p < 0.001 compared with WT; n = 20 per group.
Spontaneous antibody secretion is diminished in CKO mice
In order to assess B cell function in the absence of ATG5, spontaneous Ig secretion was assessed via ELISpot. Equal numbers of B cells isolated from spleen, BM, peritoneal cavity, mesenteric lymph node (MLN), PP and LP were plated for 24 h and Ig-specific antibody-secreting cells (ASCs) were enumerated. As shown in Figure 2A, the frequencies of splenic B cells spontaneously secreting IgM and IgG were the same in WT and CKO mice. However, in both BM and MLN, two compartments with normal B cell numbers (Fig. 1), the frequencies of IgM and IgG ASCs were significantly diminished in CKO mice (Fig. 2A). Total peritoneal B cells also showed significantly fewer IgM ASCs in the absence of ATG5, suggesting that B-1 B cells are not only fewer in number in CKO mice (Fig. 1), but a lower frequency of them are also secreting IgM (Fig. 2A). Lastly, the frequencies of IgA-secreting B cells isolated from gut-associated immune compartments (MLN, PP and LP) were significantly lower in CKO mice (Fig. 2A). Further, B cells were able to efficiently generate Ig, as surface and intracellular IgM and IgG expression levels were comparable in lymphoid tissues (Fig. 2B). Naïve B cells were also able to efficiently class-switch in vitro (Fig. 2C), corroborating our ELISpot and flow cytometry data. Thus, Atg5-deficient B cells are able to express Ig and undergo CSR, yet significantly fewer B cells spontaneously secrete antibody in the absence of ATG5.

Figure 2.Atg5-deficient B cells have decreased Ig secretion, despite the ability to express Ig and undergo CSR. (A) Purified B cells were cultured on anti-Ig coated ELISpot plates and antibody-secreting cells (ASCs) were enumerated after 24 h. Numbers of IgM ASCs (top left), IgG ASCs (top middle) and IgA ASCs (top right) are shown relative to the number of ASCs present in each WT mouse tissue. *p < 0.001; n ≥ 10 per group. Representative ELISpot wells are shown for each tissue (bottom). BM, bone marrow; PerC, peritoneal cavity; MLN, mesenteric lymph node; PP, Peyer’s patches; LP, lamina propria. (B) Total IgM and IgG expression levels were quantified by flow cytometry. Representative histograms from MLN display WT (blue line) and CKO (red line) Ig distributions in CD19+ cells. Isotype control staining is shown in gray. (C) Purified naïve B cells were induced to undergo CSR to IgG1, IgG3 or IgA. Percentages of class-switched B cells were determined via flow cytometry after 4 d. No significant differences were observed via Student’s t-test.
Atg5-deficient B cells are unable to efficiently mount antigen-specific TI and TD antibody responses in vivo
Serum Ig levels were compared by ELISA. As shown in Figure 3A, serum IgM, IgG and IgA levels in WT and CKO mice were similar, suggesting that steady-state Ig is normal in the absence of ATG5. However, steady-state Ig levels are regulated by many homeostatic mechanisms and may not accurately assess subtotal defects in B cell capacity to produce antibody. To this end, we assessed whether Atg5-deficient B cells were able to respond to classical TI and TD antigens. In vivo TI and TD B cell responses can be studied by immunizing mice with the 2,4,6-trinitrophenyl (TNP) hapten conjugated to either classical TI antigens, such as lipopolysaccharide (LPS) and Ficoll, or classical TD antigens such as chicken gamma globulin (CGG). Serum isotype-specific anti-TNP antibodies can be detected within 7 d after TI immunizations and over months after TD immunizations.

Figure 3.Atg5-deficient B cells are unable to efficiently mount antigen-specific responses. (A) Serum from age-matched littermates was harvested and IgM, IgG and IgA levels were determined by ELISA (n = 10 per group). Each mouse is individually displayed relative to the average Ig amount in WT mice. (B) Age-matched littermates were immunized i.p. with TNP-LPS. Serum was harvested on days 7 and 14 and anti-TNP IgM and IgG3 levels were determined by ELISA. *p < 0.001; n = 10 per group. (C) Age-matched littermates were immunized i.p. with TNP-Ficoll at indicated doses. Serum was harvested on days 7 and 14 and anti-TNP IgM and IgG3 levels were determined by ELISA. *p < 0.01; n = 10 per group. (D) Age-matched littermates were immunized i.p. with TNP-CGG on day 0 and day 14. Serum was harvested on days 0, 7, 14 and 28, and TNP-specific Ig levels were detected via ELISA. ELISA values plotted represent the average OD450 reading. *p < 0.01; n = 5 per group. Statistics were calculated using Student’s t-test.
Seven and 14 d after immunization with TNP-conjugated LPS or Ficoll, two TI antigens, serum TNP-specific IgM and IgG3 levels were significantly lower in CKO mice (Fig. 3B and C). In addition, Ig levels were significantly lower in CKO mice immunized with both low and high concentrations of TNP-Ficoll, suggesting an absolute defect in responding to antigen (Fig. 3C). These results indicate that expression of ATG5 in B cells is required for efficient antigen-specific antibody responses to TI antigens.
We next evaluated whether B cell expression of ATG5 is required for TD antibody responses. Serum antigen-specific Ig levels from mice immunized with TNP-conjugated CGG were monitored for 4 weeks. Similar to the TI antigen responses, CKO mice produced significantly lower levels of TNP-specific IgM, IgG and IgG1 than WT mice in response to TD antigen stimulation (Fig. 3D). Thus our data suggest that B cell ATG5 expression plays a role in antibody responses in vivo.
B cell ATG5 expression is important for appropriate antibody responses during H. polygyrus infection and intestinal inflammation
We next employed two experimental approaches to address whether the muted antibody responses observed in CKO immunized mice would translate to infectious and noninfectious disease models. Given the association of autophagy with inflammatory bowel disease and the defect in antibody secretion in gut-associated CKO B cells, we applied two intestinal in vivo models to address this question. We first used a helminth model in which mice were infected with H. polygyrus, an intestinal parasite that induces Th2-driven immune responses.32 The Th2 T cell response elicited during H. polygyrus infection is characterized by the production of high amounts of interleukin 4 (IL4), IL5 and IL10 and subsequent activation of B cells.33-35 Antigen-specific antiparasite IgE and IgG1 are secreted from the activated B cells to aid in the control of the pathogen.13,15,32 As ATG5 is intact in all cell types except B lymphocytes in CKO mice, we were able to apply this model to assess the role of ATG5 in B cell-intrinsic responses during pathogenic infection. WT and CKO mice were inoculated with H. polygyrus and serum Ig levels were monitored up to 3 weeks postinfection. Both IgE and IgG1 antibody levels were significantly lower in CKO mice at all time points, suggesting these mice are unable to mount an effective humoral Th2 response during intestinal parasite infection (Fig. 4A).

Figure 4. B cell ATG5 expression is required for appropriate antibody responses during intestinal infection and intestinal inflammation. (A) Age- and weight-matched mice were inoculated with 200 third-stage H. polygyrus larvae. Parasite-specific IgE and IgG1 were quantified by ELISA 1, 2 and 3 weeks post-infection. *p < 0.01; n = 5 per group. (B) Fecal pellets were harvested from age-matched littermates, weighed, and homogenized in PBS at volumes commensurate with the pellet weight. Supernatants were subjected to IgA-specific ELISA for analysis. *p < 0.01; n = 10 per group. Fecal IgA data points shown here correlate to the mice displayed in Figure 2A depicting total serum Ig. (C) Age- and weight-matched mice were given 3% DSS ad libitum in drinking water for 7 d, followed by 5 d of regular drinking water. At day 12, B lymphocytes were isolated from MLN, PP and LP for IgA-specific ELISpot assay analysis. IgA ASCs were enumerated after 24 h and the numbers shown are relative to the number of IgA ASCs present in each tissue from WT mice that received no DSS. *p < 0.001 compared with WT without DSS; n = 7 per group. (D) Body weights plotted from DSS-treated mice in (C). *p < 0.01. Statistics were determined using Student’s t-test.
We have demonstrated that fewer B cells from gut-associated tissues (MLN, PP and LP) spontaneously secrete IgA in the absence of ATG5 (Fig. 2A), and therefore we next assessed mucosal IgA immune responses. Although serum IgA levels were comparable between WT and CKO mice (Fig. 3A), this represents a very small pool of IgA, as the vast majority is secreted into the intestinal lumen. We therefore quantified steady-state mucosal IgA via fecal extract and found significantly lower levels of IgA present in the feces of CKO mice (Fig. 4B). Together with the ELISpot analysis (Fig. 2A), these data show that steady-state mucosal IgA production is impaired in mice lacking B cell-specific ATG5 expression.
The second in vivo system we explored was an intestinal inflammation model using dextran sulfate sodium (DSS), a chemical agent that induces intestinal inflammation by disrupting the epithelium. During DSS colitis, B lymphocytes secrete greater amounts of IgA to aid in the regulation of the altered microbial environment.10,14 In order to address the role of ATG5 in IgA responses during intestinal inflammation, WT and CKO mice were treated for 7 d with 3% DSS followed by a 5-d recovery period. As expected, purified WT B cells secreted significantly more IgA following DSS treatment in MLN, PP and LP (Fig. 4C; black and gray bars). However, CKO B cells, regardless of DSS treatment, did not secrete IgA (Fig. 4C; white and checked bars). In addition, CKO mice treated with DSS lost more weight than WT mice indicating enhanced susceptibility to intestinal inflammation in the absence of B cell ATG5 expression (Fig. 4D). Therefore, antibody production in CKO mice is impaired during homeostatic, pathogenic and inflammatory responses.
Atg5-deficient B cells are defective in PC differentiation
Given the observed lower number of ASCs (Fig. 2), impaired antigen-specific B cell responses (Figs. 3 and 4), and defective IgA production during intestinal inflammation (Fig. 4) in CKO mice, we next investigated the mechanism by which this was occurring. SDC1/CD138, also known as syndecan 1, is upregulated upon B cell activation and PC differentiation. Due to the marked decrease in the number of Ig-secreting cells in CKO mice, we assessed the frequency of SDC1-expressing cells to determine whether PC differentiation was impaired. As shown in Figure 5A, CKO mice possessed significantly fewer SDC1-expressing cells in the periphery (MLN shown). Moreover, after TD immunization, the number of SDC1-expressing cells increased approximately 3-fold in WT MLN and spleen, whereas no change was seen in CKO mice (Fig. 5B). These data suggest that ATG5 is required for appropriate upregulation of SDC1 during PC differentiation.

Figure 5.Atg5-deficient B cells are defective in plasma cell differentiation. (A) The frequency of SDC1-expressing cells was determined via flow cytometry. Representative dot plots from MLN are shown and numbers represent the percentage of CD19+ B cells expressing SDC1. (B) The number of SDC1+ cells was quantified in mesenteric lymph nodes and spleen at baseline and after TNP-CGG immunization. *p < 0.01 comparing basal to TNP-CGG immunized. (C) ATG5 deletion was confirmed by western blot. MEFs harvested from WT and Atg5-deficient mice were used as controls in lanes 1 and 2. Sorted CD19+B220+SDC1− and CD19+B220+SDC1+ populations from the mesenteric lymph nodes of WT and CKO mice were assessed. (D) CD19+B220+SDC1− cells were sorted from the MLN of WT and CKO mice and cultured with LPS (10 μg/mL) or CpG (1 μg/mL) for 48 h on ELISpot membranes. Anti-IgM antibody-secreting cells were quantified and numbers shown are relative to the number of antibody-secreting cells in WT samples. *p < 0.001; n = 6 per group. (E) Prdm1, Xbp1 and Il6 mRNA expression in sorted CD19+B220+SDC1− populations from the mesenteric lymph nodes of WT and CKO mice after 48 h of LPS stimulation. Numbers shown represent fold induction after LPS stimulation within each group. *p < 0.001 comparing fold induction in WT vs. CKO; ns = not significant; n = 6 per group.
In order to verify that ATG5 was efficiently deleted in these naïve and activated B cell compartments, we used western blotting to assess expression levels of ATG5. As our data support a decrease in CKO ASCs and activated B cells are the subset of B cells responsible for secretion, we sorted naïve CD19+B220+SDC1− and activated CD19+B220+SDC1+ B cell subsets to ensure that deletion was maintained throughout B cell maturation (sort: > 99% pure, data not shown). Using mouse embryonic fibroblasts (MEFs) from Atg5-sufficient and Atg5-deficient mice as controls, we confirmed complete deletion of ATG5 in both CD19+B220+SDC1− and CD19+B220+SDC1+ cell subsets in CKO mice (Fig. 5C). These data indicate efficient ATG5 deletion in the B cell compartment that is maintained throughout maturation.
As we found a lower frequency of SDC1-expressing cells (Fig. 5A) and impaired antibody responses in CKO mice (Fig. 2A; Figure 3B–D; Fig. 4A–D), we examined whether B cell activation was altered in the absence of ATG5. TLR agonists induce B cell activation, proliferation and differentiation to PCs.16,17 Stimulation of nonactivated CD19+B220+SDC1− B cells with LPS (TLR4 ligand) or CpG (TLR9 ligand) induced IgM secretion in WT but not CKO B cells, suggesting ATG5 is necessary for TLR-induced B cell activation (Fig. 5D). In addition, the frequency of Annexin V+ 7AAD+ cells was equal when WT and CKO naïve B cells were stimulated with LPS or CpG, suggesting that enhanced cell death during PC differentiation cannot account for the impaired number of Ig-secreting cells observed in CKO mice (data not shown). In order to understand why CKO B cell antibody secretion was significantly impaired in response to TLR ligands, we assessed PC transcription factor expression following LPS stimulation. Expression of Prdm1, a gene that encodes the PRDM1 transcriptional repressor, was significantly upregulated following LPS stimulation in WT cells (Fig. 5E). However, negligible levels of Prdm1 were expressed in CKO B cells even after LPS stimulation. Expression of Xbp1, which encodes a transcription factor induced during PC differentiation, was also upregulated in WT cells after LPS stimulation; however, no induction was detected in CKO B cells, suggesting fewer cells are terminally differentiating into antibody-secreting PCs (Fig. 5E). In addition, Il6, which is induced upon LPS stimulation, was equally expressed in WT and CKO B cells, suggesting the LPS stimulation was effective (Fig. 5E). These data suggest that CKO B cells are impaired in their ability to terminally differentiate into PCs and are therefore unable to mount effective antibody responses. Thus, the autophagy gene Atg5 plays a role in regulating this late stage of B cell differentiation.
Discussion
In this study, we demonstrated ATG5 plays a role in PC homeostasis, showing that SDC1 upregulation and B cell antibody responses during antigen-specific immunization, H. polygyrus infection and an epithelial injury model of colitis were impaired in mice lacking B cell ATG5 expression. Importantly, we found that mature B cells were able to produce Ig and undergo CSR in the absence of ATG5, suggesting B cell maturation is not impaired. Flow cytometric analysis of Ig expression suggested that these autophagy-deficient B cells were able to synthesize antibody for expression at the cell surface; however, ELISpot and in vivo data demonstrated they are impaired in Ig responses. Using several in vivo models, we demonstrated muted antibody responses and enhanced susceptibility to pathogens and intestinal inflammation in mice lacking B cell ATG5 expression. In addition, Atg5-deficient B cells were unable to respond to TLR stimulation likely due to an inability to upregulate PC transcription factors. These data imply a role for autophagy in the late stages of B cell activation and subsequent PC differentiation and provide novel insight into the pathways important during this process. While we have demonstrated defective activation and, consequently, impaired antibody secretion in Atg5-deficient B lymphocytes, autophagy may regulate B cell function in multiple stages and remains a focus for future investigation.
Through the use of rag1−/− reconstitution and conditional knockout murine models, we and others have elucidated a role for autophagy in lymphocytes.8,36,37 BECN1/Beclin 1, a protein involved in autophagosome formation, is essential for the maintenance of early lymphoid progenitors, yet dispensable for mature peripheral T and B lymphocytes.36 Likewise, we have shown that despite a developmental block at the pro-B to pre-B cell stage, Atg5-deficient B cells populate peripheral lymph organs.8 Previous studies have demonstrated that autophagy is not only important during T cell development, but is also essential for several T cell functions including survival and proliferation.37,38 We showed here that autophagy is necessary for appropriate antibody responses by B lymphocytes. These data confirm that autophagy proteins have cell type-specific effects within the lymphocyte lineage.
The data shown here support a role for autophagy in B lymphocyte differentiation to antibody-secreting PCs, building upon an emerging theme that highlights a role for autophagy in secretory cell types. Deficiencies in autophagy have been associated with impaired pancreatic β cell structure and insulin production, diminished mast cell degranulation and decreased osteoclast secretion and bone resorption.28-31 In addition, ATG5 is involved in the biogenesis and secretion of IL1B/IL-1β in macrophages.39 Intestinal Paneth cells deficient in Atg5, Atg7 or Atg16l1 possess abnormalities in secretion of antimicrobial peptides from granules.27,40 Interestingly, these Paneth cell defects mimic human disease phenotypes in Crohn disease patients harboring an ATG16L1 risk allele.27 This epithelial phenotype, together with the present data describing defective intestinal IgA secretion in B cells lacking ATG5, suggests that autophagy is important in many immune cell types and defects in this process could potentially contribute to the pathophysiology of diseases, such as Crohn disease, at multiple immune checkpoints.
We have not only shown an inherent defect in anti-helminth B cell antibody responses in the absence of ATG5 but, in doing so, have highlighted another physiological model in which autophagy deficits may enhance aberrant immune responses by compounding effects of multiple cell types. The B cell phenotype described in this model has implications in a clinical setting as more than 3 billion people worldwide are infected with helminths, which are known to induce a Th2 immune response.32,41 In the past decade, studies have demonstrated that concomitant helminth infection with viral, parasitic or bacterial pathogens leads to enhanced disease severity through helminth-driven suppression of protective Th1 responses.32 In addition to skewing T cell responses, helminth infection has also been shown to impair bacterial pathogen clearance by inhibiting autophagy in macrophages.42 This report, together with the data shown here proposing ATG5 plays a role in anti-H.polygyrus antibody responses, suggests that, during helminth infection, deficiencies in autophagy can affect several immune cell functions which may alter host immunity.
Our data propose a novel functional role for autophagy in B lymphocytes. Given the association of autophagy with a variety of diseases, the mechanisms by which this cellular process affects secretion and other biological processes must continue to be investigated. ATG5 and PRDM1/BLIMP1, the gene encoding the PRDM1 PC transcriptional regulator, exist within a locus associated with systemic lupus erythematosus, implicating autophagy in B cell-mediated autoimmunity.3,43 Our data here suggest that autophagy regulates PC differentiation, perhaps through modulating PRDM1 and XBP1 expression. Xbp1 is critical for PC differentiation, as mice deficient in this gene possess few SDC1-expressing cells and are significantly impaired in antibody secretion.44 A recent study demonstrates that the absence of XBP1 expression in B cells results in diminished, but not absent, antibody secretion in response to immunization, consistent with the phenotype we observe in mice possessing Atg5-deficient B cells.45 XBP1 is also involved in the UPR, which is regulated in large part by autophagy; therefore future studies should investigate the potential crosstalk between these pathways and its implications in PC biology.
Materials and Methods
Animals
Mice were maintained in specific-pathogen-free facilities at Massachusetts General Hospital. All animal studies were conducted under protocols approved by the Subcommittee on Research Animal Care at Massachusetts General Hospital. CD19-cre × Atg5flox/flox mice were generated as previously described.8 All mice were maintained on food and water ad libitum. Mice were used between 7 to 9 weeks of age and strains were age- and gender-matched for each experiment.
Cell isolation from immune compartments
Cells from the spleen, bone marrow, mesenteric lymph node and Peyer’s patches were harvested, filtered, and red blood cells were lysed prior to downstream applications. Peritoneal cell exudate was harvested via three 4 mL lavages and filtered prior to use. To obtain colonic lamina propria cells, colons were harvested and inverted onto polyethylene tubing (Becton Dickinson, 8050-0125). After being washed in calcium- and magnesium-free PBS, colon tissues were incubated with dithiothreitol (Sigma-Aldrich, 43817), followed by 30 mM EDTA, all at room temperature. The remaining tissue was further digested using Type IV Collagenase (108 U/mL, Sigma-Aldrich, C5138) for 90 min. Filtered cells from the digested tissue were then layered on a 45%/72% Percoll (GE Healthcare,17-0891-01) gradient and harvested at the interface after centrifugation (650 × g, 15 min).
B cell isolation
B cells were isolated from immune tissues by magnetic separation via MACS negative selection kits, per manufacturer’s protocol (Miltenyi Biotech, 130-090-862). B cell purities were 96% to 99%.
Enzyme-linked immunospot (ELISpot) assays
One × 105 purified B cells were plated on multiscreen filter plates (Millipore, S2EM004M99) coated with 1 μg/mL goat anti-mouse IgM, IgG or IgA (Southern Biotech, 1020-01, 1030-01 and 1040-01, respectively). Cells were cultured in RPMI + Glutamax supplemented with 10% FBS (complete RPMI) at 37°C with 5% CO2. After 24 h, cells were removed and bound antibody was detected via biotinylated anti-IgM, anti-IgA, anti-IgG, anti-IgG3 and anti-IgG1, as indicated (Southern Biotech, 1020-08, 1040-08, 1030-08, 1100-08 and 1070-08, respectively). HRP-conjugated streptavidin (BD PharMingen, 554066) allowed detection of secreted antibodies and plates were developed by 3-amino-9-etyhlcarbazole (AEC, Sigma-Aldrich, A6926). Antibody secreting cells (ASCs) were scanned and enumerated using an Immunospot Analyzer ELISpot reader (Cellular Technology Limited). All samples were run in triplicate.
Flow cytometry
Single-cell suspensions were prepared from tissues; after red blood cell lysis, 1 × 106 cells were incubated with 2.4G2 Mouse Fc block in PBS/FBS (BD PharMingen, 553142) for 20 min at 4°C. Cells were then washed and stained with fluorescent-conjugated antibodies for 20 min at 4°C. The following antibodies were used (BD PharMingen): IgD-FITC (553439), IgA-FITC (559354), B220-PE (553089), SDC1-PE (561070), IgM-PerCP (550881), CD19-APC (550992) and CD5-APC (550035). In addition, the following biotinylated antibodies were used (Southern Biotech): IgG-biotin (1030-08), IgG1-biotin (1070-08) and IgG3-biotin (1100-08). Biotinylated antibodies were detected after subsequent incubation with streptavidin-FITC (BD PharMingen, 554060). Fluorescently labeled cells were acquired on the FACSCalibur flow cytometer (BD Biosciences) and analyzed using FlowJo Analysis Software (Tree Star, Inc.).
In vitro class-switch recombination (CSR) assay
MACS-purified B cells were isolated from the spleen. Two × 105 cells/mL were plated in 96-well round-bottom tissue culture plates (BD Biosciences, 353077) and incubated for 4 d at 37°C with 5% CO2 in complete RPMI with CSR-inducing stimuli. B cells were incubated with 40 μg/mL LPS (InvivoGen, tlrl-pelps) and 20 ng/mL IL4(Peprotech, 214-14) to induce IgG1 CSR; 40 μg/mL LPS to induce IgG3 CSR; and 10 μg/mL CD40L (BD PharMingen, 553656), 20 ng/mL IL4 and 10 ng/mL TGFB1(Peprotech, 100-21c) to induce IgA CSR. Ig cell surface expression was assessed via flow cytometry on day 4.
ELISA
Using the same reagents as those used in the ELISpot assays, total serum and fecal IgM, IgG and IgA were detected by sandwich ELISA coated with 1 μg/mL goat anti-mouse IgM, IgG or IgA (Southern Biotech) on EIA High Bind 96-well plates (Corning, 3590). Isotype-specific biotinylated antibodies were combined with HRP-conjugated streptavidin to quantitate serum antibody levels using BD OptEIA substrate kit (BD Biosciences, 555214). TNP-specific antibody detection was achieved by coating EIA high-binding plates with 10 μg/mL TNP-BSA (Biosearch Technologies, T-5050) and subsequent detection using the same reagents described above. Samples were diluted (1:30,000 for Ig and 1:5,000 for TNP-specific antibodies). Samples were quantified using the SpectraMax M5 microplate reader (Molecular Devices), measuring absorbance at 450 nm. Samples were run in triplicate.
T cell-independent (TI) antigen immunizations
Fifty micrograms of 2,4,6-trinitrophenyl (TNP)-LPS (Biosearch Technologies, T-5065), a Type 1 TI antigen, was injected intraperitoneally (i.p.) in 200 μL of PBS. Ten μg or 50 μg of TNP-Ficoll (Biosearch Technologies, F-1300), a Type 2 TI antigen, was injected i.p. in 200 μL of PBS. Serum anti-TNP antibody levels were measured 7 and 14 d post-immunization.
T cell-dependent (TD) antigen immunization
One-hundred micrograms of TNP-chicken gamma globulin (CGG) (Biosearch Technologies, T-5052), a TD antigen, was mixed with 100 μL of Imject alum (Pierce Biotechnology, 77161) in PBS, and a total volume of 200 μL was injected i.p. Serum was harvested every week for four weeks for anti-TNP ELISA assays. Mice were boosted i.p. with the same amount of antigen on day 14.
Heligmosomoides polygyrus infection and parasite-specific ELISA
Age- and weight-matched mice were inoculated orally with 200 third-stage H. polygyrus larvae. Serum was harvested 1, 2 and 3 weeks after infection and antigen-specific ELISAs for IgE and IgG1 were performed. The parasite antigen was prepared as previously described.13 EIA High Bind 96-well ELISA plates were coated with 10 µg/mL parasite antigen and incubated at 4°C overnight. Two rows on each plate were coated with anti-mouse IgE or anti-mouse IgG1 (Southern Biotechnology) in order to generate a standard curve. Samples were diluted (1:100) and incubated at 4°C overnight. Isotype-specific biotinylated antibodies were combined with HRP-conjugated streptavidin to quantitate serum antibody levels using a BD OptEIA substrate kit (BD Biosciences, 555214). Samples were quantified using the SpectraMax M5 microplate reader (Molecular Devices), measuring absorbance at 450 nm. Samples were run in triplicate.
Fecal Ig isolation
One fecal pellet was dissolved in PBS (1 mL/100 mg feces), vortexed for 15 min at high speed, and settled at room temperature for 15 min. Samples were then centrifuged at 13,820 × g for 15 min and supernatant was harvested into a fresh tube and stored at −20°C until use.
DSS colitis
Age- and sex-matched mice were fed ad libitum 3% (w/v) dextran sulfate sodium (DSS, MP Biomedicals, 160110, MW = 36,000–50,000) dissolved in sterile, distilled drinking water for 7 d, followed by 5 d of regular drinking water. Body weight was measured daily and B cells were isolated for ELISpot analysis on day 12.
FACS sorting
Single-cell suspensions from mesenteric lymph nodes were stained with SDC1, CD19 and B220 as described above. CD19+B220+SDC1− and CD19+B220+SDC1+ cells were FACS-sorted using a MoFlo cell sorter (Beckman Coulter) and subjected to western blot analysis. Three mice were pooled for each sample in order to obtain enough cells for western blot.
Mouse embryonic fibroblasts (MEFs)
MEFs from wild-type (+/+) and atg5-deficient (−/−) mice were maintained as previously described.5
Western blotting
Cell extracts were prepared using TNN lysis buffer, pH 8 (100 mM TRIS-HCl, 100 mM NaCl, 1% NP-40, 1 mM DTT, 10 mM NaF) with protease inhibitor (Roche, 11836153001). Following SDS-PAGE (Bio-Rad, 456-9035) and transfer onto PVDF membrane (Immobilon-P, Millipore, IPVH20200), ATG12–ATG5 conjugates and actin were detected using anti-ATG5 antibody, clone D1G9 (Cell Signaling Technology, 8540) and polyclonal anti-actin antibody (Sigma Aldrich, A2066). Polyclonal goat anti-rabbit HRP (Dako, P0448) was used as secondary antibody.
TLR ligand stimulation
Sorted CD19+B220+SDC1− B cells were incubated at 37°C with 5% CO2 for 48 h in complete RPMI. Indicated samples were stimulated with 10 μg/mL LPS or 1 μg/mL CpG. ELISpots of stimulated cells were performed as described above. Cells used for isolation of RNA and subsequent transcription factor analysis were handled as described below.
Transcription factor analysis
Cells were pelleted after 48 h and RNA was extracted using the RNeasy Kit (Qiagen, 74106) according to the manufacturer’s protocol. All RNA samples were reverse-transcribed using the iScript cDNA Synthesis kit (Bio-Rad Laboratories, 170-8891). Using the iQ SYBR Green Supermix (Bio-Rad Laboratories, 1708887) for quantitative PCR, mRNA levels were determined using the iCycler with iQ5 Multicolor Real-time PCR Detection System (Bio-Rad). The reaction conditions consisted of 37 cycles of PCR with an annealing temperature of 59°C. The following primers were used: Prdm1 Fwd: GACGGGGGTACTTCTGTTCA; Prdm1 Rev: GGCATTCTTGGGAACTGTGT; Xbp1 Fwd: TCCGCAGCACTCAGACTATG; Xbp1 Rev: ACAGGGTCCAACTTGTCCAG; Il6 Fwd: GTAGCTATGGTACTCCAGAAGAC; Il6 Rev: ACGATGATGCACTTGCAGAA. The threshold cycle (CT) for each sample was determined for each gene and was normalized to the CT value of the endogenous housekeeping gene GAPDH. Data were calculated using the 2−ΔΔC(T) method.46,47
Statistical analysis
Student’s t-test was used to assess the significance of observed differences. A value of p < 0.05 was considered significant.
Acknowledgments
We acknowledge Noboru Mizushima (Tokyo Medical and Dental University, Tokyo, Japan) for providing Atg5flox/flox mice and WT and atg5−/− MEFs. We also acknowledge Natalia Nedelsky for review of the manuscript and helpful discussions. This work was supported by the National Institutes of Health Grants DK043351, DK083756, DK086502 and DK060049 (to R.J.X.), and CCFA Genetics Initiative Grant (to R.J.X./H.W.V.).
Glossary
Abbreviations:
- ASC
antibody-secreting cell
- Atg
autophagy-related gene
- BCR
B cell receptor
- BECN1
Beclin 1
- BM
bone marrow
- CGG
chicken gamma globulin
- CKO
conditional knockout
- CSR
class-switch recombination
- DSS
dextran sulfate sodium
- ELISA
enzyme-linked immunosorbent assay
- ELISpot
enzyme-linked immunospot
- Ig
immunoglobulin
- IL
interleukin
- GALT
gut-associated lymphoid tissues
- GWAS
genome-wide association studies
- LP
lamina propria
- LPS
lipopolysaccharide
- MEF
mouse embryonic fibroblast
- MLN
mesenteric lymph node
- Pax5
paired box 5
- PC
plasma cell
- PP
Peyer’s patches
- PRDM1
PR domain containing 1, with ZNF domain
- SDC1
syndecan 1
- TD
T cell-dependent
- TGFB1
transforming growth factor, beta 1
- TI
T cell-independent
- TLR
toll-like receptor
- TNP
2,4,6-trinitrophenyl
- UPR
unfolded protein response
- WT
wild-type
- XBP1
X-box binding protein 1
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/23484
References
- 1.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–35. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou XJ, Lu XL, Lv JC, Yang HZ, Qin LX, Zhao MH, et al. Genetic association of PRDM1-ATG5 intergenic region and autophagy with systemic lupus erythematosus in a Chinese population. Ann Rheum Dis. 2011;70:1330–7. doi: 10.1136/ard.2010.140111. [DOI] [PubMed] [Google Scholar]
- 4.Huett A, Xavier RJ. Autophagy at the gut interface: mucosal responses to stress and the consequences for inflammatory bowel diseases. Inflamm Bowel Dis. 2010;16:152–74. doi: 10.1002/ibd.20991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–6. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 6.Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–34. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sou YS, Waguri S, Iwata J, Ueno T, Fujimura T, Hara T, et al. The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Mol Biol Cell. 2008;19:4762–75. doi: 10.1091/mbc.E08-03-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller BC, Zhao Z, Stephenson LM, Cadwell K, Pua HH, Lee HK, et al. The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy. 2008;4:309–14. doi: 10.4161/auto.5474. [DOI] [PubMed] [Google Scholar]
- 9.Mora JR, von Andrian UH. Differentiation and homing of IgA-secreting cells. Mucosal Immunol. 2008;1:96–109. doi: 10.1038/mi.2007.14. [DOI] [PubMed] [Google Scholar]
- 10.Shimomura Y, Mizoguchi E, Sugimoto K, Kibe R, Benno Y, Mizoguchi A, et al. Regulatory role of B-1 B cells in chronic colitis. Int Immunol. 2008;20:729–37. doi: 10.1093/intimm/dxn031. [DOI] [PubMed] [Google Scholar]
- 11.Mantis NJ, Rol N, Corthésy B. Secretory IgA’s complex roles in immunity and mucosal homeostasis in the gut. Mucosal Immunol. 2011;4:603–11. doi: 10.1038/mi.2011.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Montecino-Rodriguez E, Dorshkind K. New perspectives in B-1 B cell development and function. Trends Immunol. 2006;27:428–33. doi: 10.1016/j.it.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 13.Chen CC, Louie S, McCormick B, Walker WA, Shi HN. Concurrent infection with an intestinal helminth parasite impairs host resistance to enteric Citrobacter rodentium and enhances Citrobacter-induced colitis in mice. Infect Immun. 2005;73:5468–81. doi: 10.1128/IAI.73.9.5468-5481.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fagarasan S, Kawamoto S, Kanagawa O, Suzuki K. Adaptive immune regulation in the gut: T cell-dependent and T cell-independent IgA synthesis. Annu Rev Immunol. 2010;28:243–73. doi: 10.1146/annurev-immunol-030409-101314. [DOI] [PubMed] [Google Scholar]
- 15.Finkelman FD, Shea-Donohue T, Goldhill J, Sullivan CA, Morris SC, Madden KB, et al. Cytokine regulation of host defense against parasitic gastrointestinal nematodes: lessons from studies with rodent models. Annu Rev Immunol. 1997;15:505–33. doi: 10.1146/annurev.immunol.15.1.505. [DOI] [PubMed] [Google Scholar]
- 16.Genestier L, Taillardet M, Mondiere P, Gheit H, Bella C, Defrance T. TLR agonists selectively promote terminal plasma cell differentiation of B cell subsets specialized in thymus-independent responses. J Immunol. 2007;178:7779–86. doi: 10.4049/jimmunol.178.12.7779. [DOI] [PubMed] [Google Scholar]
- 17.Gururajan M, Jacob J, Pulendran B. Toll-like receptor expression and responsiveness of distinct murine splenic and mucosal B-cell subsets. PLoS One. 2007;2:e863. doi: 10.1371/journal.pone.0000863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glimcher LH. XBP1: the last two decades. Ann Rheum Dis. 2010;69(Suppl 1):i67–71. doi: 10.1136/ard.2009.119388. [DOI] [PubMed] [Google Scholar]
- 19.Hetz C, Thielen P, Matus S, Nassif M, Court F, Kiffin R, et al. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009;23:2294–306. doi: 10.1101/gad.1830709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol. 2003;4:321–9. doi: 10.1038/ni907. [DOI] [PubMed] [Google Scholar]
- 21.Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134:743–56. doi: 10.1016/j.cell.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin KI, Angelin-Duclos C, Kuo TC, Calame K. Blimp-1-dependent repression of Pax-5 is required for differentiation of B cells to immunoglobulin M-secreting plasma cells. Mol Cell Biol. 2002;22:4771–80. doi: 10.1128/MCB.22.13.4771-4780.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shaffer AL, Lin KI, Kuo TC, Yu X, Hurt EM, Rosenwald A, et al. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity. 2002;17:51–62. doi: 10.1016/S1074-7613(02)00335-7. [DOI] [PubMed] [Google Scholar]
- 24.Shapiro-Shelef M, Lin KI, McHeyzer-Williams LJ, Liao J, McHeyzer-Williams MG, Calame K. Blimp-1 is required for the formation of immunoglobulin secreting plasma cells and pre-plasma memory B cells. Immunity. 2003;19:607–20. doi: 10.1016/S1074-7613(03)00267-X. [DOI] [PubMed] [Google Scholar]
- 25.Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity. 2004;21:81–93. doi: 10.1016/j.immuni.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 26.Chaturvedi A, Dorward D, Pierce SK. The B cell receptor governs the subcellular location of Toll-like receptor 9 leading to hyperresponses to DNA-containing antigens. Immunity. 2008;28:799–809. doi: 10.1016/j.immuni.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–63. doi: 10.1038/nature07416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DeSelm CJ, Miller BC, Zou W, Beatty WL, van Meel E, Takahata Y, et al. Autophagy proteins regulate the secretory component of osteoclastic bone resorption. Dev Cell. 2011;21:966–74. doi: 10.1016/j.devcel.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ebato C, Uchida T, Arakawa M, Komatsu M, Ueno T, Komiya K, et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 2008;8:325–32. doi: 10.1016/j.cmet.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 30.Jung HS, Chung KW, Won Kim J, Kim J, Komatsu M, Tanaka K, et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab. 2008;8:318–24. doi: 10.1016/j.cmet.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 31.Ushio H, Ueno T, Kojima Y, Komatsu M, Tanaka S, Yamamoto A, et al. Crucial role for autophagy in degranulation of mast cells. J Allergy Clin Immunol. 2011;127:1267–76, e6. doi: 10.1016/j.jaci.2010.12.1078. [DOI] [PubMed] [Google Scholar]
- 32.Wang LJ, Cao Y, Shi HN. Helminth infections and intestinal inflammation. World J Gastroenterol. 2008;14:5125–32. doi: 10.3748/wjg.14.5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Finkelman FD, Shea-Donohue T, Morris SC, Gildea L, Strait R, Madden KB, et al. Interleukin-4- and interleukin-13-mediated host protection against intestinal nematode parasites. Immunol Rev. 2004;201:139–55. doi: 10.1111/j.0105-2896.2004.00192.x. [DOI] [PubMed] [Google Scholar]
- 34.Fort MM, Cheung J, Yen D, Li J, Zurawski SM, Lo S, et al. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity. 2001;15:985–95. doi: 10.1016/S1074-7613(01)00243-6. [DOI] [PubMed] [Google Scholar]
- 35.Harnett W, Harnett MM. Molecular basis of worm-induced immunomodulation. Parasite Immunol. 2006;28:535–43. doi: 10.1111/j.1365-3024.2006.00893.x. [DOI] [PubMed] [Google Scholar]
- 36.Arsov I, Adebayo A, Kucerova-Levisohn M, Haye J, MacNeil M, Papavasiliou FN, et al. A role for autophagic protein beclin 1 early in lymphocyte development. J Immunol. 2011;186:2201–9. doi: 10.4049/jimmunol.1002223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med. 2007;204:25–31. doi: 10.1084/jem.20061303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stephenson LM, Miller BC, Ng A, Eisenberg J, Zhao Z, Cadwell K, et al. Identification of Atg5-dependent transcriptional changes and increases in mitochondrial mass in Atg5-deficient T lymphocytes. Autophagy. 2009;5:625–35. doi: 10.4161/auto.5.5.8133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 2011;30:4701–11. doi: 10.1038/emboj.2011.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cadwell K, Patel KK, Komatsu M, Virgin HW, 4th, Stappenbeck TS. A common role for Atg16L1, Atg5 and Atg7 in small intestinal Paneth cells and Crohn disease. Autophagy. 2009;5:250–2. doi: 10.4161/auto.5.2.7560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crompton DW, Nesheim MC. Nutritional impact of intestinal helminthiasis during the human life cycle. Annu Rev Nutr. 2002;22:35–59. doi: 10.1146/annurev.nutr.22.120501.134539. [DOI] [PubMed] [Google Scholar]
- 42.Su CW, Cao Y, Zhang M, Kaplan J, Su L, Fu Y, et al. Helminth infection impairs autophagy-mediated killing of bacterial enteropathogens by macrophages. J Immunol. 2012;189:1459–66. doi: 10.4049/jimmunol.1200484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gateva V, Sandling JK, Hom G, Taylor KE, Chung SA, Sun X, et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat Genet. 2009;41:1228–33. doi: 10.1038/ng.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Iwakoshi NN, Lee AH, Glimcher LH. The X-box binding protein-1 transcription factor is required for plasma cell differentiation and the unfolded protein response. Immunol Rev. 2003;194:29–38. doi: 10.1034/j.1600-065X.2003.00057.x. [DOI] [PubMed] [Google Scholar]
- 45.Taubenheim N, Tarlinton DM, Crawford S, Corcoran LM, Hodgkin PD, Nutt SL. High rate of antibody secretion is not integral to plasma cell differentiation as revealed by XBP-1 deficiency. J Immunol. 2012;189:3328–38. doi: 10.4049/jimmunol.1201042. [DOI] [PubMed] [Google Scholar]
- 46.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 47.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–8. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]