

Abstract
Objectives
To investigate the effect of nerve growth factor (NGF) on the action potential and potassium currents of non-infarcted myocardium in the myocardial infarcted rabbit model.
Methods
Rabbits with occlusion of the left anterior descending coronary artery were prepared and allowed to recover for eight weeks (healed myocardial infarction, HMI). During ligation surgery of the left coronary artery, a polyethylene tube was placed near the left stellate ganglion in the subcutis of the neck for the purpose of administering NGF 400 U/d for eight weeks (HMI + NGF group). Cardiomyocytes were isolated from regions of the non-infarcted left ventricular wall and the action potentials and ion currents in these cells were recorded using whole-cell patch clamps.
Results
Compared with HMI and control cardiomyocytes, significant prolongation of APD50 or APD90 (Action potential duration (APD) measured at 50% and 90% of repolarization) in HMI + NGF cardiomyocytes was found. The results showed that the 4-aminopyridine sensitive transient outward potassium current (Ito), the rapidly activated omponent of delayed rectifier potassium current (IKr), the slowly activated component of delayed rectifier potassium current (IKs), and the L-type calcium current (ICaL) were significantly altered in NGF + HMI cardiomyocytes compared with HMI and control cells.
Conclusions
Our results suggest that NGF treatment significantly prolongs APD in HMI cardiomyocytes and that a decrease in outward potassium currents and an increase of inward Ca2+ current are likely the underlying mechanism of action.
Keywords: Nervous growth factor, Myocardial infarction, Action potential, Cardiomyocytes
1. Introduction
Patients experiencing myocardial infarction (MI) are at an increased risk for reinfarction, remodeling, and sudden cardiac death during the post-infarction period. Healed-infarction remodeling is an important factor for triggering ventricular arrhythmias, which is thought to be due to ventricular tachycardia or fibrillation.[1],[2] The cellular and molecular mechanisms leading to healed-infarct sympathetic hyperinnervation are still unclear. This sympathetic regeneration is triggered by the release of nerve growth factor (NGF) in the non-neural cells at the non-infarcted area of the heart.
NGF is a potent growth and survival factor for sympathetic neurons.[3],[4] Previous studies showed that NGF is essential for enhanced post-infarct sympathetic sprouting and facilitated the development of high-yield ventricular arrhythmias.[5]–[7] MI tends to increase the NGF myocardial expression. The NGF originating from the non-neural cells facilitates nerve sprouting to nourish the end organs. This changes the physiological environment of the infarcted area. In dogs, NGF infusion to the left stellate ganglion after MI resulted in an increased QT interval, which is a biomarker for ventricular tachyarrhythmias. These results indicated that QT interval prolongation is causally related to the occurrence of ventricular arrhythmias and nerve sprouting after MI.[8] To test this hypothesis, we examined the effect of NGF on the repolarization of potassium currents in cardiomyocytes. We examined, the 4-aminopyridine sensitive transient outward potassium current (Ito), the rapidly activated component of delayed rectifier potassium current (IKr), the slowly activated component of delayed rectifier potassium current (IKs), and the L-type calcium current (ICaL). The non-infarcted area of the myocardium exhibits ventricular arrhythmogenic abnormalities of both conduction and repolarization. The aim of this study was to better understand the effect of NGF on the action potential and potassium currents of non-infarcted myocardium after MI.
2. Methods
All experimental procedures and protocols were carried out according to the Chinese law on animal experimentation and approved by the Animal Experimental Committee of Chinese PLA General Hospital. The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23 revised 1996).
2.1. Model of MI
A rabbit model of MI was established as previously described.[9] Briefly, New Zealand white rabbits (weighing 2.0–2.5 kg) were anesthetized with pentobarbital (30 mg/kg) and their left anterior descending coronary arteries were ligated. The animals were then allowed to recover for eight weeks (healed myocardial infarction, HMI group, n = 9). During surgery of the left anterior descending coronary artery ligation, a polyethylene tube (1.5 mm) was placed near the left stellate ganglion for administering NGF for eight weeks (HMI + NGF group, n = 8). Other animals, as control group (Ctrl group, n = 10), underwent an identical surgical procedure, but without coronary ligation or placement of the polyethylene tube.
2.2. Immunocytochemical studies
The non-infarcted region of the left ventricular wall was used for immunocytochemical studies. Five micron transmural sections were immunostatined for the nerve marker, tyrosine hydroxylase (TH), using a modified immuocytochemical ABC method.[10] Control tissues were obtained from the left ventricular wall of normal healthy rabbits. The primary antibodies used in this study were monoclonal mouse anti-rat TH (Boehringer Mannheim Biochemica, Indianapolis, IN; working concentration, 0.2 µg/mL). We analyzed three samples for each group. After staining, each slide was examined under a microscope and the nerve densities were quantified using a computer-assisted image analysis system.[11]
2.3. Isolation of ventricular cardiomyocytes
Ventricular cardiomyocytes from the non-infarcted side of the heart were isolated with the same protocol as described previously.[12] Briefly, the heart was suspended on a Langendorff perfusion apparatus, and perfused for 20 min with Tyrode's solution containing 0.33 mg/mL collagenase, 0.025 mg/mL protease E, and 1.25 mg/mL bovine serum albumin. The isolated tissue samples from the non-infarcted myocardium of the left ventricular wall were minced and sequentially digested for 20 min to 25 min in a fresh enzyme solution at 37°C. The cardiomyocytes isolated were then attached to the cover slips with cell adhesive and then incubated for 18 h for study.
2.4. Patch clamp experiments in isolated ventricular myocytes
Patch clamp experiments were performed on these isolated ventricular cardiomyocytes. Quiescent, calcium-tolerant, rod-shaped cells with clear cross striation were used for action potential recordings at 35°C. Transmembrane potentials and currents were recorded using the whole cell patch-clamp technique with a MultiClamp 700B amplifier (Axon Instruments). All signals were acquired at 5 kHz (Digidata 1322A, Axon Instruments) and analyzed by pCLAMP version 9.2 software (Axon Instruments). Whole cell currents and Action potentials (APs), obtained under voltage clamp, were filtered at 1–5 kHz and sampled at 5–50 kHz, and the series resistance was typically < 5 megaohms after about 70% compensation. The P/4 protocol was used to subtract online the leak and capacitive transients.
APs were elicited using the current-clamp mode at a rate of 5.0 Hz of 30 train suprathreshold current pulses. Cardiomyocytes were electrically stimulated by intracellular current injection through patch electrodes using depolarizing pulses with a duration of 3 ms and an amplitude of 1.5–2.5nA. Action potential duration (APD) was measured at 90% and 50% of repolarization (APD90 and APD50). Repolarization currents, including Ito, IKr, IKs, and ICaL, were recorded using the voltage-clamp mode.
For recording K+ currents, extracellular solution with tetrodotoxin inhibits Na+ current. CdCl2 inhibits Ca2+ current. Pipettes were filled with: 140 mmol/L K-aspartame acid, 4 mmol/L MgATP, 1 mmol/L MgCl2, 10 mmol/L Ethylene glycol tetraacetic acid (EGTA), 0.1 mmol/L Guanosine triphosphate (GTP), 10 mmol/L 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) mmol/L, pH adjusted to 7.3 with KOH. Myocytes were superfused with solution containing: 140 mmol/L NaCl, 4 mmol/L KCl, 1 mmol/L CaCl2, 1 mmol/L MgCl2, 10 mmol/L HEPES, 5 mmol/L glucose, pH adjusted to 7.4 with NaOH.
We used pre-pulse to -40 mV for 30 ms to inactivate INa. Ito was recorded in voltage-clamp mode with 300 ms pulses from a holding potential of -80 mV, with different test potentials increased from -40 mV to +50 mV with 10 mV steps. Steady-state activation of Ito was induced by voltage steps between -120 mV and +60 mV for 500 ms from a holding potential of -80 mV. Steady-state inactivation of Ito was induced by a condition pulse of +40 mV for 500 ms, following voltage steps between -120 mV to +30 mV, with 10 mV for 1000 ms. Time constant of close-state inactivation of Ito was induced by following depolarization pulse of +40mV for 300 ms return to -100 mV, depolarizing to -65 mV during different time pluses of 25 ms, 50 ms, 100 ms, 200 ms, 500 ms, 1000 ms, 2500 ms and 5000 ms. Time constant of inactivation was fitted by single exponential function. Voltage-dependent of the time course of recovery from inactivation was evaluated with a paired-pulse protocol: conditioning pulse was applied to +50 mV for 300 ms from holding potential of -80 mV, following test potentials of +50 mV for 300 ms during different time intervals of 40 ms, 80 ms, 160 ms, 320 ms, 640 ms, 1280 ms and 2560 ms. The time course of recovery from fast inactivation was fitted by single exponential function.
To isolate IK, we used pre-pulse to -40 mV for 30 ms to inactivate INa. The IKr was recorded in voltage-clamp mode with 250 ms pulses from a holding potential of -90 mV, to different test potentials increased from -50 mV to +50 mV with 10 mV steps following -40 mV for 1000 ms of repolarizating pulse to induce tail current. Steady-state activation of IKr was induced by voltage steps between -60 mV and +40 mV for 1000 ms, following repolarization pulse of -40 mV for 1500 mV from a holding potential of -90 mV. Steady-state inactivation of IKr was induced by a condition pulse of +40 mV for 500 ms, following voltage steps between -140 mV and +40 mV, with 20 mV steps for 20 ms, test pulse of +40 mV for 1000 ms. Time constant of fast inactivation of IKr was induced by condition pulse of +50 mV for 200 ms, following hyperpolarization pulse of -100 mV for 10 ms, test pulse range from -20 mV to +60 mV for 1500 ms. Time constant of fast inactivation was fitted by single exponential function. Voltage-dependent of the time course of recovery from fast inactivation was evaluated with a paired-pulse protocol: conditioning pulse was applied to +50 mV for 1500 ms from holding potential of -90 mV, following test potentials of -120 mV to -30 mV for 3000 ms. The time course of recovery from fast inactivation was fitted by single exponential function.
IKs was recorded in voltage-clamp mode with 4000 ms pulses from a holding potential of -40 mV, to different test potentials increased from -40 mV to +80 mV with 10 mV steps following -40 mV for 3000 ms of repolarizating pulse to induce tail current.
Steady-state activation of IKs was induced by voltage steps between -120 mV and +80 mV for 4000 ms with repolarization pulse to -40 mV for 3000 ms from a holding potential of -80 mV. Steady-state inactivation of IKs was induced by voltage steps between -120 mV to +60 mV for 2000 ms following test pulse of +60 mV for 2000 ms from a holding potential of -80 mV. The deactivation of IKs was induced by condition pulse of +60 mV for 3000 ms following test pulses from -140 mV to -20 mV for 2000 ms for 5000 ms with 20 mV step from a holding potential of -80mV. The time course of deactivation of IKs was fitted by single exponential function.
Steady-state activation (SSA) curve was described by assuming a Boltzmann function, I/Imax = A1+A2/(1+ exp((V0.5-Vrev)/k)). V1/2,act is half-maximal activation value and kact is the slope factor of activation curve. Steady- state inactivation was described by assuming a Boltzmann function, I/Imax = A1+A2/(1+exp((Vrev-V0.5)/k)). Where I is the peak current elicited by depolarizing test pulse to various potentials and Vrev is the reversal potential (obtained from the extrapolated I-V curves). Imax is the maximum of I current. V1/2,inact is half-maximal inactivation value and kinact is the slope factor of inactivation curve.
ICaL was recorded in voltage-clamp mode with 150 ms pulses from a holding potential of -40 mV, with different test potentials increased from -40 mV to +40 mV with 10 mV steps. Steady-state activation of ICaL was induced by voltage steps between -60 mV to +30 mV for 500 ms from a holding potential of -40 mV. Steady-state inactivation of ICaL was induced by test pulse of +30 mV for 500 ms after condition voltage steps between -70 mV and +30 mV, with 10 mV step for 1000 ms. The time course of recovery from inactivation of ICaL was evaluated with a paired-pulse protocol: conditioning pulse was applied to +50 mV for 150 ms from holding potential of -80 mV, following test potentials of +50 mV for 150 ms during different time interval of 40 ms, 80 ms, 160 ms, 320 ms, 640 ms, 1280 ms, 2560 ms and 5120 ms. The time course of recovery from fast inactivation was fitted by single exponential function.
2.5. Statistical analysis
Statistical analyses were performed using SPSS version 14. Data were expressed as a mean ± SE. One way ANOVA with Bonferroni post hoc test was used. P < 0.05 was considered statistically significant.
3. Results
3.1. Sympathetic nerve fibers
Sympathetic nerve fibers sprouted in the ventricles of hearts from the HMI + NGF group. The distribution of nerve fibers became less homogeneous, suggesting the presence of sympathetic hyperinnervation in the healed- infarcted ventricle after NGF treatment. The density of sympathetic nerve fibers in the HMI + NGF group was higher than those in the HMI and control groups. The density of nerve fibers calculated in the HMI + NGF group was significantly higher than that in the control group (P < 0.01). Nerve regeneration and proliferation were observed in the HMI group, but showed no significant difference when compared to the control group (Table 1, Figure 1).
Table 1. Densities of sympathetic nerve fibers in the ventricles of the three groups.
| Groups | Densities of nerve fibers (µm2/mm2) |
| Control | 11.3 ± 1.1 |
| HMI | 15.8 ± 2.8 |
| HMI + NGF | 33.8 ± 4.7# |
#P < 0.01, n = 3, vs. HMI or Control group. HMI: healed myocardial infarction; NGF: nerve growth factor.
Figure 1. The sympathetic nerve fibers in the ventricles of the three groups.
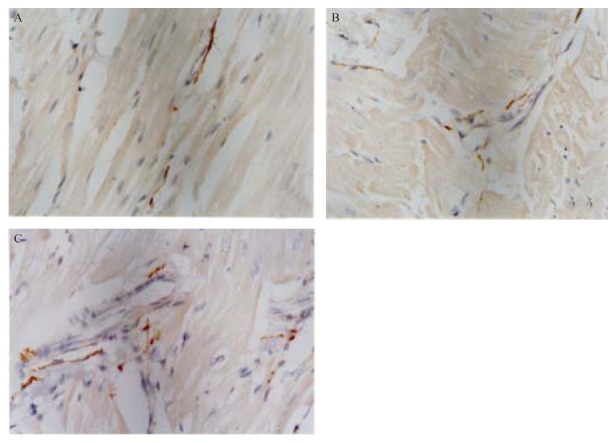
The distribution of nerve fibers in these five micron sections was determined by immunostaining for the nerve marker, tyrosine dydroxylase. Sympathetic nerve fiber density in the HMI + NGF group (A) was significantly higher than those in HMI group (B) and Ctrl group (C). Nerve regeneration and proliferation were observed in the HMI group, but they were not significantly different from the Ctrl group. Nerve fiber distribution was increased, in the HMI + NGF group, suggesting that sympathetic hyperinnervation occurred in the healed-infarct ventricle after NGF treatment. Ctrl: control; HMI: healed myocardial infarction; NGF: nerve growth factor.
Sympathetic nerve fibers sprouted in the ventricles of hearts from the HMI + NGF group. The distribution of nerve fibers became less homogeneous, suggesting the presence of sympathetic hyperinnervation in the healed-infarcted ventricle after NGF treatment. The density of sympathetic nerve fibers in the HMI + NGF group was higher than those in the HMI and control groups. The density of nerve fibers calculated in the HMI + NGF group was significantly higher than that in the control group (P < 0.01). Nerve regeneration and proliferation were observed in the HMI group, but showed no significant difference when compared to the control group (Table 1, Figure 1).
3.2. Action potentials
Action potential traces were recorded in three different groups of isolated cardiomyoctyes: the control group, the HMI group, and the HMI + NGF group. The APD50 of the HMI + NGF cardiomyocytes (233.7 ± 11.8 ms), was longer than that of the HMI (187.6 ± 10.2 ms) and control cardiomyocytes (150.3 ± 9.9 ms, P < 0.01, n = 20, Figure 2A and 2B). The APD90 was significantly different between the three groups (357.5 ± 13.5 ms in the HMI + NGF group, 272.1 ± 10.7 ms in the HMI group, and 221.7 ± 11.2 ms in the control group). These results proved the lengthening of the APD were more notable after NGF infusion (P < 0.01, n = 20, Figure 2C). In contrast, no difference was observed in the amplitude of action potentials and the resting membrane potential among the three groups (Figure 2D and E).
Figure 2. Action potentials in rabbit ventricular cardiomyocytes of the three groups.

Action potential traces were recorded in three groups and compared with the HMI and Control groups. APD50 and APD90 were markedly prolonged in HMI + NGF cardiomyocytes (A-C). No significant difference was detected in the amplitude of the action potential and the resting membrane potential among various groups (D and E). *P < 0.01 vs. Ctrl group, #P < 0.05, †P < 0.01 vs. HMI group. APD50, APD90: action potential duration measured at 50% and 90% of repolarization respectively; Ctrl: control; HMI: healed myocardial infarction; NGF: nerve growth factor.
3.3. Ito
The left inset showed current traces obtained by applying train pulses. Current amplitudes in HMI + NGF cardiomyocytes were markedly smaller than that in HMI and control cardiomyocytes (Figure 3A). At test potentials of +50mV, current densities of Ito were 16.5 ± 1.1 pA/pF in HMI + NGF cardiomyocytes, 25.9 ± 1.5 pA/pF in HMI cardiomyocytes, and 35.3 ± 2.6 pA/pF in control cardiomyocytes (P < 0.01, n = 15, Figure 3B). The current-voltage relationship showed a slower acceleration of densities (Ito) in HMI+NGF cardiomyocytes and a more positive membrane potential of +10 mV (Figure 3C).
Figure 3. Comparison of the Ito in the three groups of rabbit ventricular cardiomyocytes.

The left inset showed current traces obtained by applying train pulses from -40 mV to + 50 mV for 300 ms from holding potential of -80 mV. (A): Current amplitudes of HMI and HMI + NGF cardiomyocytes were smaller than those of Ctrl; (B): Compared with Ctrl cardiomyocytes, densities of Ito in HMI and HMI + NGF were markedly decreased, at test potentials of +50 mV; (C): Current-voltage relationship showed that densities of Ito were increased at more positive membrane potentials. *P < 0.05, #P < 0.01 vs. Ctrl group; †P < 0.05 vs. HMI group. Ctrl: control; HMI: healed myocardial infarction; Ito: transient outward potassium current; NGF: nerve growth factor.
The gating mechanism of Ito was studied. The steady- state activated curve from HMI + NGF cardiomyocytes was shifted to positive potentials (Figure 4A-C). The V1/2,act was -32.5 ± 3.2 mV in the HMI + NGF group, -62.1 ± 3.8 mV in the HMI group, and -65.4 ± 6.5 mV in control group (P < 0.01, n = 15). The kact was -18.5 ± 1.7 mV in the HMI + NGF group, -21.6 ± 1.9 mV in the HMI group, and -35.2 ± 2.4 mV in the control group. The steady-state inactivated curve exhibited no changes in both the V1/2,inact and the kinact (Figure 4D-F). The inactive, closed-state of HMI cardiomycoytes treated with NGF treatment was accelerated (Figure 4G). A slight, slow recovery from inactivation of the Ito in HMI + NGF cardiomyocytes was obtained, as evidenced by an increase in the τ value; however, no significant differences were observed when comparing the HMI cardiomyocytes (304 ± 23 ms) with the HMI group (269 ± 15 ms), (P > 0.05, Figure 4H and I).
Figure 4. Comparison of the gating mechanism of Ito in the three groups.

Steady-state activated curves of HMI and HMI + NGF cardiomyocytes were shifted to negative potentials (A-C), with significant change in V1/2,act and k. The steady-state inactivated curve was also shifted to negative potential, while k of the inactivated curve was not changed significantly (D-F). Close-state inactivation in HMI cardiomycoytes with NGF treatment was accelerated, with a shorter time constant (G). A slower recovery from inactivation of Ito in HMI cardiomyocytes with NGF treatment was obtained, with an increased τ value (H & I). *P < 0.05, #P < 0.01 vs. Ctrl; †P < 0.01 vs. HMI group. Ctrl: control; HMI: healed myocardial infarction; Ito: transient outward potassium current; NGF: nerve growth factor.
3.4. IKr
Figure 5 shows the representative IKr traces in rabbit ventricular cardiomyocytes of the three groups. Current amplitudes of the HMI cardiomyocytes with NGF treatment were smaller than those of the HMI and control cardiomyocytes (Figure 5A). At a test potential of +10 mV, tail current densities in control, HMI, and HMI + NGF groups were 44.1 ± 6.0 pA/pF, 31.3 ± 2.9 pA/pF, and 18.2 ± 1.8 pA/pF, respectively (P < 0.01, n = 12, Figure 5B). The current-voltage relationship showed that the current densities in the HMI + NGF cardiomyocytes were significantly decreased when compared with the HMI and the control cardiomyocytes with more than +10 mV difference in test potentials (Figure 5C).
Figure 5. Representative IKr traces in the groups of rabbit ventricular myocytes.

(A): Current amplitudes of the HMI cardiomyocytes with NGF treatment were significantly smaller than those of HMI and Ctrl cardiomyocytes; (B): At a test potential of 0 mV, tail current densities in the three groups were as follows: 44.1 ± 6.0 pA/pF in the Ctrl group, 31.3 ± 2.9 pA/pF in the HMI group, and 18.2 ± 1.8 pA/pF in the HMI + NGF group; (C): The current-voltage relationship showed that current densities in the HMI and the HMI + NGF cardiomyocytes were significantly decreased when compared with Ctrl. *P < 0.01 vs. Ctrl, #P < 0.01 vs. HMI group. Ctrl: control; HMI: healed myocardial infarction; IKr: rapidly activated component of delayed rectifier potassium current; Ito: transient outward potassium current; NGF: nerve growth factor.
The steady-state activated curves (IKr) from the three groups were not markedly different (Figure 6A-C), yet the steady-state inactivated curve of the HMI + NGF group was negatively shifted. The V1/2,inact shifted from -43.1 ± 3.4 mV in the HMI group to -66.3 ± 5.8 mV in the HMI + NGF group (P < 0.01, n = 12, Figure 6D-F). The time constant of the fast inactivation (IKr) in the HMI + NGF cardiomyocytes was shorter than that of the HMI or control cardiomyocytes at a test potential range of -20 mV to +40 mV (Figure 6G). In contrast, the time constant of recovery from inactivation of IKr of HMI + NGF cardiomyocytes was slightly longer than that of HMI cardiomyoctes at a range of -100 mV to -30 mV; however, this difference was not statistically significant (Figure 6H).
Figure 6. The gating mechanism of IKr in the three groups of rabbit ventricular cardiomyocytes.

The steady-state activated curve of the three groups were not markedly different (A-C), while steady-state inactivated curves for HMI and HMI + NGF cardiomyocytes were shifted negative, with a V1/2,inact shifted from -21.6 ± 3.2 mV for the Ctrl group to -46.2 ± 7.2 mV in the HMI group, -50.7 ± 6.1 mV in the HMI+NGF group (D-F). The time constant of fast inactivation (IKr) for HMI and HMI + NGF cardiomyocytes was shorter than that of Ctrl cardiomyocytes (G). In contrast, the time constant of recovery from inactivation of IKr for HMI and HMI + NGF cardiomyocytes was longer than that of Ctrl cardiomyocytes at range from -120 mV to -30 mV (H). *P < 0.01 vs. Ctrl cardiomyocytes, #P < 0.01 vs. HMI group. Ctrl: control; HMI: healed myocardial infarction; IKr: rapidly activated component of delayed rectifier potassium current; NGF: nerve growth factor.
3.5. IKs
Compared with the HMI group, the current amplitudes from NGF treated HMI cardiomyocytes were significantly smaller (Figure 7A). The peak densities of step currents of IKs in three groups were as follows: 123.15 ± 8.4 pA/pF in control cardiomyocytes, 89.7 ± 10.6 pA/pF in HMI cardiomyocytes, and 55.5 ± 5.3 pA/pF in HMI + NGF cardiomyocytes (P < 0.01, n = 15). Tail currents (IKs) in the three groups were as follows: 12.3 ± 2.4 pA/pF in the control group, 7.3 ± 0.9 pA/pF in the HMI group, and 4.9 ± 0.347 pA/pF in the HMI + NGF group (P < 0.01, n = 15, Figure 7B & C).
Figure 7. Representative IKs traces in the three groups of rabbit ventricular myocytes.

Current amplitudes of HMI cardiomyocytes with NGF treatment were significantly smaller (A). The peak densities of step and tail currents of IKs in the three groups were significantly decreased (B & C). *P < 0.01 vs. Ctrl cardiomyocytes, #P < 0.01 vs. HMI group. Ctrl: control; HMI: healed myocardial infarction; IKs: slowly activated component of delayed rectifier potassium current; NGF: nerve growth factor.
The current-voltage relationship was also tested. The SSA curves of the three groups were not markedly different (Figure 8A & B). The time constant of deactivation (IKs) in HMI + NGF cardiomyocytes was shortened compared with HMI and control cardiomyocytes at a range of -140 mV to -20 mV (Figure 8C). The steady-state inactivatation curve for HMI + NGF cardiomyocytes was negatively shifted. The V1/2,inact was -11.7 ± 1.0 mV for control cardiomyocytes, -25.4 ± 4.2 mV for HMI cardiomyoctyes, and -43.5 ± 4.3 for HMI + NGF cardiomyocytes, P < 0.01, n = 15), while the kinact was -33.3 ± 3.3 mV for control cardiomyocytes, -23.3 ± 2.3 mV for HMI cardiomyocytes, and -31.1 ± 4.3 mV for HMI + NGF cardiomyocytes, Figure 8D-F.
Figure 8. The gating mechanism of IKs in the three groups of rabbit ventricular myocytes.

The current-voltage relations also showed that current densities in HMI and HMI + NGF cardiomyocytes were significantly smaller than those in Ctrl cardiomyocytes (A). The steady-state activated curves of the three groups were not markedly different (B). The time constant of deactivation (IKs) in HMI and HMI + NGF cardiomyocytes was shortened compared with that Ctrl cardiomyocytes (C). The steady-state inactivated curves of HMI and HMI + NGF cardiomyocytes was neagitvely shifted, with significant changes in V1/2,inact and kinact (D-F). *P < 0.05, #P < 0.01 vs Ctrl cardiomyocytes; †P < 0.05, §P < 0.01 vs. HMI group. Ctrl: control; HMI: healed myocardial infarction; IKs: slowly activated component of delayed rectifier potassium current; NGF: nerve growth factor.
3.6. ICaL
ICaL in HMI + NGF cardiomyocytes were significantly increased. At 0 mV, the mean current densities were -21.9 ± 0.9 pA/pF in the HMI + NGF group, -12.0 ± 1.2 pA/pF in the HMI group, and -8.2 ± 0.7 pA/pF in the control group (P < 0.01, n = 15). The current-voltage relationship showed that current densities in HMI + NGF cardiomyocytes were significantly larger than those in the HMI and control groups, which ranged from –20 mV to +30 mV (Figure 9A&B). The steady-state activation curve of HMI + NGF cardiomyocytes was negatively shifted, with a shift in V1/2,act from -21.6 ± 3.2 mV in the control group, -28.2 ± 2.9 mV in the HMI group to -39.5 ± 3.1 mV in the HMI + NGF group (P < 0.01, n = 15), while the kact values for the three groups were -9.9 ± 1.2 mV, -17.9 ± 2.3 mV and -20.9 ± 2.7 mV, respectively. No significant differences in the steady-state inactivation curves were noted between the three groups (Figure 9C-E). Furthermore, window currents of ICaL in HMI + NGF cardiomyocytes were enhanced with a negative shift in the steady-state activation curve. Recovery from inactivation (ICaL) in HMI + NGF cardiomyocytes was faster. The average recovery time constants from inactivation were 895 ± 20 ms in the control group, 452 ± 13 ms in the HMI group, and 214 ± 12 ms in the HMI + NGF group, respectively (P < 0.01, n = 15, Figure 9F&G).
Figure 9. Representative ICaL traces in the three groups of rabbit ventricular myocytes.

(A): Current amplitudes of HMI + NGF cardiomyocytes were significantly larger than those of HMI and Control cardiomyocytes. (B): The current-voltage relations also showed that current densities in HMI + NGF cardiomyocytes were significantly increased, in the range of -20 mV and +20 mV. (C-E): The steady-state activated curves of HMI + NGF cardiomyocytes were shifted to a more negative potential, while the steady-state inactivated curves in the three groups were not significantly different. (F & G): The time course of recovery from inactivation was well fitted by biexponential relation. A faster recovery from inactivation of ICaL in HMI + NGF cardiomyocytes were obtained. *P < 0.01 vs. Ctrl cardiomyocytes, #P < 0.01 vs. HMI cardiomyocytes. Ctrl: control; HMI: healed myocardial infarction; ICaL: L-type calcium current; NGF: nerve growth factor.
4. Discussion
Previous clinical and experimental studies suggest that MI is associated with a greater risk of sudden cardiac death and is likely caused by lethal ventricular arrhythmias.[13]–[15] Ventricular arrhythmias are frequently observed after healed-infarction.[16],[17] Although the cellular mechanisms of arrhythmogenesis in this pathophysiological condition are poorly understood, they may be explained by the observation that sympathetic nerve sprouting in HMI augments the incidences of ventricular tachyarrhythmia. Beta-blockers are known to prevent and/or reduce these arrhythmias as well as sudden cardiac death.[12],[18] NGF signaling within the heart is an important regulator of sympathetic synaptic activity. In this study, we showed that the density of sympathetic nerve fibers in the HMI + NGF group was increased compared with controls. This result suggested that NGF treatment resulted in sympathetic hyperinnervation of the infarcted ventricle.
Our results from a rabbit model of HMI, also showed the cellular electrophysiological changes that were induced by the infusion of NGF to the left stellate ganglion. NGF treatment significantly increased APD, with the APD50 being increased by 24.6% and APD90 by 31.4%, respectively. Increases in the QT interval and development of arrhythmias suggested that the APD was prolonged.[19],[20] Secreted NGF by cardiomyocytes promotes cardiac sympathetic innervation.[21],[22] Infarcted myocardial cells release NGF locally causing the increased expression of neurotrophic substances, retrograde transport to the left stellate ganglion, and presumably to other thoracic ganglia. These effects trigger extensive growth of cardiac sympathetic neurons.[23],[24] In a canine healed-MI model, induction of nerve sprouting by the infusion of NGF into the left stellate ganglion resulted in increased QT intervals, ventricular tachycardia, and fibrillation.[8],[25],[26]
Prolonged APD typically occurs in heart disease when several potassium currents are reduced. Two outward voltage-dependent potassium currents are involved in the repolarization phase. One is the rapidly activating and inactivating current, Ito, which is sensitive to 4-aminopyridine, while the other is the delayed rectifier potassium current, IK. We investigated the underlying mechanisms involved in altering potassium channel currents in isolated rabbit ventricular cardiomyocytes after undergoing MI with or without the perfusion of NGF. The main depolarizing potassium current showed a significant reduction in peak current density with concomitant alterations in channel kinetics. Compared with HMI cardiomyocytes, the Ito peak density of HMI + NGF cardiomyocytes was decreased by 36.3%. Study of the channel kinetics showed a positive potential shift in the voltage dependence of steady-state activation and an augmented rate of close-state inactivation (Ito) in HMI + NGF cardiomyocytes. Ventricular arrhythmia often occurs when there is an underlying electrophysiological substrate. In addition, alterations in repolarizing potassium currents are known to contribute to arrhythmogenesis. Other studies in rats also report a significant down-regulation of channel protein expression after healed-infarct remodeling.[27],[28]
IK is responsible for the late repolarization phase of the action potential and regulates APD in many species. The drug insensitive component, IKs, shows a slow activation whereas the drug-sensitive component, IKr, activates more rapidly and exhibits a remarkable prominent inward rectification. In our study, reduction of IKr density in HMI + NGF cardiomyocytes was observed, which reduced the tail peak current by 42.2%. The steady-state inactivated curve of IKr in HMI + NGF cardiomyocytes was negatively shifted, while the time course of fast inactivation was accelerated. Since, this current contributes to the late phase of repolarization, reductions in IKr could further exacerbate action potential prolongation. In fact, the degree of prolongation at APD90 repolarization exceeded that observed at APD50.
APD is tightly regulated via alterations in IKs. IKs is enhanced at high stimulation rates due to the incomplete slow deactivation of the current. Although IKs is slow to activate, protocols involving short pulses in the physiological range indicate that IKs can provide an important contribution to action potential repolarization, particularly in epicardial and endocardial cells. Tail currents (IKs) from HMI + NGF cardiomyocytes were reduced. Analysis of the current kinetics further revealed an acceleration of activation, with a negative shift of V1/2,act, an acceleration of deactivation, and a shorter time constant of deactivation. These characteristics of IKs activation are similar to those previously reported. Jiang, et al.[29] reported a reduction of IKs in cardiomyocytes from 5-day post-MI canine hearts, which was associated with reduced mRNA for the subunits encoding IKs. IKs is considerably greater than IKr because of rectification of the latter. Thus, reduction of IKs could provide an important contribution to prolonged repolarization after post-MI.
Sanguinetti, et al.[30] showed that isoproterenol-mediated IKs augmentation could counter the action potential prolonging effect of pharmacological IKr blockade in guinea pig ventricular cardiomyocytes. When IKs availability is reduced, the effect of β-adrenergic stimulation delays rather than accelerates cardiomyocyte repolarization. The combination of pharmacological IKs blockade and β-adrenergic stimulation prolongs the action potential in canine cardiac Purkinje cells and enhances arrhythmogenicity in arterially perfused canine left ventricular wedge preparations.[31],[32]
Under normal conditions, impairment or block of one outward potassium channel cannot be expected to cause excessive and/or potentially dangerous lengthening of the APD, since the other potassium currents may provide sufficient repolarization capacity, which can be considered as a “repolarization reserve”.[33] However, in situations where the densities of one or more types of potassium channels are decreased by inheritance or remodeling,[34],[35] inhibition of other potassium channels may lead to unexpectedly augmented APD prolongation, resulting in proarrhythmic reactions. Based on our results, it seems apparent that in situations where the “repolarization reserve” is decreased, the minor loss of functional potassium channels may lead to excessive lengthening of repolarization.
In this report, we have characterized effects of HMI and HMI + NGF on ICaL. We found that current densities were markedly increased in HMI group, special treat with NGF. Electrical remodeling of cardiomyocytes might result from an increase in ICaL density, causing action potential prolongation in hyperinnervated regions. Liu et al.[36] demonstrated in rabbits that hypercholesterolemia induces proarrhythmic neural and myocardial remodeling. Nerve sprouting and sympathetic hyperinnervation were associated with changes in calcium currents, and an increased incidence of ventricular fibrillation. We found that NGF induced cardiac nerve sprouting, acted as an sympathetic hyperinnervation agonist, increased the densities of ICaL, and allowed for better cardiomyoctye recovery from inactivation.
4.1. Limitations of the present study
Finally, although we have focused on Ito, IKr, IKs, and ICaL, this does not rule out additional changes in other currents during cardiac sympathetic innervation, which may thereby impact the action potential configuration. For example, changes in the inward rectifier potassium current (IK1) can impact the action potential by influencing the resting potential and the final phase of repolarization.[37] In this article, we found that changes in the membrane potential of the three groups were minimal, suggesting that the differences in IK1 were not significant. These data suggest that our findings correlate with the data collected under more physiological conditions. Nevertheless, it is clearly possible that the mechanism underlying the changes in APD in these two conditions is different.
4.2. Conclusions
Infarcted hearts treated with NGF, reduced the three main reploarization outward potassium currents and increase inward calcium current may contribute to the action potential repolarization of cardiomyocytes far from the infarct zone. This suggests that NGF negatively regulates these outward potassium currents and positive regulates inward calcium current, prolongs the APD, and increases reentrant tachyarrhythmia.
Acknowledgments
We are grateful to the support of Dr. Nian Liu and Dr. Marco Superb for their technical assistance. This work was supported by the National Natural Science Foundation of China (No: 81170177, 30770901).
References
- 1.Aimond F, Alvarez JL, Rauzier JM, et al. Ionic basis of ventricular arrhythmias in remodeled rat heart during long-term myocardial infarction. Cardiovasc Res. 1999;42:402–415. doi: 10.1016/s0008-6363(99)00070-x. [DOI] [PubMed] [Google Scholar]
- 2.St John Sutton M, Lee D, Rouleau JL, et al. Left ventricular remodeling and ventricular arrhythmias after myocardial infarction. Circulation. 2003;107:2577–2582. doi: 10.1161/01.CIR.0000070420.51787.A8. [DOI] [PubMed] [Google Scholar]
- 3.Korsching S, Thoenen H. Nerve growth factor supply for sensory neurons: site of origin and competition with the sympathetic nervous system. Neurosci Lett. 1985;54:201–205. doi: 10.1016/s0304-3940(85)80079-3. [DOI] [PubMed] [Google Scholar]
- 4.Glebova NO, Ginty DD. Heterogeneous requirement of NGF for sympathetic target innervation in vivo. J Neurosci. 2004;24:743–751. doi: 10.1523/JNEUROSCI.4523-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou S, Jung BC, Tan AY, et al. Spontaneous stellate ganglion nerve activity and ventricular arrhythmia in a canine model of sudden death. Heart Rhythm. 2008;5:131–139. doi: 10.1016/j.hrthm.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 6.Zhou S, Paz O, Cao JM, et al. Differential beta-adrenoceptor expression induced by nerve growth factor infusion into the canine right and left stellate ganglia. Heart Rhythm. 2005;2:1347–1355. doi: 10.1016/j.hrthm.2005.08.027. [DOI] [PubMed] [Google Scholar]
- 7.Swissa M, Zhou S, Gonzalez-Gomez I, et al. Long-term subthreshold electrical stimulation of the left stellate ganglion and a canine model of sudden cardiac death. J Am Coll Cardiol. 2004;43:858–864. doi: 10.1016/j.jacc.2003.07.053. [DOI] [PubMed] [Google Scholar]
- 8.Zhou S, Cao JM, Tebb ZD, et al. Modulation of QT interval by cardiac sympathetic nerve sprouting and the mechanisms of ventricular arrhythmia in a canine model of sudden cardiac death. J Cardiovasc Electrophysiol. 2001;12:1068–1073. doi: 10.1046/j.1540-8167.2001.01068.x. [DOI] [PubMed] [Google Scholar]
- 9.Driesen RB, Verheyen FK, Dijkstra P, et al. Structural remodelling of cardiomyocytes in the border zone of infarcted rabbit heart. Mol Cell Biochem. 2007;302:225–232. doi: 10.1007/s11010-007-9445-2. [DOI] [PubMed] [Google Scholar]
- 10.Chang CM, Wu TJ, Zhou S, et al. Nerve sprouting and sympathetic hyperinnervation in a canine model of atrial fibrillation produced by prolonged right atrial pacing. Circulation. 2001;103:22–25. doi: 10.1161/01.cir.103.1.22. [DOI] [PubMed] [Google Scholar]
- 11.Cao JM, Chen LS, KenKnight BH, Ohara T, et al. Nerve sprouting and sudden cardiac death. Circ Res. 2000;86:816–821. doi: 10.1161/01.res.86.7.816. [DOI] [PubMed] [Google Scholar]
- 12.Li Y, Niu HY, Liu N, et al. Effect of imidapril on the effective refractory period and sodium current of ventricular noninfarction zone in healed myocardial infarction. Yao Xue Xue Bao. 2005;40:654–658. [PubMed] [Google Scholar]
- 13.Pascale P, Schlaepfer J, Oddo M, et al. Ventricular arrhythmia in coronary artery disease: limits of a risk stratification strategy based on the ejection fraction alone and impact of infarct localization. Europace. 2009;11:1639–1646. doi: 10.1093/europace/eup314. [DOI] [PubMed] [Google Scholar]
- 14.Billman GE. Cardiac autonomic neural remodeling and susceptibility to sudden cardiac death: effect of endurance exercise training. Am J Physiol Heart Circ Physiol. 2009;297:H1171–H1193. doi: 10.1152/ajpheart.00534.2009. [DOI] [PubMed] [Google Scholar]
- 15.Huang S, Patterson E, Yu X, et al. Proteasome inhibition 1 h following ischemia protects GRK2 and prevents malignant ventricular tachyarrhythmias and SCD in a model of myocardial infarction. Am J Physiol Heart Circ Physiol. 2008;294:H1298–H1303. doi: 10.1152/ajpheart.00765.2007. [DOI] [PubMed] [Google Scholar]
- 16.Swissa M, Zhou S, Gonzalez-Gomez I, et al. Long-term subthreshold electrical stimulation of the left stellate ganglion and a canine model of sudden cardiac death. J Am Coll Cardiol. 2004;43:858–864. doi: 10.1016/j.jacc.2003.07.053. [DOI] [PubMed] [Google Scholar]
- 17.Ducceschi V, Di Micco G, Sarubbi B, et al. Ionic mechanisms of ischemia-related ventricular arrhythmias. Clin Cardiol. 1996;19:325–331. doi: 10.1002/clc.4960190409. [DOI] [PubMed] [Google Scholar]
- 18.Ogawa M, Zhou S, Tan AY, et al. What have we learned about the contribution of autonomic nervous system to human arrhythmia? Heart Rhythm. 2009;6:S8–S11. doi: 10.1016/j.hrthm.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 19.Maczewski M, Maczewska J, Duda M. Hypercholesterolaemia exacerbates ventricular remodelling after myocardial infarction in the rat: role of angiotensin II type 1 receptors. Br J Pharmacol. 2008;154:1640–1648. doi: 10.1038/bjp.2008.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perrier E, Kerfant BG, Lalevee N, et al. Mineralocorticoid receptor antagonism prevents the electrical remodeling that precedes cellular hypertrophy after myocardial infarction. Circulation. 2004;110:776–783. doi: 10.1161/01.CIR.0000138973.55605.38. [DOI] [PubMed] [Google Scholar]
- 21.Ieda M, Fukuda K, Hisaka Y, et al. Endothelin-1 regulates cardiac sympathetic innervation in the rodent heart by controlling nerve growth factor expression. J Clin Invest. 2004;113:876–884. doi: 10.1172/JCI19480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hassankhani A, Steinhelper ME, Soonpaa MH, et al. Overexpression of NGF within the heart of transgenic mice causes hyperinnervation, cardiac enlargement, and hyperplasia of ectopic cells. Dev Biol. 1995;169:309–321. doi: 10.1006/dbio.1995.1146. [DOI] [PubMed] [Google Scholar]
- 23.Chen PS, Chen LS, Cao JM, et al. Sympathetic nerve sprouting, electrical remodeling and the mechanisms of sudden cardiac death. Cardiovasc Res. 2001;50:409–416. doi: 10.1016/s0008-6363(00)00308-4. [DOI] [PubMed] [Google Scholar]
- 24.Meloni M, Caporali A, Graiani G, et al. Nerve growth factor promotes cardiac repair following myocardial infarction. Circ Res. 2010;106:1275–1284. doi: 10.1161/CIRCRESAHA.109.210088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marban E. Cardiac channelopathies. Nature. 2002;415:213–218. doi: 10.1038/415213a. [DOI] [PubMed] [Google Scholar]
- 26.Tomaselli GF, Beuckelmann DJ, Calkins HG, et al. Sudden cardiac death in heart failure. The role of abnormal repolarization. Circulation. 1994;90:2534–2539. doi: 10.1161/01.cir.90.5.2534. [DOI] [PubMed] [Google Scholar]
- 27.Qin D, Zhang ZH, Caref EB, et al. Cellular and ionic basis of arrhythmias in postinfarction remodeled ventricular myocardium. Circ Res. 1996;79:461–473. doi: 10.1161/01.res.79.3.461. [DOI] [PubMed] [Google Scholar]
- 28.Rozanski GJ, Xu Z, Zhang K, et al. Altered K current of protein ventricular myocytes in rats with chronic myocardial infarction. Am J Physiol. 1998;274:H259–H265. doi: 10.1152/ajpheart.1998.274.1.H259. [DOI] [PubMed] [Google Scholar]
- 29.Jiang M, Cabo C, Yao J, et al. Delayed rectifier K currents have reduced amplitudes and altered kinetics in myocytes from infarcted canine ventricle. Cardiovasc Res. 2000;48:34–43. doi: 10.1016/s0008-6363(00)00159-0. [DOI] [PubMed] [Google Scholar]
- 30.Sanguinetti MC, Jurkiewicz NK, Scott A, et al. Isoproterenol antagonizes prolongation of refractory period by the class III antiarrhythmic agent E-4031 in guinea pig myocytes. Mechanism of action. Circ Res. 1991;68:77–84. doi: 10.1161/01.res.68.1.77. [DOI] [PubMed] [Google Scholar]
- 31.Abi-Gerges N, Small BG, Lawrence CL, et al. Gender differences in the slow delayed (IKs) but not in inward (IK1) rectifier K+ currents of canine Purkinje fibre cardiac action potential: key roles for IKs, beta-adrenoceptor stimulation, pacing rate and gender. Br J Pharmacol. 2006;147:653–660. doi: 10.1038/sj.bjp.0706491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Han W, Wang Z, Nattel S. Slow delayed rectifier current and repolarization in canine cardiac Purkinje cells. Am J Physiol Heart Circ Physiol. 2001;280:H1075–H1080. doi: 10.1152/ajpheart.2001.280.3.H1075. [DOI] [PubMed] [Google Scholar]
- 33.Roden DM. Repolarization reserve: a moving target. Circulation. 2008;118:981–982. doi: 10.1161/CIRCULATIONAHA.108.798918. [DOI] [PubMed] [Google Scholar]
- 34.Roden DM, Lazzara R, Rosen M, et al. Multiple mechanisms in the long-QT syndrome. Current knowledge, gaps, and future directions. The SADS Foundation Task Force on LQTS. Circulation. 1996;94:1996–2012. doi: 10.1161/01.cir.94.8.1996. [DOI] [PubMed] [Google Scholar]
- 35.Tomaselli GF, Marbán E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res. 1999;42:270–283. doi: 10.1016/s0008-6363(99)00017-6. [DOI] [PubMed] [Google Scholar]
- 36.Liu YB, Wu CC, Lu LS, et al. Sympathetic nerve sprouting, electrical remodeling, and increased vulnerability to ventricular fibrillation in hypercholesterolemic rabbits. Circ Res. 2003;92:1145–1152. doi: 10.1161/01.RES.0000072999.51484.92. [DOI] [PubMed] [Google Scholar]
- 37.Shimoni Y, Clark RB, Giles WR. Role of an inwardly rectifying potassium current in rabbit ventricular action potential. J Physiol. 1992;448:709–727. doi: 10.1113/jphysiol.1992.sp019066. [DOI] [PMC free article] [PubMed] [Google Scholar]