

Abstract
Analyzing molecular determinants of Plasmodium parasite cell death is a promising approach for exploring new avenues in the fight against malaria. Three major forms of cell death (apoptosis, necrosis and autophagic cell death) have been described in multicellular organisms but which cell death processes exist in protozoa is still a matter of debate. Here we suggest that all three types of cell death occur in Plasmodium liver-stage parasites. Whereas typical molecular markers for apoptosis and necrosis have not been found in the genome of Plasmodium parasites, we identified genes coding for putative autophagy-marker proteins and thus concentrated on autophagic cell death. We characterized the Plasmodium berghei homolog of the prominent autophagy marker protein Atg8/LC3 and found that it localized to the apicoplast. A relocalization of PbAtg8 to autophagosome-like vesicles or vacuoles that appear in dying parasites was not, however, observed. This strongly suggests that the function of this protein in liver-stage parasites is restricted to apicoplast biology.
Keywords: Plasmodium liver-stage, malaria, parasite death, autophagy-like cell death, Atg8, Atg3, Atg7, membrane expansion, apicoplast, organelle growth
Introduction
Plasmodium sporozoites injected by a female Anopheles mosquito during blood feeding invade blood vessels and travel to the liver to infect hepatocytes. Within a parasitophorous vacuole, the parasite undergoes multiple rounds of nuclear division and finally differentiates into more than 30,000 merozoites. Although a single infected hepatocyte is theoretically enough to give rise to a successful blood-stage infection, not every bite of an infected mosquito results in symptomatic infection of the mammalian host. One reason for this could be that not all parasites complete development in the hepatocyte but some die and are removed by their host cell. Although parasite cell death during liver-stage development has already been described,1 it remained to be shown whether this death is a spontaneous, nongenetically determined process or whether the parasites can indeed undergo programmed cell death.
For a long time it has been thought that programmed cell death is restricted to multicellular organisms. Recent research on protozoa has challenged this view and apoptosis-like cell death has been described in several protozoa, including parasites.2-4 In fact, we have shown previously that death of intracellular liver-stage Plasmodium parasites can be accompanied by DNA fragmentation, nuclear condensation and parasite disassembly, all typical morphological hallmarks of apoptosis.1,5 However, apart from a P. falciparum metacaspase that could be successfully linked to parasite cell death,6 genes coding for typical markers of apoptosis are absent in the genome of the parasite. Plasmodium parasites express neither BCL2-family proteins nor typical caspases.7-10
Another possibility for ordered parasite cell death is autophagic cell death. In eukaryotic cells, autophagy can allow a cell to overcome phases of starvation but is also a well-described mechanism for removal of damaged organelles and dispensable cellular material. Thus, autophagy is foremost a very powerful survival mechanism for the cell. However, if conditions do not improve, the cell can employ autophagy to induce cell death.11 Importantly, the molecular basis of autophagy and autophagic cell death is the same but the result is the polar opposite. During stage transition of Plasmodium parasites, it is very likely that dispensable cellular material accumulates but whether autophagy can be induced by the parasite to recycle this material and whether the same machinery can also be used to induce parasite cell death is not known. However, database searches reveal that the Plasmodium parasite harbors at least some homologs of autophagic machinery proteins.12-14
The molecular basis of autophagy has been elucidated using Saccharomyces cerevisiae genetic screens, by which several essential autophagy-related (ATG) genes have been identified.15 Autophagy involves complex membrane dynamics dependent on two ubiquitin-like conjugation systems. The first one is the Atg12 system, in which the Atg12 protein is conjugated via Atg7 and Atg10 to Atg5. The Atg12–Atg5 conjugate forms a complex with the Atg16 protein and is involved in the expansion of the autophagosomal membrane. In the second ubiquitin-like conjugation pathway, the cytosolic and inactive Atg8 protein is cleaved by the Atg4 protease, exposing a glycine residue. By a concerted action of Atg7 and Atg3, Atg8 is fused to phosphatidylethanolamine (PE) and then associates with the autophagosomal membrane.16 Due to the switching of Atg8 between an inactive, cytosolic and active, membrane-associated protein, it is an excellent marker for analyzing the dynamics of autophagy.17-19
The core Atg proteins are highly conserved in most eukaryotes. However, genome-wide analyses revealed that in protozoa, only subsets of the canonical protein orthologs or paralogs are present.14 In the parasitic protist Leishmania, homologs of proteins in both ubiquitin-like conjugation cascades can be found. They act similarly to those described in yeast and are important for the differentiation of one parasite stage to another.20-22 In Trypanosoma, another kinetoplastid, only members of the Atg8 ubiquitin pathway are present and no proteins of the Atg12 system are encoded in the genome.23,24 For Atg8, two homologs can be found in both T. brucei and in T. cruzi and they seem to play a role in stage differentiation and promotion of cell survival under stress conditions.
Recent studies revealed an autophagy pathway in the Plasmodium-related parasite Toxoplasma gondii.25-27 In this parasite, proteins of the Atg8-, but not of the Atg12-ubiquitin-like conjugation pathway have been identified. It was shown that under stress conditions such as starvation or drug treatment, TgAtg8 is conjugated via TgAtg3 to autophagosomal membranes. This localization was detected in extracellular parasites but also, less pronounced, in intracellular parasites.25,26 In addition, it was shown that starvation induces fragmentation of the mitochondrion, which can be blocked by 3-methylalanine, a known autophagy inhibitor. If this mitophagy is not blocked, the treated parasites die by autophagy-dependent cell death.27
In contrast to Toxoplasma, little is known about autophagic pathways in Plasmodium. A first hint that autophagy can be induced in Plasmodium parasites came from a study by Totino et al., which shows that erythrocytic stages treated with different drugs form double-membrane structures, resembling autophagosomes, in the cytoplasm.28 Since these drug treatments finally kill the parasite, it is tempting to speculate that the parasites die an autophagy-like cell death similar to what has been described for Toxoplasma.25,26 However, molecular markers for autophagy were not investigated in this study.28 In the meantime, database searches have revealed several putative ATG genes in the Plasmodium genome, further supporting the idea of an active autophagic pathway in this parasite.12,13
If the molecular mechanisms behind the observed autophagy-like cell death could be identified, they might offer powerful possibilities for inducing parasite cell death and thus open new avenues for eliminating Plasmodium parasites at any developmental stage, including the dormant stages of Plasmodium vivax.
In the present study, we distinguish different forms of cell death in P. berghei liver-stage parasites. Although we could detect an autophagy-like cell death, it seems independent of the identified P. berghei Atg8 homolog. Instead PbAtg8 was localized specifically to the apicoplast throughout liver-stage development. Thus, in this stage of parasite development, Atg8 of P. berghei does not seem to fulfill the same function as Atg8 of Toxoplasma and of other organisms.
Results
Different types of parasite cell death
At the end of exoerythrocytic development, successful P. berghei parasites differentiate to merozoites, which induce PVM rupture and death of their host cells.29,30 In vitro, the dying host cells detach and float in the culture medium.30 So far investigations of the late liver-stage have focused primarily on normally developing parasites, but since we frequently observe parasites blocked in development, we initiated a quantitative assessment of cell detachment. To our surprise the majority of parasites do not successfully complete their development in hepatocytes. We counted the number of detached and attached infected cells at 68 and 72 hpi (hours post infection) and found that only about 20% of all parasites counted successfully induced PVM breakdown and host cell detachment (Fig. 1A).

Figure 1. The majority of Plasmodium berghei liver-stage parasites do not complete development and die in an ordered way. HepG2 cells were infected with transgenic P. berghei parasites constitutively expressing mCherry. (A) Sixty-eight and 72 hpi the number of infected attached cells (arrested) and detached cells (containing fully developed merozoites) were counted. Shown are the means of five independent experiments with the corresponding SDs (standard deviation). (B) merofusosome formation: merogony starts but then merozoites fuse to the enlarging body that exhibits strong vacuolization, a hallmark of autophagy (see also Vid. S2). Scale bar: 10 µm; CLS (confocal line scanning).
Next, we investigated the fate of the parasites that did not complete their exoerythrocytic development and found that they can undergo three different types of cell death. As well as the already described apoptosis-like cell death1 (Fig. S1), we also frequently observed a necrosis-like cell death, which does not appear to be controlled by the parasite (Vid. S1). The third form of cell death was characterized by marked vacuolization and parasite shrinkage and can occur at any time during liver-stage schizogony (Fig. S2A) and merozoite development (Fig. 1B). If it happens after merozoite formation, this form of cell death results in the fusion of already-formed merozoites with a membrane-surrounded cytoplasmic mass of increasing size (Fig. 1B; Vid. S2; Fig. S3), which we named a merofusosome. Vacuolization is a hallmark of autophagy5,31 and since merofusosome formation includes vacuolization and appears to be very ordered and clearly different from apoptotic and necrotic cell death, we hypothesized that these parasites first induce autophagy and subsequently autophagy-like programmed cell death. Not all merozoites fuse with the merofusosome at the same time and we occasionally observed merozoites escaping the fusion process and being liberated from the PV (Fig. S2B). Vacuolization could also be observed in vivo in histological sections (Fig. 2A) but even more importantly by intravital imaging (Fig. 2B; Vid. S3), indicating that autophagy-like cell death is indeed a physiological event.
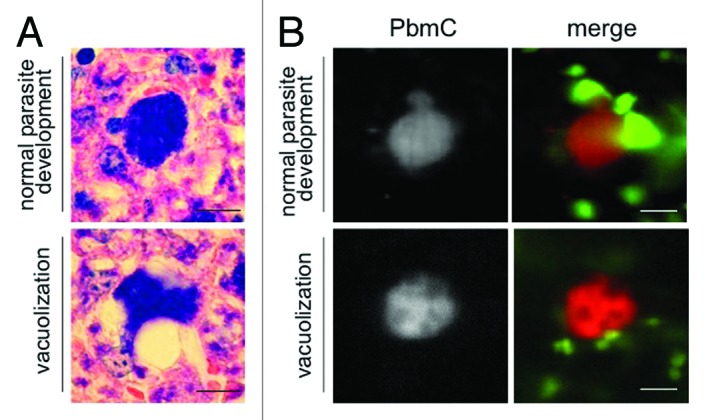
Figure 2. Vacuolization and parasite death is a physiological event that occurs also in vivo. (A) Mice were infected with P. berghei sporozoites and euthanized 38 hpi. The liver was removed, fixed and liver sections were prepared and stained with Giemsa. Parasites developing normally (top panel) are compared with parasites forming vacuoles. Scale bar: 20 µm. (B) Additionally, lys-GFP mice that express GFP mainly in neutrophils and macrophages32 were infected with mCherry-expressing parasites and intravital imaging of the anesthetized mouse was performed between 44 and 48 hpi. At this time parasites developing normally start forming merosomes (top panel, left image; PbmC: P. berghei mCherry), whereas dying parasites form vacuoles (lower panel) and finally fade (see Vid. S3). Scale bar: 20 µm; CLS.
The key morphological hallmark of autophagy is the occurrence of vesicles with two or more membranes. Transmission electron microscopy (TEM) analysis of P. berghei-infected HepG2 cells indeed revealed the presence of such vesicles in the parasite (Fig. 3), providing further evidence for our working hypothesis that the parasite can induce autophagy. Similar structures have previously been observed by Meis and Verhave who describe them as “osmiophilic membranous whorls” in early liver-stage parasites.33 Within late cytomere stage parasites, they again observed structures with several membranes, which they consider to be remnants of parasite lipid droplets. Together this could mean that the event is initially induced to support the survival of single merozoites, for example, in phases of starvation, which would again be a typical feature of autophagy. To further analyze this highly interesting type of cell death, we next searched the PlasmoDB database for putative autophagy-associated proteins.

Figure 3. Electron microscopy reveals autophagosome-like structures in P. berghei liver schizonts. P. berghei-infected HepG2 cells were fixed 48 hpi and analyzed by electron microscopy. P, parasite; N, host cell nucleus. Arrows indicate autophagic-like vesicle with multiple membranes, arrowheads indicate the parasite plasma membrane. Boxed areas are displayed at a higher magnification in the right panel. Scale bar: 2 µm.
Database analysis
The putative autophagy-related proteins identified by database searching were predominantly homologs of proteins pertaining to the Atg8 ubiquitin-like conjugation pathway, which is normally responsible for autophagosomal membrane expansion and completion (Table 1; Table S1). Homologs of Atg12–Atg5 ubiquitin-like conjugation pathway proteins are largely absent.
Table 1. Putative homologs of autophagy-related proteins have been identified in the genome of P. berghei.
| Autophagy-related proteins | P. berghei homologs | Expectation value | % pairwise identity with yeast | Reciprocal BLAST hit No. | |
|---|---|---|---|---|---|
| Expansion and completion |
Atg8 |
PBANKA_050401 |
2.2e-27 |
45 |
1 |
|
Atg4 |
PBANKA_102540 |
3.5e-7 |
11 |
1 |
|
|
Atg3 |
PBANKA_041570 |
1.4e-38 |
29 |
1 |
|
|
Atg7 |
PBANKA_092220 |
3.3e-50 |
21 |
1 |
|
|
Atg12 |
PBANKA_133320 |
1.2e-7 |
15 |
9 |
|
| Atg10 |
PBANKA_101190 |
0.14 |
9 |
1 |
|
| Atg5 |
PBANKA_101430 |
0.03 |
11 |
- |
|
| Atg16 | PBANKA_082930 | 0.0001 | 24 | - |
Apart from the Atg4 protein (underlined) all members of the Atg8 ubiquitin-like conjugation pathway are highly conserved in P. berghei (bold). Proteins of the Atg12 ubiquitin-like conjugation system either show a low conservation (underlined) or are not present at all. S. cerevisiae Atg protein sequences were used to search for P. berghei homologs using the BLAST function of PlasmoDB (expectation values of this search are presented). Percentage of pairwise identity with yeast was acquired via Needleman-Wunsch alignment. The hit number of reciprocal BLAST search against UniProtKB/Swiss-Prot(swissprot) database of the organism S. cerevisiae is indicated.
Interestingly, whereas most members of the Atg8 ubiquitin-like conjugation pathway, such as PbAtg8, PbAtg3 and PbAtg7, all have a reasonably high similarity to the respective homologous yeast proteins, the putative PbAtg4, which belongs to the same pathway, has a much lower similarity. Comparison of Atg8 proteins of different organisms revealed that unusually, Plasmodium Atg8 exposes a C-terminal glycine (Fig. 4A).12 In yeast and most other eukaryotes, Atg8 needs to be processed at its C terminus by the Atg4 protease to expose the glycine residue, which is then critical for lipidation. Since the C terminus of Plasmodium Atg8 needs no processing, a typical Atg4 protease is not necessary to expose the glycine residue for lipidation and PbAtg4 might therefore have diverged during evolution to perform a different role. Based on the bioinformatic observation that Plasmodium Atg8 proteins continually expose a glycine residue at the C terminus, it can be predicted that there is no cytoplasmic pool of Atg8 but that it is always lipidated and thus is constitutively associated with membranes.

Figure 4. Atg8 is evolutionarily conserved and the P. berghei ATG8 gene is constitutively transcribed. (A) Comparison of the Atg8 amino acid sequence of P. berghei, S. cerevisiae and Homo sapiens. Conserved amino acids are indicated with gray boxes. Note that the C-terminal glycine residue of Atg8 is exposed only for P. berghei. (B) The PbATG8 gene is transcribed in all parasites stages tested. Total RNA was prepared from P. berghei-infected red blood cells, mosquito midguts and HepG2 cells at indicated time points. RT-PCR was performed using a primer pair specific for PbATG8. To detect genomic DNA contamination (negative control), samples lacking reverse transcriptase were processed in parallel (-RT). As a positive control for each sample, a cDNA fragment of constitutively transcribed PbTubulin β chain, putative was amplified by RT-PCR.
PbAtg8 localizes to the apicoplast
Because Atg8 serves as an excellent marker for autophagy in many organisms,17,19 we focused on this protein for further characterizing the observed autophagy-like processes, including the vacuolization of merofusosomes. Transcription of the ATG8 gene was analyzed by RT-PCR. Transcripts could be detected throughout the Plasmodium life cycle (Fig. 4B). To analyze the intracellular distribution of PbAtg8, we generated transgenic parasites expressing GFP-PbAtg8 under a liver stage-specific promoter34 and constitutively expressing cytoplasmic mCherry (Fig. 5A). N-terminal GFP-tagging of PbAtg8 was chosen to exclude masking of the glycine residue at the C terminus.

Figure 5. GFP-PbAtg8 localizes to the apicoplast. (A) Schematic representation of the pLSGFP-PbAtg8CmCherry plasmid. Expression of GFP-PbAtg8 was under the control of a liver stage-specific promoter (LS);34 expression of mCherry under the control of the constitutive eef1α promoter (Con). (B) HepG2 cells were infected with transgenic P. berghei parasites expressing GFP-PbAtg8. 48 and 56 hpi, the living parasites were analyzed by fluorescence microscopy. Scale bar: 10 µm, widefield (WF). In addition, infected HepG2 cells were fixed 56 hpi and used for IFA. The parasites were stained with an anti-PbACP antiserum (ACP) to label the apicoplast (C) or an anti-TgHSP70 antiserum (HSP70) to label the mitochondrion (D). Parasites were co-stained with anti-GFP (GFP-PbAtg8) and DNA was labeled with DAPI. Areas containing details additionally displayed at a higher magnification are highlighted in the merged picture. Scale bar: 10 µm; CPS (confocal point scanning).
Live imaging at 48 and 56 hpi showed that GFP-PbAtg8 localized to structures that resembled either the apicoplast or mitochondrion (Fig. 5B). To analyze further the localization of GFP-PbAtg8, we fixed GFP-PbAtg8-infected cells and stained them with antibodies against PbACP (putative acyl carrier protein, ACP)/(PBANKA_030560), a protein localizing to the apicoplast (Fig. 5C) and HSP70, a mitochondrial protein35 (Fig. 5D). Clearly, GFP-PbAtg8 localized to the apicoplast and not to the mitochondrion.
To rule out mislocalization of the fusion protein, we sought to detect the endogenous PbAtg8 protein. We therefore generated a specific anti-PbAtg8 antiserum (Fig. S4) and used it to stain HepG2 cells infected with parasites expressing GFP targeted either to the apicoplast (PbGFPAPICO) or to the mitochondrion (PbGFPMITO)36 (Fig. 6A and B). In addition, we co-stained parasites with anti-PbAtg8 and either anti-PbACP or anti-TgHSP70 (Fig. S5). This clearly confirmed the previous experiment, showing that PbAtg8 localizes to the apicoplast and not to the mitochondrion. We analyzed different developmental stages of the parasite in HepG2 cells and found PbAtg8 to be exclusively associated with the apicoplast (data not shown). Although it was predicted that the exposed glycine residue results in constitutive lipidation and membrane association, the localization to the apicoplast was not expected.

Figure 6. Endogenous PbAtg8 localizes to the apicoplast. (A and B) HepG2 cells were infected with transgenic P. berghei parasites expressing GFP fusion proteins targeted to either the apicoplast (ApicoGFP) (A) or to the mitochondrion (MitoGFP) (B).36 56 hpi the cells were fixed and stained with anti-PbAtg8 (PbAtg8) and anti-GFP. DNA was labeled with DAPI. Areas containing details additionally displayed at a higher magnification are highlighted in the merged picture. Scale bar: 10 µm; CPS. The C-terminal glycine residue of PbAtg8 is essential for lipidation and apicoplast membrane association. (C and D) HepG2 cells were infected with transgenic P. berghei parasites expressing either GFP-PbAtg8 (C) or GFP-PbAtg8G124A (D) under a liver stage-specific promoter and mCherry under a constitutive promoter. 48 hpi parasites were analyzed by live fluorescence microscopy. Scale bar: 10 µm; WF.
Lipidation of PbAtg8 is important for localization to the apicoplast
To confirm that constitutive lipidation is necessary for the association of PbAtg8 to the apicoplast, we next generated P. berghei parasites where the terminal glycine was exchanged for alanine. For this, a GFP-PbAtg8G124A mutant parasite strain was generated by site-directed mutagenesis of the plasmid encoding wild-type GFP-PbAtg8. A single-point mutation was introduced, which leads to an amino acid substitution from glycine to alanine at position 124 of PbAtg8. GFP fused to the N terminus of PbAtg8G124A and the additional cassette for cytoplasmic mCherry expression allowed live imaging analysis. Transgenic parasites expressing the mutant GFP-PbAtg8 fusion protein under the liver stage-specific promoter showed a weak cytoplasmic distribution of the fusion protein, similar to that of mCherry (Fig. 6D), confirming the importance of the C-terminal glycine residue for lipidation and membrane association.
PbAtg8 is not involved in vesicle generation in dying liver-stage parasites
Since PbAtg8 localizes to the apicoplast in normally developing liver-stage parasites, it was now important to determine whether it localizes to autophagosome-like vesicles during parasite death. Such vesicles could, for example, be the vacuoles observed in merofusosomes but might also be a distinct set of vesicles not observed so far. At 60 hpi, we could clearly see many parasites arrested in development, some of them undergoing extensive vacuolization, indicating ongoing autophagy-like cell death (see also Fig. 1B; Vid. S2). To investigate if PbAtg8 changes localization during this type of cell death, we co-stained the parasites with antibodies against PbAtg8 and PbACP. Interestingly, we could not observe relocalization of PbAtg8 to autophagosomes; it always remained associated with the apicoplast (Fig. 7A). Even when the apicoplast was destroyed during cell death, PbAtg8 was still found associated with the remnants of this organelle. To further study PbAtg8 distribution during parasite autophagy, we treated P. berghei-infected HepG2 cells with rapamycin. Although the growth of the parasite (Fig. S6A) and the development of the apicoplast (Fig. S6B) were clearly affected by this treatment, we did not see a redistribution of PbAtg8 to classical autophagosomes. Together, in liver-stage parasites, PbAtg8 appears not to be involved in the described autophagy-like cell death of P. berghei or at least does not associate with the autophagosome-like vesicles described in this work.

Figure 7. PbAtg8 does not localize to autophagosomes in dying parasites (A) and cannot complement yeast Atg8 (B). (A) HepG2 cells were infected with P. berghei parasites constitutively expressing mCherry (mCherry). 60 hpi the cells were fixed and stained with an anti-PbACP antiserum (ACP) to label the apicoplast and an anti-PbAtg8 antiserum (PbAtg8) to monitor the localization of this protein during parasite cell death. DNA was labeled with DAPI. A representative parasite showing strong vacuolization is depicted. A higher magnification of some important details is presented in the merged image. Scale bar: 10 µm: CPS. (B) Scatg8Δ and wt strains were transformed with empty pFL61 plasmid or the same plasmid containing PbAtg8 or ScAtg8 as a positive control. Western blot analysis was performed with the transformed strains using anti-aminopeptidase I antibodies. Transport of prApe1 to the vacuole where it matures (Ape1) only takes place in the presence of a functional autophagy pathway. The prApe1 and the Ape1 bands are marked with arrows.
PbAtg8 cannot complement a yeast ATG8 knockout
To further investigate this rather unexpected result, we wanted to know whether PbAtg8 could support autophagy in a heterologous system. In a complementation assay, a S. cerevisiae strain deficient for Atg8 (Scatg8Δ) was transfected with a yeast expression vector encoding PbAtg8. The resulting yeast cell line was tested for its ability to process the vacuolar hydrolase aminopeptidase I (Ape1) from its inactive precursor (prApe1) to its mature form (Ape1), a process that is defective in Scatg8Δ mutants. Western blot analysis revealed that in the PbAtg8-complemented Scatg8Δ strain, prApe1 was not processed, indicating that the P. berghei Atg8 was not able to complement for the deletion of yeast ATG8 (Fig. 7B). In contrast, when we complemented the Scatg8Δ strain with ScAtg8, using the same yeast expression vector as above, we clearly detected processing of prApe1, confirming the validity of the assay. Together these data suggest that PbAtg8 is not a functional homolog of S. cerevisiae Atg8.
PbAtg7 and PbAtg3 localize to the mitochondrion
As Atg8 in liver-stage parasites do not appear to have a functional role in autophagy, we sought to analyze the localization of the other members of the putative Atg8 ubiquitination system, PbAtg7 and PbAtg3. We generated transgenic parasite lines expressing either Atg7-GFP (Fig. S7) or Atg3-GFP (Fig. S8) fusion proteins and additionally successfully raised antibodies against Atg7 (Fig. S7B). In liver-stage parasites, both molecules clearly localized to the mitochondrion (Figs. S7 and S8), suggesting that they are not functionally linked to PbAtg8 in this phase of parasite development. Recently it has been suggested that P. falciparum Atg8 interacts with Atg3.37 However, evidence of this interaction was restricted to structural analysis of PfAtg8 and a short Atg3 motif. Still, it is feasible that in other parasite stages apart from the asexual blood-stage and the liver-stage, Atg3 is needed to link Atg8 to membranes.
Discussion
Three major forms of cell death have been described in multicellular organisms: necrosis, apoptosis and autophagic cell death, all of which are genetically determined.5,38 Whereas necrosis results in an immediate plasma membrane breakdown provoking inflammatory immune responses, apoptosis and autophagy are the most common types of programmed cell death and are immunologically comparatively silent. Morphologically, apoptosis is characterized by cell shrinkage, membrane blebbing, altered plasma membrane composition, loss of mitochondrial integrity, chromatin condensation and DNA fragmentation. In contrast, the morphological hallmarks of autophagy are the appearance of autophagosomes and a strong vacuolization.5,31
Different forms of cell death have been postulated for drug-treated P. falciparum blood-stage parasites.39,40 Our data indicate that all three types of cell death can also occur in untreated parasites, suggesting the existence of physiological pathways underlying different manifestations of cell death. If we can identify such pathways, we may be able to specifically trigger them and, in this way, eliminate the parasite. In Plasmodium parasites, the existence of an apoptosis-like cell death has already been described30,41,42 but little is known about autophagy and autophagy-like cell death. For molecular characterization, we focused on this type of cell death because, in contrast to the other cell death types, genes coding for substantial numbers of autophagy marker proteins have been identified in the genomes of Plasmodium parasites (Table 1; Table S1).12,13 Why and when would a parasite need to employ autophagy and when an autophagic cell death? The parasite, during its life cycle, undergoes several extensive remodeling processes, and autophagy could be used to remove dispensable cellular material, similar to what has been suggested for Toxoplasma gondii parasites.3,43,44 Under starvation conditions or under other unfavorable circumstances, the parasite might also use autophagy to support the survival of at least some daughter parasites. However, if autophagy is not halted, it might then progress to a form of cell death that is immunologically relatively silent and thus favorable for the parasite population in future Plasmodium infections.
We consider the observed cell death in exoerythrocytic Plasmodium parasites to be autophagy-like cell death because it exhibits some important morphological and biological hallmarks of this type of cell death. Morphologically, vesicles with multiple membranes and parasites with extensive vacuolization have been observed. Biologically, the process occasionally allows single merozoites to survive and thus clearly differs from apoptosis and necrosis.
Although the autophagy-like cell death of exoerythrocytic parasites was observed at different developmental stages, the most interesting observation was the occurrence of merofusosomes. Merozoite formation is based on repeated invagination of the parasite membrane and merofusosomes appear to almost be a reversal of this process, whereby formed merozoites re-fuse with an enlarging cytoplasmic mass. If merofusosome formation is indeed the reversal of mechanisms behind merozoite formation, it is likely that the molecular mechanisms behind merozoite generation and destruction are also related. Thus the study of merozoite formation should lead the way for investigating the observed autophagy-like cell death and the appearance of merofusosomes and vice versa.
In this study, we followed the working hypothesis that autophagy-like cell death of Plasmodium liver-stage parasites and the typical autophagy marker protein Atg8 are functionally linked. To our surprise, in liver-stage parasites PbAtg8 appears not to be involved in autophagosome formation, as it associates with the apicoplast throughout liver-stage parasite development, even during parasite cell death. While our manuscript was under review, it was published that P. falciparum Atg8 also localizes to the apicoplast of asexual blood-stage parasites,45 confirming our observation on liver-stage parasites. In the study by Kitamura et al., the authors have used immunoelectron microscopy to confirm the unexpected Atg8 localization. The Atg8-positive structure identified as the apicoplast by Kitamura et al., looks very similar to the structures recently described by Sinai and Roepe46 and proposed to be autophagosomes of starved P. falciparum parasites. Clearly, further investigations are needed to determine whether PfAtg8 indeed re-localizes to autophagosomes in starved parasites or whether the observed structures are remnants of a disintegrated or already divided apicoplast.
Even if Plasmodium Atg8 is not involved in autophagic events during liver-stage development, we speculate that autophagy plays an important role during stage conversion to recycle obsolete cellular material from converting parasites, similar to what has been observed in other parasite species, including the closely related parasite T. gondii.25,26 It would be particularly interesting to analyze Atg8 expression and localization during male gametocyte formation, during which the apicoplast is removed.47 Since other components of the Atg8 pathways, Atg3 and Atg7, have been identified in the genome of Plasmodium parasites12-14 it will now be very interesting to investigate their function in greater detail and in other parasite stages. Our own results in liver-stage parasites indicate that they are associated with the parasite mitochondrion, suggesting that they also have a function in organelle biology rather then in the generation of autophagosomes but again, this might be different in other life-cycle stages of the parasite. Interestingly, genetic targeting of Atg3 of Toxoplasma had a severe effect on mitochondrial morphology suggesting that the localization and function of Atg3 might indeed be related to the mitochondria,26 not just in Plasmodium parasites but also in other apicomplexa.
An indication that PbAtg8 might not be a typical autophagy marker came from the fact that PbAtg8 constitutively exposes a glycine residue at the C terminus. Other Atg8 proteins need a processing step, executed by the Atg4 protease, for exposure of the glycine residue, subsequent lipidation, and membrane association.19 In fact, this is the reason why nonprocessed Atg8 is found in the cytoplasm and is only incorporated into autophagosomes after processing and lipidation. It was therefore expected that PbAtg8 would not be found in a cytoplasmic pool but would rather be constantly lipidated and incorporated into membranes, although other mechanisms to mask the C-terminal glycine might exist.42 Interestingly, T. gondii also expresses an Atg8 ortholog (TgAtg8) with an exposed C-terminal glycine residue. However, TgAtg8 still switches between the cytoplasmic, nonlipidated form and the lipidated form associated with autophagosomes,26,42 suggesting its C-terminal glycine must be masked by other means. Although the TgAtg3-TgAtg7-TgAtg8 system appears to be involved in autophagy, it obviously has an important mitochondrial function.27 In fact, TgAtg8 was partly localized to the mitochondria.
The fact that the membrane association of PbAtg8 is restricted to the apicoplast strongly suggests that PbAtg8 must possess an unknown motif directly or indirectly responsible for apicoplast association. Since lipidation allows incorporation into membranes but not transport through membranes, one can predict that PbAtg8 incorporation is restricted to the outer leaflet of the apicoplast membrane and it is therefore not surprising that normal apicoplast targeting signals for import into the organelle have not been identified. The assumption that Atg8 incorporates into the outer leaflet of the apicoplast membrane is supported by the immunoelectron analysis of Kitamura et al.45 As well as for processing prior to lipidation, Atg4 proteases are also needed to release Atg8 from membranes to control autophagosome-membrane formation.48 The fact that we never observed any release of Atg8 from the apicoplast membrane supports our assumption that PbAtg4 has diverged to fulfill an alternative function.
One question now is why Plasmodium liver-stage parasites incorporate PbAtg8 exclusively into the apicoplast, but Toxoplasma parasites do not. The answer might come from the differing biology of these parasites during their intracellular development. Whereas Toxoplasma produces daughter cells by endodyogeny, by which organelle-fission events occur prior to each of the repeated phases of cytokinesis, Plasmodium liver-stage parasites undergo schizogony, with an enormous expansion of the apicoplast and the mitochondrion to extensively branched but single organelles, with organelle fission occurring shortly before daughter cell formation.36 This impressive and very fast organelle growth requires extensive resources for membrane expansion and PbAtg8 could indeed have a role in membrane recruitment. During autophagy in other organisms, Atg8 is involved in membrane expansion and so evolving to use a similar mechanism but instead for organelle expansion appears not to be such a leap. A genetic knockout of PbAtg8 would help to further characterize its function but it appears to be essential for Plasmodium blood-stages, preventing a knockout parasite line being produced.13 This is actually a very interesting observation since a lethal phenotype would not necessarily be expected for an autophagy-restricted function but is commonly observed when genes encoding proteins with a functional role in the apicoplast are genetically targeted.47,49
If PbAtg8 is involved in apicoplast membrane expansion, why would the mitochondrion be able to grow to a similar extent independently of PbAtg8? The reason might be that growth and branching of the two organelles do not rely on identical molecular machineries. The observed extensive branching of the apicoplast47 is restricted to a subset of apicomplexan parasites, whereas branching of mitochondria is a very ancient process found in most protozoa and multicellular organisms. In fact, mammalian mitochondria in some cell types exhibit huge branching and Atg8 has never been associated with these organelles. Similar to this, Plasmodium parasites might employ the mitochondrion-specific branching machinery without the help of Atg8. Clearly more research is needed to explore the exact function of PbAtg8 during the liver-stage but also in other parasite stages and to understand the mechanistic differences between the expansion of the apicoplast and the mitochondrion.
Since PbAtg8 appears not to be directly involved in autophagy, the molecular details of the observed autophagy-like cell death in Plasmodium liver-stage parasites still remain to be explored. This is the most interesting aspect of studying parasite cell death as an understanding of the underlying molecular mechanisms might allow the triggering of parasite death at any given time during development, including in the dormant stages of P. vivax.
Materials and Methods
Animal work statement
Mice used in the experiments were between 6 and 10 weeks of age and were bred in the central animal facility of the University of Bern or in-house at the Bernhard Nocht Institute. Experiments were conducted with strict accordance to the guidelines of the Swiss Tierschutzgesetz (TSchG; Animal Rights Laws) and European regulations and approved by local authorities in Bern and Hamburg.
Database searches and sequence analysis
Plasmodium berghei putative orthologs of proteins involved in autophagy were identified using the BLAST functions of the Plasmodium Genomics Resource webpage (www.plasmodb.org). Percentage pairwise identities with yeast sequences were acquired by alignment using the Needleman-Wunsch algorithm. Hit numbers of reciprocal BLAST search against the UniProtKB/Swiss-Prot (swissprot) database S. cerevisiae were acquired via the National Center for Biotechnology Information (NCBI) webpage (www.ncbi.nlm.nih.gov). Sequence analysis was performed using MacVector.
Generation of transgenic P. berghei parasites
All parasite strains used in the paper have a P. berghei ANKA background. PbcmCherry parasites express mCherry under the constitutive eef1αa promoter and show cytosolic localization of the fluorescent protein (pcmCherry).50 PbcGFPMITO parasites and PbcGFPAPICO parasites express GFP targeted to the mitochondrion and apicoplast, respectively, and under the constitutive eef1α promoter.36 For generation of parasites expressing GFP-PbAtg8 (PBANKA_050401), cDNA was amplified using the primer pair 5′-GGTCTAGAATGCCATCATTAAAAG-3′ and 5′-GGTCTAGATTATCCAAAACAAC-3′ and ligated into a modified version of the pGFP103464 plasmid,34 where the stop codon had been eliminated, to generate the pLSGFP-PbAtg8 plasmid. The expression cassette for constitutive mCherry expression was amplified by PCR from the pCmCherry plasmid using the primers 5′-CGTAGGTACCAGCTTAATTCTTTTCGAGCTCTTT-3′, binding to the 5′ of the eef1α promoter and 5′-ACTGGGTACCCGAAATTGAAGGAAAAAACATCATTTG-3′, binding to the 3′ of the 3′UTR of the pbdhfr/ts. The cassette was cloned into the pLSGFP-PbAtg8 plasmid via KpnI (New England Biolabs, R0142) to generate the pLSGFP-PbAtg8CmCherry plasmid. The same procedure was performed with the primer pair 5′-GGTCTAGAATGCCATCATTAAAAG-3′ and 5′- GGTCTAGATTATGCAAAACAACTTTCACAAC -3′ using to generate the pLSGFP-PbAtg8G124ACmCherry plasmid. Primer pair 5′-GGGGATCCATGCACAAAATAGGAGATGC-3′ and 5′-GGGGATTCTATATATTTATCTTTATATG-3′ and the unmodified pGFP103464 plasmid was used to generate pLSPbAtg3-GFPCmCherry. pLSPbAtg7-GFPCmCherry was generated using the same method but with gDNA as template and using primer pair 5′-GGGGATTCATGAATTCTTTAAAATATGAAATAATTCC-3′ and 5′-GGGGATTCCTCAAAAATTATTACATCTTTTTCATCGGC-3′. The generated plasmids were transfected into P. berghei blood-stage parasites, using previously published methods.51 Plasmids allow integration into the c- and d-ssu-rrna locus by single crossover.
Generation of mammalian expression vectors
PbATG8 was amplified from cDNA using primer pair 5′-CCCTCGAGCCATGCCATCATTAAAAG-3′ and 5′-CCGGATCCTTATCCAAAACAACTTTCACAAC-3′ and ligated into pEGFP-C1 with appropriate restriction enzymes (New England Biolabs, R0146, R0136).
Culture and in vitro infection of HepG2 cells
HepG2 cells were purchased from the European Cell Culture Collection and kept in Minimum Essential Medium with Earle’s salts supplemented with 10% FCS, 1% penicillin/streptomycin and 1% L-glutamine (all from PAA Laboratories, E15-024, A15-101, P11-010, M11-004). They were cultured at 37°C and 5% CO2 and split twice a week using Accutase (PAA Laboratories, L11-007). For infection, either 3 or 6 × 105 cells were seeded on coverslips in 24-well plates or on glass-bottom dishes. HepG2 cells were infected with sporozoites prepared from the salivary glands of female Anopheles stephensi mosquitoes infected with the aforementioned P. berghei parasites. Sporozoites were incubated with HepG2 cells for 2 h and cells were then washed and incubated with MEM medium described above containing Amphotericin B (PAA Laboratories, P11-001) at 37°C and experiments performed as described below. For autophagy induction, cells were treated from 24 hpi with 0.5 µM Rapamycin (LC Laboratories, R-5000). After 48 hpi the area of the parasites was quantified using ImageJ.
Transfection of HepG2 cells
HepG2 cells were harvested by Accutase treatment and 2 × 106 cells were pelleted by centrifugation at 160 g. They were resuspended in Nucleofector V solution (Lonza, VVCA-1003) and transfected with 3 µg pEGFP-N1 plasmid (Clontech, 6085-1) using program T-28 of the Nucleofector transfection device according to the manufacturer’s instructions.
Quantification of infected attached and detached cells
To quantify the number of parasites that are able to complete liver-stage development successfully, HepG2 cells were infected as described above. Sixty-eight and 72 hpi the culture supernatant containing the detached cells and merosomes was combined in a fresh well of a 24-well plate, nuclei were stained using 1 µg/ml Hoechst 33342 (Sigma, B2261) and detached cells were counted using a Leica DMI6000B widefield epifluorescence microscope. In addition the number of infected, but still attached, cells was counted. The quantitative data are presented as mean ± SD.
RT-PCR analysis
Total RNA was isolated from 0.05% saponin-treated (Sigma, 47036) infected red blood cells, 20 infected mosquito midguts day 10 after infection and infected HepG2 cells 24, 48 and 56 h after infection using the NucleoSpin RNA Extract Kit II (Macherey-Nagel, 740955.50). Random-primed cDNA synthesis was performed in a reverse transcriptase reaction; the resulting cDNA was then used as a template in PCR reactions with primer pair 5′-TGTGTGAAAAAGCGCATAGATCAA-3′ and 5′- TTTTTGGAATAATATTATTGACAAATAAATAT-3′. As an internal control, P. berghei tubulin β chain, putative (PBANKA_120690) cDNA was amplified using primer pair 5′-TGGAGCAGGAAATAACTGGG-3′ and 5′-ACCTGACATAGCGGCTGAAA-3′.
Protein expression and antibody purification
For antibody generation, the coding sequence of PbATG8 was amplified from cDNA using primer pair 5′-GGGGATCCATGCCATCATTAAAAG-3′ and 5′-GGGGTCGACTTATCCAAAACAAC-3′. PbACP (PBANKA_030560) was amplified without the apicoplast targeting sequence using primer pair 5′-ATGAATTCTTCAAAAATATGAGCAACCATGCC-3′ and 5′-ATCTCGAGTTATGCATCAGGCTTTTTATTTTTTTCTAT-3′. The PCR products were cloned into pGEX-6P-1 using appropriate restriction enzymes (New England Biolabs, R0138, R0136, R0101, R0146). GST-PbAtg8 and GST-PbACP were expressed in the BL21 (D3) RIG E. coli strain and purified using glutathione-agarose resin (Sigma, G4510). For generation of GST-PbAtg8 antisera, 20 µg of the purified protein was mixed with one volume of Freund’s adjuvant complete (Sigma, F5881) and intraperitoneally injected into an NMRI mouse. After two weeks, the mouse was boosted with the same amount of protein mixed with Freund’s adjuvant incomplete (Sigma, F5506) followed by a second boost two weeks later. The immunized mouse was sacrificed, blood was collected and antiserum was obtained after centrifugation of the coagulated blood. The GST-PbAtg8 antiserum was affinity-purified using blotted MBP-PbAtg8 as follows. The coding sequence of PbATG8 was amplified from cDNA using primer pair 5′-GGGGATCCATGCCATCATTAAAAG-3′ and 5′-GGGGTCGACTTATCCAAAACAAC-3′ and cloned into pMAL-cRI . MBP-PbAtg8 was expressed in the BL21 (D3) RIG E. coli strain and purified using Amylose Resin (New England Biolabs, E8021S). 200 µg protein were loaded on a SDS-Gel and blotted onto a PVDF membrane. Upon Ponceau-staining for visualization, the band corresponding to the antigen was cut out. After blocking with TBS/2%BSA, the band was incubated with the GST-PbAtg8 antiserum. After three washing steps the bound antibodies were eluted with glycine buffer, pH 2.8 and in a last step neutralized with 1M Tris base to a pH of 7.5. For GST-PbACP, 80 µg of purified protein was mixed with one volume of Gerbu adjuvant 100 (GerbuBiotechnik GmbH, 3100) and subcutaneously injected into a Wistar rat, followed by two boosts at two week intervals. The immunized rat was sacrificed after confirming the presence of specific antibody in preliminary experiments and the blood was collected. Antisera was obtained after centrifugation of the blood.
For PbAtg7 antibody production, a part of the coding sequence of PbATG7 (PBANKA_092220) was amplified using primer pair 5′- GGGGATCCAATTCACGAGTTTCTTTTTC-3′ and 5′- GGGGTCGACTTATTCATCTATCGTTCTATAAC-3′ and cloned into pMAL-cRI and pGEX-6P-1. MBP-PbAtg7 and GST-PbAtg7 were expressed in the BL21 (D3) RIG E. coli strain and purified using Amylose Resin or glutathione agarose beads, respectively. The immunization of a rat with MBP-PbAtg7 was performed at Eurogentec. The antiserum obtained after three rounds of immunization was used directly in IFAs and western blot analysis.
Indirect immunofluorescence analysis
HepG2 cells were infected as described above. After the indicated time periods, cells were fixed with 4% paraformaldehyde in PBS (20 min, room temperature) and permeabilized with ice-cold methanol (10 min). After washing with PBS, unspecific binding sites were blocked by incubation in 10% FCS/PBS for 1 h at room temperature followed by incubation with primary antibody (mouse anti-PbAtg8; rabbit anti-TgHSP70; rabbit anti-GFP, Invitrogen, A-11122), and subsequently with fluorescently labeled secondary antibodies (anti-rabbit HiLyteFluorTM 647, AnaSpec, 81255; anti-mouse Alexa488 Molecular Probes, A11001). For co-staining with mouse anti-PbAtg8 and rat anti-PbACP, sequential staining was performed. The first staining was performed as described above. After an additional 10 min of fixation with 4% paraformaldehyde, a second staining step with rat anti-PbACP and subsequently with anti-rat Alexa 647 (Molecular probes®, A21247) was performed. DNA was visualized by staining with 10 µg/ml DAPI (Sigma). Labeled cells were mounted on microscope slides with Dako Fluorescent Mounting Medium (Dako, S3023) and analyzed by confocal point scanning (CPS) microscopy using the Leica TCS SP5. Images were acquired using a Leica HCX PL APO CS 63× 1.2 water objective and the Leica LAS AF Software, Version 2.6.0.7266.
Live cell imaging and time-lapse microscopy
Live cell imaging and time-lapse microscopy was performed using the Leica DMI6000B widefield epifluorescence microscope (Wetzlar). Images were acquired using a Leica HCX PL APO 100× 1.4 oil objective and the Leica LAS AF Software, Version 2.6.0.7266. In addition, time-lapse microscopy was also performed with a Zeiss Observer, Z1 inverted microscope, integrated into an LSM5 Live imaging setup. Images were acquired by confocal line scanning (CLS) microscopy using a Zeiss Plan-Apochromat 63×/1.4 oil objective and the Zeiss Efficient Navigation 2008 and 2009 software (LSM). During imaging, cells were kept in 5% CO2 at 37°C. Intravital microscopy was performed as previously described52 using the LSM 510 Zeiss microscope in the LSM 5 live mode. Images were acquired by CLS microscopy using a Zeiss Plan-Apochromat 63×/1.40 oil DIC M27 objective and the Zeiss LSM 5 Duo Release Version 4.2. Lys-GFP mice that express GFP mainly in neutrophils and macrophages32 were infected with mCherry-expressing parasites and intravital imaging of the anesthetized mouse was performed between 44 and 48 hpi. Image processing was performed using ImageJ.
Yeast complementation
Genes coding for PbATG8 and ScATG8 were amplified from cDNA using primer pairs 5′- GGCGGCCGCATGCCA TCATTAAAAG-3′ and 5′- GGGCGGCCGCTTATCCAAAACAACTTTC3′ and 5′-GGCGGCCGCCTACCTGCCAAATGTATTTTC-3′ and 5′-GGGCGGCCGCTAGCCTACCTGC CAAATGTAT TTTC-3, respectively. The fragments were cloned into the yeast expression vector pFL6153 via NotI (New England Biolabs, R0189) and the resulting plasmids used to transform Scatg8Δ WCG strains of S. cerevisiae by the acetate method.54 In addition, the Scatg8Δ WCG strain and the wild-type WCG strain were transformed with the empty vector. Transformant cultures were grown in yeast nitrogen base media containing 0.5% ammonium sulfate and containing amino acids but lacking uracil, until an optical density of 1 at 600 nm was reached. At an OD of 7.5 yeast strains were centrifuged and resuspended in 100 µl Laemmli’s sample buffer each. By addition of 200 µl glass beads (0.45 to 0.50 mm, Braun Biotechnology, 8541701) and an incubation in the FastPrep® FP120 Cell Disrupter (Qbiogene, Inc.) at 4.0 m/sec for 45 sec, the yeast cells were lysed. The extraction was performed five times, with cells incubated on ice. Before analysis by western blotting, the lysate was incubated at 98°C for 7 min.
Western blotting
Parasite lysate was generated by lysing P. berghei-infected red blood cells with 0.2% Saponin for 20 min at room temperature. After centrifugation, the parasite pellet was resuspended in 2× Laemmli’s sample buffer and incubated at 98°C for 5 min. Parasite lysates, proteins or yeast lysates were separated on a 12% or 14% SDS gel and blotted onto an Immobilon® FL Transfer membrane (MilliporeTM, IPFL00010). After blocking for 1 h at room temperature in 5% milk powder/TBS, the appropriate primary antibody was applied in 5% milk powder/0.05% Tween-20/TBS at 4°C overnight. The next day, the blot was washed three times with 0.05% Tween-20/TBS and the secondary antibody (Li-COR, 926-32210, 926-32219, 926-32211) was applied in 5% milk powder/0.05% Tween-20/TBS/0,02% SDS for 1 h at room temperature in the dark. Bands were visualized using the Odyssey Infrared Imaging System (Li-COR).
Histology of P. berghei-infected liver
NMRI mice between 8 and 10 weeks of age were infected with 1 × 106 P. berghei ANKA sporozoites obtained from salivary glands of infected Anopheles stephensi mosquitoes. After 38 hpi, livers were removed and fixed overnight in 4% buffered formaldehyde solution and embedded in paraffin using standard methods. Sections (5-µm thick) were cut and stained with Giemsa.
Electron microscopy
HepG2 cells were infected with PbGFPcon parasites. Cells were FACS-sorted to enrich for infected cells and seeded on Thermanox coverslips. 48 hpi cells were washed twice with PBS, fixed with 2% glutaraldehyde in sodium-cacodylate buffer, pH 7.2 and postfixed with 1% osmium tetroxide. Samples were dehydrated at increasing ethanol concentrations and propylene oxide. Cells were embedded in an epoxy resin (Epon). Ultrathin sections were made using Ultra Cut E (Reichert/Leica) and stained with uranyl acetate and lead citrate. Sections were examined with a Tecnai Spirit (Fei) at an acceleration voltage of 80 kV.
Supplementary Material
Acknowledgments
We are grateful to Daniel Klionsky for providing the anti-Ape1 antiserum and to Dominique Soldati for the anti-HSP70 antiserum. We thank Michael Thumm for the WCG wild-type and the Scatg8Δ mutant yeast strains and for the ScATG8 cDNA. Stefanie Graewe is thanked for providing Video S2. This work was supported by grants from DFG (HE 4497/1-2), the Indo Swiss Joint Research Programme and the EviMalaR EU consortium.
Glossary
Abbreviations:
- ACP
acyl carrier protein
- Ape1
vacuolar hydrolase aminopeptidase 1
- APICO
apicoplast
- Atg
autophagy-related
- Atg12
autophagy-related 12
- Atg16
autophagy-related 16
- Atg3
autophagy-related 3
- Atg4
autophagy-related 4
- Atg5
autophagy-related 5
- Atg7
autophagy-related 7
- Atg8
autophagy-related 8
- BCL2
B-cell lymphoma 2
- CLS
confocal line scanning
- con
constitutive
- CPS
confocal point scanning
- DAPI
4',6-diamidino-2-phenylindole
- DNA
deoxyribonucleic acid
- GFP
green-fluorescent protein
- GST
glutathione S-transferase
- hpi
hours post infection
- HSP70
heat shock protein 70
- IFA
immunofluorescence analysis
- MAP1LC3/LC3
microtubule-associated protein 1 light chain 3
- Pb
Plasmodium berghei
- PbAtg3
Plasmodium berghei autophagy-related 3
- PbAtg4
Plasmodium berghei autophagy-related 4
- PbAtg7
Plasmodium berghei autophagy-related 7
- PbAtg8
Plasmodium berghei autophagy-related 8
- PBS
phosphate-buffered saline
- PCR
polymerase chain reaction
- PE
phosphatidylethanolamine
- Pf
Plasmodium falciparum
- PfAtg8
Plasmodium falciparum autophagy-related 8
- prApe1
precursor of vacuolar hydrolase aminopeptidase 1
- PV
parasitophorous vacuole
- PVM
parasitophorous vacuole membrane
- RT
reverse transcriptase
- Sc
Saccharomyces cerevisiae
- ScAtg8
Saccharomyces cerevisiae autophagy-related 8
- SD
standard deviation
- TBS
tris-buffered saline
- TEM
transmission electron microscopy
- Tg
Toxoplasma gondii
- TgAtg3
Toxoplasma gondii autophagy-related 3
- TgAtg7
Toxoplasma gondii autophagy-related 7
- TgAtg8
Toxoplasma gondii autophagy-related 8
- TgHSP70
Toxoplasma gondii heat shock protein 70
- WF
widefield
- wt
wild-type
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/23689
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/23689
References
- 1.van de Sand C, Horstmann S, Schmidt A, Sturm A, Bolte S, Krueger A, et al. The liver stage of Plasmodium berghei inhibits host cell apoptosis. Mol Microbiol. 2005;58:731–42. doi: 10.1111/j.1365-2958.2005.04888.x. [DOI] [PubMed] [Google Scholar]
- 2.Bruchhaus I, Roeder T, Rennenberg A, Heussler VT. Protozoan parasites: programmed cell death as a mechanism of parasitism. Trends Parasitol. 2007;23:376–83. doi: 10.1016/j.pt.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 3.Lüder CG, Stanway RR, Chaussepied M, Langsley G, Heussler VT. Intracellular survival of apicomplexan parasites and host cell modification. Int J Parasitol. 2009;39:163–73. doi: 10.1016/j.ijpara.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 4.van Zandbergen G, Lüder CG, Heussler V, Duszenko M. Programmed cell death in unicellular parasites: a prerequisite for sustained infection? Trends Parasitol. 2010;26:477–83. doi: 10.1016/j.pt.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 5.Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. Nomenclature Committee on Cell Death 2009 Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meslin B, Beavogui AH, Fasel N, Picot S. Plasmodium falciparum metacaspase PfMCA-1 triggers a z-VAD-fmk inhibitable protease to promote cell death. PLoS One. 2011;6:e23867. doi: 10.1371/journal.pone.0023867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carlton JM, Angiuoli SV, Suh BB, Kooij TW, Pertea M, Silva JC, et al. Genome sequence and comparative analysis of the model rodent malaria parasite Plasmodium yoelii yoelii. Nature. 2002;419:512–9. doi: 10.1038/nature01099. [DOI] [PubMed] [Google Scholar]
- 9.Nedelcu AM. Comparative genomics of phylogenetically diverse unicellular eukaryotes provide new insights into the genetic basis for the evolution of the programmed cell death machinery. J Mol Evol. 2009;68:256–68. doi: 10.1007/s00239-009-9201-1. [DOI] [PubMed] [Google Scholar]
- 10.Deponte M, Becker K. Plasmodium falciparum--do killers commit suicide? Trends Parasitol. 2004;20:165–9. doi: 10.1016/j.pt.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 11.Tsujimoto Y, Shimizu S. Another way to die: autophagic programmed cell death. Cell Death Differ. 2005;12(Suppl 2):1528–34. doi: 10.1038/sj.cdd.4401777. [DOI] [PubMed] [Google Scholar]
- 12.Brennand A, Gualdrón-López M, Coppens I, Rigden DJ, Ginger ML, Michels PA. Autophagy in parasitic protists: unique features and drug targets. Mol Biochem Parasitol. 2011;177:83–99. doi: 10.1016/j.molbiopara.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 13.Duszenko M, Ginger ML, Brennand A, Gualdrón-López M, Colombo MI, Coombs GH, et al. Autophagy in protists. Autophagy. 2011;7:127–58. doi: 10.4161/auto.7.2.13310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rigden DJ, Herman M, Gillies S, Michels PA. Implications of a genomic search for autophagy-related genes in trypanosomatids. Biochem Soc Trans. 2005;33:972–4. doi: 10.1042/BST20050972. [DOI] [PubMed] [Google Scholar]
- 15.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–77. doi: 10.1016/S1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 16.Klionsky DJ, Baehrecke EH, Brumell JH, Chu CT, Codogno P, Cuervo AM, et al. A comprehensive glossary of autophagy-related molecules and processes (2nd edition) Autophagy. 2011;7:1273–94. doi: 10.4161/auto.7.11.17661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–75. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mizushima N. Methods for monitoring autophagy. Int J Biochem Cell Biol. 2004;36:2491–502. doi: 10.1016/j.biocel.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 20.Besteiro S, Williams RA, Morrison LS, Coombs GH, Mottram JC. Endosome sorting and autophagy are essential for differentiation and virulence of Leishmania major. J Biol Chem. 2006;281:11384–96. doi: 10.1074/jbc.M512307200. [DOI] [PubMed] [Google Scholar]
- 21.Williams RA, Smith TK, Cull B, Mottram JC, Coombs GH. ATG5 is essential for ATG8-dependent autophagy and mitochondrial homeostasis in Leishmania major. PLoS Pathog. 2012;8:e1002695. doi: 10.1371/journal.ppat.1002695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Williams RA, Woods KL, Juliano L, Mottram JC, Coombs GH. Characterization of unusual families of ATG8-like proteins and ATG12 in the protozoan parasite Leishmania major. Autophagy. 2009;5:159–72. doi: 10.4161/auto.5.2.7328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alvarez VE, Kosec G, Sant’Anna C, Turk V, Cazzulo JJ, Turk B. Autophagy is involved in nutritional stress response and differentiation in Trypanosoma cruzi. J Biol Chem. 2008;283:3454–64. doi: 10.1074/jbc.M708474200. [DOI] [PubMed] [Google Scholar]
- 24.Li FJ, Shen Q, Wang C, Sun Y, Yuan AY, He CY. A role of autophagy in Trypanosoma brucei cell death. Cell Microbiol. 2012;14:1242–56. doi: 10.1111/j.1462-5822.2012.01795.x. [DOI] [PubMed] [Google Scholar]
- 25.Besteiro S. Role of ATG3 in the parasite Toxoplasma gondii: autophagy in an early branching eukaryote. Autophagy. 2012;8:435–7. doi: 10.4161/auto.19289. [DOI] [PubMed] [Google Scholar]
- 26.Besteiro S, Brooks CF, Striepen B, Dubremetz JF. Autophagy protein Atg3 is essential for maintaining mitochondrial integrity and for normal intracellular development of Toxoplasma gondii tachyzoites. PLoS Pathog. 2011;7:e1002416. doi: 10.1371/journal.ppat.1002416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ghosh D, Walton JL, Roepe PD, Sinai AP. Autophagy is a cell death mechanism in Toxoplasma gondii. Cell Microbiol. 2012;14:589–607. doi: 10.1111/j.1462-5822.2011.01745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Totino PR, Daniel-Ribeiro CT, Corte-Real S, de Fátima Ferreira-da-Cruz M. Plasmodium falciparum: erythrocytic stages die by autophagic-like cell death under drug pressure. Exp Parasitol. 2008;118:478–86. doi: 10.1016/j.exppara.2007.10.017. [DOI] [PubMed] [Google Scholar]
- 29.Graewe S, Stanway RR, Rennenberg A, Heussler VT. Chronicle of a death foretold: Plasmodium liver stage parasites decide on the fate of the host cell. FEMS Microbiol Rev. 2012;36:111–30. doi: 10.1111/j.1574-6976.2011.00297.x. [DOI] [PubMed] [Google Scholar]
- 30.Sturm A, Amino R, van de Sand C, Regen T, Retzlaff S, Rennenberg A, et al. Manipulation of host hepatocytes by the malaria parasite for delivery into liver sinusoids. Science. 2006;313:1287–90. doi: 10.1126/science.1129720. [DOI] [PubMed] [Google Scholar]
- 31.Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23:2891–906. doi: 10.1038/sj.onc.1207521. [DOI] [PubMed] [Google Scholar]
- 32.Faust N, Varas F, Kelly LM, Heck S, Graf T. Insertion of enhanced green fluorescent protein into the lysozyme gene creates mice with green fluorescent granulocytes and macrophages. Blood. 2000;96:719–26. [PubMed] [Google Scholar]
- 33.Meis JF, Verhave JP. Exoerythrocytic development of malarial parasites. Adv Parasitol. 1988;27:1–61. doi: 10.1016/S0065-308X(08)60352-8. [DOI] [PubMed] [Google Scholar]
- 34.Helm S, Lehmann C, Nagel A, Stanway RR, Horstmann S, Llinas M, et al. Identification and characterization of a liver stage-specific promoter region of the malaria parasite Plasmodium. PLoS One. 2010;5:e13653. doi: 10.1371/journal.pone.0013653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pino P, Aeby E, Foth BJ, Sheiner L, Soldati T, Schneider A, et al. Mitochondrial translation in absence of local tRNA aminoacylation and methionyl tRNA Met formylation in Apicomplexa. Mol Microbiol. 2010;76:706–18. doi: 10.1111/j.1365-2958.2010.07128.x. [DOI] [PubMed] [Google Scholar]
- 36.Stanway RR, Mueller N, Zobiak B, Graewe S, Froehlke U, Zessin PJ, et al. Organelle segregation into Plasmodium liver stage merozoites. Cell Microbiol. 2011;13:1768–82. doi: 10.1111/j.1462-5822.2011.01657.x. [DOI] [PubMed] [Google Scholar]
- 37.Hain AU, Weltzer RR, Hammond H, Jayabalasingham B, Dinglasan RR, Graham DR, et al. Structural characterization and inhibition of the Plasmodium Atg8-Atg3 interaction. J Struct Biol. 2012;180:551–62. doi: 10.1016/j.jsb.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–20. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ch’ng JH, Kotturi SR, Chong AG, Lear MJ, Tan KS. A programmed cell death pathway in the malaria parasite Plasmodium falciparum has general features of mammalian apoptosis but is mediated by clan CA cysteine proteases. Cell Death Dis. 2010;1:e26. doi: 10.1038/cddis.2010.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Engelbrecht D, Durand PM, Coetzer TL. On Programmed Cell Death in Plasmodium falciparum: Status Quo. J Trop Med. 2012;2012:646534. doi: 10.1155/2012/646534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arambage SC, Grant KM, Pardo I, Ranford-Cartwright L, Hurd H. Malaria ookinetes exhibit multiple markers for apoptosis-like programmed cell death in vitro. Parasit Vectors. 2009;2:32. doi: 10.1186/1756-3305-2-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Besteiro S. Which roles for autophagy in Toxoplasma gondii and related apicomplexan parasites? Mol Biochem Parasitol. 2012;184:1–8. doi: 10.1016/j.molbiopara.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 43.Besteiro S. Role of ATG3 in the parasite Toxoplasma gondii: autophagy in an early branching eukaryote. Autophagy. 2012;8:435–7. doi: 10.4161/auto.19289. [DOI] [PubMed] [Google Scholar]
- 44.Coppens I. Metamorphoses of malaria: the role of autophagy in parasite differentiation. Essays Biochem. 2011;51:127–36. doi: 10.1042/bse0510127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kitamura K, Kishi-Itakura C, Tsuboi T, Sato S, Kita K, Ohta N, et al. Autophagy-related Atg8 localizes to the apicoplast of the human malaria parasite Plasmodium falciparum. PLoS One. 2012;7:e42977. doi: 10.1371/journal.pone.0042977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sinai AP, Roepe PD. Autophagy in Apicomplexa: a life sustaining death mechanism? Trends Parasitol. 2012;28:358–64. doi: 10.1016/j.pt.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stanway RR, Witt T, Zobiak B, Aepfelbacher M, Heussler VT. GFP-targeting allows visualization of the apicoplast throughout the life cycle of live malaria parasites. Biol Cell. 2009;101:415–30, 5, 430. doi: 10.1042/BC20080202. [DOI] [PubMed] [Google Scholar]
- 48.Yu ZQ, Ni T, Hong B, Wang HY, Jiang FJ, Zou S, et al. Dual roles of Atg8-PE deconjugation by Atg4 in autophagy. Autophagy. 2012;8:883–92. doi: 10.4161/auto.19652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McFadden GI. The apicoplast. Protoplasma. 2011;248:641–50. doi: 10.1007/s00709-010-0250-5. [DOI] [PubMed] [Google Scholar]
- 50.Graewe S, Retzlaff S, Struck N, Janse CJ, Heussler VT. Going live: a comparative analysis of the suitability of the RFP derivatives RedStar, mCherry and tdTomato for intravital and in vitro live imaging of Plasmodium parasites. Biotechnol J. 2009;4:895–902. doi: 10.1002/biot.200900035. [DOI] [PubMed] [Google Scholar]
- 51.Janse CJ, Franke-Fayard B, Mair GR, Ramesar J, Thiel C, Engelmann S, et al. High efficiency transfection of Plasmodium berghei facilitates novel selection procedures. Mol Biochem Parasitol. 2006;145:60–70. doi: 10.1016/j.molbiopara.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 52.Thiberge S, Blazquez S, Baldacci P, Renaud O, Shorte S, Ménard R, et al. In vivo imaging of malaria parasites in the murine liver. Nat Protoc. 2007;2:1811–8. doi: 10.1038/nprot.2007.257. [DOI] [PubMed] [Google Scholar]
- 53.Minet M, Dufour ME, Lacroute F. Complementation of Saccharomyces cerevisiae auxotrophic mutants by Arabidopsis thaliana cDNAs. Plant J. 1992;2:417–22. doi: 10.1111/j.1365-313x.1992.00417.x. [DOI] [PubMed] [Google Scholar]
- 54.Goldshmidt H, Matas D, Kabi A, Carmi S, Hope R, Michaeli S. Persistent ER stress induces the spliced leader RNA silencing pathway (SLS), leading to programmed cell death in Trypanosoma brucei. PLoS Pathog. 2010;6:e1000731. doi: 10.1371/journal.ppat.1000731. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.