

Abstract
AIM: To investigate whether hepatitis B virus (HBV) exacerbates hepatic cholesterol accumulation, and explore the underlying mechanisms.
METHODS: HepG2 cells were infected with adenovirus (Ad) containing 1.3-fold overlength HBV genome. Real-time polymerase chain reaction and Western blotting were used to measure mRNA and protein expression of target genes. Cholesterol accumulation was measured by fluorescence microscopy. Cell toxicity due to Ad-HBV treatment was determined by the mitochondrial tetrazolium assay. The protein levels of toll-like receptors (TLRs) were determined by Western blotting.
RESULTS: Ad-HBV increased hepatic cholesterol accumulation and enhanced the mRNA and protein levels of low-density lipoprotein receptor (LDLR) and 3-hydroxy-3-methylglutharyl-coenzyme A reductase (HMGCoAr) mRNA and protein expression in HepG2 cells. In addition, these inductive effects were partly offset by suppressing TLR2 expression levels by small interfering RNA in HepG2 cells.
CONCLUSION: Ad-HBV increases LDLR and HMGCoAr expression, resulting in exacerbated cholesterol accumulation in HepG2 cells, which was mediated via the TLR2 pathway.
Keywords: Hepatitis B virus, Toll-like receptors, Low-density lipoprotein receptor, 3-hydroxy-3-methylglutharyl-coenzyme A reductase
Core tip: This study investigated whether hepatitis B virus (HBV) exacerbates hepatic cholesterol accumulation and explored the underlying mechanisms. The authors found that adenovirus HBV increased low-density lipoprotein receptor and 3-hydroxy-3-methylglutharyl-coenzyme A reductase expression, resulting in exacerbated cholesterol accumulation in HepG2 cells, which was mediated via the toll-like receptor 2 pathway. These results may also have implications in the treatment of atherosclerosis.
INTRODUCTION
Hepatitis B virus (HBV) infection is a major public health problem worldwide[1]. A possible role for infections in atherosclerosis has been deeply scrutinized since the demonstration that herpes virus induced atherosclerosis in chickens in 1978[2]. It has been shown that the incidence of hepatic steatosis in HBeAg-negative chronic hepatitis B patients is about 32%[3]. A published study from a health-screening test cohort showed that there was a strong association between hepatitis virus carriers and carotid atherosclerosis[4,5].
It is still controversial as to whether HBV-induced inflammation correlates with disease in organs other than the liver. To date, there are few data available to prove the association between HBV infection and atherosclerosis. Kiechl et al[6] found no significant association between chronic hepatitis and the development of new carotid atheromatous plaques, although they did not specify the type of hepatitis virus. However, another study in Japan demonstrated an increased prevalence of carotid atherosclerosis in HBV carriers[7].
Research has revealed that HBV-induced inflammation correlates with disease in organs other than the liver[8]. A previous report indicated that inflammation plays an important role in atherosclerosis[9]. In addition, this adverse impact of virus infection is partly, if not all, mediated by toll-like receptors (TLRs)[10-13]. For instance, Zhang et al[14] found that TLR2/4 signaling involved in the adaptive immune response plays a role in chronic HBV infection.
However, the role of HBV infection in hepatic cholesterol accumulation is still unclear. Therefore, the aims of the present investigation were to test: (1) whether HBV affects the expression of genes related to cholesterol metabolism such as low-density lipoprotein receptor (LDLR) and 3-hydroxy-3-methylglutharyl-coenzyme A reductase (HMGCoAr) in hepatocytes; and (2) whether TLRs are involved in lipid metabolism disorders caused by HBV.
MATERIALS AND METHODS
Cell culture
HepG2 and AD293 cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 1% (v/v) penicillin-streptomycin at 37 °C in a humid atmosphere of 5% CO2. HepG2 cells were switched to serum-free medium 24 h before treatment.
Amplification and quantification of adenoviral vector-hepatitis B virus
In this study, adenoviral vectors were designed, which initiated HBV replication from a 1.3-fold overlength HBV genome. A control adenoviral vector (Ad) was also included. AD293 cells were infected with 1 × 105 to 1 × 107 copies of Ad-HBV for 72 h. Cells were harvested and counted under the microscope. An equal number of cells were maintained in phosphate buffered saline (PBS) and underwent three freeze-thaw cycles. Lysates were cleared by centrifugation at 14 000 × g, divided into equal volumes and used for real-time polymerase chain reaction (PCR) and infecting HepG2 cells. The HBV DNA was quantified using the BIO-RAD iCycler real-time PCR system and the qPCR Master Mix (Da An Gene Co., Ltd, Guangzhou, China). The copy of viral genome equivalents was determined using a calibration curve containing known amounts of HBV DNA. Ad was amplified in AD293 cells. To quantify Ad, the infected efficiency of Ad in AD293 cells was measured and normalized to the infected efficiency of Ad-HBV.
Cytotoxicity assay
Cell toxicity due to Ad-HBV treatment was determined by the mitochondrial tetrazolium assay (MTT). HepG2 cells were grown in media with Ad-HBV at various concentrations for 4 d before addition of the MTT agent. Optical density was read at 570 nm using the BiotekElx-800 plate reader. Cells treated with vehicle served as controls, and the cell viability of the Ad-HBV treated group was normalized to that of the control group.
Quantitative RT-PCR
Total RNA was isolated from cultured HepG2 cells treated with different concentration of Ad-HBV (1 × 105, 1 × 106, 1 × 107 copies) for 24 h as well as HepG2 cells incubated for 0-24 h with 1 × 106 copies Ad-HBV using the guanidinium-phenol-choloroform method[15]. Total RNA (500 ng) was used as a template for RT-PCR. The RT reaction was set up using a kit from TaKaRa (PrimeScriptTM RT reagent Kit, Dalian, China). Following synthesis, cDNA was split for the separate amplification of target genes using specific primers as shown in Table 1. All primers were designed using the following website (www.idtdna.com/Primerquest/). Real-time PCR was performed in a BIROD using SYBR Premix Ex TaqTM (TaKaRa, Dalian, China) according to the manufacturer’s protocol. After PCR, a dissociation curve (melting curve) was constructed at the temperature ranging from 60 °C to 95 °C. Ct values were averaged and normalized to GAPDH. Relative expression was determined by the ∆∆Ct comparative threshold method.
Table 1.
Primers for real-time polymerase chain reaction
| Gene | Primers for real-time polymerase chain reaction |
| LDLR | Sense: 5’-TCAACACACAACAGCAGATGGCAC-3’ |
| Antisense: 5’-AAGGCTAACCTGGCTGTCTAGCAA-3’ | |
| HMGCoAr | Sense: 5’-TATGTGCTGCTTTGGCTGCATGTC-3’ |
| Antisense: 5’-ATACCAAGGACACACAAGCTGGGA-3’ | |
| TLR2 | Sense: 5’-ACCTGTCCAACAACAGGATCACCT-3’ |
| Antisense: 5’-TGTTCAAGACTGCCCAGGGAAGAA-3’ | |
| TLR4 | Sense: 5’-GCCGAAAGGTGATTGTTGTGGTGT-3’ |
| Antisense: 5’-ACTGCCAGGTCTGAGCAATCTCAT-3’ | |
| GAPDH | Sense: 5’-AGGAGTAAGAAACCCTGGAC-3’ |
| Antisense: 5’-CTGGGATGGAATTGTGAG-3’ |
LDLR: Low-density lipoprotein receptor; TLR2: Toll-like receptor 2; TLR4: Toll-like receptor 4. HMGCoAr: 3-hydroxy-3-methylglutharyl-coenzyme A reductase; GAPDH: Glyceraldehyde-3-phosphate dehydrogenase.
TLRs-siRNA
Sequences of TLR2/TLR4 siRNA were purchased from Santa Cruz (United States. The transfection of siRNA was performed using a Lipofectamine kit (Invitrogen, United States) according to the manufacturer’s instructions. The medium was changed 6 h after transfection, and the cells were incubated with Ad-HBV or Ad for a further 24 h. The cells were then harvested and the protein levels of TLRs were determined by Western blotting. Cholesterol uptake measurements were then carried out.
Western blotting
Cells were washed with PBS, scraped into lysis buffer (Tris-EDTA + Complete protease inhibitor; Roche, United States) and mechanically homogenized. Total protein samples (40 μg per well) were electrophoresed on 8% SDS-polyacrylamide gel and transferred to polyvinylidene difluoride (PVDF) membranes at 100V for 100 min. Membranes were incubated overnight with anti-LDLR (1:2000, Millipore), anti-HMGCoAr (1:1000, Millipore), anti-TLR2 (1:1000, Millipore), anti-TLR4 (1:1000, Santa Cruz), or anti-GAPDH (1:10 000, Sigma). Anti-mouse secondary antibody conjugated with horseradish peroxidase (Promega, United States) and SuperSignal West Pico Chemiluminescent Substrate (Pierce, United States) were used for detection.
Modified LDL uptake measurements
HepG2 cells were incubated with human 1’-dioctadecyl-1-3,3,3’,3’-tetramethyling-docarbocyanine perchlorate (Dil)-labeled acetylated low density lipoprotein (Dil-acl-LDL) (10 μg/mL) for 4 h, DAPI staining was used to detect the nucleus. The two color images were visualized under a fluorescence microscope. ZEN 2008 and Image software were used to analyze the quantity of Dil-acl-LDL.
Statistical analysis
Results were shown as mean ± SE, and all experiments were run in triplicate. The statistical significance of differences between groups was determined using the Student t test. aP < 0.05, bP < 0.01, vs the control group.
RESULTS
Cell toxicity of Ad-HBV
The toxicity of Ad-HBV against HepG2 cells used for propagation of viral infections was measured. Both Ad and Ad-HBV below 107 copies/mL did not exhibit either toxic or proliferative effects on HepG2 cells, while 1 × 108 copies/mL of the virus reduced HepG2 cell survival rates (Figure 1).
Figure 1.
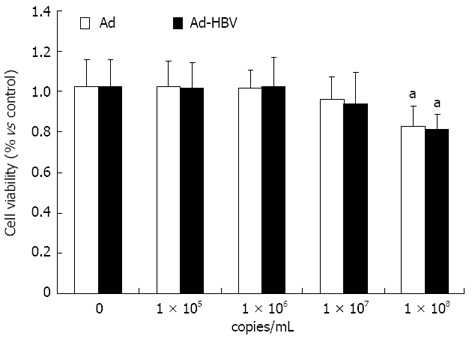
Cell toxicity by adenovirus hepatitis B virus. The data are presented as means ± SE. aP < 0.05 vs control group, n = 6. Ad: Adenovirus; HBV: Hepatitis B virus.
Ad-HBV changes the mRNA levels of genes related to cholesterol metabolism in HepG2 cells
Whether HBV leads to cholesterol metabolism disorders in the liver is still controversial. Therefore, we determined the effects of Ad-HBV on the mRNA levels of genes related to cholesterol metabolism in HepG2 cells. As shown in Figure 2A, Ad-HBV at 1 × 105 and 1 × 106 copies/mL significantly augmented the mRNA levels of LDLR and HMGCoAr. When the concentration of Ad-HBV reached 107 copies/mL, the mRNA expression was suppressed. Ad-HBV at 1 × 106 copies/mL significantly induced mRNA expression of LDLR (2.56 ± 0.33 vs 1.03 ± 0.25, P < 0.01, n = 3) and HMGCoAr (2.98 ± 0.25 vs 1.01 ± 0.18, P < 0.01, n = 3). Ad-HBV upregulated the mRNA levels of LDLR and HMGCoAr in a time-dependent manner. HepG2 cells were maintained with 1 × 106 copies/mL of Ad-HBV for different time periods and values were expressed as fold changes relative to the controls. After 24 h of incubation the mRNA expression of LDLR (2.56 ± 0.33 vs 1.03 ± 0.34, P < 0.01, n = 3) and HMGCoAr (2.99 ± 0.25 vs 1.01 ± 0.18, P < 0.01, n = 3) reached a peak (Figure 2B).
Figure 2.
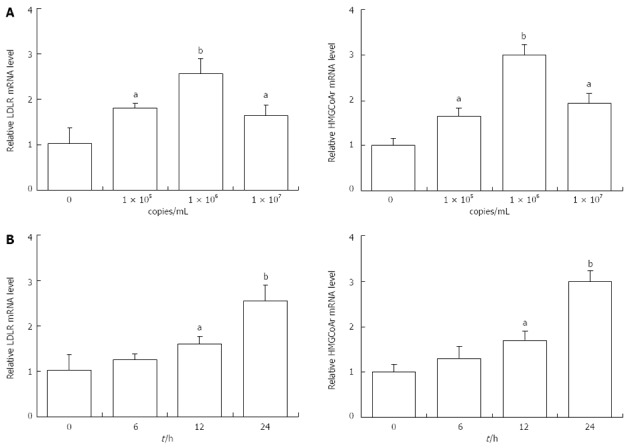
Effects of adenovirus hepatitis B virus treatment on the mRNA levels of genes related to cholesterol metabolism in HepG2 cells. A: HepG2 cells were treated with different concentration of adenovirus-hepatitis B virus (Ad-HBV) (1 × 105, 1 × 106, 1 × 107 copies) for 24 h; B: HepG2 cells were incubated for 0-24 h with 1 × 106 copies Ad-HBV. aP < 0.05, bP < 0.01 vs control group. LDLR: Low-density lipoprotein receptor.
Ad-HBV changes the mRNA levels of TLRs in HepG2 cells
Researchers recently showed that TLRs were involved in viral infection and its downstream effects. To investigate whether Ad-HBV upregulated the mRNA levels of genes related to cholesterol metabolism in HepG2 cells, TLRs mRNA expression was determined. Ad-HBV at 1 × 105 copies/mL and 1 × 106 copies/mL significantly induced the mRNA levels of TLR2. Values were expressed as fold changes relative to the controls. When the concentration of Ad-HBV was increased to 1 × 107 copies/mL, mRNA expression was suppressed (Figure 3A). These changes were consistent with the changes in cholesterol metabolism-related genes. The mRNA levels of TLR4 were increased in HepG2 cells treated with Ad-HBV in a dose-dependent manner (Figure 3B).
Figure 3.
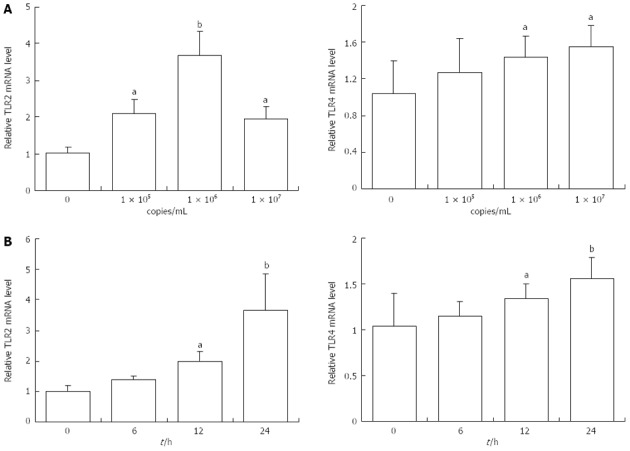
Effects of adenovirus hepatitis B virus treatment on toll-like receptors mRNA levels in HepG2 cells. A: Adenovirus-hepatitis B virus (Ad-HBV) at 105 and 106 copies profoundly augmented the mRNA levels of toll-like receptor 2 (TLR2). While the concentration of Ad-HBV up to 107 copies, the mRNA expression were suppressed instead. B: The mRNA of TLR4 were increased in HepG2 cells treated with Ad-HBV, which was in a dose-dependent manner. Results were representative of three similar experiments. aP < 0.05, bP < 0.01 vs control group.
TLR2 mediated the effects of Ad-HBV on the expression of proteins related to cholesterol metabolism and intake of cholesterol in HepG2 cells
To clarify the mechanism of Ad-HBV-upregulated mRNA levels of proteins related to cholesterol metabolism in HepG2 cells, we used siRNA-mediated downregulation of TLR2/TLR4 to confirm our hypothesis that TLR2/TLR4 may participate in this process. To evaluate the involvement of TLR2/TLR4 in the effects of Ad-HBV, we attenuated the expression of TLR2 using human TLR2-SiRNA, which suppressed TLR2 protein level by up to 80% (Figure 4). Transient transfection of HepG2 cells with TLR2-siRNA substantially abolished Ad-HBV-mediated upregulation of LDLR and HMGCoAr and the uptake of Dil-acl-LDL (Figure 4). TLR4-SiRNA did not change the expression of proteins related to cholesterol metabolism during Ad-HBV infection (data not shown).
Figure 4.
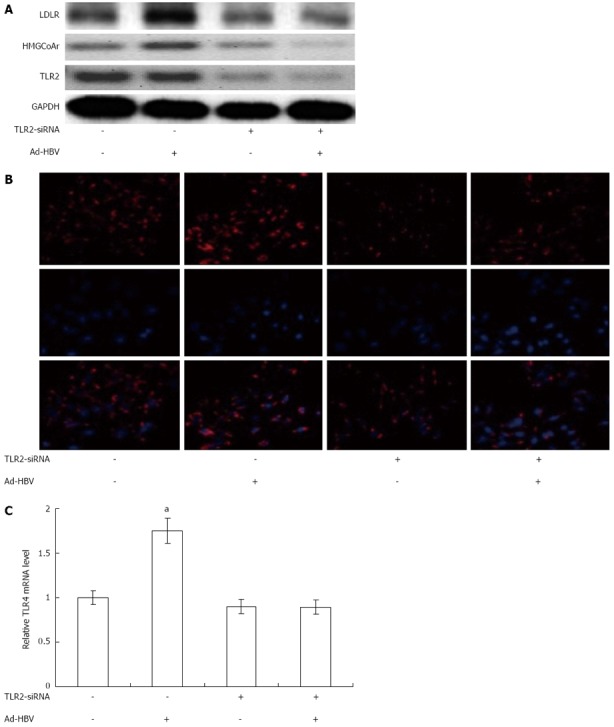
Effects of toll-like receptor 2-SiRNA on the expression of proteins related to cholesterol metabolism and intake of cholesterol by HepG2 cells treated with adenovirus hepatitis B virus. A: The protein expression of low-density lipoprotein receptor (LDLR) and 3-hydroxy-3-methylglutharyl-coenzyme A reductase (HMGCoAr) were assayed; B: HepG2 Cells were exposed for 4 h to Dil-acl-LDL (10 μg/mL), after thorough washing with phosphate buffered solution, fluorescence of Dil-acl-LDL was detected in cytoplasm of cells by fluorescence microscopy; C: Relative quantity of Dil-acl-LDL in HepG2 cells were analyzed by software. Results were representative of three similar experiments. aP < 0.05, bP < 0.01 vs control group. TLR2: Toll-like receptor 2; LDLR: Low-density lipoprotein receptor; LDL: Low density lipoprotein; Ad-HBV: Adenovirus hepatitis B virus.
DISCUSSION
There is increasing evidence to indicate that hepatic lipid accumulation is related to hepatic fibrosis and inflammation, resulting in cell apoptosis and cancer[16,17]. In particular, it is assumed that lipid accumulation is a prerequisite for subsequent events leading to liver injury in nonalcoholic fatty liver disease[18]. In addition, it was recently shown that hepatic steatosis may be a factor in HCV-induced liver pathogenesis and may impair the response to interferon-based therapy[19,20]. Due to the importance of lipid accumulation, the mechanism by which nonalcoholic fatty liver disease and HCV infection cause hepatic steatosis has been studied intensively[4]. However, the molecular mechanisms by which HBV infection causes hepatic steatosis have been poorly investigated.
According to current knowledge, liver LDLR is the most important receptor of binding and internalization of plasma-derived LDL-cholesterol and regulates plasma LDL concentration. Changes in receptor activity alter the rates of LDL uptake by the liver with a corresponding increase or decrease in plasma LDL levels[21,22]. Our results showed that the mRNA and protein levels of LDLR were increased following infection of HepG2 cells with Ad-HBV.
3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCoAr) is an enzyme which catalyzes the conversion of HMGCoA to mevalonate, the rate limiting step in cholesterol biosynthesis. Normally, in mammalian cells this enzyme is suppressed by cholesterol and its derivatives from the LDLR mediated-internalization of LDL as well as oxidized species of cholesterol. Competitive inhibition of the reductase upregulates the expression of LDLR in the liver, which in turn increases the catabolism of plasma LDL and lowers the plasma concentration of cholesterol, an important determinant of atherosclerosis[23-25].
HBV, like many other microorganisms that contribute to the pathogenesis of atherosclerosis, can colonize the vascular tissues[26], induce vasculitis[27], and stimulate inflammatory and immune responses that may lead to vascular damage and precipitate atherosclerosis. It was reported in one Japanese study that HBV infection can be atherogenic in otherwise healthy subjects with preserved liver function[7]. We reasoned that HBV would be a rational candidate pathogen among the stimuli that contribute to atherosclerosis. The mechanism behind this association is still unclear, and additional studies are required.
TLRs have been reported to play an important role in liver damage after infection with HBV, and the mechanisms may involve virus-induced immune modulation[14,28,29]. TLRs are also involved in other immune diseases mediated by HBV. TLR4 had been reported to not only inhibit HBV replication, but also to induce immune injury in cells[30]. Consistent with previous studies, we found that Ad-HBV increased the expression of TLR2/4. Most importantly, we observed that the upregulation of LDLR and HMGCoAr via Ad-HBV can be partially blocked by silencing TLR2, but not TLR4. Taken together, these results suggest that Ad-HBV infection-induced cholesterol accumulation in hepatocytes is mediated by TLR2.
In conclusion, our data indicate that HBV is able to induce the gene expression of TLR2, thereby causing hepatic lipid accumulation by increasing genes related to cholesterol absorption and metabolism. Because increased lipid deposition is involved in the progression of severe liver injury such as hepatitis and hepatocellular carcinoma, our results provide important information in understanding the development and progression of HBV-induced pathogenesis.
COMMENTS
Background
Cholesterol accumulation plays an important role in the progression of atherosclerosis. This study was undertaken to investigate whether hepatitis B virus (HBV) exacerbates hepatic cholesterol accumulation and the underlying mechanisms were examined.
Research frontiers
Previous studies have mainly focused on the potential effect of HBV in the progression of atherosclerosis. The authors hypothesized that HBV increases low-density lipoprotein receptor and 3-hydroxy-3-methylglutharyl-coenzyme A reductase expression resulting in exacerbated cholesterol accumulation in HepG2 cells, which was mediated via the toll-like receptor 2 (TLR2) pathway.
Innovations and breakthroughs
To further clarify the potential effect of HBV in the progression of atherosclerosis, the authors examined the effects of adenovirus hepatitis B virus (Ad-HBV) in the progression of atherosclerosis which is partly mediated via the TLR2 pathway.
Applications
The results show that Ad-HBV up-regulates the expression of genes related to cholesterol metabolism via the TLR2 pathway. Further studies are required to evaluate the mechanism by which HBV regulates the TLR2 pathway.
Terminology
There are some associations between hepatitis virus and carotid atherosclerosis. Hepatitis virus causes liver and even systemic inflammatory reactions, and inflammation is one the pathophysiological changes in atherosclerosis.
Peer review
This manuscript presented that Ad-HBV up-regulated the expression of genes related to cholesterol metabolism in HepG2 cells. Furthermore, the atherosclerosis effect of Ad-HBV is via TLR2 pathway. The experiment seems to be correct. The study appears well conducted and the results discussed with honesty, and caution.
Footnotes
Supported by The Youth Foundation of Hunan Provincial Education Department, No. 11B110; the Graduate Innovation Project of Hunan Provincial Education Department, No. CX2010B382
P- Reviewer Driscoll D S- Editor Wen LL L- Editor A E- Editor Zhang DN
References
- 1.Nelson PK, Mathers BM, Cowie B, Hagan H, Des Jarlais D, Horyniak D, Degenhardt L. Global epidemiology of hepatitis B and hepatitis C in people who inject drugs: results of systematic reviews. Lancet. 2011;378:571–583. doi: 10.1016/S0140-6736(11)61097-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fabricant CG, Fabricant J, Litrenta MM, Minick CR. Virus-induced atherosclerosis. J Exp Med. 1978;148:335–340. doi: 10.1084/jem.148.1.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bondini S, Kallman J, Wheeler A, Prakash S, Gramlich T, Jondle DM, Younossi ZM. Impact of non-alcoholic fatty liver disease on chronic hepatitis B. Liver Int. 2007;27:607–611. doi: 10.1111/j.1478-3231.2007.01482.x. [DOI] [PubMed] [Google Scholar]
- 4.Targher G, Bertolini L, Padovani R, Rodella S, Arcaro G, Day C. Differences and similarities in early atherosclerosis between patients with non-alcoholic steatohepatitis and chronic hepatitis B and C. J Hepatol. 2007;46:1126–1132. doi: 10.1016/j.jhep.2007.01.021. [DOI] [PubMed] [Google Scholar]
- 5.Kim K, Kim KH, Kim HH, Cheong J. Hepatitis B virus X protein induces lipogenic transcription factor SREBP1 and fatty acid synthase through the activation of nuclear receptor LXRalpha. Biochem J. 2008;416:219–230. doi: 10.1042/BJ20081336. [DOI] [PubMed] [Google Scholar]
- 6.Kiechl S, Egger G, Mayr M, Wiedermann CJ, Bonora E, Oberhollenzer F, Muggeo M, Xu Q, Wick G, Poewe W, et al. Chronic infections and the risk of carotid atherosclerosis: prospective results from a large population study. Circulation. 2001;103:1064–1070. doi: 10.1161/01.cir.103.8.1064. [DOI] [PubMed] [Google Scholar]
- 7.Ishizaka N, Ishizaka Y, Takahashi E, Toda Ei E, Hashimoto H, Ohno M, Nagai R, Yamakado M. Increased prevalence of carotid atherosclerosis in hepatitis B virus carriers. Circulation. 2002;105:1028–1030. doi: 10.1161/hc0902.105718. [DOI] [PubMed] [Google Scholar]
- 8.Wang JY, Liu P. Abnormal immunity and gene mutation in patients with severe hepatitis-B. World J Gastroenterol. 2003;9:2009–2011. doi: 10.3748/wjg.v9.i9.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bzowska M, Nogieć A, Skrzeczyńska-Moncznik J, Mickowska B, Guzik K, Pryjma J. Oxidized LDLs inhibit TLR-induced IL-10 production by monocytes: a new aspect of pathogen-accelerated atherosclerosis. Inflammation. 2012;35:1567–1584. doi: 10.1007/s10753-012-9472-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 11.Wei XQ, Guo YW, Liu JJ, Wen ZF, Yang SJ, Yao JL. The significance of Toll-like receptor 4 (TLR4) expression in patients with chronic hepatitis B. Clin Invest Med. 2008;31:E123–E130. doi: 10.25011/cim.v31i3.3469. [DOI] [PubMed] [Google Scholar]
- 12.Chen Z, Cheng Y, Xu Y, Liao J, Zhang X, Hu Y, Zhang Q, Wang J, Zhang Z, Shen F, et al. Expression profiles and function of Toll-like receptors 2 and 4 in peripheral blood mononuclear cells of chronic hepatitis B patients. Clin Immunol. 2008;128:400–408. doi: 10.1016/j.clim.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 13.Broering R, Lu M, Schlaak JF. Role of Toll-like receptors in liver health and disease. Clin Sci (Lond) 2011;121:415–426. doi: 10.1042/CS20110065. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Y, Lian JQ, Huang CX, Wang JP, Wei X, Nan XP, Yu HT, Jiang LL, Wang XQ, Zhuang Y, et al. Overexpression of Toll-like receptor 2/4 on monocytes modulates the activities of CD4(+)CD25(+) regulatory T cells in chronic hepatitis B virus infection. Virology. 2010;397:34–42. doi: 10.1016/j.virol.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 15.Wang YH, Chen YF, Chen SR, Chen X, Chen JW, Shen XY, Mou YG, Liu PQ. Aspirin increases apolipoprotein-A-I-mediated cholesterol efflux via enhancing expression of ATP-binding cassette transporter A1. Pharmacology. 2010;86:320–326. doi: 10.1159/000321727. [DOI] [PubMed] [Google Scholar]
- 16.Powell EE, Jonsson JR, Clouston AD. Steatosis: co-factor in other liver diseases. Hepatology. 2005;42:5–13. doi: 10.1002/hep.20750. [DOI] [PubMed] [Google Scholar]
- 17.Ha HL, Shin HJ, Feitelson MA, Yu DY. Oxidative stress and antioxidants in hepatic pathogenesis. World J Gastroenterol. 2010;16:6035–6043. doi: 10.3748/wjg.v16.i48.6035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004;114:147–152. doi: 10.1172/JCI22422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guedj H, Guedj J, Negro F, Lagging M, Westin J, Bochud PY, Bibert S, Neumann AU. The impact of fibrosis and steatosis on early viral kinetics in HCV genotype 1-infected patients treated with Peg-IFN-alfa-2a and ribavirin. J Viral Hepat. 2012;19:488–496. doi: 10.1111/j.1365-2893.2011.01569.x. [DOI] [PubMed] [Google Scholar]
- 20.Roingeard P. Hepatitis C virus diversity and hepatic steatosis. J Viral Hepat. 2013;20:77–84. doi: 10.1111/jvh.12035. [DOI] [PubMed] [Google Scholar]
- 21.Go GW, Mani A. Low-density lipoprotein receptor (LDLR) family orchestrates cholesterol homeostasis. Yale J Biol Med. 2012;85:19–28. [PMC free article] [PubMed] [Google Scholar]
- 22.Hofmann SL, Russell DW, Brown MS, Goldstein JL, Hammer RE. Overexpression of low density lipoprotein (LDL) receptor eliminates LDL from plasma in transgenic mice. Science. 1988;239:1277–1281. doi: 10.1126/science.3344433. [DOI] [PubMed] [Google Scholar]
- 23.Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 24.Geelen MJ, Gibson DM, Rodwell VW. Hydroxymethylglutaryl-CoA reductase--the rate-limiting enzyme of cholesterol biosynthesis. A report of a meeting held at Nijenrode Castle, Breukelen, The Netherlands, August 24, 1985. FEBS Lett. 1986;201:183–186. doi: 10.1016/0014-5793(86)80604-4. [DOI] [PubMed] [Google Scholar]
- 25.Medina MW, Gao F, Ruan W, Rotter JI, Krauss RM. Alternative splicing of 3-hydroxy-3-methylglutaryl coenzyme A reductase is associated with plasma low-density lipoprotein cholesterol response to simvastatin. Circulation. 2008;118:355–362. doi: 10.1161/CIRCULATIONAHA.108.773267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mason A, Wick M, White H, Perrillo R. Hepatitis B virus replication in diverse cell types during chronic hepatitis B virus infection. Hepatology. 1993;18:781–789. doi: 10.1002/hep.1840180406. [DOI] [PubMed] [Google Scholar]
- 27.Guillevin L. Virus-associated vasculitides. Rheumatology (Oxford) 1999;38:588–590. doi: 10.1093/rheumatology/38.7.588. [DOI] [PubMed] [Google Scholar]
- 28.Wu J, Lu M, Meng Z, Trippler M, Broering R, Szczeponek A, Krux F, Dittmer U, Roggendorf M, Gerken G, et al. Toll-like receptor-mediated control of HBV replication by nonparenchymal liver cells in mice. Hepatology. 2007;46:1769–1778. doi: 10.1002/hep.21897. [DOI] [PubMed] [Google Scholar]
- 29.Lian JQ, Wang XQ, Zhang Y, Huang CX, Bai XF. Correlation of circulating TLR2/4 expression with CD3+/4+/8+ T cells and Treg cells in HBV-related liver cirrhosis. Viral Immunol. 2009;22:301–308. doi: 10.1089/vim.2009.0039. [DOI] [PubMed] [Google Scholar]
- 30.Zhou Y, Zhu N, Wang X, Wang L, Gu LJ, Yuan WJ. The role of the toll-like receptor TLR4 in hepatitis B virus-associated glomerulonephritis. Arch Virol. 2013;158:425–433. doi: 10.1007/s00705-012-1508-3. [DOI] [PubMed] [Google Scholar]