

Abstract
Increasing evidence suggests oxidative damage as a key factor contributing to the failure of the conventional outflow pathway tissue to maintain appropriate levels of intraocular pressure, and thus increase the risk for developing glaucoma, a late-onset disease which is the second leading cause of permanent blindness worldwide. Autophagy is emerging as an essential cellular survival mechanism against a variety of stressors, including oxidative stress. Here, we have monitored, by using different methodologies (LC3-I to LC3-II turnover, tfLC3, and Cyto ID), the induction of autophagy and autophagy flux in TM cells subjected to a normobaric hyperoxic model of mild chronic oxidative stress. Our data indicate the MTOR-mediated activation of autophagy and nuclear translocation of TFEB in oxidatively stressed TM cells, as well as the role of autophagy in the occurrence of SA-GLB1/SA-β-gal. Concomitant with the activation of the autophagic pathway, TM cells grown under oxidative stress conditions displayed, however, reduced cathepsin (CTS) activities, reduced lysosomal acidification and impaired CTSB proteolytic maturation, resulting in decreased autophagic flux. We propose that diminished autophagic flux induced by oxidative stress might represent one of the factors leading to progressive failure of cellular TM function with age and contribute to the pathogenesis of primary open angle glaucoma.
Keywords: TFEB, autophagy, cathepsin, glaucoma, lysosomes, oxidative stress, senescence-associated-β-galactosidase, trabecular meshwork
Introduction
The trabecular meshwork (TM), a tiny tissue located in the anterior segment of the eye between the cornea and the sclera, is one of the tissues involved in maintaining appropriate levels of IOP. Elevated IOP occurs when the amount of aqueous humor entering the anterior chamber of the eye cannot exit through the TM conventional outflow pathway.1 Resistance to aqueous humor outflow increases with aging, although the molecular mechanisms responsible are not clear yet.2,3 Evidence suggests that acceleration in the production of reactive oxygen species (ROS) causes oxidative damage to the TM with aging, and that this might contribute to the observed loss in TM tissue functionality in ocular hypertension and in primary open angle glaucoma (POAG). Supporting this hypothesis, a number of studies show increased expression of oxidative markers, including oxidative DNA damage and peroxidized lipids, as well as diminished overall antioxidant potential in the glaucomatous TM tissue.4-13 Consistent with this is the particular higher sensitivity of the TM to oxidative radicals compared with other tissues in the anterior chamber of the eye.14
One of the potential cellular responses to ROS and oxidative damage is induction of autophagy.15-17 Autophagy is an evolutionarily conserved mechanism that allows for the degradation of long-lived proteins and organelles within lysosomes by lysosomal hydrolases. Three different types of autophagy have been described in mammalian cells based on the delivery route of the cytoplasmic material to the lysosomal lumen. Among them, macroautophagy (referred to here as autophagy) is the most extensively studied. Autophagy is a dynamic process that involves (1) the formation of autophagosomes, a double membrane-bound organelle that engulfs material targeted for degradation; (2) the maturation of autophagosomes into autolysosomes, thereby acquiring lysosomal enzymes and acidic pH; and (3) the degradation of the inner limiting membrane of the autophagosome and cargo material by lysosomal hydrolases. All these steps are highly regulated by a number of evolutionary conserved autophagy related genes (ATG genes) and ubiquitin-like conjugation systems.18
Autophagy occurs constitutively at basal levels, and it is rapidly upregulated by stress conditions (i.e., starvation, oxidative stress), playing an active role in maintaining normal cellular homeostasis and assisting in the clearance of misfolded proteins and damaged organelles.19,20 The importance of autophagy is highlighted by an increased number of studies linking dysfunction in the autophagy pathway with several human diseases, from infectious diseases to cancer and neurodegeneration.21 Moreover, a decline in autophagy has been observed in most tissues with aging and has been considered responsible, at least in part, for the accumulation of damaged cellular components in almost all tissues of aging organisms.22,23
Our laboratory has recently demonstrated that chronic exposure of TM cells to oxidative stress, as an in vitro model of aging, causes profound changes in the lysosomal system, including increased lysosomal mass and content of autophagic vacuoles, accumulation of intralysosomal oxidized material and damaged mitochondria, as well as decreased cathepsin L activity.24 Together with these changes, oxidatively stressed cultures show elevated senescence-associated-β-galactosidase (SA-GLB1/SA-β-gal), which is also elevated in the TM from glaucoma donors compared with age-matched controls.25 While some of these findings indicate the activation of the lysosomal degradative pathway in response to oxidative damage in TM cells, others suggest impaired lysosomal function and decreased degradative capacity in the stressed cultures. In order to clarify these potentially conflicting results, we have further investigated here the effect of chronic oxidative stress in the autophagic function in TM cells. For this, we have monitored, by different methodologies, the induction of autophagy and autophagy flux in TM cells subjected to mild, chronic oxidative stress. Our data indicate the induction of autophagy in chronically stressed TM cells. Concomitant with the activation of the autophagic pathway, TM cells grown under oxidative stress displayed reduced lysosomal acidification and impaired cathepsin proteolytic maturation, resulting in decreased autophagic flux.
Results
Levels of the autophagic marker LC3-II in oxidatively stressed TM cells
The conjugation of LC3-I to phosphatidylethanolamine (PE) to form LC3–PE conjugate (LC3-II) constitutes the only known autophagosome marker and is the most useful tool to monitor autophagy. LC3-II is recruited to the autophagosomal membrane and remains associated with it until fusion with the lysosome, thus serving as a bona fide marker of autophagosome number.26,27
Porcine TM cells were subjected to chronic oxidative stress as indicated in Materials and Methods. To suppress lysosomal proteolysis, the protease inhibitor leupeptin (Leup, 10 μg/mL) was added to the culture media twice per week. As shown in Figure 1A, cells grown under 40% O2 displayed increased steady-state protein levels of the autophagosome marker LC3-II compared with cells grown under physiological conditions. Protein levels of LC3-I were not significantly altered. While leupeptin did not affect the amount of LC3-II in the cells grown at 5% O2, the presence of the lysosomal inhibitor slightly increased LC3-II in the cells grown at 40% O2. To discriminate whether the increase in LC3-II in oxidatively stressed cultures resulted from induction of autophagy or decreased autophagic flux, lysosomal degradation was completely blocked by treating the cells with the lysosomotropic basifying agent chloroquine (CQ, 30 nM) for 3 h at the end of the two-week period. Lysosomal basification significantly increased the amount of LC3-II in cells grown both at 5% O2 and 40% O2 conditions (Fig. 1B). A slightly greater increase was observed in the cells grown under hyperoxic conditions. These results indicated augmented LC3-II synthesis in the oxidatively stress cultures. Whether this occurs simultaneously with decreased lysosomal degradation cannot be inferred from these experiments.28 We did not observe any variation in the protein levels of SQSTM1/p62, a protein that becomes incorporated into the completed autophagosome and is degraded in autolysosomes, thus serving as a readout of autophagic degradation.29
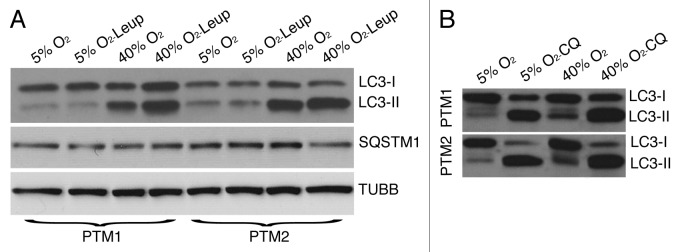
Figure 1. LC3-I to LC3-II turnover in oxidatively stressed TM cells. (A) Protein expression levels of LC3-I, LC3-II, and SQSTM1 in TM cells grown under physiological 5% O2 or oxidative 40% O2 conditions for two weeks in the presence or absence of Leup (10 μM, twice per week), evaluated by WB analysis. TUBB/β-tubulin was used as loading control. (B) Expression levels of LC3-I and LC3-II in TM cells grown under 5% O2 or 40% O2 conditions for two weeks followed by a three h treatment with CQ (30 nM). Blots are representative from three independent experiments.
Quantification of autophagy by flow cytometry in oxidatively stressed TM cells
Quantification of autophagy by flow cytometry was conducted using the commercially available kit, Cyto-IDTM Autophagy Detection Kit (Enzo Life Science). Since this novel tool has still not been extensively tested in the literature, and the supplier does not provide any information about the chemical, we first validated its usefulness in detecting autophagy in TM cells. For this, we treated porcine TM cells with the autophagic inductor trehalose for 24 h. Autophagy was monitored in parallel by flow cytometry and WB (LC3-I to LC3-II conversion). As shown in Figure 2A, trehalose treatment significantly increased the levels of LC3-II, evaluated by WB. As expected, a greater amount of LC3-II was found when autophagic degradation was blocked by addition of bafilomycin A1 (BafA1, 10 nM) during the last three h of culture. Flow cytometry analysis also showed increased levels of CytoID fluorescence in the cultures treated with trehalose (45.134 ± 3.14 RFU in trehalose-treated cells compared with 24.180 ± 0.308 RFU in control cells, p = 0.0003, n = 3) (Fig. 2B). Slightly decreased rather than increased CytoID fluorescence was observed in the BafA1-treated cultures using this technique, suggesting that while this chemical seems to be useful for quantifying net autophagic induction, it cannot be used to monitor autophagic flux. Cells grown under 40% O2 oxidative stress conditions demonstrated higher autophagic activity compared with those ones grown under physiological conditions (40.295 ± 1.117 RFU vs 28.993 ± 0.551 RFU, p < 0.0001, n = 4) (Fig. 2C).
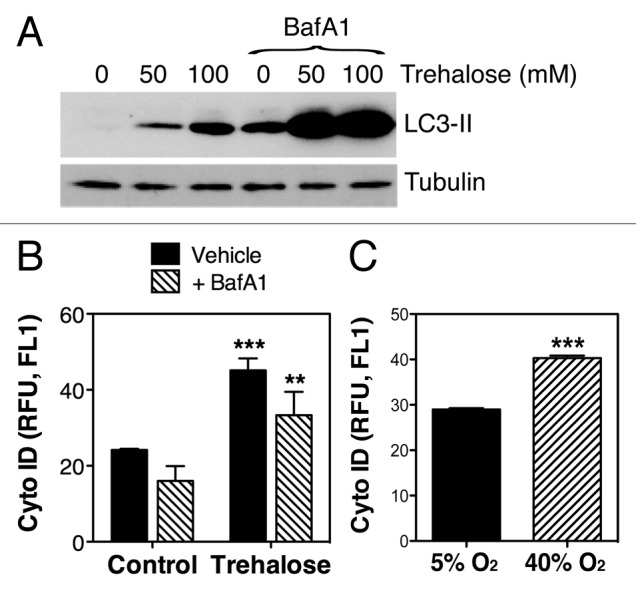
Figure 2. Quantification of autophagy by flow cytometry using CytoID. (A) Expression levels of LC3-II in TM cells treated for 24 h with increasing concentrations of trehalose (0 mM, 50 mM, 100 mM) in the absence or presence of BafA1 (10 nM). Blots are representative from three independent experiments. (B) Flow cytometry quantification of CytoID fluorescence in TM cells treated for 24 h with trehalose (50 mM) in the absence or presence of BafA1 (10 nM). (C) Flow cytometry quantification of CytoID fluorescence in TM cells grown for two weeks under 5% O2 or 40% O2 conditions. Data are means ± SD, n = 3, *** p < 0.0001, **p < 0.001.
Autophagy dynamics in oxidatively stressed TM cells using tfLC3
To monitor autophagic flux we used the tfLC3 method.30 This method relies on the different sensitivity to the acidic and/or proteolytic conditions of the lysosome lumen between GFP and RFP. The GFP fluorescence signal is sensitive, whereas mRFP is more stable. Therefore, colocalization of both GFP and RFP fluorescence indicates a compartment that has not fused with a lysosome, such as the phagophore or an autophagosome. In contrast, an mRFP signal without GFP corresponds to an amphisome or autolysosome. Since primary cultures of TM cells do not transfect well, and our experiments required expression of the transgene for up to two weeks, we generated a replication-deficient adenovirus harboring tfLC3 (AdtfLC3) as indicated in Materials and Methods. To verify the specificity of AdtfLC3 in our system, we infected porcine TM cells with AdtfLC3 (m.o.i = 10 pfu/cell). At 3 d.p.i., cells were treated for 24 h with rapamycin (Rap, 1 μM) or BafA1 (10 nM). Under steady-state conditions, porcine TM cells showed some degree of basal autophagy as demonstrated by the red/yellow puncta staining (Fig. 3). The number of red (autolysosomes) and yellow (autophagosomes) puncta per cell significantly increased in the presence of rapamycin. Bafilomycin A1 induced an accumulation of yellow puncta representing autophagosomes or autolysosomes with impaired lysosomal degradation due to the increased in lysosomal pH. These results validate the use of AdtfLC3 to monitor autophagic dynamics in primary cultures of TM cells. Experiments were then conducted in our experimental model of chronic oxidative stress. As shown in Figure 4, cells grown under a 40% O2 environment displayed a higher number of red and yellow puncta per cell compared with control cultures (red puncta: 143.81 ± 100.2 puncta/cell at 40% O2 vs. 55.8 ± 47.46 puncta/cell at 5% O2, p = 0.0002, n = 30; yellow puncta: 31.1 ± 25.47 puncta/cell at 40% O2 vs. 9.12 ± 6.79 puncta/cell at 5% O2, p < 0.0001, n = 30).

Figure 3. Monitoring autophagy flux in TM cells using AdtfLC3. Porcine TM cells were transduced with AdtfLC3 (m.o.i = 10 pfu/cell). At 3 d.p.i., cells were treated for 24 h with rapamycin (Rap, 1 μM) or BafA1 (10 nM). Confocal images were taken, and red and yellow puncta per cell counted. Data are means ± SD of three independent experiments, ten cells/experiment, n = 30, *** p < 0.0001, **p < 0.001.

Figure 4. Autophagy flux in oxidatively stressed TM cells. Porcine TM cells were transduced with AdtfLC3 (m.o.i = 10 pfu/cell) and grown for two weeks under 5% O2 or 40% O2 conditions. Confocal images were then taken, and red and yellow puncta per cell counted. Data are means ± SD of three independent experiments, ten cells/experiment, n = 30, *** p < 0.0001, **p < 0.001.
Comparative analysis of gene expression levels of components of the autophagic molecular machinery in nonstressed and oxidatively stressed TM Cells
We monitored, by qPCR analysis, the mRNA expression levels of several genes involved in either induction of autophagy (BECN1, ATG4B, ATG5, ATG7, LC3A, LC3B) or autophagic flux (LAMP1 and several V-ATPases), as well as those of LAMP2A, essential for chaperone-mediated autophagy (Fig. 5A). No significant differences in fold change were observed in the mRNA content of BECN1, ATG4, ATG5, LC3A/B, ATP6V0A2, or ATP6V1G1. However, TM cells cultured under hyperoxic conditions demonstrated a significant downregulation of ATG7 (0.214 ± 0.122 fold, p = 0.001, n = 4) and upregulation of ATP6V1B2 (1.424 ± 0.15 fold, p = 0.011, n = 4), ATP6V1C1 (1.438 ± 0.28 fold, p = 0.05, n = 4), and ATP6V1G2 (1.719 ± 0.42 fold, p = 0.04, n = 4). Also, although it did not reach statistical significance, LAMP1 and LAMP2A were found to be upregulated under chronic oxidative stress in all the cell lines tested (LAMP1: 1.849 ± 0.345 fold, p = 0.091, n = 4; LAMP2A: 1.987 ± 0.392 fold, p = 0.08, n = 4).
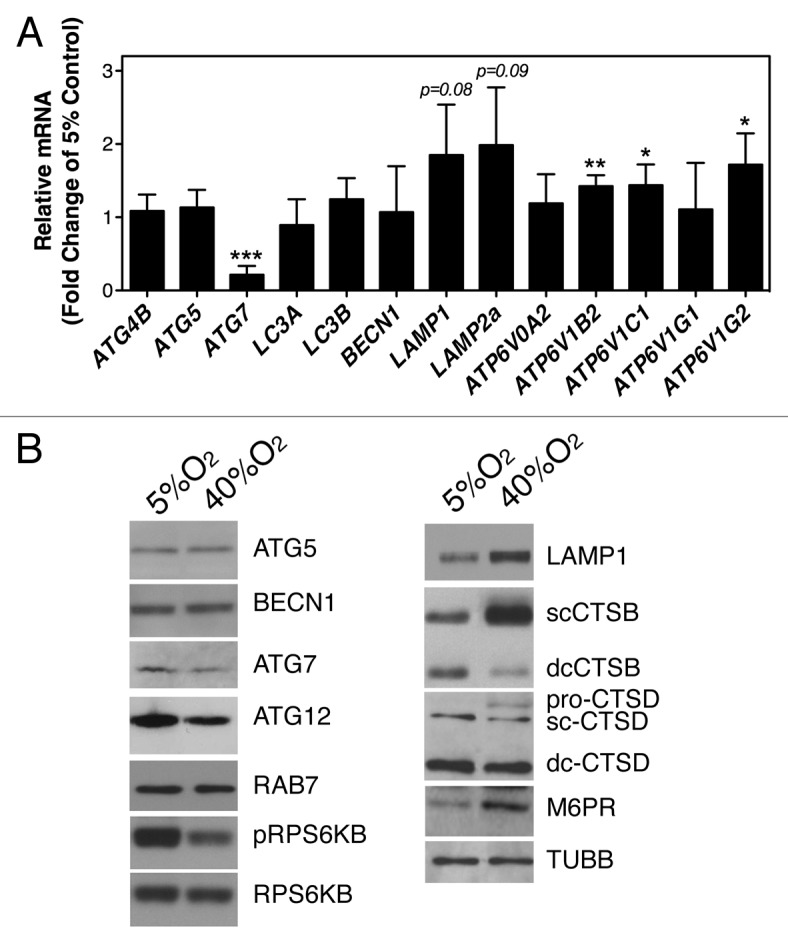
Figure 5. Expression levels of autophagy- and lysosomal-related genes in oxidatively stressed TM cells quantified by (A) qPCR analysis, and (B) immunoblot. Data are means ± SD, n = 3, *** p < 0.0001, **p < 0.001, *p < 0.05. Blots are representative from three independent experiments.
Potential changes in protein levels were also evaluated by immunoblot, based on antibody availability (Fig. 5B). As shown in Figure 5B, a decrease in ATG7 was also confirmed at the protein level, together with a decrease in ATG12. No significant changes were observed in the amounts of ATG5 or RAB7A, a protein essential for fusion of autophagosomes to lysosomes. Western blot analysis revealed a generalized increased in the levels of proteins from the lysosomal machinery, including the lysosomal membrane protein LAMP1, the lysosomal hydrolases cathepsin B (CTSB), as well as the mannose-6-phosphate receptor (M6PR), involved in the transport of the lysosomal hydrolases from the Golgi to the lysosomes. A slight increase in pro-CTSD, but not in mature CTSD, was also observed.
Inhibition of the mechanistic target of rapamycin (MTOR) pathway is the best-characterized signaling pathway known to activate autophagy, in particular in response to starvation. Oxidatively stressed cultures showed decreased phosphorylated levels of p70S6 Kinase (RPS6KB/p70S6K), a downstream target of MTOR, indicating that activation of autophagy by mild chronic oxidative stress is mediated through the AKT-MTOR pathway.
Effect of oxidative stress on transcription factor EB (TFEB) activity
Very recently, TFEB has been identified as a master transcription factor regulator of lysosomal biogenesis and autophagy during starvation and in lysosomal storage disorders.31,32 We wondered whether chronic oxidative stress could also modulate TFEB activity. For this, and based on the low efficacy of available antibodies in recognizing endogenous TFEB, we constructed a recombinant adenovirus expressing human TFEB tagged to flag.31 As shown in Figure 6A, TFEB-Flag was located in the cytosol of AdTFEB-FLAG-infected TM cells under control conditions, as detected by immunofluorescence using an anti-Flag antibody. Upon induction of autophagy either with sucrose or trehalose treatment, TFEB-Flag translocated to the nuclei, thus confirming the usefulness of AdTFEB-Flag to monitor TFEB dynamics in TM cells. We then proceeded to monitor the cellular location of TFEB-Flag in TM cells grown under physiological or oxidative stress conditions. While TFEB-Flag was primarily found in the cytosol of cells grown at 5% O2, with very low or almost undetectable amounts of the transcription factor in their nuclei, TM cells grown at 40% O2 displayed a significantly higher and substantial amount of TFEB-Flag in the nuclear fraction (Fig. 6B). No cross-contamination between the cytosolic and nuclear fractions were observed using anti-TUBB and anti-LMNA antibodies. As reported in other cell types, overexpression of TFEB led to increased levels of LC3-II in TM cells grown under both 5% and 40% oxygen atmosphere.

Figure 6. Effect of oxidative stress on TFEB activity. (A) Immunofluorescence confocal microscopy image using anti-Flag antibody of TM cells transduced with AdTFEB-Flag (m.o.i = 20 pfu/cell) and treated at 3 d.p.i., with either sucrose (100 mM) or trehalose (50 mM) for 24 h. (B) Western blot analysis of TFEB and LC3-I/II in cytosolic and nuclear fractions of TM cells transduced with AdTFEB-Flag (m.o.i = 20 pfu/cell) and grown for two weeks under 5% O2 or 40% O2 conditions. Anti-TUBB and anti-LMNA were used to confirm purity of obtained fractions and as loading controls. Blots are representative from three independent experiments.
Lysosomal function in oxidatively stressed TM cells
Previous work reported by our laboratory showed decreased lysosomal activity per lysosomal mass in oxidatively stress TM cells.24 In these studies, cathepsin activities and cathepsin protein levels were quantified using an omni-cathepsin substrate and a pan-cathepsin antibody, respectively. To gain more insight into this observation, we repeated these experiments using different substrates to monitor the activity of several cathepsins. As observed in Figure 7A, cells under oxidative stress conditions showed decreased cathepsin activities with all the substrates tested compared with control cultures (z-FR: 50.31 ± 12.93%, p = 0.0218; z-RR: 39.06 ± 12.15%, p = 0.0130; z-GPR: 52.224 ± 6.29, p = 0.0057; z-VVR: 53.19 ± 5.25, p = 0.0042; CTSD/E: 68.72 ± 9.04%, p = 0.0267, n = 3).

Figure 7. Lysosomal function in oxidatively stressed TM cells. (A) Lysosomal enzyme activity determined in cell lysates using the following fluorogenic substrates: Z-FR-AMC, Z-RR-AMC, Z-GPR-AMC, Z-VVR-AMC, and CTSD/E. Relative fluorescence units (RFU) were normalized by total proteins. Graphs represent the percentage of increase compared with the 5% O2 condition. (B) Representative immunoblot showing expression levels of LAMP1, CTSB, and CTSD in purified lysosomal-enriched fractions of TM cells grown under 5% O2 or 40% O2 conditions. (C) Lysosomal pH in cells grown under physiological and oxidative stress conditions as quantified using LysoSensor Yellow-Blue dextran. (D) Levels of cellular proteolysis in TM cells grown for two weeks under 5% O2 or 40% O2 conditions quantified using [3H]-leu. Graph on the left represents cellular proteolysis as calculated by dividing the soluble radioactivity cpm found in the culture media (right graph) by the total cellular radioactivity cpm (graph in center). Data are the means ± SD, n = 3, *p < 0.05, **p < 0.001.
The protein levels of CTSB and CTSD were evaluated by WB (Fig. 5B). The antibody against CTSD recognized three different bands corresponding to pre-CTSD (52 kDa), single-chain mature CTSD (48 kDa), and the heavy chain of the double-chain CTSD (32 kDa). No significant changes in the amount of any of the CTSD forms were observed in the stressed cultures. The antibody against CTSB recognized three different bands: 47 kDa, corresponding to pre-CTSB (not shown in this blot, but apparent in leupeptin-treated cells); 30 kDa, corresponding to mature single-chain CTSB (scCTSB); and 22 kDa, corresponding to the heavy chain of the mature-double chain CTSB (dcCTSB). Cells grown at 40% O2 displayed higher protein levels of scCTSB, but lower levels of dcCTSB. Similar results were obtained using lysosomal-enriched fractions (Fig. 7B). Altogether, these data suggest a defect in the proteolytic activation of CTSB in TM cells under chronic oxidative stress. Based on these results, we next examined the lysosomal pH using LysoSensor Yellow/Blue dextran. A statistically significant decrease in the 535:430 ratio was observed in the cells exposed to oxidative stress, indicating less acidic lysosomal pH in these cultures (Fig. 7C).
We evaluated the effect of lysosomal basification and decreased cathepsin activities in cellular proteolysis. As shown in Figure 7D, despite the activation of autophagy, cells grown under oxidative stress did not display higher proteolysis levels compared with those grown under physiological conditions. Moreover, the amount of [3H]-leu in the intracellular fraction, which correlates with total protein synthesis, was significantly reduced in the cells at 40% O2 (3275.1 ± 103.9 cpm vs. 6420.7 ± 1280.5 cpm, p = 0.013, n = 3) suggesting lower protein turnover in these cultures.
Effect of autophagy on SA-GLB1 activity
Our laboratory previously reported an increase in SA-GLB1 activity in the glaucomatous outflow pathway compared with age-matched control donors.25 More recently, higher SA-GLB1 activity was also observed in TM cells exposed to chronic oxidative stress and found it to correlate with increased lipofuscin and lysosomal content.24 Although SA-GLB1 was first described in senescent cells and has been widely used as marker for cellular senescence and aging, SA-GLB1 has also been found in other cellular conditions, including high confluency or starvation. SA-GLB1 has been hypothesized to be a surrogate marker of high lysosomal content or activity and reflect, at least in part, stress-response activation of autophagy.33-36
To investigate whether SA-GLB1 activity requires induction of autophagy, confluent cultures of porcine TM cells were grown either at 5% O2 or 40% O2 in the presence of increasing concentrations of the autophagy inhibitor 3-MA, added twice per week to the culture media. As seen in Figure 8A, 3-MA significantly reduced, in a dose-dependent manner, the increase in LC3-II observed in the stressed cultures, further confirming that the higher levels of LC3-II in the cells at 40% O2 are at least partly mediated by induction of autophagy. In addition, blocking autophagosome formation significantly diminished the percentage of increase in autofluorescence (340.15 ± 34.12% vs. 520.23 ± 57.32%, p = 0.009, n = 3), lysosomal mass (210.74 ± 26.16% vs. 394.45 ± 32.11%, p = 0.001, n = 3) and SA-GLB1 (220.85 ± 21.87% vs. 427.63 ± 5.78%, p < 0.0001, n = 3) observed in TM cells under chronic oxidative stress when compared with those grown under physiological conditions (Fig. 8B).
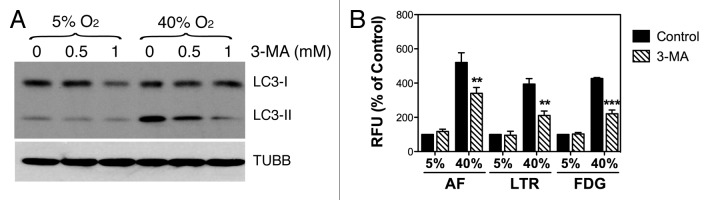
Figure 8. Autophagy inhibition in oxidatively stress TM cells. (A) Protein expression levels of LC3-I and LC3-II in TM cells grown under physiological 5% O2 or oxidative 40% O2 conditions for two weeks in the presence of 3-MA (0 mM, 0.5 mM, 1 mM added twice per week), evaluated by WB analysis. TUBB was used as loading control. Blots are representative from three independent experiments. (B) Effects of 3-MA in lipofuscin content, lysosomal mass, and SA-GLB1 in TM cells grown at 5% O2 and 40% O2. Graphs represent the percentage of change compared with the 5% O2 condition. Data are the means ± SD, n = 3, **p < 0.001, ***p < 0.0001.
To further confirm a relationship between the activation of the autophagic lysosomal pathway and SA-GLB1, we treated TM cells with the autophagy inducers rapamycin (500nM, MTOR dependent) or trehalose (Treh, 50 mM, MTOR independent) for two weeks. Elevated SA-GLB1 could only be observed, however, in trehalose- (520.8 ± 71.19 RFU compared with 227.96 ± 52.34 RFU, p = 0.0006, n = 4) but not in rapamycin-treated cells (236.99 ± 66.72 RFU) (Fig. 9A). As shown in Figure 9B, both drug treatments led to a significant increase in lysosomal mass, as quantified by LAMP1 immunoblotting.

Figure 9. Autophagy and SA-GLB1 activity. (A) SA-GLB1 activity and (B) protein expression levels of LAMP1 and LC3B-I/II in TM cells treated for two weeks with either rapamycin (500 nM, twice/week) or trehalose (50 mM, twice/week). (C) SA-GLB1 activity. (D) mRNA) and (E) protein expression levels of LC3 and LAMP1 in TM cells transduced for two weeks with AdTFEB-Flag (20 pfu/cell). Data are the means ± SD, n = 3, **p < 0.001, ***p < 0.0001. Blots are representative from three independent experiments.
Next, we explored whether induction of lysosomal biogenesis via TFEB overexpression could increase SA-GLB1 activity. For this, porcine TM cells were infected with either AdNull or AdTFEB-Flag (20 pfu/cell). SA-GLB1 was evaluated at two weeks post-infection. As illustrated in Figure 9C, no significant differences in SA-GLB1 activity were observed in the cultures infected with AdTFEB-Flag compared with AdNull-infected cells. AdTFEB-Flag transduction significantly increased the mRNA and protein expression levels of LC3B and LAMP1 (Fig. 9D and E), indicating induction of autophagy and lysosomal biogenesis.
Conclusion
Although it has been traditionally thought that autophagy was a catabolic process involving lysosomal degradation in response to lack of nutrients, autophagy is emerging as a cellular survival mechanism against a variety of stressors. In this study, we report the MTOR-mediated activation of macroautophagy and the nuclear translocation of TFEB in TM cells when exposed to mild, chronic oxidative stress. Moreover, our results show reduced acidification of the lysosomal compartment and the defective proteolytic activation of CTSB in TM cells subjected to chronic oxidative stress. Finally, we provide here novel evidence supporting a role of autophagy in the induction of SA-GLB1 activity.
A key event required for activation of autophagy is the lipidation of LC3-I to LC3-II. LC3 is synthesized as a precursor form that is cleaved by the protease ATG4B, resulting in the cytosolic isoform LC3-I. Upon induction of autophagy, LC3-I is conjugated to phosphatidylethanolamine (PE) to form LC3-II. LC3-II is incorporated to the nascent and elongating autophagosome membrane and remains on the autophagosomes until fusion with the lysosomes, after which LC3-II found on the lumenal surface of autophagosomes is degraded, while LC3-II on the cytoplasmic face of autolysosomes is delipidated by ATG4 and recycled.18 western blot analysis showed a significant steady-state increased in the levels of LC3-II in TM cells grown at 40% O2 conditions, compared with those grown under physiological conditions. As already addressed by others, increased LC3-II does not necessarily result from induction of autophagy, but also from diminished autophagy flux or, although less frequently discussed in the literature, by reduced delipidation and recycling of LC3-II by ATG4.29,37 When lysosomal degradation was blocked by addition of the basifying agent chloroquine, we observed a further increase in the amount of LC3-II in the oxidatively stressed cultures. This increase was slightly greater compared with that observed in cells grown under physiological conditions. These data indicated that the increased LC3-II in the stressed cultures primarily resulted from activation of autophagy rather than decreased autophagic flux, although a low level of impaired autophagic flux could not be discarded.28
Similar conclusions were reached when autophagy was monitored using AdtfLC3.30 In agreement with the results obtained by WB analysis, TM cells grown under oxidative stress conditions displayed a higher autophagic activity as demonstrated by the increased number of red puncta (autolysosomes) and yellow puncta (autophagosomes/defective autolysosomes) per cell. The number of red puncta per cell was significantly higher than the number of yellow puncta per cell, suggesting no deficient maturation of autophagosomes into autolysosomes in the stressed cultures.
Induction of autophagy in oxidatively stressed TM cells was additionally confirmed using a commercially available kit recently launched to the market (Cyto-ID® Autophagy Detection Kit, Enzo Life Science), which allows quantification of autophagy by flow cytometry. The probe included in the kit is a cationic amphiphilic tracer that selectively labels vacuoles associated with the autophagy pathway, including phagophores, autophagosomes and autolysosomes, while minimally staining the lysosomes. Although this dye seems to be useful for detection and quantification of autophagy, in our hands it did not prove to be an efficient tool for monitoring autophagic flux.
Despite the induction of autophagy in TM cells exposed to chronic oxidative stress, we did not observe higher mRNA or protein expression levels in any of the autophagy-related genes analyzed in these cultures. In contrast, we did find a significant downregulation of ATG7, which was confirmed at the protein level, as well as a modest decrease in ATG5 and ATG12 proteins in the oxidatively stressed cultures. This finding was unexpected since ATG5, ATG7, and ATG12 are all essential for LC3-II formation, as well as elongation and expansion of the phagophore to form the autophagosome.38-40 At the moment, we do not know the reason for this downregulated expression of ATG7, ATG5, and ATG12 in the oxidatively stressed TM cultures. There is no information regarding how these genes are regulated, or the fate of the protein complexes after dissociation from the autophagosome. Downregulated expression of ATG7 and ATG12–ATG5, together with increased LC3-II, has been also reported in primary cultures of senescent fibroblasts.41 Moreover, increased LC3-II levels were also found in the ATG7- or ATG12-knocked down cultures. It is possible that another LC3 lipidation pathway is yet to be discovered, or that very low amounts of ATG7 and ATG12–ATG5 are required for formation of LC3-II. In this line of thought, Nishida et al. have recently identified an ATG5-ATG7-independent alternative autophagy pathway; however, lipidation of LC3-II does not occur in this type of autophagy.42 Very interestingly, the phenotype described in Atg7- and Atg12-knocked down cells showed a remarkable similarity to that reported by our laboratory in TM cells exposed to chronic oxidative stress, including: increased autofluorescence, increased intracellular ROS production, increased lysosomal and mitochondrial content, decreased mitochondrial membrane potential, increased SA-GLB1, and decreased cathepsin activity.24
For efficient degradation of the cargo contained within the autophagosomes, activation of autophagy must be coupled with the lysosomal system. Recently, the transcription factor TFEB has been identified as a master gene that controls major steps of the autophagic pathway during starvation, including autophagosome formation, autophagosome-lysosome fusion, and substrate degradation by driving expression of autophagy and lysosomal genes.32 Our results here show that, in addition to starvation, exposure to chronic oxidative stress also promotes nuclear localization of TFEB and increased LC3-II levels. During the preparation of this manuscript, two independent groups have reported a novel mechanism by which lysosomes sense and regulate their function determined by MTOR-mediated TFEB phosphorylation.43,44 Interestingly, we did observe decreased levels of phosphorylated RPS6KB, a downstream target of MTOR, in the oxidatively stressed cultures, indicating at least partial inhibition of the MTOR pathway. Based on these novel studies, it is plausible that inhibition of MTOR under oxidative stress conditions might trigger nuclear translocation of TFEB. Ongoing studies are directed to confirm this, and investigate the precise role of TFEB on regulating the autophagic and lysosomal pathways in response to oxidative stress.
Consistent with the nuclear translocation of TFEB, induction of autophagy was accompanied by the upregulation of the lysosomal machinery in TM cells grown at 40% O2 conditions, characterized by increased expression levels of LAMP1, LAMP2, several v-ATPases, as well as several cathepsins (CTSB, CTSD). However, in agreement with our preliminary findings, the stressed cultures showed a decrease in serine, cysteine, and aspartyl protease activities, which suggested diminished lysosomal degradative potential. Similar to other proteases, cathepsins are synthesized as inactive precursors, which are activated upon arrival to the endosome by proteolytic removal of the propeptide to yield the mature single-chain form. Once in the lysosomes, cathepsins are further cleaved rendering the double-chain cathepsin, composed of a heavy-chain and a light chain linked by a disulfide bridge. Proteolytic activation of cathepsins can be facilitated either by autocatalytic activation at acidic pH, by activation by other proteases, or both.45 Our data indicated lysosomal basification in the oxidatively stressed TM cells. Since lysosomal proteases are optimally active in the acidic pH, such an increase in lysosomal pH could certainly explain the overall decrease in cathepsin activities in these cultures, either by directly affecting the autocatalytic activation or indirectly by interfering with the activation of other proteases required for proteolytic cleavage. Another possibility is their inhibition by endogenous protein inhibitors (cystatins). Unfortunately, none of the antibodies tested recognized porcine cystatins, so we could not confirm any differential expression between TM cells grown under physiological and oxidative stress conditions. Finally, lysosomal cathepsins can also be directly inactivated by oxidative stress.46
Intriguingly, although TM cells grown at 40% O2 atmosphere displayed much higher amounts of mature scCTSB compared with cells grown under 5% O2, the levels of dcCTSB were drastically reduced, indicating impaired proteolytic cleavage of CTSB to the mature double-chain form in the cells grown under chronic oxidative stress conditions. Defective proteolytic processing of CTSD, however, was not noticed. At this moment we are not certain of the mechanisms underlying this improper maturation of CTSB and why the processing of CTSD is not affected. As mentioned earlier, this proteolytic cleavage occurs in the lysosomes, but the protease responsible for it has not been characterized yet.47 One possibility is that the activity of this still unknown protease is affected by the increased lysosomal pH. Alternatively, CTSB might be mistargeted to other cellular compartment, i.e., plasma membrane, in the oxidatively stressed cultures. However, defective CTSB maturation was also found in enriched lysosomal fractions.
Both improper CTSB maturation and decreased cathepsin activities are likely to be responsible for the unexpected finding of no increase in cellular proteolysis despite activation of autophagy in the oxidatively stressed cultures. The higher number of autolysosomes and the increased presence of cargo material within them, while keeping the same cellular degradative capacity, might result in the accumulation of undegraded material within residual bodies and compromise cellular components turnover and cell function. Very importantly, a pathological effect of the intralysosomal accumulation of substances in TM tissue physiology is highlighted by the observation that ocular hypertension and glaucoma are clinical manifestations associated with a number of lysosomal storage disorders, including Hurler Disease, Morquio syndrome and Maroteaux–Lamy syndrome.48,49
An important finding in our studies is the connection between autophagy and SA-GLB1. We have previously shown an increased number of cells displaying SA-GLB1 activity in the glaucomatous outflow pathway compared with age-matched control tissue.25 Moreover, we have demonstrated a correlation between increased lysosomal content and detectable SA-GLB1 in TM cells grown under chronic oxidative stress, thus providing the first link between oxidative stress, altered lysosomal function, and POAG.24 The nature of this abnormal SA-GLB1 activity, which is not exclusive but can be observed in other conditions others than senescence, is still unknown. In our experimental model, when application of chronic oxidative stress was conducted in the presence of the autophagy inhibitor 3-MA, we partially blocked the increase in SA-GLB1 activity, as well as that in lipofuscin and lysosomal mass observed in the stressed cultures, suggesting that the occurrence of SA-GLB1 activity is mediated by LC3-dependent autophagy. However, while trehalose treatment led to increased SA-GLB1 in TM cells, treatment with rapamycin or TFEB overexpression did not. Activation of autophagy and increased lysosomal mass were observed in all three cases. One might argue that the higher LAMP1 and LC3-II levels found in trehalose-treated cells might account for the induction of SA-GLB1; however, the amounts of LC3-II and LAMP1 in rapamycin-treated and TFEB overexpressing cells were very similar to those observed in the cultures grown at 40% O2, which showed elevated SA-GLB1. Altogether, these data indicate that although activation of autophagy is needed for induction SA-GLB1 activity, contrary to what some investigators have proposed,35,36 SA-GLB1 is not a surrogate marker for increased lysosomal mass. In addition, our results suggest that SA-GLB1 activity is not MTOR-mediated, but it is triggered by other signaling pathways or mechanisms activated during trehalose treatment and oxidative stress. In this sense, Narita et al.50 have recently reported the colocalization of SA-GLB1 activity within autolysosomes together with LC3, SQSTM1, and LAMP2 in the TOR-autophagy spatial coupling compartment in a model of RAS-induced senescence. It is possible that the occurrence of SA-GLB1 activity results from reduced autophagic flux within autolysosomes. This is consistent with the results obtained using AdtfLC3, and our own previous studies, which show the accumulation of autolysosomes in the oxidatively stressed TM cells.
In summary, our results indicate that while TM cells respond to oxidative challenge by activation of the autophagic lysosomal degradative pathway, chronic exposure to oxidative stress leads to lysosomal basification and defective proteolytic activation of lysosomal enzymes with subsequent decrease of autophagic flux. Given the critical role of lysosomal function in cellular and tissue homeostasis, including the degradation of phagocytosed material, we propose that diminished autophagic flux induced by oxidative stress might represent one of the factors leading to progressive failure of cellular TM function with age and contribute to the pathogenesis of POAG.
Materials and Methods
Reagents
3-Methyladenine (3-MA, Sigma-Aldrich, M9281), bafilomycin A1 (Sigma-Aldrich, B1793), leupeptin (Sigma-Aldrich, L8511), chloroquine (Sigma-Aldrich, C6628), sucrose (Sigma-Aldrich, S7903), trehalose (Sigma-Aldrich, T9531), and rapamycin (Calbiochem, 553210).
Cell culture
Primary cultures of porcine TM cells were prepared from porcine cadaver eyes, obtained from a local abattoir less than five h postmortem, and maintained as previously described.24 Briefly, the TM was dissected and digested with 2 mg/mL of collagenase (Sigma-Aldrich, C2674) for 1 h at 37°C. The digested tissue was placed in gelatin-coated 35-mm dishes and cultivated in low-glucose Dulbecco’s Modified Eagle Medium (DMEM) with L-glutamine and 110 mg/L sodium pyruvate (Gibco, 11885), supplemented with 10% fetal bovine serum (FBS, Gibco, 10082-147), 100 mM nonessential amino acids (Gibco, 11140-050), 100 units/ml penicillin and 100 mg/mL streptomycin sulfate and 0.25 mg/ml amphotericin B (Gibco, 15240-062). Cells were maintained and propagated until passage three at 37°C in a humidified air with 5% CO2 incubator. Cell lines were subcultivated 1:2 when confluent.
Experimental model of chronic oxidative stress
Chronic oxidative stress was induced by subjecting porcine TM cells to normobaric hyperoxic conditions.24 For this, confluent cultures of porcine TM cells at passage 4 were grown for 2 weeks at 40% O2 and 5% CO2. Control cultures were grown under physiological oxygen conditions (5% O2, 5% CO2) in a triple-gas incubator (SANYO, MCO-5M, Panasonic Healthcare Company of North America). When indicated, autophagy inhibitors or activators were added to the culture media twice per week.
Obtention of protein lysates
For whole-cell lysates, cells were washed twice in PBS and lysated in 20 mM HEPES, 2 mM EGTA, 5 mM EDTA, and 0.5% NP-40 containing protease inhibitor cocktail (Thermo Scientific, 1858566) and phosphatase inhibitor cocktail (Thermo Scientific, 78420). After sonication for one min on ice, the lysates were clarified by centrifugation. Cytoplasmic and nuclear lysates were obtained as follows. Cells were washed and incubated on ice for 15 min in Triton X-100 lysis buffer (50 mM TRIS-HCl pH 7.5, 0.5% Triton X-100, 137.5 mM NaCl, 10% Glycerol, with protease and phosphatase inhibitors). Insoluble nuclei were separated by centrifugation at 13,000 rpm for 15 min at 4°C. The supernatant, containing the cytoplasmic/membrane fraction, was collected. The nuclear pellet was rinsed with lysis buffer, resuspended in Triton X-100 lysis buffer containing 0.5% SDS and protease/phosphatase inhibitors, and clarified by centrifugation at 13,000 rpm for 15 min at 4°C. Protein concentration was determined with a protein assay kit (Micro BCA, Thermo Scientific, 23235).
Western blot analysis
Protein samples (10 μg) were separated by 10% polyacrylamide SDS-PAGE gels (15% polyacrylamide for LC3 detection) and transferred to PVDF membranes (Bio-Rad, 162-0177). The membranes were blocked with 5% nonfat dry milk and incubated overnight with the specific primary antibodies. The bands were detected by incubation with a secondary antibody conjugated to horseradish peroxidase and chemiluminescence substrate (ECL Plus, GE Healthcare, RPN2132). Antibodies used are listed in Table 1.
Table 1. List of primary antibodies used.
| Target protein | Company | Catalog number | Dilution |
|---|---|---|---|
| LC3A/B |
Abcam |
ab58610 |
1:500 |
| LC3B |
Cell Signaling |
3868S |
1:1000 |
| SQSTM1 |
Sigma-Aldrich |
P0067 |
1:1000 |
| TUBB |
Sigma-Aldrich |
T5293 |
1:1000 |
| ATG5 |
Cell Signaling |
8540P |
1:1000 |
| BECN1 |
Cell Signaling |
3495P |
1:1000 |
| ATG7 |
Cell Signaling |
2631P |
1:1000 |
| ATG12 |
Cell Signaling |
4180P |
1:1000 |
| RAB7A |
Abcam |
ab50533 |
1:2000 |
| LAMP1 |
Abcam |
ab24170 |
1:1000 |
| CTSB |
Abcam |
ab58802 |
1:1000 |
| CTSD |
Santa Cruz Biotechnology |
SC-6494 |
1:500 |
| M6PR |
Hybridoma Bank |
22d4 |
1:1000 |
| Flag |
Sigma-Aldrich |
F1804 |
1:1000 |
| LMNA |
Santa Cruz Biotechnology |
SC-6215 |
1:500 |
| RPS6KB |
Santa Cruz Biotechnology |
SC-230 |
1:1000 |
| p-RPS6KB | Santa Cruz Biotechnology | SC-7984-R | 1:1000 |
RNA isolation and quantitative real-time PCR
Total RNA from porcine TM primary cultures was isolated (RNeasy kit, Qiagen, 74104) according to the manufacturer’s protocol and treated with DNase I (Qiagen, 79254). RNA yields were determined by Nanodrop spectrophotometry (NanoDrop 1000, Thermo Scientific, Waltham, MA). First-strand cDNA was synthesized from total RNA (1 μg) by reverse transcription using oligo(dT) primers and reverse transcriptase (SuperScript First Strand System, Invitrogen, 11904018). Real-time PCR was performed in a 20 μL mixture containing 1 μL of the cDNA preparation diluted five times, 10 μL master mix (SsoFast EvaGreen Supermix, Bio-Rad, 1725201), and 500 nM of each primer in a thermocycler system (iCycler iQ; Bio-Rad, using the following PCR parameters: 95°C for 5 min, followed by 50 cycles of 95°C for 15 sec, 60°C for 15 sec, and 72°C for 15 sec. The fluorescence threshold value (Ct) was calculated using the thermocycler system software. The absence of nonspecific products was confirmed by both the analysis of the melt curves and by electrophoresis in 3% acryl-agarose gels. ACTB/β-Actin served as an internal standard of mRNA expression. The change (x-fold) was calculated with the Equation 2−ΔΔCt, where ΔCt = Ctgene − CtAct, and ΔΔCt = ΔCtExp−ΔCtCon. The sequences of the primers used for the amplifications are shown in Table 2.
Table 2. Primer sequences used for qPCR analysis.
| Gene symbol | GenBank | Forward | Reverse |
|---|---|---|---|
|
ATG4B |
NM_001190283 |
AAGGACGAGATCTTGGCTGA |
GCGTGCAGGACACTGAAGTA |
|
ATG5 |
HM046510 |
TCACAAGCAACTCTGGATGG |
GAAGCCACAGGACGAAAGAG |
|
ATG7 |
NM_001190285 |
TTAGCCCAGTACCCTGGATG |
CTTTGGGACAATCTGGGCTA |
|
LC3A |
NM_001170827 |
CATGAGCGAGTTGGTCAAGA |
AGGAAGCCATCCTCATCCTT |
|
LC3B |
NM_001190290.1 |
CCACCGCCTCTTGGAATCGCA |
ACTCTCTGTTCGAAGGTTCGG |
|
BCN1 |
NM_001044530 |
AGGAGCTGCCGTTGTACTGT |
CACTGCCTCCTGTGTCTTCA |
|
LAMP1 |
NM_001011507 |
AGAAATGCCACCCGTTACAG |
CGCACCTGTACTTGGTGTTG |
|
LAMP2a |
XM_001926458 |
TTCAGCCCTTCAGTGTGATG |
GGCGCTTGAGACCAATAAAG |
|
ATP6V0A2 |
XM_003359098.2 |
ATCCCAGACTCAGTCAGTCACAGG |
CCGGAGCCAAAGAGGTTGACGG |
|
ATP6V1B2 |
XM_003483379.1 |
GGCTGGCAGCTGCTCCGAATC |
AGCATCGGGCCAGCGATGAA |
|
ATP6V1C1 |
NM_001190205.1 |
CTGCCCTCGTGTCGGTGTCG |
ACGGGAGACTGCGCTGAGAA |
|
ATP6V1G1 |
NM_001190194.1 |
GACACCTCGGAGTTGCTGCCG |
GACACCTCGGAGTTGCTGCCG |
| ATP6V1G2 | EU282339.1 | CACCCGGTGCAGAAGGCTGAAA | TACCAGACGGCTCTCCCCCAC |
Determination of autophagy by flow cytometry
Autophagy was quantified by flow cytometry using the Cyto-IDTM Autophagy Detection Kit (Enzo Life Science, ENZ-51031-K200) following the manufacturer’s instructions. The mean green fluorescence of 10,000 cells was recorded (FL-1 channel) and quantified (CellQuest software; BD Biosciences). Nonstained control cells were included to evaluate the baseline fluorescence.
Construction of replication-deficient recombinant adenovirus AdtfLC3 and AdTFEB-Flag
Generation of recombinant adenoviruses AdtfLC3 and AdTFEB-Flag was performed using the ViraPowerTM Adenoviral Expression System (Invitrogen, K4930-00). For this, the RFP-EGFP-LC3 (2300 nt) and the human TFEB-Flag (1431 nt) DNA fragment sequences were amplified by high-fidelity PCR (Advantage-HF 2 PCR kit, Clontech, 639123) using the tfLC3 (obtained from Dr. Yoshimori) and the TFEB-3xFlag (obtained from Dr. Ballabio) plasmid constructs as template, respectively, and the following specific primers: tfLC3-F: CACCATGGCCTCCTCCGAGGACGTCAT; tfLC3-R: CTGTATGTCTGTCACAAGCATGGCTCTC; TFEB-F: CACCATGGCGTCACGCATAGGGTTG; TFEB-R: TCACTACTTGTCATCGTCATC. The PCR was performed at 94°C for 5 min followed by 35 cycles of 94°C for 30 sec, 60°C for 30 sec, and 72°C for 4 min. The PCR products were purified and cloned into the pENTRTM/D-TOPO cloning vector (Invitrogen, K243520); the absence of mutations potentially introduced by the DNA polymerase was confirmed by sequencing. The pENTR-tfLC3 and pENTR-TFEB-Flag generated were then recombined with the pAd/CMV/V5-DEST vector (Invitrogen, V493-20) by Gateway® technology using the LR ClonaseTM II enzyme mix (Invitrogen, 11791020) to create the pAdCMV-tfLC3 and pAdCMV-TFEB-Flag. The pAdCMV-tfLC3 and pAdCMV-TFEB-Flag were then linearized by Pac I (New England Biolabs, RO547S) digestion and transfected into 293A cells using Effectene transfection reagent (Qiagen, 301425) to produce the recombinant adenoviruses AdtfLC3 and AdTFEB-Flag. High-titer viral stocks were purified with the Adeno MINI Purification Virakit (Virapur, 003059), and then titered using the BD Adeno-X Rapid Titer kit (Clontech, 632250).
Transduction of TM cells with replication-deficient adenoviruses
Viral suspensions diluted in a small volume of serum-free media were allowed to adsorb to the cell surface membrane by incubation during 90 min at 37°C, 5% CO2 with shaking every 15 min to get a homogeneous distribution of the viral particles in the plate. After the adsorption period, regular culture medium was added to plates. The multiplicity of infection (m.o.i.) is indicated in each particular experiment.
Counting of red and yellow puncta in AdtfLC3-transduced cells
Cells were washed in PBS and fixed for 10 min at room temperature in 4% paraformaldehyde. Images were taken by confocal microscopy in a Zeiss LSM 510 upright microscope using a 63×/1.0 dipping objective (Carl Zeiss Microscopy). Images from five different fields were taken in each individual experiment. Red and yellow puncta were counted in a masked manner both manually and automatically using the Green and Red Puncta Colocalization ImageJ Plugin, using the by two independent investigators. A total of two cells per field were counted (10 cells per experiment). Experiments were repeated three times using different cell lines (total cells counted per condition = 30). To facilitate manual counting the Find Edges process in Image J was applied to the images.
Immunofluorescence
Cells were grown onto gelatin-coated coverslips and treated as indicated. After treatment, cells were fixed with 4% paraformadehyde in PBS for 10 min, washed with PBS, and blocked in 20% goat serum for 30 min at room temperature. Cells were washed and incubated first with a specific anti-Flag antibody diluted 1:500 in 20% goat serum for 90 min at room temperature, and then with AlexaFluor 568 Goat anti-mouse IgG (H+L) (Life Technology, A-11004) for one h at room temperature. Cells were counterstained with DAPI and mounted with Fluoromount (Sigma-Aldrich, F4680). Images were taken by confocal microscopy (Nikon Eclipse 90i, Nikon Instruments).
Assessment of lysosomal enzyme activity
Cells grown in a 24-well plate were washed in PBS and lysated for 30 min at 4°C with shaking in 100 μL of 50 mM sodium acetate (pH 5.5), 0.1 M NaCl, 1 mM EDTA, and 0.2% Triton X-100. Lysates were clarified by centrifugation and immediately used for determination of proteolytic activity. For this, 1 μL of cell lysates was incubated at 37°C for 30 min in lysis buffer (100 μL) in the presence of the appropriate fluorogenic substrate. The following cathepsin substrates were used in this study: z-FR-AMC (20 μM; Santa Cruz Biotechnology, SC-3136), z-RR-AMC (20 μM, Enzo Life Sciences, P-137), z-VVR-AMC (20 μM, Enzo Life Sciences, P-199), z-GPR-AMC (20 μM, Enzo Life Sciences, P-142), and Cathepsin D and E substrate (10 μM, Enzo Life Sciences, P-145). The AMC released as a result of proteolytic activity was quantified with a microtiter plate reader (exc: 380 nm; em: 440 nm), and normalized by total protein content. The AMC released as a result of CTSD and CTSE proteolytic activity was read at 340 nm (exc) and 420 nm (em).
Measurement of endogenous cellular autofluorescence
Endogenous cellular autofluorescence was detected under the FITC filter by fluorescence microscopy and quantified by flow cytometry (FACSCalibur; BD Biosciences). For this, the fluorescence emitted by 10,000 cells in the FL-2 channel (563 to 607 nm wavelength band) was recorded and analyzed.
Quantification of lysosomal cellular content
Cells were incubated for 15 min at 37°C in fresh culture medium containing Lysotracker Red (LTR, 500 nM; Invitrogen, L7528). Specific lysosomal labeling was confirmed by fluorescence microscopy and quantified by flow cytometry in the red spectrum (FL-3 channel).
Flow cytometry determination of SA-GLB1 activity
Quantification of SA-GLB1 activity was performed by flow cytometry with the fluorogenic substrate C12FDG (Invitrogen, F1179). First, alkalinization of the lysosomal compartment was induced by treating cell monolayers with 300 μM chloroquine for 1 h at 37°C under 5% CO2. The cells (1 × 106) were then trypsinized and incubated for 1 min at 37°C in 50 μL of prewarmed PBS containing C12FDG (33 μM). C12FDG uptake was stopped by adding 500 μL ice-cold PBS. The mean green fluorescence of 10,000 cells was recorded (FL-1 channel) 30 min later and quantified (CellQuest software; BD Biosciences). Nonstained control cells were included, to evaluate the baseline fluorescence. The laser intensity settings were adjusted to the lowest level so as not to detect autofluorescence.
Determination of lysosomal pH
Lysosomal pH was monitored using LysoSensor Yellow/Blue dextran (Invitrogen), which exhibits a pH-dependent dual-emission spectra in living cells. In acidic organelles the probe has predominantly yellow fluorescence, and in less acidic organelles it has blue fluorescence. Briefly, cells grown in 24-well plates were subjected to chronic oxidative stress as described above, and then loaded overnight with 1 mg/ mL of LysoSensor Yellow/Blue dextran (Invitrogen, L22460). Cells were then washed, trypsinized and resuspended in 200 μL of PBS. The fluorescence from the acidic compartments in the labeled cells was quantified with a fluorescence microplate reader at an emission wavelength of 530/430 nm with excitation at 340 nm. Lower 535/430 ratio indicates less acidic lysosomal pH.
Proteolysis assay
Following exposure to chronic oxidative stress, cells were labeled with 2 μCi/mL [3H]-leucine (MP Biomedicals, 20036E) for 48 h to maximize labeling of long-lived proteins. At the end of the labeling period, cells were washed five times with PBS and incubated with chase medium containing 50 times the molar concentration of leucine for two h to chase-out short-lived proteins. Chase medium was then replaced, and cells were incubated with fresh chase medium for 16 h. Culture media containing free [3H]-leucine released upon intracellular degradation was collected, clarified by centrifugation at 500 g × 5 min and precipitated overnight at 4°C in a final concentration of 10% trichloroacetic acid. Cells were washed and lysates generated in solubilization buffer (0.1 N NaOH, 0.1% sodium deoxycholate) at 37°C for 2 h. Radioactivity was counted in the acid-soluble fraction and in the solubilized cells. Proteolysis was calculated by dividing the soluble radioactivity cpm by the total cellular radioactivity cpm.
Statistical analysis
All experimental procedures were repeated at least three times in independent experiments with different cell lines. The percentage of increase of the experimental conditions compared with the control was calculated and averaged. Data are represented as the mean ± SD and were analyzed with Student’s t-test. p < 5% was considered statistically significant.
Acknowledgments
The authors would like to thank Dr. Tamotsu Yoshimori (Osaka University, Japan) and Dr. Andrea Ballabio (Telethon Institute of Genetics and Medicine, Italy) for kindly providing the plasmids constructs of tfLC3 and TFEB-3XFlag, respectively. This work was supported by grants from the National Eye Institute of Health (R01EY020491, R21EY019137, P30EY005722), American Health Assistance Foundation ( AHAF-G2012022) and Research to Prevent Blindness.
Glossary
Abbreviations:
- IOP
intraocular pressure
- POAG
primary open angle glaucoma
- TM
trabecular meshwork
- ROS
reactive oxygen species
- ATG
autophagy-related genes
- LAMP
lysosome-associated membrane protein
- SA-GLB1/SA-β-gal
senescence-associated-β-galactosidase
- PE
phosphatidylethanolamine
- tfLC3
tandem fluorescence LC3
- GFP
green fluorescence protein
- RFP
red fluorescence protein
- m.o.i
multiplicity of infection
- LTR
lysotracker red
- M6PR
mannose-6-phosphate receptor (cation dependent)
- TFEB
transcription factor EB
- CTSB
cathepsin B
- CTSD
cathepsin D
- scCTSB
single chain CTSB
- dcCTSB
double chain CTSB
- scCTSD
single chain CTSD
- dcCTSD
double chain CTSD
- Leup
leupeptin
- CQ
chloroquine
- BafA1
bafilomycin A1
- MTOR
mechanistic target of rapamycin
- d.p.i.
days postinfection
- Rap
rapamycin
- RFU
relative fluorescence units
- Leu
leucine
- cpm
counts per min
- 3-MA
3-methyladenine
- AF
autofluorescence
- C12FDG/FDG
di-β-D- galactopyranoside
- WB
western blot
- Treh
trehalose
- SQSTM1
sequestosome 1
- TUBB
β-tubulin
- BECN1
beclin 1
- LMNA
lamin A/C
- RPS6KB
ribosomal protein S6 kinase
- p-RPS6KB
phosphorylated ribosomal protein S6 kinase
- ATP6V0A2
ATPase, H+ transporting, lysosomal V0 subunit a2
- ATP6V1B2
ATPase, H+ transporting, lysosomal V1 subunit b2
- ATP6V1C1
ATPase, H+ transporting, lysosomal V1 subunit c1
- ATP6V1G1
ATPase, H+ transporting, lysosomal V1 subunit g1
- ATP6V1G2
ATPase, H+ transporting, lysosomal V1 subunit g2
- min
minutes
- h
hours
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/23568
References
- 1.Stamer WD, Acott TS. Current understanding of conventional outflow dysfunction in glaucoma. Curr Opin Ophthalmol. 2012;23:135–43. doi: 10.1097/ICU.0b013e32834ff23e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Toris CB, Yablonski ME, Wang YL, Camras CB. Aqueous humor dynamics in the aging human eye. Am J Ophthalmol. 1999;127:407–12. doi: 10.1016/S0002-9394(98)00436-X. [DOI] [PubMed] [Google Scholar]
- 3.Gabelt BT, Kaufman PL. Changes in aqueous humor dynamics with age and glaucoma. Prog Retin Eye Res. 2005;24:612–37. doi: 10.1016/j.preteyeres.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 4.Zanon-Moreno V, Marco-Ventura P, Lleo-Perez A, Pons-Vazquez S, Garcia-Medina JJ, Vinuesa-Silva I, et al. Oxidative stress in primary open-angle glaucoma. J Glaucoma. 2008;17:263–8. doi: 10.1097/IJG.0b013e31815c3a7f. [DOI] [PubMed] [Google Scholar]
- 5.Aslan M, Cort A, Yucel I. Oxidative and nitrative stress markers in glaucoma. Free Radic Biol Med. 2008;45:367–76. doi: 10.1016/j.freeradbiomed.2008.04.026. [DOI] [PubMed] [Google Scholar]
- 6.Ferreira SM, Lerner SF, Brunzini R, Evelson PA, Llesuy SF. Oxidative stress markers in aqueous humor of glaucoma patients. Am J Ophthalmol. 2004;137:62–9. doi: 10.1016/S0002-9394(03)00788-8. [DOI] [PubMed] [Google Scholar]
- 7.Saccà SC, Pascotto A, Camicione P, Capris P, Izzotti A. Oxidative DNA damage in the human trabecular meshwork: clinical correlation in patients with primary open-angle glaucoma. Arch Ophthalmol. 2005;123:458–63. doi: 10.1001/archopht.123.4.458. [DOI] [PubMed] [Google Scholar]
- 8.Saccà SC, Izzotti A. Oxidative stress and glaucoma: injury in the anterior segment of the eye. Prog Brain Res. 2008;173:385–407. doi: 10.1016/S0079-6123(08)01127-8. [DOI] [PubMed] [Google Scholar]
- 9.Kumar DM, Agarwal N. Oxidative stress in glaucoma: a burden of evidence. J Glaucoma. 2007;16:334–43. doi: 10.1097/01.ijg.0000243480.67532.1b. [DOI] [PubMed] [Google Scholar]
- 10.Saccà SC, Izzotti A, Rossi P, Traverso C. Glaucomatous outflow pathway and oxidative stress. Exp Eye Res. 2007;84:389–99. doi: 10.1016/j.exer.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 11.Ziangirova GG, Antonova OV. [Lipid peroxidation in the pathogenesis of primary open-angle glaucoma] Vestn Oftalmol. 2003;119:54–5. [PubMed] [Google Scholar]
- 12.He Y, Leung KW, Zhang Y-H, Duan S, Zhong X-F, Jiang R-Z, et al. Mitochondrial complex I defect induces ROS release and degeneration in trabecular meshwork cells of POAG patients: protection by antioxidants. Invest Ophthalmol Vis Sci. 2008;49:1447–58. doi: 10.1167/iovs.07-1361. [DOI] [PubMed] [Google Scholar]
- 13.De La Paz MA, Epstein DL. Effect of age on superoxide dismutase activity of human trabecular meshwork. Invest Ophthalmol Vis Sci. 1996;37:1849–53. [PubMed] [Google Scholar]
- 14.Izzotti A, Sacca S, Longobardi M, Cartiglia C, Izzotti A, Saccà SC, Longobardi M, Cartiglia C. Sensitivity of ocular anterior chamber tissues to oxidative damage and its relevance to the pathogenesis of glaucoma. Invest Ophthalmol Vis Sci. 2009;50:5251–8. doi: 10.1167/iovs.09-3871. [DOI] [PubMed] [Google Scholar]
- 15.Kiffin R, Bandyopadhyay U, Cuervo AM. Oxidative stress and autophagy. Antioxid Redox Signal. 2006;8:152–62. doi: 10.1089/ars.2006.8.152. [DOI] [PubMed] [Google Scholar]
- 16.Menardo J, Tang Y, Ladrech S, Lenoir M, Casas F, Michel C, et al. Oxidative stress, inflammation, and autophagic stress as the key mechanisms of premature age-related hearing loss in SAMP8 mouse Cochlea. Antioxid Redox Signal. 2012;16:263–74. doi: 10.1089/ars.2011.4037. [DOI] [PubMed] [Google Scholar]
- 17.Moore MN. Autophagy as a second level protective process in conferring resistance to environmentally-induced oxidative stress. Autophagy. 2008;4:254–6. doi: 10.4161/auto.5528. [DOI] [PubMed] [Google Scholar]
- 18.Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–73. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 19.Kaushik S, Cuervo AM. Autophagy as a cell-repair mechanism: activation of chaperone-mediated autophagy during oxidative stress. Mol Aspects Med. 2006;27:444–54. doi: 10.1016/j.mam.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mariño G, Madeo F, Kroemer G. Autophagy for tissue homeostasis and neuroprotection. Curr Opin Cell Biol. 2011;23:198–206. doi: 10.1016/j.ceb.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 21.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rubinsztein DC, Mariño G, Kroemer G. Autophagy and aging. Cell. 2011;146:682–95. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 23.Cuervo AM, Bergamini E, Brunk UT, Dröge W, Ffrench M, Terman A. Autophagy and aging: the importance of maintaining “clean” cells. Autophagy. 2005;1:131–40. doi: 10.4161/auto.1.3.2017. [DOI] [PubMed] [Google Scholar]
- 24.Liton PB, Lin Y, Luna C, Li G, Gonzalez P, Epstein DL. Cultured porcine trabecular meshwork cells display altered lysosomal function when subjected to chronic oxidative stress. Invest Ophthalmol Vis Sci. 2008;49:3961–9. doi: 10.1167/iovs.08-1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liton PB, Challa P, Stinnett S, Luna C, Epstein DL, Gonzalez P. Cellular senescence in the glaucomatous outflow pathway. Exp Gerontol. 2005;40:745–8. doi: 10.1016/j.exger.2005.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004;117:2805–12. doi: 10.1242/jcs.01131. [DOI] [PubMed] [Google Scholar]
- 28.Rubinsztein DC, Cuervo AM, Ravikumar B, Sarkar S, Korolchuk V, Kaushik S, et al. In search of an “autophagomometer”. Autophagy. 2009;5:585–9. doi: 10.4161/auto.5.5.8823. [DOI] [PubMed] [Google Scholar]
- 29.Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–5. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 30.Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–60. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- 31.Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–7. doi: 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- 32.Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–33. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Young ARJ, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JFJ, et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23:798–803. doi: 10.1101/gad.519709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Young AR, Narita M. Connecting autophagy to senescence in pathophysiology. Curr Opin Cell Biol. 2010;22:234–40. doi: 10.1016/j.ceb.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 35.Kurz DJ, Decary S, Hong Y, Erusalimsky JD. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci. 2000;113:3613–22. doi: 10.1242/jcs.113.20.3613. [DOI] [PubMed] [Google Scholar]
- 36.Lee BY, Han JA, Im JS, Morrone A, Johung K, Goodwin EC, et al. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell. 2006;5:187–95. doi: 10.1111/j.1474-9726.2006.00199.x. [DOI] [PubMed] [Google Scholar]
- 37.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–75. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383–435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- 39.Geng J, Klionsky DJ. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep. 2008;9:859–64. doi: 10.1038/embor.2008.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell. 2008;19:2092–100. doi: 10.1091/mbc.E07-12-1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kang HT, Lee KB, Kim SY, Choi HR, Park SC. Autophagy impairment induces premature senescence in primary human fibroblasts. PLoS One. 2011;6:e23367. doi: 10.1371/journal.pone.0023367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, et al. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature. 2009;461:654–8. doi: 10.1038/nature08455. [DOI] [PubMed] [Google Scholar]
- 43.Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal. 2012;5:ra42. doi: 10.1126/scisignal.2002790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peña-Llopis S, Vega-Rubin-de-Celis S, Schwartz JC, Wolff NC, Tran TAT, Zou L, et al. Regulation of TFEB and V-ATPases by mTORC1. EMBO J. 2011;30:3242–58. doi: 10.1038/emboj.2011.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Repnik U, Stoka V, Turk V, Turk B. Lysosomes and lysosomal cathepsins in cell death. Biochim Biophys Acta. 2012;1824:22–33. doi: 10.1016/j.bbapap.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 46.O’Neil J, Hoppe G, Sayre LM, Hoff HF. Inactivation of cathepsin B by oxidized LDL involves complex formation induced by binding of putative reactive sites exposed at low pH to thiols on the enzyme. Free Radic Biol Med. 1997;23:215–25. doi: 10.1016/S0891-5849(96)00612-0. [DOI] [PubMed] [Google Scholar]
- 47.Nishimura Y, Tsuji H, Kato K, Sato H, Amano J, Himeno M. Biochemical properties and intracellular processing of lysosomal cathepsins B and H. Biol Pharm Bull. 1995;18:829–36. doi: 10.1248/bpb.18.829. [DOI] [PubMed] [Google Scholar]
- 48.Ashworth JL, Biswas S, Wraith E, Lloyd IC. The ocular features of the mucopolysaccharidoses. Eye (Lond) 2006;20:553–63. doi: 10.1038/sj.eye.6701921. [DOI] [PubMed] [Google Scholar]
- 49.Biswas J, Nandi K, Sridharan S, Ranjan P. Ocular manifestation of storage diseases. Curr Opin Ophthalmol. 2008;19:507–11. doi: 10.1097/ICU.0b013e32831215c3. [DOI] [PubMed] [Google Scholar]
- 50.Narita M, Young ARJ, Arakawa S, Samarajiwa SA, Nakashima T, Yoshida S, et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science. 2011;332:966–70. doi: 10.1126/science.1205407. [DOI] [PMC free article] [PubMed] [Google Scholar]