

Abstract
Mammals express ~20 different connexins, the main gap junction forming proteins in mammals, and 3 pannexins, homologues of innexins, the main gap junction forming proteins in invertebrates. In both classes of gap junction, each channel is formed by two hemichannels, one contributed by each of the coupled cells. There is now general, if not universal, agreement that hemichannels of both classes can open in response to various physiological and pathological stimuli when they are not apposed to another hemichannels and face the external milieu. Connexin (and likely pannexin) hemichannel permeability is consistent with that of the cell-cell channels and open hemichannels can be a release site for relatively large molecules such as ATP and glutamate, which can serve as transmitters between cells. Here we describe three experimental paradigms in which connexin and pannexin hemichannel signaling occurs. 1) In cultures of spinal astrocytes FGF-1 causes the release of ATP, and ATP causes opening of pannexin hemichannels, which then release further ATP. Subsequently, several hours later, connexin hemichannels are also opened by an unknown mechanism. Release of ATP appears to become self sustaining through action of P2X7 receptors to open pannexin hemichannels and then connexin hemichannels, both of which are ATP permeable. 2) Spinal cord injury by dropping a small weight on the exposed cord is followed by release of ATP in the region surrounding the primary lesion. This release is greatly reduced in a mouse in which Cx43 is knocked down in the astrocytes. Application of FGF-1 causes a similar release of ATP in the uninjured spinal cord, and an inhibitor of the FGF-1 receptor, PD173074, inhibits both FGF-1 and injury-induced release. Reduction in ATP release is associated with reduced inflammation and less secondary expansion of the lesion. 3) Cortical astrocytes in culture are permeabilized by hypoxia, and this effect is increased by high or low glucose. The mechanism of permeabilization is opening of Cx43 hemichannels, which can lead to cell death. Activated microglia secrete TNF-α and IL-1β, which open connexin hemichannels in astrocytes. Astrocytes release ATP and glutamate which can kill neurons in co-culture through activation of neuronal pannexin hemichannels. These studies implicate two kinds of gap junction hemichannel in inflammatory responses and cell death.
Introduction
As most of us know, gap junctions are intercellular structures that directly connect the interiors of adjacent cells by a pathway not open to the extracellular space. The gene family encoding most if not all gap junctions in vertebrates is different from that in most invertebrates, although there are remarkable convergences between the families in basic structure, physiological properties, and pharmacological sensitivity. The vertebrate family, the connexins, first appear in the biome in ascidians, which are procordates, and thus invertebrates, and they do express a few innexins/pannexins (Sasakura et al., 2003). Amphioxus (Branchiostoma), the lancelet, looks more like a vertebrate precursor, but has only the invertebrate innexins/pannexins and lacks connexins (Holland et al., 2008). The larval ascidian, or tadpole, does have a notochord and dorsal nerve cord, but these features are lost in metamorphosis to the sessile, adult form.
Let’s make a little nomenclatural aside. Some time ago Panchin and colleagues detected orthologs of innexins in the human and mouse genomes, then recently published (mouse pannexin 1, mPanx1, and human pannexin 2, hPanx2, Panchin et al., 2000; Baranova et al., 2004, added Panx3). They proposed to call the combined family pannexins, because they were everywhere (that is in both invertebrates and vertebrates), a nomenclature that appears quite reasonable to us. (There is also a rumor that Panchin was naming the family for himself, but we have seen no substantiation of that allegation. Pannexin is certainly better than an alternative, ubiquinexin.) Subsequently, Monyer and colleagues cloned the three innexin homologs from mouse and rat and termed them pannexin 1 – 3. They used Px1, Px2 and Px3 as the protein names, which seemed, by analogy to connexins, quite satisfactory. We will have to use Panx1, etc., for the gene names, because of gene nomenclature agreements, but Px1, etc., are still defensible for the proteins, although usage now favors the longer form (Sosinsky et al., 2011).
Another nomenclatural aside about hemichannels v. connexons and pannexons. By “hemichannel” we mean half a gap junction cell-cell channel, one hemichannel (hemichannel) of which is provided by each of the joined cells. Now, if one of these hemichannels is not connected with a hemichannel in another membrane and opens to the exterior, is it still a hemichannel? Or has it become a channel? Clearly it is a channel, but we prefer not to change the name. Dan Goodenough coined the term connexon for the hexameric connexin hemichannels in cell-cell channels, a reasonable usage. However, it does lead to difficulties in verbal communication in that connexon and connexin sound rather much alike unless one enunciates carefully. The term hemichannel is out there, and for this 5 presentation, we will stick with it. The issue with pannexins is a bit more complex. Pannexins generally do not form gap junctions between mammalian cells, although coupling may develop between Px1 transfected cells of mammalian lines (Vanden Abeele et al., 2006; Lai et al., 2007) and also between Xenopus oocytes exogenously expressing Px1 (Bruzzone et al., 2005). It is early to conclude that no mammalian cells make gap junctions with pannexins, and the exogenous expression systems (almost) provide proof of principle. So if mammalian pannexins don’t make gap junctions, can one call their single membrane channels hemichannels? In this presentation we will continue to do that. Since the evolutionarily related innexins/pannexins in invertebrates do make both cell-cell channels and active hemichannels in unapposed membranes, one can argue that use of hemichannel for mammalian pannexin channels is justifiable, while admitting the potential value of the term, pannexon.
Background, probably unnecessary here: Gap junction channels are composed of two hemichannels, one provided by each of the joined cells. Each hemichannel is a hexamer of connexin or pannexin. Not all monomers in a hemichannel need be of the same type; heteromeric, as opposed to homomeric, hemichannels do exist. Not all hemichannels of one family are compatible, that is capable of forming a cell-cell channel, with all other hemichannels members of that family, and connexins don’t form channels with pannexins. Each monomer is a tetraspan protein, i.e., with four transmembrane domains, and the N- and C- termini are cytoplasmic. The transmembrane domains are connected by two extracellular loops, E1 between TM1 and TM2 and E2 between TM3 and TM4. A cytoplasmic loop, CL, connects TM2 and TM3. For connexins at least, the cytoplasmic loop and C-terminus are the most variable regions of the proteins. The extracellular loops have highly conserved cysteines that may stabilize the structure through intramolecular cysteine/cysteine bonds.
Gap junction hemichannels that open to the exterior?
In the beginning there was reluctance to think that a hemichannel could open when it was not joined to another hemichannel in an apposed membrane. First, if the hemichannel had the same permeability characteristics as the cell-cell channel and opened to the external medium, it would let in Na+ and Ca2+ and let out K+, ATP, glutamate, and other important cytoplasmic constituents. Second, there were limited casual measurements of macroscopic conductance, suggesting that any junction precursors in the membranes were not open to the medium before the junctions formed. The clincher came later when Bukauskas and colleagues showed at the single channel level that, when the first cell- cell channel opened after moving cells into contact, the non-junctional conductance did not change, and therefore that the (hemi)channels forming the cell-cell channel had not been open to the exterior before joining together (Bukauskas, 2001, Bukauskas and Weingart, 1993). Also, no opening and closing of hemichannels, seen as steps in conductance to a reversal potential near zero, were seen, although subsequent studies demonstrated their existence. The argument against hemichannels was strengthened by the observation that Cx46 makes “connexons (hemichannels)” in Xenopus oocytes (Paul et al., 1991). This result might be considered a rule proving exception [with the reservation that exceptions don’t prove rules but negate them], and it does support the prediction that open hemichannels would be deleterious. In any case, in moderate Ca2+ saline, high levels of Cx46 expression kill the oocytes, leakage indeed being stressful. [Ficoll in the bathing medium rescues them, for a while at least.]
Other early observations indicated that Cx43 hemichannels could open to the exterior. Isolated cells expressing Cx43 could take up dyes to which biological membranes were usually impermeable, but which did permeate gap junctions (Li et al., 1996). Moreover, uptake depended on expression level and was reduced by gap junction blockers. Subsequently, hemichannel activity was recorded in single cells (Contreras et al., 2003). When HeLa cells transfected with Cx43 were made inside positive by ~40 – 60 mV, channels opened. When the potential was returned to, say, −20 mV the channels closed. The conductance for Cx43 was ~220 pS, about twice the conductance of the cell-cell channels, as one might expect. Of course, it didn’t have to be that way; the channel geometry could be different at the extracellular end or farther in, when it was not apposed to another hemichannel. Moreover, in the simplest account, access resistance should be a larger fraction of the total resistance of a hemichannel than of a cell-cell channel. Another feature confirmed the connexin mediation of the channel activity; Cx43 wildtype (WT) gap junctions have both fast gating to a substate and slow gating between the fully open and fully closed states. (We think it is fully closed, because the cell conductance can be small compared to the fully open single channel conductance, and there appear to be many closed hemichannels in parallel.) The fast gate is missing in gap junctions formed of Cx43-EGFP where EGFP is attached to the C terminus. The same is true of the hemichannels. If EGFP is attached to the N terminus, plaques will form between cells but with no functioning channels; similarly EGFP-Cx43 gets to the cell surface, but there are no functional hemichannels. Finally, there is pharmacology. Agents, such as octanol and carbenoxolone that block gap junctions, also block hemichannels. Dye uptake measurements had suggested that La3+ blocked Cx43 hemichannels, and it did block the single channel activity (as does Gd3+)(Contreras et al., 2002, 2003). More recently, several “connexin mimetic” peptides with the same sequence as segments of the extracellular loops were shown to block hemichannels (Deplantez et al., 2012).
There were a few problems. The dye uptake occurred at the resting potential, a potential where very little hemichannel opening was observed in whole cell recordings. However, we could not conclude that the upper limit on opening was insufficient to mediate the observed dye uptake (Contreras et al., 2003). Opening at inside negative resting potentials has been observed in cytokine treated astrocytes (Retamal et al., 2007) Another possible discrepancy was that the substate in hemichannels tended to be of greater conductance than the substate in cell-cell channels (Bennett et al., 2003). Nonetheless, the weight of evidence indicates that connexins form “unapposed” hemichannels that can open to the extracellular medium.
In this presentation, we will consider three interrelated but distinct stories. We will describe how application of FGF-1 to spinal astrocytes in culture causes opening of pannexin and connexin hemichannels and release of ATP. We will summarize data from spinal cord traumatic injury (by dropping a small weight on the cord) indicating that ATP is released in the perilesion region, presumably by astrocytes, and causes recruitment of microglia and macrophages to this region and increased secondary expansion of the lesion (Wang et al., 2004). This release of ATP is markedly reduced by PD172074, an inhibitor of the FGF-1 receptor (FGFR)(Garré et al, submitted). FGF-1 applied to the uninjured spinal cord causes ATP release, which is greatly reduced by the FGFR inhibitor. Application of the inhibitor immediately after a weight drop lesion reduces recruitment of inflammatory cells and expansion of the lesion. In a conditional Cx43 KO in astrocytes, ATP release, recruitment of microglia and macrophages following injury are reduced, and recovery of function is improved; these findings implicate Cx43 in mediation of ATP release and the destructive inflammatory response (Huang et al., 2012; but see Iglesias et al., 2009, material considered below). Finally, we will show how stressors including hypoxia, high glucose, and the toxic peptide, Aβ25–35, act on microglia and astrocytes to release ATP and glutamate that together kill neurons in culture; in these experiments the release of toxic molecules by astrocytes is mediated by connexin hemichannels, and these molecules lead to opening of pannexin hemichannels in neurons and cell death (Orellana et al, 2010, 2011). The systems used were mixed cultures and conditioned media allowing analysis of interactions between microglia, astrocytes, and neurons. The basic theme is that inflammatory interactions between cells of different types are important in neuropathology. This review is parochial, and for a wider perspective, other contributions to this special issue Electrical Synapses should be consulted, particularly those of the Editors, DC Spray and R Dermietzel.
Spinal astrocytes in culture express Px1 and Cx43 hemichannels, which open in response to FGF-1 treatment
Spinal astrocytes in culture are activated by treatment with acidic fibroblast growth factor, FGF-1. FGF-1 binds to the FGF receptor, which is a tyrosine kinase. (Although not relevant to this study, the liganded receptor is internalized and transported to the nucleus.) The astrocytes become permeable to ethidium (Etd+, applied as ethidium bromide) and Lucifer yellow (LY2−) within minutes of FGF-1 application (Garré et al., 2010). The maximum observed uptake rate (of LY2−) is after 4 – 7 h FGF-1 treatment. At ~2 h uptake is mediated entirely by Px1 hemichannels. At ~7 h, dye uptake is mediated by both Px1 and Cx43 hemichannels.
What’s happening? See Fig. 1. The study is described in the format Conclusion. (Evidence. Published in Garré, 2010, if not otherwise indicated.)
Fig. 1.

Proposed reactions initiated by FGF-1 in spinal astrocytes in culture. (1) FGF-1 binds to its (dimeric) receptor, likely causing a rise in cytoplasmic Ca2+. (2) ATP is released from vesicles, an action blocked by BoTN A. (3) The ATP released acts on P2X7Rs, which allows Ca2+ to enter. (4) Activation of P2X7Rs leads to opening of Px1 hemichannels (green arrow), allowing ATP release and Ca2+ (and Etd+) entry. (5) Cx43 hemichannels are opened hours later, either through an action of FGF-1 or of P2X7Rs and Px1 hemichannels. (6) Gap junctional communication is reduced. (From Garré et al., 2010)
FGF-1 acts on the FGF receptor (FGFR), a tyrosine kinase, to cause dye uptake. (Dye uptake is blocked by the FGFR inhibitor, PD173074.)
Activation of FGFRs increases cytoplasmic free Ca2+….. (Fura-2 measurement. Unpublished.)
And causes the release of ATP from vesicles through activation of phospholipase C. (Direct measurement of ATP in the medium. ATP release is blocked by BoNT A, which cleaves the SNARE protein, SNAP-25, necessary for exocytosis in many cells. Dye uptake is blocked by the PLC inhibitor, U73122, but not its inactive analog, U73343 (unpublished).)
ATP activates P2X7Rs, presumably by both autocrine and paracrine pathways. (Dye uptake is blocked by bath applied apyrase, an ecto-nucleotidase, and by P2X7R antagonists.)
P2X7Rs mediate further influx of Ca2+ (Samways et al., 2008.)
And opening of Px1 hemichannels which release ATP. (E.g., Iglesias et al. 2008.)
And also permit dye influx. (After 2 h FGF-1 treatment, dye uptake is blocked by 0.1 mM CBX, a concentration that blocks both Px1 and Cx43 hemichannels. There is no effect of octanol, which blocks Cx43 hemichannels. Also, at this time dye uptake is reduced by prior treatment of the astrocytes with siRNA-Px1; at this time dye uptake by astrocytes from Cx43 KO mice is not significantly different from uptake by wild type astrocytes.)
The release of ATP likely becomes self sustaining, and vesicular release may no longer contribute. (ATP and benzoyl ATP permeabilize cells in the same way as does FGF-1.)
By 7 h, uptake is mediated by both Px1 and Cx43 hemichannels. (Permeability is partially blocked by Cx43 hemichannel blockers (octanol, heptanol, La3+, Gd3+) and completely blocked by carbenoxolone. Also, flux through both classes of hemichannels is indicated by reduced levels in cells treated with siRNA-Px1 and in cells from Cx43 KO mice. Although cytoplasmic ATP levels are reduced in the Cx43 KO (Iglesias et al., 2009), basal ATP release and that evoked after 2h FGF-1 treatment were little affected in the Cx43 KO.)
FGF-1 may cause some increase in permeability independent of extracellular ATP, but the effect is not as large at that mediated by ATP. (FGF-1 application can produce an increase in permeability after Px1 siRNA pretreatment or in the presence of apyrase or purinergic blockers.)
As will be seen in the next section, the action of FGF-1 on spinal astrocytes in situ differs from that reported here. Moreover, cortical astrocytes in culture do not show permeabilization like that of spinal astrocytes in culture. Both mechanism and functional significance of these differences require further investigation.
Spinal cord injury, ATP, and FGF-1
Spinal cord trauma modeled by weight drop injury can cause death at the site of contact followed by secondary expansion of the lesion site over time. Previous studies by Nedergaard and colleagues indicated a role for P2X7 receptors in the sequelae of a weight drop lesion in rat (Wang et al., 2004; Peng et al., 2009). ATP is released in the region surrounding the focal injury as shown by imaging of luciferin-luciferase light emission (Fig. 2). As the secondary enlargement of the lesion progresses, signs of inflammation include increased expression of GFAP and invasion of activated microglia and macrophages. Antagonists of P2X7Rs reduce inflammation and lesion expansion, and improve functional recovery. ATP and benzoylATP strongly excite motoneurons in spinal cord slices; the activity is not affected by AMPA and NMDA receptor antagonists, but is blocked by oATP and other P2X7R antagonists, thus implicating P2X receptors. ATP also causes a rise in intracellular Ca2+, and it was suggested that ATP is an excitotoxin (Wang et al., 2004). Translational relevance was added by the observation that Brilliant blue G, a P2X7 antagonist that is related to a food dye, crosses the (intact) blood brain barrier and is neuroprotective; the expectation is that this compound or a close relative will be well tolerated and possibly useful in treating spinal cord injury (Peng et al., 2009). Our observations with astrocytes in culture may relate to these findings.
Fig. 2.

Weight drop-induced spinal cord injury (SCI): Release of ATP in the perilesion region is reduced in the astrocyte Cx43 KO. A. Schematics of the weight drop mechanism and ATP imaging. Luciferin-luciferase solution is applied to the dorsal surface of the exposed spinal cord. B. Bright field (BF) image of the lesion site (inner dotted line) and merged BF and bioluminescence (BL, in red) images. There is extensive luminescence in the perilesion region between inner and outer dotted lines. C. In the Cx43 KO there is little luminescence around the lesion (dotted line). The arrow head indicates the mid-dorsal vein. Modified from Huang et al., 2012.
A subsequent study showed that lesion-induced ATP release (Fig. 2) and inflammation is reduced in a Cx43 KO restricted to astrocytes (Huang et al., 2012). The KO is of a floxed Cx43 with Cre expression under the control of the hGFAP promoter (Theis et al., 2003). In culture at least, the Cx43 KO spinal astrocytes have moderately (~50%) reduced levels of cytoplasmic ATP (Iglesias et al., 2009), but the observed decreased in release in the in situ spinal cord appears greater than can be accounted for by that difference. To exclude the possibility of compensatory upregulation of Cx30, which is another connexin expressed in astrocytes, the Cre-mediated Cx43 KO is on a background of a germ line Cx30 KO, and the controls are WT Cx43 on the same background (Wallraff et al., 2006). In these double KO mice, the lesion is smaller, there are fewer signs of inflammation in the perilesion region, nerve conduction past the lesion and motor function show more rapid and greater recovery (measured by the Basso Mouse Scale (BMS) of motor function, Basso et al., 2006), and the secondary expansion of the lesion is reduced (Fig. 3). These data do not distinguish between absence of hemichannels and of gap junctions in the neuroprotective role. Cx43 hemichannels could be releasing ATP, and coupling between astrocytes could allow ATP to diffuse from non-releasing cells into releasing cells, thus increasing the amount of ATP available for efflux.
Fig. 3.

Recovery of conduction across the lesion and of motor performance after spinal cord injury (SCI) is greater in the Cx43 KO, and lesion volume is reduced. A, B. Compound action potentials propagated across the lesion 7 d after SCI at different stimulus strengths. Also shown in A are Luxol blue stained cross sections of the cord at 7 d post-SCI. In the Cx43 KO, CAPs are greater in amplitude, and there is less disruption of the nerve fibers. D. Recovery over time measured by the Basso Mouse Score (BMS) of motor function is greater in the Cx43 KO. E. Lesion volume is greater 8 wk after injury in WT (n = 8) than the Cx43 KO (n = 6). * p < 0.05. Modified from Huang et al., 2012.
This work was extended to test whether FGF-1 is involved in the post-lesion changes (submitted for publication). We observed that the FGF-1 inhibitor, PD173074, greatly reduces the ATP release following weight drop injury, and there is less recruitment of inflammatory cells and lesion expansion, implicating FGF-1 in the injury response. Also, FGF-1 application to the intact spinal cord causes ATP release, consistent with FGF-1 release from injured neurons causing ATP release from nearby intact astrocytes. We have not yet determined whether FGF-1 application without injury leads neurodegeneration.
The findings with in vitro astrocytes are somewhat disparate from those obtained in vivo. FGF-1 is active in both situations and causes ATP release, and in both, as expected, ATP release is blocked by PD173074. In vivo, ATP release is greatly reduced in the Cx43 KO. Our first thought is that the major source of ATP is opening of Cx43 hemichannels by FGF-1, although the decreased release of ATP could be contributed to by reduced cytoplasmic ATP in the Cx43 KO as seen in spinal astrocytes in culture (Iglesias et al., 2009). In contrast in vitro, initial FGF-1 induced release is vesicular as indicated by BoNT A sensitivity, and then Px1 hemichannels are opened, although relative contributions of vesicular release and release through Px1 hemichannels have not been determined. Cx43 hemichannels open only after more than 2 h FGF-1 treatment and should then contribute to ATP release. Certainly, properties of astrocytes in vitro and in vivo differ (see for example, Wilhelm et al., 2004) and also differ in specific regions of the cord (Tsai et al., 2012); furthermore, the Cre-mediated Cx43 KO, although relatively late in onset, may change their properties.
Other in vitro systems with astrocytes, microglia and neurons
As illustrated in the previous section, astrocytes along with many other functions participate in CNS inflammatory responses, both as instigator and effector. The major point to be illustrated in this section is that inflammatory stimuli of different kinds can summate to cause connexin and pannexin hemichannels to open. The data are from published studies (Orellana et al., 2010, 2011).
Hypoxia glucose permeabilizes cortical astrocytes in culture by opening connexin hemichannels, and this effect is greater in abnormal glucose. In these experiments the cells are subjected to different periods of hypoxia in glucose concentrations ranging from zero to normal (5 mM) to high (27 – 37 mM) followed by reoxygenation, that is restoration of oxygenated normal saline solution with normal glucose (corresponding to “reperfusion” in situ). Here there are two stressors, hypoxia and abnormal glucose during the hypoxia. Injury is measured as uptake of ethidium ions (Etd+) through opening of connexin hemichannels, or after greater stress, uptake of labeled dextran, ascribed to membrane breakdown in dying or dead cells. The medium during hypoxia is an “ischemic saline”, which mimics the ionic milieu during cessation of blood flow in the CNS and is lower in Na+ and Ca2+ and higher in K+ than normal medium (Bondarenko and Chesler, 2001) [and normal medium during the hypoxia prevents most or all of the effects to be described]. Astrocytes subject to 3 h hypoxia in normal (5mM) glucose show no signs of damage after reoxygenation (Fig. 4A, C, I, 6A). After 3 h hypoxia in high glucose, astrocytes are transiently permeable to Etd+ after 1 h reoxygenation and are fully recovered after 3 h (Fig. 4I, 6A). The uptake is through Cx43 hemichannels, because the permeability increase is absent in astrocytes from (germ line) Cx43 KO mice and is reduced by connexin but not pannexin hemichannel blockers (Gap26, Fig. 4A, E, Gap27 or La3+, but not 10panx1 or the purinergic blocker, oATP). (The point of the purinergic blocker is that activation of P2X7 receptors can lead to opening of pannexin hemichannels.) If the hypoxic exposure is for 6 h in normal glucose, there is still little change during the hypoxic period, but uptake increases to a plateau at 1 to 2 h reoxygenation (Fig. 4J). 6 h hypoxia in high glucose produces some increase during the hypoxic period and then rise to a higher plateau during reoxygenation. In each case hypoxia in low glucose causes permeabilization that is greater than in normal glucose and not so great as in high glucose (Fig. 4I, J). The relation between glucose concentration during hypoxia and dye uptake rate after 1 h reoxygenation is U-shaped (Fig. 4K). With a 3 h hypoxic period, elevated glucose for 24 h before or 1h after has no effect on the Etd+ uptake (not illustrated); thus, the effect of abnormal glucose is initiated during the hypoxic period and does not require it before or after.
Fig. 4.

Permeabilization of astrocytes after 3 h hypoxia in high or normal glucose in “ischemic saline” followed by reoxygenation in normal glucose in normal saline.
Hypoxia–reoxygenation increases rate of Etd+ uptake by rat cortical astrocytes in culture, an effect potentiated by high glucose. A. Time-lapse measurements of Etd+ uptake under control conditions and starting at 1 h reoxygenation after 3 h hypoxia in 5 mM or 27 mM glucose. Gap 26, a Cx43 hemichannel blocker, applied after 12 min of Etd+ uptake measurement greatly reduces uptake after hypoxia in 27 mM glucose. B–D. Fluorescence micrographs of Etd+ uptake (10 min exposure to dye) under control conditions (B) and at 1 h reoxygenation after 3 h hypoxia in 5 mM glucose (C) or 27 mM glucose (D). E. Time-lapse measurements of Etd+ uptake as in A under control conditions and starting at 1 h reoxygenation after 6 h hypoxia in 5 mM or 27 mM glucose. F–H. Fluorescence micrographs of Etd+ uptake (10 min exposure to dye) under control conditions (F) and at 1 h reoxygenation after 6 h hypoxia in 5 mM (G) or 27 mM glucose (H). I. Averaged data normalized to control of Etd+ uptake rate measured from the beginning of reoxygenation (time 0) following 3 h hypoxia in 0 mM, 5 mM, or 27 mM glucose. *** P < 0.001, 27 vs. 5 mM glucose; ††† p < 0.001, 27 vs. 0 mM glucose; £££ p < 0.001, 0 vs. 5 mM glucose. (J) Etd+ uptake rate as in I following 6 h hypoxia in 0 mM, 5 mM, or 27 mM glucose. *** p < 0.001, ** p < 0.005, * p < 0.05, 27 vs. 5 mM glucose; ††† p < 0.001, †† p < 0.005, † p < 0.05, 27 vs. 0 mM; £ p < 0.05, 0 vs. 5 mM. K. Etd+ uptake rate at 1 h reoxygenation following 3 h or 6 h of hypoxia in different glucose concentrations. *** p < 0.001, * p < 0.05, 27 vs. 0 mM glucose. Each value corresponds to mean ± SE of 20 cells in a representative of five experiments. Bar = 60 μm. From Orellana et al., 2010.
Fig. 6.

Influence of microglia on hypoxia/reoxygenation-induced permeabilization of astrocytes. A. 3h hypoxia in high (but not normal) glucose transiently increases Etd+ uptake during reoxygenation. B. Co-culture with microglia prevents the increase in Etd+ uptake seen in A. C. Co-culture of astrocytes with microglia (MG) plus Aβ25–35 for 24 h before 3 h hypoxia followed by reoxygenation causes a large and prolonged increase in Etd+ uptake, more so in high than in normal glucose. D. Hypoxia and reoxygenation with Aβ25–35 in the medium has no additional effect compared to hypoxia and reoxygenation alone as in A. E, F. 24 h pretreatment with conditioned medium from microglia + Aβ25–35 or with TNF-α+ IL-1β causes comparable changes to those in 24 h co-cultures with microglia + Aβ25–35 (C). ** p < 0.01, *** p < 0.001 27 vs. 5 mM glucose. From Orellana et al., 2011.
As would be expected, prolonging the hypoxia further stresses cortical astrocytes. Although they survive for at least 6 h reoxygenation after 3 h hypoxia in high glucose, 6 h hypoxia and 6 h reoxygenation kills a fraction of the cells in normal glucose and many more of the cells in high glucose (Fig. 5D, E, G). Cx43 hemichannels are implicated in cell death, because the cells are protected by connexin hemichannel blockers and not by pannexin hemichannel blockers (Fig. 5H). An inhibitor of p38 MAPK, SB202190, applied before the hypoxia in high glucose, reduces dye uptake after 6 h reoxygenation and prevents cell death (Fig. 5H) and reduction in dye coupling (not shown); this fragmentary observation points to molecular mechanisms ultimately leading to cell death.
Fig. 5.
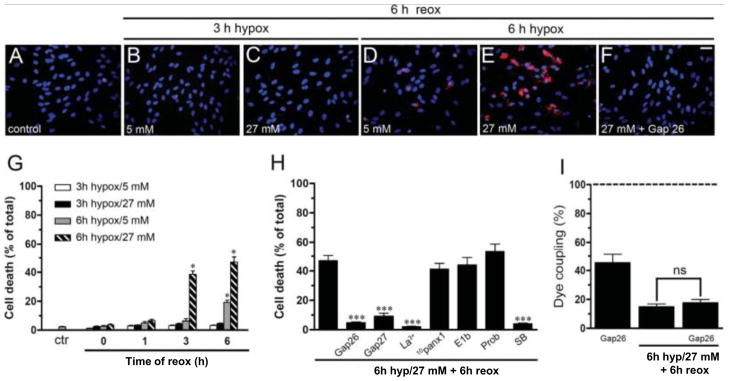
Prolonged hypoxia causes death of astrocytes with greater mortality after hypoxia in high glucose. A – F. Micrographs of astrocyte cultures subjected to the indicated periods of hypoxia and concentrations of glucose followed by 6 h reoxygenation in normal glucose. Hoechst 33462 nuclear stain and Rhodamine B dextran are used to identify cells and indicate membrane break down. D. Significant death occurs after 6h hypoxia in 5 mM glucose and 6 h reoxygenation. E. More death occurs when the hypoxia is in 27 mM glucose. F. The connexin hemichannel blocker Gap26 prevents cell death. G. Cell death quantified as a function of duration of hypoxia, glucose concentration, and time of reoxygenation. H. Connexin hemichannel blockers (Gap26, Gap27, and La3+) are protective, as is the p38 MAPK inhibitor, SB202190, applied before the hypoxia. Pannexin hemichannel blockers (10panx1, E1b, and probenecid (Prob)) are not protective. I. The connexin hemichannel blocker, Gap26, applied during the reoxygenation period reduces coupling in control cells, presumably by preventing formation of new cell-cell channels that would compensate for internalization of existing channels. Gap26 did not affect the reduction in dye coupling produced by 6h hypoxia in high glucose followed by 6 h reoxygenation. * p < 0.05 hypoxia/reoxygenation vs. control, *** p < 0.001 hypoxia/reoxygenation without vs. with addition of indicated connexin hemichannel blockers or SB202190. From Orellana et al., 2010.
The increases in dye uptake are associated with increase in surface expression of Cx43 as determined by biotinylation and western analysis, while total Cx43 is unchanged (Orellana et al., 2010, 2011). [The recovery of permeability following the transient increase in uptake caused by 3 h hypoxia in high glucose is associated with return to basal surface expression.] The increase in surface expression (to ~3 times control) is not quite sufficient to account for the increased uptake (to ~6 times control), and an increase in open probability and/or increase in permeability per open channel may occur. In astrocytes subjected to metabolic inhibition, insertion of hemichannels measured by biotinylation without change in open probability can account for the entire increase in dye uptake rate (Retamal et al., 2006). The loss of coupling in the insulted astrocytes is associated with reduction in the number of large Cx43 immunoreactive plaques, presumptive gap junctions, and increase in the number of small immunoreactive puncta likely to represent trafficking vesicles associated with internalization, as well as supplying new hemichannels for cell-cell channel formation (Orellana et al., 2010). The loss of large Cx43 plaques after hypoxia/reoxygenation suggests that the decrease in coupling is contributed to by internalization of junctions, but there could also be a contribution from closing of the cell-cell channels. With the Cx43 antibody used, we did not visualize the increased surface expression indicated by biotinylation; the distribution in the membrane could well have been too diffuse to detect with our methods. Although the increase of surface permeability is paralleled by decrease in coupling, the change in permeability is unlikely to be due to separation of cell-cell channels to form hemichannels connected to the external milieu. In general connexin cell-cell channels do not split into two hemichannels facing the extracellular space (although innexin/pannexin channels may do so, Lane and Swales, 1978), but small clusters of channels are internalized into one cell as double walled vesicles that are transported to lysosomes for degradation; the vesicles include both cell membranes and a small amount of cytoplasm from the apposed cell (“annular gap junctions”, Gaietta et al., 2002).
Connexin hemichannel blockers have no discernable effect on the ischemia-induced decrease in coupling indicating that increased leak of dye through these hemichannels does not cause the decrease in dye coupling (see Fig. 5I). (The > 50% decrease in dye coupling associated with 6 h Gap26 treatment without hypoxia likely results from block of cell-cell channel formation with continuing internalization of existing channels. The decrease caused by Gap26 treatment does not add to the decrease caused by 6 h hypoxia in high glucose followed by 6 h reoxygenation; this observation raises the possibility of occlusion in the uncoupling processes.)
Dye uptake after 6 h hypoxia in high glucose and 1 h reoxygenation is reduced within minutes by DTT applied during dye application (Orellana et al., 2010), consistent with previous results with metabolic inhibition (Retamal et al., 2007a) and suggesting oxidation of the hemichannels or an associated molecule as a result of hypoxia/reoxygenation. [DTT increases dye uptake in control cells and little effect after 3 h hypoxia in high glucose and 1 h reoxygenation.] DTT does not reverse the reduction in dye coupling over the same time course. The differential effect of DTT on permeability and dye coupling is consistent with internalization of the gap junctions as the cause of uncoupling. The inhibitor of p38 MAPK, SB202190, applied before hypoxia in high glucose, reduces dye uptake after 1 h reoxygenation and but does not prevent reduction in dye coupling between cells, again indicating different mechanisms.
To address the question of whether the action of hypoxia/reoxygenation on Cx43 in astrocytes was cell type specific, we examined Etd+ uptake in HeLa cells transfected with Cx43-EGFP (Orellana et al., 2010). As observed previously (Contreras et al., 2002), rate of uptake in control conditions is proportional to amount of Cx43-EGFP expressed. Rate of uptake is increased by 6 h hypoxia, and the degree of permeabilization is greater in 27 mM than in 5 mM glucose. The magnitude is ~5 fold and comparable to the effects in astrocytes. However, the permeability increase is maximal immediately after reoxygenation and presumably develops during the hypoxia. Moreover, the permeability returns to near normal in ~2 h reoxygenation, in contrast to the delayed and longer lasting permeability increases in astrocytes. The resilience of HeLa cells has been well documented and may reflect their tumor origin and years of selection in laboratories (Anderson et al., 2006; Skloot, 2010)
Now we introduce microglia. Co-culture of cortical astrocytes with microglia for 24 h prior to 3h hypoxia in high glucose prevents the transient rise in astrocyte permeability that occurs in the absence of microglia; the microglia appear to protect the astrocytes (Fig. 6A, B). Contrasting results are obtained when astrocytes are co-cultured for 24 h with microglia and a toxic peptide fragment of amyloid precursor protein, Aβ25–35, prior to 3h hypoxia in high glucose; Etd+ uptake after reoxygenation is greatly increased and prolonged (Fig. 6C). If the hypoxia is in normal glucose, uptake is also increased, but to a lesser extent. Aβ25–35 is another potential stressor (Pike et al., 1995), but it is acting through the microglia and has little direct effect on the astrocytes (compare Fig. 6D to Fig. 6A). Medium conditioned by 24 h culture of microglia in the presence of Aβ25–35 (CM-Aβ) has the same effect as co-culture with microglia and the peptide (Fig. 6E). The conclusion is that Aβ causes microglia to release a toxic substance or substances that sensitize astrocytes to the hypoxia in hyperglycemic conditions. Activated microglia are known to release TNF-α and IL-1β (Retamal, 2007b), and 24 h treatment with these agents mimics the effect of microglia plus Aβ or of CM-Aβ (Fig. 6F). Moreover, blocking agents indicate that the entire effect of the activated microglia is due to the summated actions of TNF-α and of IL-1β alone. Not one of the 24 h pretreatments by itself permeabilizes the cells (not shown).
Again, the permeabilization is due to opening of Cx43 hemichannels. None of the protocols increase permeability in astrocytes from Cx43 KO mice. For CM-Aβ and TNF-α + IL-1β treatments, connexin hemichannel blockers prevent the increases in permeability, as well as reduce the basal permeability in control cells. None of the Px1 hemichannel blockers are effective.
As above, the increases in astrocyte dye uptake are associated with increased surface expression of Cx43.
As above, in all of the conditions in which astrocytes are permeabilized, dye coupling is decreased with a similar time course and magnitude. Moreover, block of hemichannels does not increase the dye coupling, indicating that the reduced coupling is not due to leak from hemichannels.
Permeabilized astrocytes are bad for neurons
Now we address the question of the effects of leaky astrocytes on neurons. When pure neuronal cultures are subjected to 3h hypoxia in normal glucose (and ischemic saline as before) and their survival measured after reoxygenation, there is very little neuronal death at 12 h but about 50% death at 24 h (Fig. 7C, histogram not shown). Pretreatment with medium conditioned by microglia + Aβ for 24 h (CM-Aβ) before hypoxia has no additional effect, indicating that the neurons are insensitive to TNF-α+ IL-1β (and whatever else microglia secrete into CM-Aβ)(Fig. 7E, F). Astrocytes protect neurons from hypoxia; in co-culture 3 h hypoxia in normal glucose followed by reoxygenation, neuronal death at 24 h is minimal (Fig. 7I). However, in co-cultures pretreated with CM-Aβ for 24 h followed by 3 h hypoxia in normal glucose, many neurons, more than 50%, die within one hour of reoxygenation (Fig. 7K, L). (Few die before reoxygenation and extending reoxygenation up to 24 h has little further effect. The neuronal population may have more and less sensitive fractions.) The combined effect of astrocytes and CM-Aβ is absent with Cx43 KO astrocytes and greatly reduced by Cx43 hemichannel blockers applied during reoxygenation. These data suggest that astrocytes acted on by CM-Aβ release neurotoxic molecules through Cx43 hemichannels that open when the cells are subjected to the additional stress of hypoxia/reoxygenation. Further evidence for this hypothesis is provided by astrocyte conditioned medium prepared by treating astrocytes for 24 h with CM-Aβ, subjecting them to 3h hypoxia in normal glucose followed by 1 h reoxygenation in normal medium. This posthypoxia conditioned medium, CM-Ast, when applied to pure neuronal cultures for 1 h in normal glucose (and oxygen) greatly increases neuronal death (Fig. 8B). However, astrocyte conditioned medium made with Cx43 KO astrocytes or in the presence of Cx43 hemichannel blockers during the reoxygenation period is not neurotoxic, consistent with release from astrocytes through Cx43 hemichannels opened during reoxygenation (see Fig. 7N, O).
Fig. 7.

Pretreatment with conditioned medium from microglia cultured for 24 h with Aβ25–35 (CM-Aβ) followed by hypoxia-reoxygenation causes dendritic beading and death of neurons in co-culture with astrocytes. Representative confocal micrographs of immunofluorescence of MAP-2 staining (green) and Etd+ uptake (red) by neurons (N) cultured without and with astrocytes (Ast). A – C. Neurons under control conditions or after 3 h hypoxia followed by 1 or 24 h reoxygenation. B. After 3 h hypoxia and 1 h reoxygenation neurons appear normal. C. After 24 h reoxygenation many neurons show beading of neurites, but there is no Etd+ uptake indicating that neuronal membranes are intact. D – F. Neurons incubated for 24 h with CM-Aβ prior to 3 h hypoxia and reoxygenation show beading comparable to that in control medium. G – I. Neuron-astrocyte cocultures show no beading or Etd+ uptake when subject to hypoxia-reoxygenation as in B – C; astrocytes are protective. J – L. After 24 h pretreatment with CM-Aβ followed by 3 h hypoxia, cocultures of astrocytes and neurons show marked Etd+ uptake by astrocytes and by dying neurons at 1 h and 24 h reoxygenation. M – O. Neuronal death and astrocyte Etd+ uptake in cocultures caused by CM-Aβ pretreatment and hypoxia/reoxygenation is prevented by the connexin hemichannel blocker, Gap26, applied during reoxygenation. From Orellana et al., 2011.
Fig. 8.
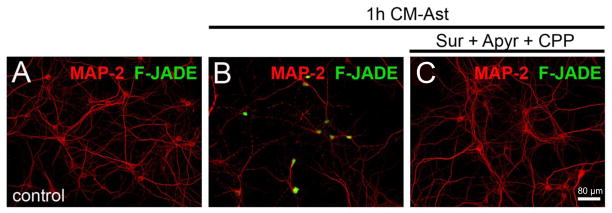
Neuronal death is induced by exposure to medium conditioned by activated astrocytes subjected to hypoxia/reoxygenation (CM-Ast); death is reduced by inhibition of NMDA and P2X receptors. Representative confocal micrographs of immunofluorescence of MAP-2 (red) to identify neurons and Fluoro Jade (F-Jade, green) to indicate neuronal death. A. Control conditions. B. After exposure to CM-Ast for 1 h many neurons are Fluoro Jade positive. C. Treatment with 200 μM suramin, 10 U/ml apyrase and 20 μM CPP prior to CM-Ast protects the neurons. From Orellana et al., 2011.
Neuronal Etd+ uptake and death caused by CM-Ast are completely blocked by the pannexin hemichannel blockers, probenecid and 10panx1 (Orellana et al., 2011). Uptake and death are partially reduced by P2X blockers and apyrase, a soluble ATPase, consistent with submaximal opening of pannexin hemichannels by ATP acting through P2X receptors. NMDA receptors may also contribute to pannexin hemichannel opening by increasing intracellular Ca2+, and the combination of the NMDA antagonist, CPP [3-(2-carboxypiperazin-4-yl)propyl-1-phosphonic acid], and the ATP antagonists is as protective as the pannexin hemichannel blockers (see Fig. 8C). We interpret these data to mean that activation of NMDA receptors and of P2X7 receptors each leads to opening of Px1 hemichannels, more in combination than independently, and that these hemichannels mediate dye uptake and cell death. Astrocyte conditioned medium does contain elevated levels of both glutamate and ATP, and comparable levels of glutamate and ATP act together to permeabilize and kill neurons in pure culture. Connexin hemichannel blockers do not provide any neuroprotection against CM-Ast. It is likely that neuronal death is preceded by high frequency activity, depolarization and Ca2+ overload.
The mechanisms explored here are diagrammed in Fig. 8. Microglia release TNF-α and IL-1β in response to stimulation with Aβ25–35. TNF-α and IL-1β act on astrocytes stressed by hypoxia and abnormal glucose to release glutamate and ATP. Glutamate acting through NMDA receptors and ATP acting through P2X receptors cause opening of neuronal pannexin hemichannels, which admit Ca2+ as well as release ATP leading to neuronal death. Although not indicated, dying neurons may release FGF-1, possibly leading to a vicious cycle.
Coda
Microglia and astrocytes can be neuroprotective or through inflammatory responses can lead to neuronal death. The mechanisms of release of toxic molecules depend on the cell subtype and on the conditions exemplified here by in vivo and in vitro preparations subjected to trauma, inflammatory agents, and stressors such as hypoxia and abnormal glucose. Further knowledge of the mechanisms will suggest additional therapeutic interventions.
Fig. 9.
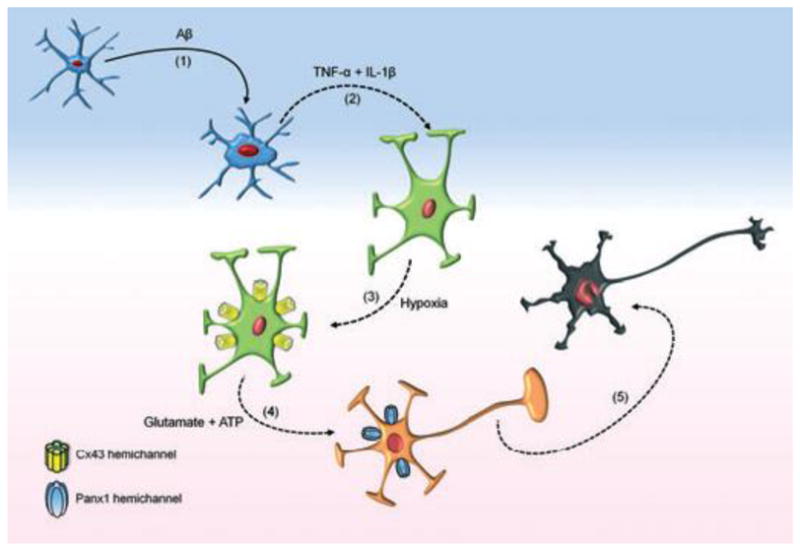
The inflammatory interactions from activated microglia to astrocytes to neurons. Release of FGF-1 from stressed or dying neurons is not indicated. From Orellana et al., 2011.
Acknowledgments
This study was supported by grants from the National Institutes of Health NS55363 to M.V.L.B, who is the Sylvia and Robert S. Olnick Professor of Neuroscience, NS72238 and HL84464 to F.F.B., NS075177 and NS078304 and grants from the W. M. Keck Foundation and the Dana Foundation to M. N., a grant from CONICYT 24080055 to J.A.O.; and grants from FONDECYT 1070591, ANILLO ACT 71, and FONDEF DO7I1086 to J.C.S.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson KM, Tsui P, Guinan P, Rubenstein M. The proliferative response of hela cells to 2-deoxy-D-glucose under hypoxic or anoxic conditions: An analogue for studying some properties of in vivo solid cancers. Anticancer Res. 2006;26:4155–4162. [PubMed] [Google Scholar]
- Baranova A, Ivanov D, Petrash N, Pestova A, Skoblov M, Kelmanson I, Shagin D, Nazarenko S, Geraymovych E, Litvin O, Tiunova A, Born TL, Usman N, Staroverov D, Lukyanov S, Panchin Y. The mammalian pannexin family is homologous to the invertebrate innexin gap junction proteins. Genomics. 2004;83:706–16. doi: 10.1016/j.ygeno.2003.09.025. [DOI] [PubMed] [Google Scholar]
- Basso DM, Fisher LC, Anderson AJ, Jakeman LB, McTigue DM, Popovich PG. Basso Mouse Scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J Neurotrauma. 2006;23:635–659. doi: 10.1089/neu.2006.23.635. [DOI] [PubMed] [Google Scholar]
- Bennett MVL, Contreras JE, Bukauskas FF, Saez JC. New roles for astrocytes: Gap junction hemichannels have something to communicate. Trends Neurosci. 2003;26:610–617. doi: 10.1016/j.tins.2003.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondarenko A, Chesler M. Glia. 2001;34:134–142. doi: 10.1002/glia.1048. [DOI] [PubMed] [Google Scholar]
- Bruzzone R, Hormuzdi SG, Barbe MT, Herb A, Monyer H. Pannexins, a family of gap junction proteins expressed in brain. Proc Natl Acad Sci USA. 2003;100:13644–13649. doi: 10.1073/pnas.2233464100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruzzone R, Barbe MT, Jakob NJ, Monyer H. Pharmacological properties of homomeric and heteromeric pannexin hemichannels expressed in Xenopus oocytes. J Neurochem. 2005;92:1033–1043. doi: 10.1111/j.1471-4159.2004.02947.x. [DOI] [PubMed] [Google Scholar]
- Bukauskas FF. Inducing de novo formation of gap junction channels. Methods Mol Biol. 2001;154:379–393. doi: 10.1385/1-59259-043-8:379. [DOI] [PubMed] [Google Scholar]
- Bukauskas FF, Weingart R. Multiple conductance states of newly formed single gap junction channels between insect cells. Pflugers Arch. 1993;423:152–154. doi: 10.1007/BF00374973. [DOI] [PubMed] [Google Scholar]
- Contreras JE, Sanchez HA, Eugenin EA, Speidel D, Theis M, Willecke K, Bukauskas FF, Bennett MVL, Saez JC. Metabolic inhibition induces opening of unapposed connexin 43 gap junction hemichannels and reduces gap junctional communication in cortical astrocytes in culture. Proc Natl Acad Sci USA. 2002;99:495–500. doi: 10.1073/pnas.012589799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras JE, Sáez JC, Bukauskas FF, Bennett MVL. Gating and regulation of connexin 43 (Cx43) hemichannels. Proc Nat Acad Sci USA. 2003;100:11388–11393. doi: 10.1073/pnas.1434298100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desplantez T, Verma V, Leybaert L, Evans WH, Weingart R. Gap26, a connexin mimetic peptide, inhibits currents carried by connexin43 hemichannels and gap junction channels. Pharmacol Res. 2012;65:546–552. doi: 10.1016/j.phrs.2012.02.002. [DOI] [PubMed] [Google Scholar]
- Garré JM, Retamal MA, Cassina MP, Barbeito L, Bukauskas FF, Sáez JC, Bennett MVL, Abudara V. FGF-1 induces ATP release from spinal astrocytes in culture and opens pannexin and connexin hemichannels. Proc Natl Acad Sci USA. 2010;107:22659–22664. doi: 10.1073/pnas.1013793107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaietta G, Deerinck TJ, Adams SR, Bouwer J, Tour O, Laird DW, Sosinsky GE, Tsien RY, Ellisman MH. Multicolor and electron microscopic imaging of connexin trafficking. Science. 2002;296:503–507. doi: 10.1126/science.1068793. [DOI] [PubMed] [Google Scholar]
- Holland LZ, Albalat R, Azumi K, Benito-Gutiérrez E, Blow MJ, Bronner-Fraser M, Brunet F, Butts T, Candiani S, Dishaw LJ, Ferrier DE, Garcia-Fernàndez J, Gibson-Brown JJ, Gissi C, Godzik A, Hallböök F, Hirose D, Hosomichi K, Ikuta T, Inoko H, Kasahara M, Kasamatsu J, Kawashima T, Kimura A, Kobayashi M, Kozmik Z, Kubokawa K, Laudet V, Litman GW, McHardy AC, Meulemans D, Nonaka M, Olinski RP, Pancer Z, Pennacchio LA, Pestarino M, Rast JP, Rigoutsos I, Robinson-Rechavi M, Roch G, Saiga H, Sasakura Y, Satake M, Satou Y, Schubert M, Sherwood N, Shiina T, Takatori N, Tello J, Vopalensky P, Wada S, Xu A, Ye Y, Yoshida K, Yoshizaki F, Yu JK, Zhang Q, Zmasek CM, de Jong PJ, Osoegawa K, Putnam NH, Rokhsar DS, Satoh N, Holland PW. The amphioxus genome illuminates vertebrate origins and cephalochordate biology. Genome Res. 2008;18:1100–11. doi: 10.1101/gr.073676.107. Erratum in: Genome Res. 2008;18:1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Han X, Li X, Lam E, Peng W, Lou N, Torres A, Yang M, Garré JM, Tian GF, Bennett MV, Nedergaard M, Takano T. Critical role of connexin 43 in secondary expansion of traumatic spinal cord injury. J Neurosci. 2012;32:3333–3338. doi: 10.1523/JNEUROSCI.1216-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias R, Dahl G, Qiu F, Spray DC, Scemes E. Pannexin 1: the molecular substrate of astrocyte “hemichannels”. J Neurosci. 2009;29:7092–7097. doi: 10.1523/JNEUROSCI.6062-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias R, Locovei S, Roque A, Alberto AP, Dahl G, Spray DC, Scemes E. P2X7 receptor-Pannexin1 complex: pharmacology and signaling. Am J Physiol Cell Physiol. 2008;295:C752–C760. doi: 10.1152/ajpcell.00228.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CP, Bechberger JF, Thompson RJ, MacVicar BA, Bruzzone R, Naus CC. Tumor-suppressive effects of pannexin 1 in C6 glioma cells. Cancer Res. 2007;67:1545–1554. doi: 10.1158/0008-5472.CAN-06-1396. [DOI] [PubMed] [Google Scholar]
- Lane NJ, Swales LS. Changes in the blood brain barrier of the central nervous system in the blowfly during development, with special reference to the formation and disaggregation of gap and tight junctions. II. Pupal development and adult flies. Dev Biol. 1978;62:415–431. doi: 10.1016/0012-1606(78)90225-7. [DOI] [PubMed] [Google Scholar]
- Li H, Liu TF, Lazrak A, Peracchia C, Goldberg GS, Lampe PD, Johnson RG. Properties and regulation of gap junctional hemichannels in the plasma membranes of cultured cells. J Cell Biol. 1996;134:1019–1030. doi: 10.1083/jcb.134.4.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orellana JA, Froger N, Ezan P, Jiang JX, Bennett MVL, Naus CC, Giaume C, Sáez JC. ATP and glutamate released via astroglial connexin 43 hemichannels mediate neuronal death through activation of pannexin 1 hemichannels. J Neurochem. 2011;118:826–840. doi: 10.1111/j.1471-4159.2011.07210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orellana JA, Hernández DE, Ezan P, Velarde V, Bennett MVL, Giaume C, Sáez JC. Hypoxia in high glucose followed by reoxygenation in normal glucose reduces the viability of cortical astrocytes through increased permeability of connexin 43 hemichannels. Glia. 2010;58:329–343. doi: 10.1002/glia.20926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchin Y, Kelmanson I, Matz M, Lukyanov K, Usman N, Lukyanov S. A ubiquitous family of putative gap junction molecules. Curr Biol. 2000;10:R473–R474. doi: 10.1016/s0960-9822(00)00576-5. [DOI] [PubMed] [Google Scholar]
- Parpura V, Heneka MT, Montana V, Oliet SH, Schousboe A, Haydon PG, Stout RF, Jr, Spray DC, Reichenbach A, Pannicke T, Pekny M, Pekna M, Zorec R, Verkhratsky A. Glial cells in (patho)physiology. J Neurochem. 2012;121:4–27. doi: 10.1111/j.1471-4159.2012.07664.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul DL, Ebihara L, Takemoto LJ, Swenson KI, Goodenough DA. Connexin46, a novel lens gap junction protein, induces voltage-gated currents in nonjunctional plasma membrane of Xenopus oocytes. J Cell Biol. 1991;115:1077–1089. doi: 10.1083/jcb.115.4.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng W, Cotrina ML, Han X, Yu H, Bekar L, Blum L, Takano T, Tian GF, Goldman SA, Nedergaard M. Systemic administration of an antagonist of the ATP-sensitive receptor P2X7 improves recovery after spinal cord injury. Proc Natl Acad Sci USA. 2009;106:12489–12493. doi: 10.1073/pnas.0902531106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike CJ, Walencewicz-Wasserman AJ, Kosmoski J, Cribbs DH, Glabe CG, Cotman CW. Structure-activity analyses of beta-amyloid peptides: contributions of the beta 25–35 region to aggregation and neurotoxicity. J Neurochem. 1995;64:253–265. doi: 10.1046/j.1471-4159.1995.64010253.x. [DOI] [PubMed] [Google Scholar]
- Retamal MA, Cortés CJ, Reuss L, Bennett MVL, Sáez JC. S-nitrosylation and permeation through connexin 43 hemichannels in astrocytes: Induction by oxidant stress and reversal by reducing agents. Proc Natl Acad Sci USA. 2006;103:4475–4480. doi: 10.1073/pnas.0511118103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retamal MA, Froger N, Palacios-Prado N, Ezan P, Sáez PJ, Sáez JC, Giaume C. Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. J Neurosci. 2007a;27:13781–13792. doi: 10.1523/JNEUROSCI.2042-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retamal MA, Schalper KA, Shoji KF, Bennett MV, Sáez JC. Opening of connexin 43 hemichannels is increased by lowering intracellular redox potential. Proc Natl Acad Sci USA. 2007b;104:8322–8327. doi: 10.1073/pnas.0702456104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samways DS, Migita K, Li Z, Egan TM. On the role of the first transmembrane domain in cation permeability and flux of the ATP-gated P2X2 receptor. J Biol Chem. 2008;283:5110–5117. doi: 10.1074/jbc.M708713200. [DOI] [PubMed] [Google Scholar]
- Sasakura Y, Shoguchi E, Takatori N, Wada S, Meinertzhagen IA, Satou Y, Satoh N. A genomewide survey of developmentally relevant genes in Ciona intestinalis. X. Genes for cell junctions and extracellular matrix. Dev Genes Evol. 2003;213:303–313. doi: 10.1007/s00427-003-0320-1. [DOI] [PubMed] [Google Scholar]
- Skloot R. The immortal life of Henrietta Lacks. Crown Publishing Group, Random House, Inc; New York: 2010. [Google Scholar]
- Sosinsky GE, Boassa D, Dermietzel R, Duffy HS, Laird DW, MacVicar B, Naus CC, Penuela S, Scemes E, Spray DC, Thompson RJ, Zhao HB, Dahl G. Pannexin channels are not gap junction hemichannels. Channels (Austin) 2011;5:193–197. doi: 10.4161/chan.5.3.15765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theis M, Jauch R, Zhuo L, Speidel D, Wallraff A, Döring B, Frisch C, Söhl G, Teubner B, Euwens C, Huston J, Steinhäuser C, Messing A, Heinemann U, Willecke K. Accelerated hippocampal spreading depression and enhanced locomotory activity in mice with astrocyte-directed inactivation of connexin43. J Neurosci. 2003;23:766–776. doi: 10.1523/JNEUROSCI.23-03-00766.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai HH, Li H, Fuentealba LC, Molofsky AV, Taveira-Marques R, Zhuang H, Tenney A, Murnen AT, Fancy SP, Merkle F, Kessaris N, Alvarez-Buylla A, Richardson WD, Rowitch DH. Regional astrocyte allocation regulates CNS synaptogenesis and repair. Science. 2012;337:358–362. doi: 10.1126/science.1222381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Abeele F, Bidaux G, Gordienko D, Beck B, Panchin YV, Baranova AV, Ivanov DV, Skryma R, Prevarskaya N. Functional implications of calcium permeability of the channel formed by pannexin 1. J Cell Biol. 2006;174:535–546. doi: 10.1083/jcb.200601115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallraff A, Köhling R, Heinemann U, Theis M, Willecke K, Steinhäuser C. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J Neurosci. 2006;26:438–5447. doi: 10.1523/JNEUROSCI.0037-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Arcuino G, Takano T, Lin J, Peng WG, Wan P, Li P, Xu Q, Liu QS, Goldman SA, Nedergaard M. P2X7 receptor inhibition improves recovery after spinal cord injury. Nat Med. 2004;10:821–827. doi: 10.1038/nm1082. [DOI] [PubMed] [Google Scholar]
- Wilhelm A, Volknandt W, Langer D, Nolte C, Kettenmann H, Zimmermann H. Localization of SNARE proteins and secretory organelle proteins in astrocytes in vitro and in situ. Neurosci Res. 2004;48:249–257. doi: 10.1016/j.neures.2003.11.002. [DOI] [PubMed] [Google Scholar]