

Abstract
Rationale
The endoplasmic reticulum (ER) is a major intracellular Ca2+ store in endothelial cells (ECs). The Ca2+ concentration in the ER greatly contributes to the generation of Ca2+ signals that regulate endothelial functions. Many proteins, including stromal interaction molecule 1/2 (STIM1/2), Orai1/2/3, and sarcoplasmic/ endoplasmic reticulum Ca2+-ATPase 3 (SERCA3), are involved in the ER Ca2+ refilling after store depletion in ECs.
Objective
This study is designed to examine the role of Ca2+ in the ER in coronary endothelial dysfunction in diabetes.
Methods and Results
Mouse coronary ECs (MCECs) isolated from diabetic mice exhibited (1) a significant decrease in the Ca2+ mobilization from the ER when the cells were treated by SERCA inhibitor, and (2) significant downregulation of STIM1 and SERCA3 protein expression in comparison to the controls. Overexpression of STIM1 restored (1) the increase in cytosolic Ca2+ concentration due to Ca2+ leak from the ER in diabetic MCECs, (2) the Ca2+ concentration in the ER, and (3) endothelium-dependent relaxation that was attenuated in diabetic coronary arteries.
Conclusions
Impaired ER Ca2+ refilling in diabetic MCECs, due to the decrease in STIM1 protein expression, attenuates endothelium-dependent relaxation in diabetic coronary arteries, while STIM1 overexpression has a beneficial and therapeutic effect on coronary endothelial dysfunction in diabetes.
Keywords: diabetic complications, vascular relaxation, cyclopiazonic acid, Ca2+ homeostasis, endothelial dysfunction
Ischemic heart disease is a major risk for mortality in diabetic patients. Coronary blood flow and coronary vascular resistance are tightly controlled by vascular tone and vascular density. Endothelial cells (ECs) serve as a major player in the regulation of vascular tone and the formation of new vessels. Thus, endothelial dysfunction is considered to be a risk factor for cardiovascular complications in many diseases.1–4 In diabetic patients, coronary endothelial dysfunction and vascular rarefaction in the heart cause cardiac ischemia and heart failure due to a shortage of vascular supply versus heart demand.5–10 We have recently demonstrated that capillary density in the left ventricle is significantly decreased in the heart of diabetic mice and that endothelium-dependent relaxation is significantly attenuated in diabetic coronary arteries (CAs) compared with control CAs.11
Endothelial function depends, to various extents, on the changes in cytosolic Ca2+ concentration ([Ca2+]cyt). [Ca2+]cyt is controlled by Ca2+ mobilization from intracellular stores coupled to Ca2+ influx from external medium. In ECs, the endoplasmic reticulum (ER) accounts for approximately 75% of the total intracellular Ca2+ stores and the [Ca2+] in the ER significantly determines the generation of important Ca2+ signals that regulates vascular tone.12 The ER membrane constitutes Ca2+ pumps (sarco/endoplasmic reticulum ATPase [SERCA]) and several classes of intracellular Ca2+-releasing channels, including the inositol triphosphate receptors (IP3Rs) and the ryanodine receptors (RyRs). ECs predominantly express SERCA3 (and also express low level of SERCA2b).13 Depletion of Ca2+ from the ER activates Ca2+-permeable channels in the plasma membrane and induces store-operated Ca2+ entry (SOCE), which ensures long-term signaling. Stromal interaction molecule (STIM) and Orai were identified recently as essential proteins for SOCE.14,15 Recent reports demonstrate that STIM1 serves as a functional sensor for SOCE and therefore contributes to Ca2+ refilling into the ER after store depletion.16–18 However, the role of STIM1 in coronary endothelial dysfunction in diabetes is unexplored.
In this study, we demonstrate that the increase in [Ca2+]cyt due to Ca2+ leak from the ER induced by a SERCA blocker (an indirect way to measure the Ca2+ concentration in the ER) is significantly inhibited in mouse coronary ECs (MCECs) isolated from diabetic mice compared with ECs from control mice. Protein expression of STIM1 and SERCA3 is significantly lower in MCECs from diabetic mice than in MCECs from control mice. Not only does STIM1 overexpression in diabetic MCECs increase the amount of Ca2+ leakage from the ER and raise [Ca2+]ER toward the control level, but it also restores endothelium-dependent relaxation that was attenuated in diabetic CAs. These data suggest that the restoration of an increase in [Ca2+]cyt due to Ca2+ leak from the ER by STIM1 overexpression has a beneficial effect on coronary endothelial function, which may subsequently decrease the incidence of cardiac ischemia in diabetes.
Methods
An expanded Methods section is available in the Online Data Supplement.
Animal Preparation
All investigations conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Publication No. 85–23, Revised 1985). This study was conducted in accordance with the guidelines established by the Institutional Animal Care and Use Committee in the University of Illinois at Chicago. Six-week-old male C57BL6 mice were purchased from Harlan Laboratories (Madison, WI); mice in the diabetic group received a single injection of streptozotocin (133 mg/kg, dissolved in citrate buffer, IV). All data were obtained from mice 6 weeks after injection. Plasma glucose levels were 10.1±0.6 mmol/L in control mice and 32.2±0.6 mmol/L in diabetic mice.
Statistical Analysis
Values are expressed as mean±SEM. Bonferroni tests for multiple statistical comparisons and Student t test for unpaired samples were carried out to identify significant differences. Differences were considered to be statistically significant when P<0.05.
Results
Hyperglycemia Attenuates the Rise in [Ca2+]cyt Due to Ca2+ Leakage From the ER
Ca2+ is an essential signaling element for endothelial functions, including endothelium-dependent vascular relaxation by activating the endothelial nitric oxide synthase (a Ca2+-dependent enzyme)19,20 and Ca2+-activated K+ channels in ECs (which leads to hyperpolarization).21–23 Ca2+ in the ER contributes greatly to Ca2+-dependent endothelial function. 24–28 We first tested whether MCECs isolated from diabetic mice altered Ca2+ leak from the ER after stimulation by cyclopiazonic acid (CPA, a SERCA inhibitor, 10 μmol/L) in the absence of extracellular Ca2+. The rise in [Ca2+]cyt during CPA treatment in ECs superfused with Ca2+-free solution is often referred to as an indirect estimation of [Ca2+]ER. As shown in Figure 1B and 1C (1st ΔF/F0 and 1st AUC), the rise in [Ca2+]cyt due to CPA-mediated Ca2+ leak was significantly attenuated in MCECs isolated from diabetic mice (red tracings) compared with ECs from control mice (black tracings). The resting level of [Ca2+]cyt (referred as F0 in the graph) was not significantly different between control ECs and diabetic ECs (control, 1.87±0.06; diabetic, 1.79±0.05, P=0.33). We then tested the increase in [Ca2+]cyt through SOCE by adding extracellular Ca2+ in the presence of CPA. As shown in Figure 1C (described as 2nd ΔF/F0 and 2nd AUC), there was no significant difference in SOCE between control and diabetic MCECs. The exposure of ECs to high glucose (HG) significantly attenuated the Ca2+ leak from the ER compared with ECs treated with normal glucose (NG) (Figure 2). These data suggest that hyperglycemia leads to a decrease in [Ca2+]ER in ECs.
Figure 1. Hyperglycemia significantly inhibits the rise in [Ca2+]cyt due to Ca2+ release/leakage from the ER during cyclopiazonic acid (CPA) treatment in coronary ECs.
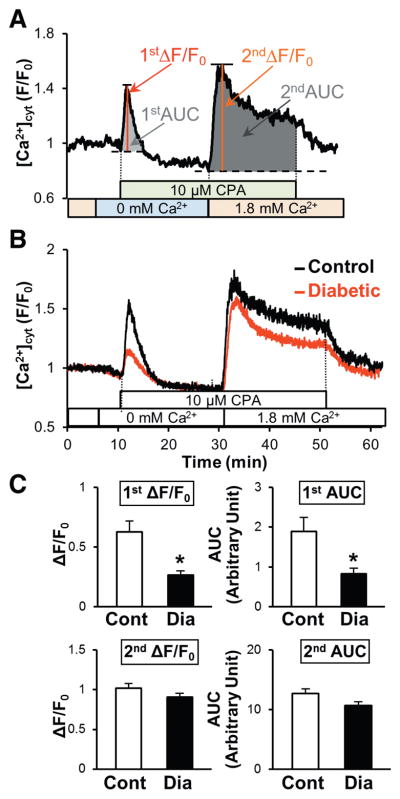
A, Typical record of the change in [Ca2+]cyt in coronary ECs and the parameters used for the statistical analysis. First peak describes the rise in [Ca2+]cyt by CPA treatment in the absence of extracellular Ca2+ (indirect indicator of [Ca2+]ER) and 2nd peak indicates store-operated Ca2+ entry (SOCE). B, Averaged record of the change in [Ca2+]cyt in coronary ECs isolated from control (black) and diabetic mice (red). Three days after EC isolation, ECs were used for [Ca2+]cyt measurement with Fura-2-AM. Thirty minutes after preincubation of cells with physiological salt solution with Ca2+ (Ca2+-PSS), extracellular solution was switched to Ca2+ free-PSS; 10 μmol/L CPA was added in Ca2+-free-PSS to determine the [Ca2+]ER indirectly. Data are described as a normalized ratio (F/F0, F=I340/I380, F0=average of F during first 5 minutes recording in Ca2+-PSS). C, Summarized data of ΔF/F0 and area under the curve (AUC). Control (Cont, open bars); n=18 cells, diabetic (Dia, solid bars); n=34 cells. Data are mean±SEM. P*<0.05 versus Cont.
Figure 2. High-glucose treatment over 48 hours significantly inhibits the rise in [Ca2+]cyt due to Ca2+ release/leakage from the ER during CPA treatment in coronary ECs.

Summarized data of the rise in [Ca2+]cyt due to Ca2+ release/leakage from the ER (1st ΔF/ F0 and 1st area under the curve [AUC]) and SOCE (2nd ΔF/F0 and 2nd AUC). Normal glucose (NG, open bars); n=22 cells, high glucose (HG, solid bars); n=17 cells. Data are mean±SEM. *P<0.05 versus NG.
Hyperglycemia Decreases the Protein Expression of STIM1 and SERCA3
[Ca2+]ER is regulated by the activity of Ca2+ pump (SERCA) and several classes of Ca2+ release channels (IP3Rs and RyRs) and by SOCE (eg, STIM1/2, Orai1–3). We used freshly isolated MCECs to determine STIM1 and SERCA3 protein expression. MCECs from diabetic mice exhibited significantly lower protein expression of STIM1 and SERCA3 than control MCECs (Figure 3A). HG-treatment in ECs ex vivo not only downregulated STIM1 and SERCA3 protein expression (Figure 3B), but also inhibited the coupling of STIM1-SERCA3 in comparison to ECs treated with NG (Figure 3C). The attenuated rise of [Ca2+]cyt due to Ca2+ leak from the ER during CPA stimulation, shown in Figure 1, might be due to the attenuated ER refilling resulting from the decrease in SERCA3 and STIM1 protein expression in diabetic MCECs.
Figure 3. Hyperglycemia downregulates protein expression of STIM1 and SERCA3 in coronary ECs.
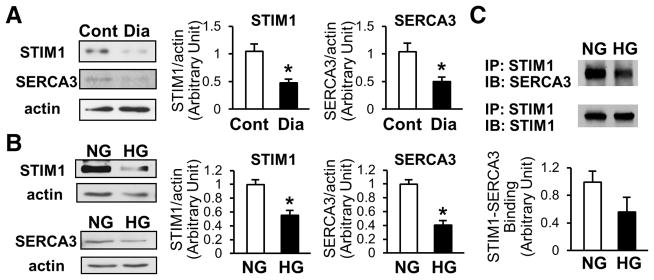
A, Freshly isolated coronary ECs from control mice (Cont, open bars) or diabetic mice (Dia, solid bars) were used to determine the protein concentration of STIM1 and SERCA3 by Western blot. Data are normalized by the signal of actin. STIM1; n=4 in each group, SERCA3; n=6 in each group. Data are mean±SEM. *P<0.05 versus Cont. B, Two days after the exposure of ECs to NG (open bars) or HG (solid bars), cells were lysed and protein concentration of STIM1 and SERCA3 was measured. STIM1, n=7 in each group; SERCA3, n=3 in each group. Data are mean±SEM. *P<0.05 versus NG. C, Immunoprecipitation (IP) of SERCA3 with STIM1. After NG or HG treatment, cells were lysed and IP was performed using anti-STIM1 N-terminus antibody. Immunoblotting (IB) was determined using anti-SERCA3 antibody. The same membrane was used for IB with anti-STIM1 C-terminus to normalize the data. For each group, n=3. Data are mean±SEM. *P<0.05 versus NG.
Overexpression of STIM1 Significantly Enhances the Rise of [Ca2+]cyt Due to Ca2+ Leak From the ER During CPA Stimulation and Increases the [Ca2+]ER in Diabetic MCECs
An increase in [Ca2+]cyt in ECs leads to vascular relaxation, thus restoring the level of Ca2+ in the ER of diabetic MCECs helps improve endothelium-dependent relaxation. Enhanced SOCE and a subsequent increase in [Ca2+]cyt in smooth muscle cells by upregulated STIM1 and/or STIM2 are, however, one of the important causes for systemic29 and pulmonary30 hypertension. Thus, we designed an adenovirus that encoded STIM1 positive mutant with a Tie2 promoter, which can be selectively expressed in ECs.31,32 As shown in Figure 4, overexpression of the STIM1 restored the level of Ca2+ leak from the ER during CPA stimulation in diabetic MCECs toward the control level. In addition, direct measurement of [Ca2+]ER demonstrates that STIM1 overexpression in diabetic MCECs increases the resting level of [Ca2+]ER and the amount of Ca2+ released from the ER after CPA treatment toward the level seen in control MCECs (Figure 5). These data suggest that STIM1 regulates Ca2+ refilling into the ER in diabetic ECs.
Figure 4. STIM1 overexpression restores the attenuated increase in [Ca2+]cyt due to Ca2+ release/leakage from the ER during CPA treatment in MCECs isolated from diabetic mice.

A, Construction of STIM1-positive mutant with the Tie2 promoter in an adenoviral vector. B, STIM1 adenovirus (Adv) infection in HCECs upregulates STIM1 protein concentration determined by Western blot. C, STIM1 protein expression level in MCECs after Adv infection. Representative images showing STIM1 protein expression level determined by immunofluorescence in control MCECs infected with control-Adv (Cont EC–Cont Adv), diabetic MCECs infected with control-Adv (Dia EC–Cont Adv), and diabetic MCECs infected with STIM1 Adv (Dia EC–STIM1 Adv). Dark dots in the cells are beads used for cell isolation. Bar=20 μm. Lower panel shows summarized data of STIM1 expression level (intensity). Cont EC–Cont Adv, n=104 cells; Dia EC–Cont Adv, n=143 cells; Dia EC–STIM1 Adv, n=151 cells. Data are mean±SEM. *P<0.05 versus Cont EC–Cont Adv. #P<0.05 versus Dia EC–Cont Adv. D, STIM1 overexpression in MCECs isolated from diabetic mice significantly increased the rise in [Ca2+]cyt due to Ca2+ leakage but not by SOCE toward the level of control MCECs. Summarized data of the rise in [Ca2+]cyt due to Ca2+ release/leakage from the ER (1st ΔF/F0 and 1st area under the curve [AUC]) and SOCE (2nd ΔF/F0 and 2nd AUC). Control ECs infected with control Adv (Cont EC–Cont Adv, open bars), n=18; diabetic ECs infected with control Adv (Dia EC–Cont Adv, solid bars), n=22; and diabetic ECs infected with STIM1 Adv (Dia EC–STIM1 Adv, hatched bars), n=25. Data are mean±SEM. *P<0.05 versus Cont EC–Cont Adv. #P<0.05 versus Dia EC–Cont Adv. ANOVA was performed to test the statistical difference between the groups.
Figure 5. STIM1 overexpression increases the [Ca2+]ER in MCECs isolated from diabetic mice.

A, Typical record of the change in [Ca2+]ER in coronary ECs and the parameters used for the statistical analysis. B, Averaged record of the change in [Ca2+]ER in control MCECs infected with control-Adv (Cont EC–Cont Adv, black tracing), diabetic MCECs infected with control-Adv (Dia EC–Cont Adv, red tracing), and diabetic MCECs infected with STIM1 Adv (Dia EC–STIM1 Adv, blue tracing). C, Summarized data of resting level of [Ca2+]ER, ΔPeak, and the area under the curve (AUC; the change in [Ca2+]ER after CPA treatment). Cont EC–Cont Adv (open bars), n=25; Dia EC–Cont Adv (solid bars), n=21; and Dia EC–STIM1 Adv (hatched bars), n=30. Data are mean±SEM. *P<0.05 versus Cont EC–Cont Adv. #P<0.05 versus Dia EC–Cont Adv. ANOVA was performed to test the statistical difference between the groups.
It must be emphasized that in these ex vivo experiments, we did not overexpress STIM1 in diabetic MCECs above the level seen in control cells. Because STIM1 is significantly downregulated in coronary endothelial cells in diabetic mice, we actually restored the expression level of STIM1 in diabetic MCECs to the level similar to the STIM1 expression level in control MCECs. Furthermore, our experiments demonstrated that overexpression of STIM1 in HEK293 cells had no inhibitory effect on Orai1 expression and function; STIM1 overexpression actually enhances the increase in [Ca2+]cyt due to Ca2+ influx through SOC/Orai1 channels and augments Ca2+ currents through SOC/Orai1 channels (Online Figure VII).
Overexpression of STIM1 Restores Endothelium-Dependent Vascular Relaxation That Was Attenuated in Diabetic CAs
Diabetic CAs exhibit attenuated endothelium-dependent relaxation (assessed by acetylcholine [ Ach]-induced relaxation) but not smooth muscle cell–dependent relaxation (assessed by SNP).11 STIM1-Adv or control-Adv was infected in CAs dissected from control or diabetic mice, and isometric tension experiments were performed 24 hours after infection. Figure 6A demonstrates that 10 μmol/L CPA-induced relaxation was significantly attenuated in diabetic CAs infected with control-Adv compared with control CAs infected with control-Adv. However, STIM1 overexpression in diabetic CAs significantly augmented CPA-induced vascular relaxation close to the level shown in control CAs (Figure 6A). ACh-induced relaxation was significantly attenuated in diabetic CAs compared with control CAs (Figure 6B), whereas 10−4 mol/L SNP-induced vascular relaxation was not significantly different between control and diabetic CAs (control CAs, 99.7±3.5; diabetic CAs, 103.3±8.2; P=0.70). STIM1 overexpression significantly increased ACh-induced vascular relaxation in diabetic CAs, whereas there was no difference in SNP-induced relaxation between diabetic CAs infected with control Adv and diabetic CAs infected with STIM1-Adv (Figure 6C). In addition, we measured and compared nitric oxide production in control and diabetic MCECs. As shown in Figure 6D and 6E, nitric oxide production at the resting condition and during CPA treatment in the absence of extracellular Ca2+ is decreased in diabetic MCECs compared with control MCECs, which is partially but significantly restored toward the control level by STIM1 overexpression in diabetic MCECs. These data suggest that downregulated STIM1 protein expression in coronary ECs leads to a decrease in Ca2+ release from the ER, and subsequently attenuates endothelium-dependent relaxation in diabetic CAs.
Figure 6. Overexpression of STIM1 restores CPA- and ACh-induced relaxation in diabetic CAs and increases nitric oxide (NO) production in diabetic MCECs.

A, After precontraction of the CAs, CPA-induced (10 μmol/L) vascular relaxation was observed. Relaxation was calculated versus the magnitude of the contraction induced by PGF2α and described as percentage. Control CAs infected with control Adv (Cont CA–Cont Adv, open bar), n=4; diabetic CAs infected with control Adv (Dia CA–Cont Adv, solid bar), n=5; diabetic CAs infected with STIM1 Adv (Dia CA–STIM1 Adv, hatched bar), n=3. Data are mean±SEM. *P<0.05 versus Cont CA– Cont Adv. #P<0.05 versus Dia CA–Cont Adv. B, Endothelium-dependent relaxation was determined by ACh-induced relaxation. After preconstruction of CAs, ACh was administrated with a dose-dependent manner. Cont CA–Cont Adv (open circles), n=4; Dia CA–Cont Adv (solid circles), n=5; Dia CA–STIM1 Adv (solid triangles), n=4. Data are mean±SEM. *P<0.05 versus Cont CA–Cont Adv. #P<0.05 versus Dia CA–Cont Adv. C, Endothelium-independent relaxation was determined by SNP-induced relaxation. After preconstruction of CAs, SNP was administrated with a dose-dependent manner. Dia CA–Cont Adv (solid circles), n=3; Dia CA–STIM1 Adv (solid triangles), n=3. Data are mean±SEM. D, Resting level of DAF intensity was obtained from the average intensity of first 2 to 4 minutes during Ca2+ PSS perfusion. Cont EC–Cont Adv (open bar), n=44; Dia EC–Cont Adv (solid bar), n=49; Dia EC–STIM1 Adv (hatched bar), n=44. Data are mean±SEM. *P<0.05 versus Cont EC–Cont Adv. #P<0.05 versus Dia EC–Cont Adv. E, NO production due to Ca2+ release/leakage from the ER during CPA treatment in coronary ECs. Left graph shows a typical record of DAF-FM intensity change indicated as F/F0. The slope between time 15 (t15) and time 24 (t24) (total 9 minutes) was calculated (gray line) and used as an indication of NO production in response to CPA [d(F/F0)/dt]. Right panel shows the summarized data of d(F/F0)/dt during CPA treatment (t15–t24). Cont EC–Cont Adv (open bar), n=44; Dia EC–Cont Adv (solid bar), n=49; Dia EC–STIM1 Adv (hatched bar), n=44. Data are mean±SEM. *P<0.05 versus Cont EC–Cont Adv. #P<0.05 versus Dia EC–Cont Adv. ANOVA was performed to test the statistical difference between the groups.
Inhibition of STIM1 Attenuates the Rise in [Ca2+]cyt Due to Ca2+ Release/Leakage From the ER During CPA Treatment and Decreases [Ca2+]ER in Coronary ECs
Two days after STIM1 siRNA transfection in coronary ECs by electroporation, Ca2+ release/leakage from the ER during CPA treatment, SOCE, and [Ca2+]ER were examined. Our data demonstrate that STIM1 downregulation not only decreases the SOCE but also attenuates the increase in [Ca2+]cyt due to Ca2+ leak from the ER and significantly decreases [Ca2+]ER in ECs (Figure 7).
Figure 7. STIM1 downregulation attenuates the rise in [Ca2+]cyt due to Ca2+ release/leakage from the ER during CPA treatment and decreases [Ca2+]ER in coronary ECs.

A, STIM1 siRNA transfection in HCECs downregulates STIM1 protein expression determined by Western blot. Values are mean±SEM (n=2). *P<0.05 versus control siRNA. B, Summarized data of the rise in [Ca2+]cyt due to Ca2+ release/leakage from the ER (1st ΔF/F0 and 1st area under the curve [AUC]) and SOCE (2nd ΔF/F0 and 2nd AUC). Control siRNA (open bars), n=25 cells; STIM1 siRNA (solid bars), n=27 cells. Data are mean±SEM. *P<0.05 versus control siRNA. C, Summarized data of resting level of [Ca2+]ER, ΔPeak, and the AUC (the change in [Ca2+]ER after CPA treatment). Control siRNA (open bars), n=24; STIM1 siRNA (solid bars), n=30. Data are mean±SEM. *P<0.05 versus control siRNA.
Treatment of Free Fatty Acid Significantly Decreases STIM1 mRNA and Protein Levels but HG Decreases STIM1 Protein Expression Without Changing the mRNA Level in Mouse Coronary ECs
In diabetes, plasma glucose and free fatty acid (FFA) levels are both significantly increased, and these changes trigger and progress vascular complications. We tested whether FFA or HG affects STIM1 mRNA levels and found that only FFA decreases STIM1 mRNA, however both HG and FFA significantly decrease STIM1 protein expression in ex vivo (Figure 8).
Figure 8. High glucose (HG) and free fatty acid (HF) treatment downregulate protein expression of STIM1, whereas only HF decreases mRNA level of STIM1, in mouse coronary ECs.

A and B, STIM1 protein (A, n=5 in each group) and mRNA (B, n=6 in each group) levels were measured in ECs treated with NG (open bars) or HG (solid bars) for 48 hours. Data are mean±SEM. *P<0.05 versus NG. C and D, STIM1 protein (A, n=3 in each group) and mRNA (B, n=6 in each group) levels were measured in ECs treated with vehicle (NF, open bars) or HF (solid bars) for 24 hours. Data are mean±SEM. *P<0.05 versus NF.
Discussion
The major findings of the current study are that (1) downregulation of STIM1 protein expression in diabetic MCECs leads to attenuated coronary vascular relaxation due to the decrease in stored Ca2+ in the ER, and (2) STIM1 expression in diabetic MCECs restores the endothelial function, which may subsequently decrease the incidence of cardiac ischemia in diabetes.
Inhibition of SERCA (by CPA or thapsigargin) causes a transient increase in [Ca2+]cyt of ECs due to Ca2+ leakage from the ER and induces endothelium-dependent relaxation (EDR). Prolonged treatment of ECs with CPA or thapsigargin (eg, for 10–15 minutes) depletes Ca2+ from the ER and thus inhibits ACh-induced vasodilation.24–27 These observations suggest that a rise in [Ca2+]cyt in ECs due to Ca2+ release from the ER is crucial for EDR. In addition to inducing EDR, the sufficient [Ca2+]ER and the rise in [Ca2+]cyt, due to Ca2+ release from the ER also contribute to stimulating EC migration and proliferation.33,34 Taken together, Ca2+ released from the ER plays an important role in endothelial function. In other words, the attenuated Ca2+ release from the ER (eg, by a decreased level of [Ca2+] in the ER) exerts a maladaptive effect on physiological functions of endothelium. Coronary ECs isolated from diabetic mice exhibit a significant attenuation of the rise in [Ca2+]cyt during CPA treatment compared with coronary ECs isolated from control mice, whereas Ca2+ influx via SOC is not significantly different between control and diabetic MCECs (Figure 1). HG treatment in normal ECs significantly lowers CPA-mediated Ca2+ leak from the ER compared with NG-treated ECs (Figure 2). These data imply that decreased Ca2+ release from the ER in coronary ECs by hyperglycemia is one of the causes for attenuated coronary vascular relaxation in diabetes.
STIM, a functional sensor of stored [Ca2+] in the ER, and Orai, a pore-forming subunit of SOC, were identified as essential proteins for SOCE.14,15 In ECs, STIM1 and Orai1 are both highly expressed.16 When the ER Ca2+ store is depleted, STIM1 undergoes a conformational change, which allows it to multimerize, translocate to the ER-plasma membrane junction (or puncta), bind with Orai1 tetramers on the plasma membrane, activate SOC, and induce SOCE. The STIM1-mediated SOCE not only contributes to a sustained rise in [Ca2+]cyt but also contributes to Ca2+ refilling into the ER by interacting with SERCA (Online Figure V).17,18 Our data demonstrate that protein expressions of STIM1 and SERCA3 are significantly decreased in MCECs isolated from diabetic mice compared with control MCECs (Figure 3A). Ex vivo experiments using HG reveal that HG treatment not only lowers STIM1 and SERCA3 protein expression (Figure 3B) but also decreases the coupling of STIM1-SERCA3 (Figure 3C). Further experiments are required in order to define the detailed mechanisms that cause the decrease in STIM1-SERCA3 coupling. Although STIM2 is not predominant among STIM subtypes, MCECs express STIM2, and its function in diabetic MCECs also must be examined.
One would expect to see that the decreased STIM1 protein expression in diabetic MCECs should attenuate SOCE-mediated [Ca2+]cyt increase. However, contrary to our expectations, our data showed that there was no significant difference in the increase of [Ca2+]cyt due to SOCE between control MCECs and diabetic MCECs (Figure 1C, lower panels). It has been reported that STIM1 regulates Ca2+ refilling into the ER not only via the SOC-mediated indirect pathway but also via direct interaction with SERCA in other cell types.17,18 The indirect refilling mechanism is mainly caused by the uptake of increased cytosolic Ca2+ due to SOCE via SERCA on the ER membrane. The direct refilling mechanism is that the Ca2+ ions entered cell via SOC can be “directly” sequestered by the SERCA without slowly diffusing into the cytosol due to the STIM1-SERCA interaction, and this mechanism may be more efficient than the indirect mechanism in refilling Ca2+ into the ER. We hypothesize that the degree of decrease in STIM1 protein expression (as shown in diabetic MCECs) is sufficient to affect the level of [Ca2+]ER, but insufficient to regulate SOCE-mediated global increase in [Ca2+]cyt. In addition, it is possible that other STIM1-independent mechanisms for SOCE (eg, TRPC1) may be compensatorily affected in diabetic MCECs, which makes the amplitude of SOCE comparable between control and diabetic MCECs.
In addition to the store depletion-mediated STIM1 interaction with Orai1,16 direct interaction between SERCA3 and TRPC channels (eg, human TRPC1 and TRPC6) can be stimulated by extracellular ligand and store depletion to form the STIM/SERCA3/TRPC complex.35–37 In human platelets, SERCA3 forms the macromolecular protein complex with TRPC1/6 and IP3R that is activated by store depletion or thrombin to mediate SOCE. In pulmonary vascular endothelial cells, Orai1 interacts with TRPC and regulates the ion selectivity and activation kinetics of store-operated channels. 38,39 Physical interaction among TRPC/Orai, SERCA and STIM in vascular endothelial cells would significantly enhance the efficiency for Ca2+ refilling into the ER (eg, via a direct refilling mechanism without a global increase in [Ca2+]cyt) and maintaining the normal endothelial function. The data from our study indicate that downregulated STIM1 (and SERCA3) in diabetic MCECs leads to a decrease in [Ca2+]ER and an inhibition of endothelium-dependent coronary vasodilation. It is possible that dysfunctional (or inhibited) interaction among STIM1, SERCA3, and TRPC is also involved in the attenuated Ca2+ refilling into the ER in diabetic ECs.
The increase in [Ca2+]cyt in smooth muscle cells (SMCs) causes smooth muscle contraction and SMC proliferation, which lead to the increase in vascular tension and resistance. Indeed, upregulated STIM1/2 in SMCs has been implicated in several cardiovascular diseases.29,30,40–42 In contrast, recent reports have highlighted the importance of STIM1 in endothelial physiological functions.16,39,43 This current study is the first report to demonstrate the pathophysiological role of STIM1 in diabetic ECs. Since STIM1 overexpression must be selective in ECs, we designed the adenovirus that carries the Tie2 promoter so that STIM1 is only expressed in ECs31,32 (Figure 4A through C and Online Figures IV and VI). After infection of STIM1-Adv in diabetic MCECs, the CPA-induced rise in [Ca2+]cyt due to Ca2+ leak from the ER (Figure 4D) and the [Ca2+]ER (Figure 5) were significantly increased toward the level similar to control MCECs. These data suggest that STIM1 regulates [Ca2+]ER in normal ECs and that overexpression of STIM1 (or restoration of the expression level of STIM1) in diabetic MCECs helps to increase the Ca2+ release from the ER and thus endothelium-dependent coronary vasodilatation.
We also examined the effect of STIM1 on Ca2+ leak from the ER in control MCECs (Online Figure I); overexpression of STIM1-Adv in control MCECs did not increase the rise in [Ca2+]cyt after CPA treatment. It might be because that (1) the endogenous level of STIM1 protein is very high in control MCECs, therefore overexpression of exogenous STIM1 is unable to further enhance its function, and (2) the amount of SERCA in control MCECs is low and insufficient to bind to extra STIM1 to enhance Ca2+ uptake or refilling.
Endothelial dysfunction is considered to be a major risk factor for cardiovascular complications in diabetes.44–46 The main physiological roles of ECs are the regulation of vascular tone, new vessel formation, and vascular wall permeability. It has been reported that in Type1 diabetic animal models as well as in human diabetic patients, capillary density in the heart is progressively decreased,11,47–50 endothelium-dependent relaxation is significantly attenuated in the coronary artery,6,9,11,51,52 and endothelial cell permeability is increased.53 Augmented tension and increased resistance in the coronary artery lead to an insufficient delivery of oxygen to the cardiac myocyte and causes cardiac ischemia, a leading cause of mortality and mobility in diabetes. Therefore, a decrease in vascular contractility in coronary arteries should have beneficial effects on coronary vascular complications in diabetes. ACh-induced relaxation was significantly decreased in CAs in diabetic mice compared with control CAs. STIM1 overexpression not only restored CPA-induced vascular relaxation but also augments ACh-induced relaxation to the level shown in control CAs through increasing nitric oxide production (Figure 6). These data suggest that a decrease in Ca2+ release from the ER due to downregulated STIM1 protein expression in ECs is one of the causes that lead to attenuated endothelium-dependent relaxation in CAs in diabetes.
In coronary arterial SMCs, the protein expression level of STIM1 is slightly (but statistically significant) upregulated in diabetic mice in comparison to control mice. However, the 40K+-mediated coronary vasoconstriction was actually decreased in coronary arteries isolated from diabetic mice (Online Figure III). We will further examine the function of STIM1 in diabetic SMCs in the future.
We have shown the lower expression level of SERCA3 protein in diabetic MCECs than in control MCECs (Figure 3). These data imply that overexpression of SERCA3 may also have beneficial effects on coronary endothelial dysfunction in diabetes. In Figure 7, we examined the effect of STIM1 knockdown on the Ca2+ leak from the ER, SOCE, and [Ca2+]ER in normal ECs. Downregulation of STIM1 with siRNA significantly inhibited the CPA-induced increase in [Ca2+]cyt due to Ca2+ leakage from the ER and the CPA-mediated increases in [Ca2+]cyt due to SOCE and significantly decreased [Ca2+]ER in ECs. Furthermore, we demonstrate that STIM1 overexpression fully restores not only [Ca2+]ER in diabetic MCECs but also endothelium-dependent relaxation in diabetic coronary artery in Figures 4 to 6. These data suggest that STIM1 may play a prominent role in coronary endothelial dysfunction in diabetes. Further studies are required to determine the role of SERCA3 and its interaction with STIM1 and TRPC channels in diabetes.
We are not sure why and how STIM1 protein expression level is altered in diabetic coronary endothelial cells. In diabetes, the plasma levels of glucose and FFA are both elevated. In our ex vivo experiments, we showed that exposure of EC to high glucose and FFA significantly decreases the protein expression of STIM1, whereas only FFA decreases the mRNA expression of STIM1 (Figure 8). These observations suggest that increased protein degradation (eg, ubiquitinylation) and microRNA-mediated posttranscriptional modulation are potential mechanisms involved in the HG-mediated decrease in STIM1 protein level.
Taken together, our data suggest that the restoration of effective and sufficient Ca2+ release from the ER by STIM1 overexpression improves endothelium-dependent relaxation in diabetic CAs, and may be a novel strategy to develop a therapeutic approach for cardiac ischemia in diabetes.
Supplementary Material
Novelty and Significance.
What Is Known?
Coronary vascular endothelial dysfunction is implicated in the development and progression of cardiac ischemia and heart failure due to a decrease in coronary blood flow.
The Ca2+ concentration in the endoplasmic reticulum (ER) is important for generating critical Ca2+ signals to mediate endothelium-dependent vasodilation.
Stromal interaction molecule (STIM) protein (eg, STIM1) is an important regulator that activates Ca2+-permeable channels in the plasma membrane after depletion of Ca2+ from the ER and refill Ca2+ into the ER.
What New Information Does This Article Contribute?
Protein expression of STIM1 is significantly downregulated and the Ca2+ concentration in the ER is markedly decreased in coronary endothelial cells in diabetes in comparison to coronary endothelial cells isolated from controls.
Downregulated STIM1 in diabetic coronary endothelial cells contributes to the decreased Ca2+ concentration in the ER and results in significant inhibition of endothelium-dependent coronary vasodilation.
Restoration of STIM1 protein expression in coronary endothelial cells has a beneficial effect on coronary endothelial dysfunction in diabetes.
Attenuated endothelium-dependent coronary vasodilation is an important cause for cardiac ischemia and cardiovascular complications in diabetes. This study provides compelling evidence that the decreased Ca2+ concentration in the endoplasmic reticulum and the downregulated expression of STIM proteins in coronary endothelial cells play an important pathogenic role in diabetic endothelial dysfunction. Overexpression (or restoration of) of STIM1 protein in diabetic coronary endothelial cells increases (or restores) Ca2+ concentration in the ER, enhances nitric oxide production, and restores endothelium-dependent relaxation in diabetic coronary arteries. These findings yield critical information on the new targets that can be used to develop novel therapeutic approaches for cardiovascular complications and cardiac ischemia in diabetes.
Acknowledgments
Sources of Funding
This work was supported in part by grants DK083506 and HL115578 (A.M.) and HL066012 and HL098053 (J.Y.) from the National Institutes of Health.
Non-standard Abbreviations and Acronyms
- ACh
acetylcholine
- CA
coronary artery
- EC
endothelial cell
- ER
endoplasmic reticulum
- IP3R
inositol triphosphate receptor
- MCEC
mouse coronary endothelial cell
- RyR
ryanodine receptor
- SERCA
sarcoplasmic/endoplasmic reticulum Ca2+-ATPase
- SOCE
store-operated Ca2+ entry
- STIM1
stromal interaction molecule
Footnotes
The online-only Data Supplement is available with this article at http://circres.ahajournals.org/lookup/suppl/doi:10.1161/CIRCRESAHA.112.275743/-/DC1.
Disclosures: None.
References
- 1.Chrissobolis S, Miller AA, Drummond GR, Kemp-Harper BK, Sobey CG. Oxidative stress and endothelial dysfunction in cerebrovascular disease. Front Biosci. 2011;16:1733–1745. doi: 10.2741/3816. [DOI] [PubMed] [Google Scholar]
- 2.Arora S, Vaishya R, Dabla PK, Singh B. NAD(P)H oxidases in coronary artery disease. Adv Clin Chem. 2010;50:65–86. doi: 10.1016/s0065-2423(10)50004-0. [DOI] [PubMed] [Google Scholar]
- 3.Luksha L, Agewall S, Kublickiene K. Endothelium-derived hyperpolarizing factor in vascular physiology and cardiovascular disease. Atherosclerosis. 2009;202:330–344. doi: 10.1016/j.atherosclerosis.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 4.Bian K, Doursout MF, Murad F. Vascular system: role of nitric oxide in cardiovascular diseases. J Clin Hypertens. 2008;10:304–310. doi: 10.1111/j.1751-7176.2008.06632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mizuno R, Fujimoto S, Saito Y, Nakamura S. Exercise-induced delayed onset of left ventricular early relaxation in association with coronary microcirculatory dysfunction in patients with diabetes mellitus. J Card Fail. 2010;16:211–217. doi: 10.1016/j.cardfail.2009.10.024. [DOI] [PubMed] [Google Scholar]
- 6.Nahser PJ, Jr, Brown RE, Oskarsson H, Winniford MD, Rossen JD. Maximal coronary flow reserve and metabolic coronary vasodilation in patients with diabetes mellitus. Circulation. 1995;91:635–640. doi: 10.1161/01.cir.91.3.635. [DOI] [PubMed] [Google Scholar]
- 7.Borgquist R, Nilsson PM, Gudmundsson P, Winter R, Leosdottir M, Willenheimer R. Coronary flow velocity reserve reduction is comparable in patients with erectile dysfunction and in patients with impaired fasting glucose or well-regulated diabetes mellitus. Eur J Cardiovasc Prev Rehabil. 2007;14:258–264. doi: 10.1097/HJR.0b013e328021072b. [DOI] [PubMed] [Google Scholar]
- 8.Strauer BE, Motz W, Vogt M, Schwartzkopff B. Impaired coronary flow reserve in NIDDM: a possible role for diabetic cardiopathy in humans. Diabetes. 1997;46:S119–S124. doi: 10.2337/diab.46.2.s119. [DOI] [PubMed] [Google Scholar]
- 9.Strauer BE, Motz W, Vogt M, Schwartzkopff B. Evidence for reduced coronary flow reserve in patients with insulin-dependent diabetes: a possible cause for diabetic heart disease in man. Exp Clin Endocrinol Diabetes. 1997;105:15–20. doi: 10.1055/s-0029-1211722. [DOI] [PubMed] [Google Scholar]
- 10.Samuel SM, Akita Y, Paul D, Thirunavukkarasu M, Zhan L, Sudhakaran PR, Li C, Maulik N. Coadministration of adenoviral vascular endothelial growth factor and angiopoietin-1 enhances vascularization and reduces ventricular remodeling in the infarcted myocardium of type 1 diabetic rats. Diabetes. 2010;59:51–60. doi: 10.2337/db09-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Makino A, Platoshyn O, Suarez J, Yuan JX, Dillmann WH. Downregulation of connexin40 is associated with coronary endothelial cell dysfunction in streptozotocin-induced diabetic mice. Am J Physiol Cell Physiol. 2008;295:C221–C230. doi: 10.1152/ajpcell.00433.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tran QK, Ohashi K, Watanabe H. Calcium signalling in endothelial cells. Cardiovasc Res. 2000;48:13–22. doi: 10.1016/s0008-6363(00)00172-3. [DOI] [PubMed] [Google Scholar]
- 13.Szewczyk MM, Davis KA, Samson SE, Simpson F, Rangachari PK, Grover AK. Ca2+-pumps and Na2+-Ca2+-exchangers in coronary artery endothelium versus smooth muscle. J Cell Mol Med. 2007;11:129–138. doi: 10.1111/j.1582-4934.2007.00010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006;443:230–233. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- 15.Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, Cahalan MD. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abdullaev IF, Bisaillon JM, Potier M, Gonzalez JC, Motiani RK, Trebak M. Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ Res. 2008;103:1289–1299. doi: 10.1161/01.RES.0000338496.95579.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lopez JJ, Jardin I, Bobe R, Pariente JA, Enouf J, Salido GM, Rosado JA. STIM1 regulates acidic Ca2+ store refilling by interaction with SERCA3 in human platelets. Biochem Pharmacol. 2008;75:2157–2164. doi: 10.1016/j.bcp.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 18.Jousset H, Frieden M, Demaurex N. STIM1 knockdown reveals that store-operated Ca2+ channels located close to sarco/endoplasmic Ca2+ ATPases (SERCA) pumps silently refill the endoplasmic reticulum. J Biol Chem. 2007;282:11456–11464. doi: 10.1074/jbc.M609551200. [DOI] [PubMed] [Google Scholar]
- 19.Busse R, Mulsch A. Calcium-dependent nitric oxide synthesis in endothelial cytosol is mediated by calmodulin. FEBS Lett. 1990;265:133–136. doi: 10.1016/0014-5793(90)80902-u. [DOI] [PubMed] [Google Scholar]
- 20.Forstermann U, Pollock JS, Schmidt HH, Heller M, Murad F. Calmodulin-dependent endothelium-derived relaxing factor/nitric oxide synthase activity is present in the particulate and cytosolic fractions of bovine aortic endothelial cells. Proc Natl Acad Sci U S A. 1991;88:1788–1792. doi: 10.1073/pnas.88.5.1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nishiyama M, Hashitani H, Fukuta H, Yamamoto Y, Suzuki H. Potassium channels activated in the endothelium-dependent hyperpolarization in guinea-pig coronary artery. J Physiol. 1998;510:455–465. doi: 10.1111/j.1469-7793.1998.455bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ling BN, O’Neill WC. Ca2+-dependent and Ca2+-permeable ion channels in aortic endothelial cells. Am J Physiol. 1992;263:H1827–H1838. doi: 10.1152/ajpheart.1992.263.6.H1827. [DOI] [PubMed] [Google Scholar]
- 23.Feletou M, Vanhoutte PM. EDHF: an update. Clin Sci. 2009;117:139–155. doi: 10.1042/CS20090096. [DOI] [PubMed] [Google Scholar]
- 24.Edwards DH, Chaytor AT, Bakker LM, Griffith TM. Modulation of gap-junction-dependent arterial relaxation by ascorbic acid. J Vasc Res. 2007;44:410–422. doi: 10.1159/000104254. [DOI] [PubMed] [Google Scholar]
- 25.Zheng XF, Guan YY, Kwan CY. Cyclopiazonic acid causes endothelium-dependent relaxation in rat aorta. Zhongguo Yao Li Xue Bao. 1993;14:21–26. [PubMed] [Google Scholar]
- 26.Yamashita S, Miyagawa K, Ohashi M, Sugiyama M, Sato K, Ueda R, Dohi Y. Altered effect of cyclopiazonic acid on endothelium-dependent relaxation in femoral arteries from hypertensive rats. J Cardiovasc Pharmacol. 2002;40:220–227. doi: 10.1097/00005344-200208000-00007. [DOI] [PubMed] [Google Scholar]
- 27.Kamata K, Umeda F, Kasuya Y. Possible existence of novel endothelium-derived relaxing factor in the endothelium of rat mesenteric arterial bed. J Cardiovasc Pharmacol. 1996;27:601–606. doi: 10.1097/00005344-199604000-00022. [DOI] [PubMed] [Google Scholar]
- 28.Taniguchi H, Hirano H, Tanaka Y, Tanaka H, Shigenobu K. Possible involvement of Ca2+ entry and its pharmacological characteristics responsible for endothelium-dependent, NO-mediated relaxation induced by thapsigargin in guinea-pig aorta. J Pharm Pharmacol. 1999;51:831–840. doi: 10.1211/0022357991773032. [DOI] [PubMed] [Google Scholar]
- 29.Giachini FR, Chiao CW, Carneiro FS, Lima VV, Carneiro ZN, Dorrance AM, Tostes RC, Webb RC. Increased activation of stromal interaction molecule-1/Orai-1 in aorta from hypertensive rats: a novel insight into vascular dysfunction. Hypertension. 2009;53:409–416. doi: 10.1161/HYPERTENSIONAHA.108.124404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song MY, Makino A, Yuan JX. STIM2 Contributes to enhanced store-operated Ca2+ entry in pulmonary artery smooth muscle cells from patients with idiopathic pulmonary arterial hypertension. Pulm Circ. 2011;1:84–94. doi: 10.4103/2045-8932.78106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schlaeger TM, Bartunkova S, Lawitts JA, Teichmann G, Risau W, Deutsch U, Sato TN. Uniform vascular-endothelial-cell-specific gene expression in both embryonic and adult transgenic mice. Proc Natl Acad Sci U S A. 1997;94:3058–3063. doi: 10.1073/pnas.94.7.3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Palma M, Venneri MA, Naldini L. In vivo targeting of tumor endothelial cells by systemic delivery of lentiviral vectors. Hum Gene Ther. 2003;14:1193–1206. doi: 10.1089/104303403322168028. [DOI] [PubMed] [Google Scholar]
- 33.Kimura C, Oike M, Koyama T, Ito Y. Alterations of Ca2+ mobilizing properties in migrating endothelial cells. Am J Physiol Heart Circ Physiol. 2001;281:H745–H754. doi: 10.1152/ajpheart.2001.281.2.H745. [DOI] [PubMed] [Google Scholar]
- 34.Shukla N, Freeman N, Gadsdon P, Angelini GD, Jeremy JY. Thapsigargin inhibits angiogenesis in the rat isolated aorta: studies on the role of intracellular calcium pools. Cardiovasc Res. 2001;49:681–689. doi: 10.1016/s0008-6363(00)00269-8. [DOI] [PubMed] [Google Scholar]
- 35.Redondo PC, Jardin I, Lopez JJ, Salido GM, Rosado JA. Intracellular Ca2+ store depletion induces the formation of macromolecular complexes involving hTRPC1, hTRPC6, the type II IP3 receptor and SERCA3 in human platelets. Biochim Biophys Acta. 2008;1783:1163–1176. doi: 10.1016/j.bbamcr.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 36.Redondo PC, Salido GM, Pariente JA, Sage SO, Rosado JA. SERCA2b and 3 play a regulatory role in store-operated calcium entry in human platelets. Cell Signal. 2008;20:337–346. doi: 10.1016/j.cellsig.2007.10.019. [DOI] [PubMed] [Google Scholar]
- 37.Salido GM, Sage SO, Rosado JA. TRPC channels and store-operated Ca2+ entry. Biochim Biophys Acta. 2009;1793:223–230. doi: 10.1016/j.bbamcr.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 38.Cioffi DL, Wu S, Chen H, Alexeyev M, St Croix CM, Pitt BR, Uhlig S, Stevens T. Orai1 determines calcium selectivity of an endogenous TRPC heterotetramer channel. Circ Res. 2012;110:1435–1444. doi: 10.1161/CIRCRESAHA.112.269506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li J, Cubbon RM, Wilson LA, et al. Orai1 and CRAC channel dependence of VEGF-activated Ca2+ entry and endothelial tube formation. Circ Res. 2011;108:1190–1198. doi: 10.1161/CIRCRESAHA.111.243352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Edwards JM, Neeb ZP, Alloosh MA, Long X, Bratz IN, Peller CR, Byrd JP, Kumar S, Obukhov AG, Sturek M. Exercise training decreases store-operated Ca2+ entry associated with metabolic syndrome and coronary atherosclerosis. Cardiovasc Res. 2010;85:631–640. doi: 10.1093/cvr/cvp308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Varga-Szabo D, Braun A, Kleinschnitz C, Bender M, Pleines I, Pham M, Renne T, Stoll G, Nieswandt B. The calcium sensor STIM1 is an essential mediator of arterial thrombosis and ischemic brain infarction. J Exp Med. 2008;205:1583–1591. doi: 10.1084/jem.20080302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Agrotis A, Koulis C. STIM1: a new therapeutic target in occlusive vascular disease? Cardiovasc Res. 2009;81:627–628. doi: 10.1093/cvr/cvp018. [DOI] [PubMed] [Google Scholar]
- 43.Hirano K, Hirano M, Hanada A. Involvement of STIM1 in the proteinase-activated receptor 1-mediated Ca2+ influx in vascular endothelial cells. J Cell Biochem. 2009;108:499–507. doi: 10.1002/jcb.22279. [DOI] [PubMed] [Google Scholar]
- 44.Farhangkhoee H, Khan ZA, Kaur H, Xin X, Chen S, Chakrabarti S. Vascular endothelial dysfunction in diabetic cardiomyopathy: pathogenesis and potential treatment targets. Pharmacol Ther. 2006;111:384–399. doi: 10.1016/j.pharmthera.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 45.Yang G, Lucas R, Caldwell R, Yao L, Romero MJ, Caldwell RW. Novel mechanisms of endothelial dysfunction in diabetes. J Cardiovasc Dis Res. 2010;1:59–63. doi: 10.4103/0975-3583.64432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chan NN, Vallance P, Colhoun HM. Nitric oxide and vascular responses in Type I diabetes. Diabetologia. 2000;43:137–147. doi: 10.1007/s001250050022. [DOI] [PubMed] [Google Scholar]
- 47.Tasca C, Stefaneanu L, Vasilescu C. The myocardial microangiopathy in human and experimental diabetes mellitus: a microscopic, ultrastructural, morphometric and computer-assisted symbolic-logic analysis. Endocrinologie. 1986;24:59–69. [PubMed] [Google Scholar]
- 48.Thompson EW. Quantitative analysis of myocardial structure in insulin-dependent diabetes mellitus: effects of immediate and delayed insulin replacement. Proc Soc Exp Biol Med. 1994;205:294–305. doi: 10.3181/00379727-205-43710. [DOI] [PubMed] [Google Scholar]
- 49.Warley A, Powell JM, Skepper JN. Capillary surface area is reduced and tissue thickness from capillaries to myocytes is increased in the left ventricle of streptozotocin-diabetic rats. Diabetologia. 1995;38:413–421. doi: 10.1007/BF00410278. [DOI] [PubMed] [Google Scholar]
- 50.Yoon YS, Uchida S, Masuo O, Cejna M, Park JS, Gwon HC, Kirchmair R, Bahlman F, Walter D, Curry C, Hanley A, Isner JM, Losordo DW. Progressive attenuation of myocardial vascular endothelial growth factor expression is a seminal event in diabetic cardiomyopathy: restoration of microvascular homeostasis and recovery of cardiac function in diabetic cardiomyopathy after replenishment of local vascular endothelial growth factor. Circulation. 2005;111:2073–2085. doi: 10.1161/01.CIR.0000162472.52990.36. [DOI] [PubMed] [Google Scholar]
- 51.El-Remessy AB, Tawfik HE, Matragoon S, Pillai B, Caldwell RB, Caldwell RW. Peroxynitrite mediates diabetes-induced endothelial dysfunction: possible role of Rho kinase activation. Exp Diabetes Res. 2010:247861. doi: 10.1155/2010/247861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Clements RT, Sodha NR, Feng J, Boodhwani M, Liu Y, Mieno S, Khabbaz KR, Bianchi C, Sellke FW. Impaired coronary microvascular dilation correlates with enhanced vascular smooth muscle MLC phosphorylation in diabetes. Microcirculation. 2009;16:193–206. doi: 10.1080/10739680802461950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hadi HA, Carr CS, Al Suwaidi J. Endothelial dysfunction: cardiovascular risk factors, therapy, and outcome. Vasc Health Risk Manag. 2005;1:183–198. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.