

Abstract
PTH increases urinary Pi excretion by reducing expression of two renal cotransporters [NaPi-IIa (Npt2a) and NaPi-IIc (Npt2c)]. In contrast to acute transporter regulation that is cAMP/protein kinase A dependent, long-term effects require phospholipase C (PLC) signaling by the PTH/PTHrP receptor (PPR). To determine whether the latter pathway regulates Pi through Npt2a and/or Npt2c, wild-type mice (Wt) and animals expressing a mutant PPR incapable of PLC activation (DD) were tested in the absence of one (Npt2a−/− or Npt2c−/−) or both phosphate transporters (2a/2c-dko). PTH infusion for 8 days caused a rapid and persistent decrease in serum Pi in Wt mice, whereas serum Pi in DD mice fell only transiently for the first 2 days. Consistent with these findings, fractional Pi excretion index was increased initially in both animals, but this increase persisted only when the PPR Wt was present. The hypophosphatemic response to PTH infusion was impaired only slightly in PPR Wt/Npt2c−/− or DD/Npt2c−/− mice. Despite lower baselines, PTH infusion in PPR Wt/Npt2a−/− mice decreased serum Pi further, an effect that was attenuated in DD/Npt2a−/− mice. Continuous PTH had no effect on serum Pi in 2a/2c-dko mice. PTH administration increased serum 1,25 dihydroxyvitamin D3 levels in Wt and DD mice and increased levels above the elevated baseline with ablation of either but not of both transporters. Continuous PTH elevated serum fibroblast growth factor 23 and blood Ca2+ equivalently in all groups of mice. Our data indicate that PLC signaling at the PPR contributes to the long-term effect of PTH on Pi homeostasis but not to the regulation of 1,25 dihydroxyvitamin D3, fibroblast growth factor 23, or blood Ca2+.
PTH plays a critical role in phosphate and calcium homeostasis through the activation of the PTH/PTHrP receptor (PPR), which couples to several signaling pathways, most notably the Gsα-linked adenylyl cyclase (AC)-protein kinase A (PKA) cascade and the Gq/11-linked phospholipase C (PLC)-protein kinase C (PKC) pathway (1). In the renal proximal convoluted tubules, which account for 70–80% of the reabsorption of filtered phosphate, PTH decreases the brush border membrane abundance of NaPi-IIa (Npt2a) and NaPi-IIc (Npt2c) (2–6), the two major sodium-phosphate cotransporters that facilitate reabsorption of this mineral in the proximal renal tubules. Npt2a is the dominant transporter for reclaiming phosphate from the glomerular filtrate, and inactivation of the gene encoding this transporter causes an approximately 70% decrease in phosphate transport (7). Npt2c is developmentally regulated in rodents and was thought to be of limited importance in adult animals. In fact, ablation of Npt2c in mice does not appear to attenuate phosphate homeostasis (5). However, 1,25 dihydroxyvitamin D3 [1,25(OH)2D3] levels are increased in Npt2c-null animals (8), and double mutants lacking both transporters show more profound hypophosphatemia than Npt2a-null mice, indicating that Npt2c does contribute significantly to phosphate regulation in this species (5).
Consistent with these findings, humans with homozygous or compound heterozygous Npt2c mutations develop hereditary hypophosphatemic rickets with hypercalciuria (9, 10), a disorder characterized by an increase in urinary phosphate excretion that is PTH and fibroblast growth factor 23 (FGF23) independent and associated with elevated 1,25(OH)2D3 levels and hypercalciuria. In addition to promoting renal phosphate excretion, PTH is essential for maintaining blood ionized calcium within narrow limits. It furthermore stimulates 1,25(OH)2D3 synthesis in the proximal renal tubules and enhances calcium reabsorption in the distal convoluted tubule and the collecting duct by increasing expression and functional properties of the epithelial transient receptor potential cation channel, subfamily V, member 5 (TRPV5) (11–13).
In the proximal tubules, PPR is expressed at both the apical and basolateral membranes of the cell (14–16). Activation of apical or basolateral PPRs induces a strong and rapid down-regulation of Npt2a due to retrieval of the protein from the brush border membrane and its subsequent lysosomal degradation (3). Ex vivo and in vitro studies using PTH(3–34) suggested that the apical PPR predominantly couples to the phosphatidylcholine-dependent phospholipase C (PC-PLC) and diacylglycerol/PKC pathway, whereas the basolateral PPR activates the AC/PKA pathway (3, 17, 18). However, PTH(3–34) does not activate phosphatidylinositol-specific phospholipase C (PI-PLC) to stimulate the generation of inositol trisphosphate (IP3) and diacylglycerol/PKC through activation of Gq/11 in vitro (19); consequently, this peptide should be referred to as a PC-PLC but not PI-PLC agonist.
Although some ex vivo evidence suggests that defective coupling of apical PPR to PC-PLC/PKC prevents internalization of Npt2a in Na+/H+ exchanger regulatory factor1-deficient mice (17), the role of this signaling pathway or the more studied PI-PLC/PKC pathway through the PPR in Npt2a regulation and phosphate homeostasis in vivo has not been fully elucidated. In contrast, an important in vivo role of the PTH-activated AC/PKA signaling pathway in the acute down-regulation of Npt2a appears to be established (20); this is consistent with the findings in patients affected by pseudohypoparathyroidism type Ia, who lack Gsα in the proximal renal tubules and thus fail to increase urinary phosphate excretion after PTH administration (21–23).
Although the PTH-dependent regulation of Npt2a and Npt2c is well established, the in vivo contribution of the distinct signaling pathways used by the PPR to regulate Pi homeostasis have been studied less extensively (4, 5, 24–28). The aim of this study was to determine whether PI-PLC signaling via the PPR is essential for Pi homeostasis through the regulation of Npt2a and/or Npt2c expression. To investigate these questions, we used genetically manipulated knock-in mice (here named DD) expressing only a mutant PPR, in which the wild-type residues EKKY in the second intracellular loop were replaced with DSEL; this modified receptor activates AC normally but fails to activate PI-PLC (29, 30). We have previously shown that serum Pi is dramatically reduced in wild-type but not in DD mice undergoing continuous infusion of PTH(1–34) for 2 weeks or receiving a calcium-deficient diet for 3 weeks (31). This surprising observation suggested that the PI-PLC signaling pathway downstream of the activated PPR is critical for maintaining a sustained phosphaturic effect in response to elevated PTH. DD mice were crossed with the mice deficient for one or both of the type II sodium-dependent phosphate cotransporters expressed in the kidney, namely Npt2a and/or Npt2c (5, 27). We examined the dynamic effect of continuous PTH infusion on serum Pi and blood Ca2+ in these various mutant mice and demonstrated that the PPR stimulated PI-PLC signaling pathway contributes to the long-term PTH-dependent regulation of Pi homeostasis but not to the regulation of blood calcium levels.
Materials and Methods
Animals
Wild-type (Wt), DD (both PPR alleles carry the DSEL mutation), Npt2a−/−, and Npt2c−/−mice used in the present experiments had all been crossed into the C57/B6 background for more than 10 generations; only males were used. Genotyping by PCR amplification of genomic DNA was performed as described previously (5, 30). The following genotypes were generated through appropriate crossings: Npt2a−/−, Npt2c−/−, Npt2a−/−/Npt2c−/− (2a/2c-dko) and DD/Npt2a−/−, DD/Npt2c−/−, and DD/2a/2c-dko; Wt and DD mice served as controls. All animals were maintained in facilities operated by the Center for Comparative Research of the Massachusetts General Hospital, and all experimental procedures were approved by the institution's Subcommittee on Research Animal Care.
PTH infusion
Human PTH(1–34) (referred to as PTH) was infused into mice beginning at 7 weeks of age. The peptide was reconstituted in a solution containing 150 mM NaCl, 1 mM HCl, and 2% heat-inactivated mouse serum and loaded into Alzet osmotic minipumps (model 1002; DURECT Corp, Cupertino, California), which had been equilibrated in 0.9% NaCl at 37°C for 2 hours and then implanted into an interscapular sc pocket under Avertin anesthesia. Mice were infused with vehicle (control) or PTH at a dose of 40 μg/kg·d for 8 days, with 6–8 mice in each group; this dose of PTH was chosen after documenting in preliminary studies that higher doses led to more severe hypercalcemia and a high frequency of premature death.
Serum and urine biochemistry
Tail vein blood was collected immediately prior to and at different times after implantation of minipumps. A spot urine was collected just before tail vein bleeding. Blood ionized Ca2+ was measured with a Ca2+/pH analyzer (model 634; Chiron Diagnostics, Los Angeles, California). Serum and urine inorganic phosphorus were measured spectroscopically using a kit (Stanbio Laboratory, Boerne, Texas). Serum creatinine and urine creatinine were assessed, respectively, with a serum creatinine assay kit from BioVision (model K625–100; Mountain View, California) and a urine creatinine assay kit from BioAssay Systems (DICT-500; Hayward, California). Serum 1,25(OH)2D3 concentrations were measured using an enzyme immunoassay kit from Immunodiagnostic Systems, Inc. (Fountain Hills, Arizona). Serum FGF23 was measured with a mouse ELISA kit from Immutopics, Inc. (San Clemente, California) that measures intact as well as C-terminal FGF23; the ELISA kit for measuring human PTH (hPTH)(1–34) was from the same company (Immutopics Inc.). The fractional excretion index for Pi (FEIPi) was calculated as follows: urine Pi/(urine creatinine × serum Pi) (7).
Quantitative real-time PCR
Total RNA was extracted from kidneys of mice with a continuous infusion of PTH or vehicle for 8 days using Trizol reagent (Invitrogen, Carlsbad, California) and was reverse transcribed into cDNA with SuperscriptRT II (Invitrogen, Grand Island, New York). SYBRGreen universal master mix (Applied Biosystems, Foster City, California) was used for quantitative, real-time PCR, as described previously (32). The PCR primer sequences used are as follows: Npt2a forward, 5′-aacctctgtcaccaacaccat-3′, reverse, 5′-aaggaagcaaccacaagc-3′; Npt2c forward, 5′-aagaacgctgaccaactgaaa-3′, reverse 5′-ttgctgcctagtagctggaaa-3′; PPR forward, 5′-gcactgcacgcgcaactaca-3′, reverse 5′-acctgcgcgatgatatgcaac-3′; 1α-hydroxylase forward, 5′-aagtcactgtccagagcgctg-3′, reverse, 5′-gttgtccagagttccagcata-3′; 24α-hydroxylase forward, 5′-tcaatgaggtcttggctgatt-3′, reverse, 5′-aaggtcagggcttcttcctct-3′; and gapdh forward, 5′-tggagtggtgtcttcactact-3′, reverse, 5′-aagcagttggtggtgcaggat-3′. Relative expression was calculated for each gene by the 2-ΔΔCT method with Gapdh for normalization.
Statistics
All experimental values are expressed as mean ± SEM. Statistical analyses were performed using unpaired, 2-tailed Student's t tests for comparisons between 2 groups and 1-way ANOVA and Fisher's protected least significant difference tests for comparison of more than 2 groups.
Results
Continuous PTH caused a persistent decrease in serum Pi in Wt but not DD mice
To explore the role of PI-PLC signaling in regulating Pi homeostasis after activation of the PPR by PTH, we assessed the temporal response of serum Pi and urinary Pi excretion to a continuous infusion of PTH. Male mice at the age of 7 weeks received continuous infusions of vehicle (control) or hPTH(1–34) at a dose of 40 μg/kg·d for 8 days, which resulted in equally elevated hPTH(1–34) levels in each genetic background (Supplemental Figure 1, published on The Endocrine Society's Journals Online web site at http://endo.endojournals.org). Serum Pi was measured before and at 1, 2, 4, and 8 days after starting the infusions. As shown in Figure 1A, baseline serum Pi was normal in DD mice, suggesting that PI-PLC signaling by the activated PPR is not critical for maintaining serum Pi under basal conditions. Continuous infusion of PTH caused a rapid and persistent decrease in serum Pi in Wt mice, whereas in the PTH-infused DD mice serum Pi decreased only transiently (for the first 2 days) and then returned to basal level; this suggested that PPR-activated PI-PLC signaling is essential for the prolonged but not for the short-term response of serum Pi to continuous PTH. To determine whether the surprising alteration of serum phosphate response observed in the PTH-infused DD mice might be partly due to a blunted decrease in the tubular reabsorption of phosphate in response to PTH, we next measured the FEIPi. PTH infusion resulted in a striking and equivalent initial increase in the fractional excretion of phosphate in both Wt and DD mice, but this phosphaturic response then was significantly attenuated in DD mice beyond the first 2 days of PTH infusion (increased by 3.5-fold in Wt vs 1.4-fold in DD on day 8) (Figure 1B). The reduction in fractional excretion of phosphate was not due to impaired renal function in DD mice (see below and Supplemental Figure 2).
Figure 1.

Effect of continuous PTH on serum Pi and urine FEIPi. Wt and DD mice received continuous infusion of hPTH(1–34) (PTH) (40 μg/kg·d) or vehicle (Veh), as indicated, for 8 days. Each group contained 6–8 mice. Serum phosphate, urine phosphate, and urine creatinine were measured prior to and 1, 2, 4, and 8 days after starting the infusion. A, Serum phosphate measurements. B, FEIPi assessments. Data were expressed as mean ± SEM. a, P < .05 (PTH-DD vs PTH-PPR Wt mice at corresponding time points, n = 6); b, P < .05 (PTH vs vehicle of the same genotype, n = 6).
Absence of Npt2a dramatically impaired PTH-mediated Pi homeostasis
Because Npt2a mediates most of the renal tubular Pi transport, we next determined whether the attenuated persistence of the phosphaturic response to PTH infusion seen in DD mice could occur in the absence of Npt2a. As shown in Figure 2A, mice lacking Npt2a exhibited significantly lower basal levels of serum Pi in comparison with Wt mice (see Figure 1). Nonetheless, these animals showed a readily detectable hypophosphatemic response to PTH, albeit attenuated in magnitude. Thus, the maximum reduction was 0.39 ± 0.06 mmol/L in PPR Wt/Npt2a−/−, compared with 1.23 ± 0.06 mmol/L in Wt mice. However, the PTH effect appeared to be more sustained in the PPR Wt/Npt2a−/− mice than in DD/Npt2a−/− animals because the maximal reduction in serum Pi levels occurred on day 4 in the PPR Wt/Npt2a−/− mice, and even by day 8, serum Pi levels were still significantly lower than in vehicle-infused PPR Wt/Npt2a−/− or DD/Npt2a−/− mice.
Figure 2.
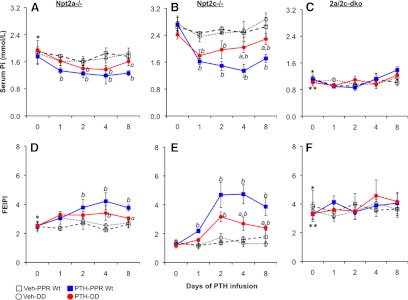
Ablation of Npt2a and/or Npt2c attenuated the effects of continuous PTH on Pi metabolism. Mice deficient for Npt2a (Npt2a−/−) (A and D) or Npt2c (Npt2c−/−) (B and E) or lacking both transporters (2a/2c-dko) (C and F) expressing either the Wt PPR or the DD mutant, as indicated, received a continuous infusion of hPTH(1–34) (PTH) (40 μg/kg·d) or vehicle (Veh) for 8 days. Serum phosphate, urine phosphate, and urine creatinine were measured prior to and 1, 2, 4, and 8 days after starting the infusion. Each group contained 6–8 mice. A–C represent serum phosphate measurements, and D–F represent the determinations of FEIPi. Data were expressed as mean ± SEM. a, P < .05 (PTH-DD vs PTH-PPR Wt mice at corresponding time points, n = 6); b, P < .05 (PTH vs vehicle of the same genotype, n = 6). *P < .05 [compared with Wt basal levels seen in Figure 1 (day 0), n = 12]; **P < .05 [compared with Npt2a−/−basal levels (day 0), n = 12].
In contrast, Wt animals had by that time already recovered substantially from hypophosphatemia and showed a reduction in urinary phosphate excretion. Consistent with the lowered basal serum Pi, the urine Pi excretion index was significantly increased at baseline in the PPR Wt/Npt2a−/− compared with the Wt mice (2.51 ± 0.29 vs 1.68 ± 0.27, respectively) and increased further by PTH infusion in the PPR Wt/Npt2a−/− mice. The increased excretion of urine Pi after PTH infusion was much more modest in the PPR Wt/Npt2a−/− mice (on day 4 FEIPi was increased by PTH over basal levels up to 3.8-fold in the Wt mice vs 1.5-fold in the PPR Wt/Npt2a−/− animals) (Figures 1B and 2D). In the DD/Npt2a−/− mice, we also observed a similar increase in basal urine Pi excretion (2.47 ± 0.23 in the DD/Npt2a−/− mice vs 1.52 ± 0.31 in the DD mice). PTH infusion modestly increased urine Pi excretion in this group of mice as well (Figure 2D). These observations further confirm that Npt2a mediates the major effect of PTH on Pi homeostasis and also suggest that Npt2a is a major mediator of both the acute (cAMP/PKA dependent) and sustained (PI-PLC dependent) phosphaturic effects of PTH.
Npt2c deficiency modestly attenuated the effect of continuous PTH on Pi homeostasis
Because PTH also decreases the brush border membrane abundance of Npt2c in the proximal tubules, we next determined whether ablation of Npt2c influenced regulation of Pi homeostasis by continuous PTH in both mice expressing the PPR Wt and the DD mutant. Unlike Npt2a ablation, removal of Npt2c did not cause a significant alteration in the basal levels of serum Pi in mice that are either Wt or DD for the PPR. In contrast to PPR Wt/Npt2a−/− mice (see Figure 2A), PPR Wt/Npt2c−/− mice showed a robust decrease in serum Pi levels that were only slightly less pronounced than the decline observed in Wt animals (Figures 1A and 2B). Thus, continuous PTH persistently decreased serum Pi by 1.37 ± 0.19 mmol/L and increased urine Pi excretion by 2.9-fold in the PPR Wt/Npt2c−/− mice (Figure 2, B and E). The persistent effect of PTH also was attenuated in the DD mice without Npt2c, ie, the DD/Npt2c−/− mice, demonstrating that the remaining Npt2a transporter requires PTH-induced PI-PLC signaling for its full effect after chronic administration of PTH. Also, the diminished long-term effect of PTH observed in the DD mouse was less dramatically attenuated in the DD/Npt2c−/− mice, suggesting that Npt2c may mediate some effect of the PPR-stimulated PI-PLC signaling pathway on Pi metabolism.
Complete ablation of both transporters abolished PTH-mediated Pi homeostasis
Because both Npt2a and Npt2c mediated the effects of continuous PTH on serum Pi homeostasis, we further explored whether continuous PTH could still affect serum Pi in the absence of both transporters. Not surprisingly, the levels of serum Pi were profoundly reduced at baseline in the double-mutant PPR Wt/2a/2c-dko and in the triple mutant DD/2a/2c-dko mice (1.12 ± 0.10 and 1.02 ± 0.05 mmol/L, respectively) when compared with either Wt or DD mice (2.54 ± 0.10 or 2.57 ± 0.23 mmol/L, respectively) (see Figures 1A and 2C). The serum Pi levels were even lower than in PPR Wt/Npt2a−/− or DD/Npt2a−/− mice (1.82 ± 0.26 and 1.91 ± 0.10 mmol/L, respectively) (Figure 2, A and C). The FEIPi was much higher at baseline in the mice lacking both transporters, PPR Wt/2a/2c-dko and DD/2a/2c-dko mice (3.56 ± 0.7 and 3.41 ± 0.36, respectively), compared with the Wt or DD mice (1.68 ± 0.27 and 1.52 ± 0.31, respectively), even higher than in Npt2a−/− mice with or without DD mutant PPR (2.51 ± 0.29 or 2.47 ± 0.23, respectively) (Figures 1B and 2, D and F). In the absence of both Npt2a and Npt2c, continuous infusion of PTH exerted no further effect on serum Pi or on urine Pi excretion in mice that are either Wt or DD for the PPR (Figure 2, C and F), indicating that both transporters, Npt2a and to a lesser extent Npt2c, are key mediators of PTH-regulated phosphate homeostasis.
PTH-induced hypercalcemia was not attenuated in DD mice or mice deficient for Npt2a and/or Npt2c
PTH is also a key regulator of serum calcium homeostasis, and sustained PTH elevations via continuous administration of PTH or primary hyperparathyroidism causes hypercalcemia in both humans and animals. To determine whether the PPR/PI-PLC signaling pathway is essential for PTH-regulated serum calcium homeostasis and responses in vivo, we examined the dynamic effect of PTH on blood ionized calcium during continuous PTH infusion in mice that are PPR Wt or DD. As shown in Figure 3, PTH infusion caused a sustained elevation of blood ionized calcium in both PPR Wt and DD mice, suggesting that the PPR-activated PI-PLC signaling pathway is not required for the hypercalcemic response to continuous PTH. Moreover, continuous PTH infusion elevated blood-ionized calcium in all the mice that were deficient for either one or both phosphate transporters, regardless of the elevated baseline calcium level observed in Npt2a−/− mice or the much higher basal levels seen in the mice lacking Npt2a and Npt2c (Figure 3).
Figure 3.

Effect of continuous PTH on blood-ionized calcium. Sixteen groups of mice from 8 different genotypes received a continuous infusion of PTH or vehicle for 8 days, as indicated (Npt-Wt: wild-type for both Npt2a and Npt2c and more as described in Figure 2). Blood-ionized calcium was measured prior to and 1, 2, 4, and 8 days after starting the infusion. Data were expressed as mean ± SEM. b, P < .05 (PTH vs vehicle of the same genotype, n = 6). *P < .05 [compared with Npt2-Wt basal levels (day 0), n = 12]; **P < .05 [compared with Npt2a−/− basal levels (day 0), n = 12].
Effect of continuous PTH on serum FGF23 in DD and Npt2 mutant mice
Because FGF23 is a key regulator of serum phosphate homeostasis through its action on Npt2a protein in the brush border membrane of renal proximal tubule cells, we next measured the serum FGF23 to determine whether the altered response of serum phosphate in the PTH-infused DD mice might be due to a reduction in FGF23 in response to continuous PTH. Whereas PTH infusion for 24 hours significantly lowered the serum phosphate, there was no effect on serum FGF23 in either PPR Wt or DD mice at that point in time (Figure 4A). Interestingly, a continuous infusion of PTH for 8 days significantly increased the levels of serum FGF23 in all the mice, regardless of whether the animals were deficient in the PI-PLC signaling pathway downstream of the activated PPR or were ablated for Npt2a or Npt2c; a modestly attenuated response to a continuous PTH was, however, seen in the DD/Npt2a−/− mice (Figure 4B).
Figure 4.
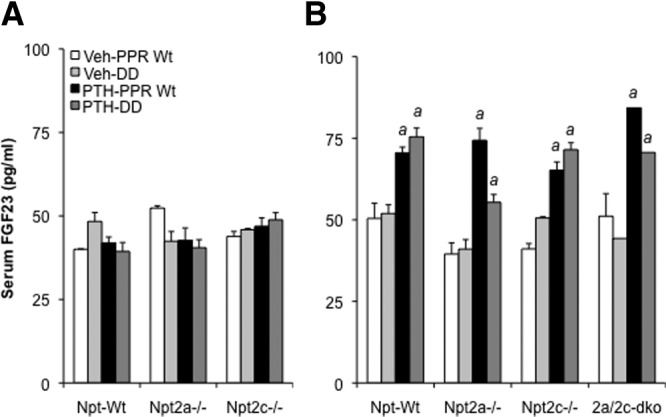
Effect of continuous PTH on serum FGF23. All the mice received a continuous infusion of PTH or vehicle for 8 days, as described in Figure 1. Serum FGF23 of the infused mice was measured after infusion for 24 hours (A) and for 8 days (B). Data were expressed as mean ± SEM (n = 6). a, P < .05 (compared with vehicle infused mice with the same genotype).
Effect of continuous PTH on serum 1,25(OH)2D3 in DD and Npt2 mutant mice
1,25(OH)2D3 plays a role in serum phosphate homeostasis by modestly increasing intestinal phosphate absorption. Because elevated PTH is expected to increase levels of serum 1,25(OH)2D3, we next measured serum 1,25(OH)2D3 to determine whether the missing persistent response of serum phosphate to PTH seen in DD mice might be due to a defective 1,25(OH)2D3 response in those mice. Continuous PTH infusion caused a dramatic increase in serum levels of 1,25(OH)2D3 in both PPR Wt and DD mice, however (Figure 5). This PTH-dependent increase in serum 1,25(OH)2D3 was also observed in PPR Wt and DD mice with ablation of either Npt2a or Npt2c despite the elevated basal levels in these single knockouts. In contrast, a continuous infusion of PTH did not further elevate serum 1,25(OH)2D3 in mice null for both Npt2a and Npt2c, which exhibited much higher basal levels of serum 1,25(OH)2D3.
Figure 5.
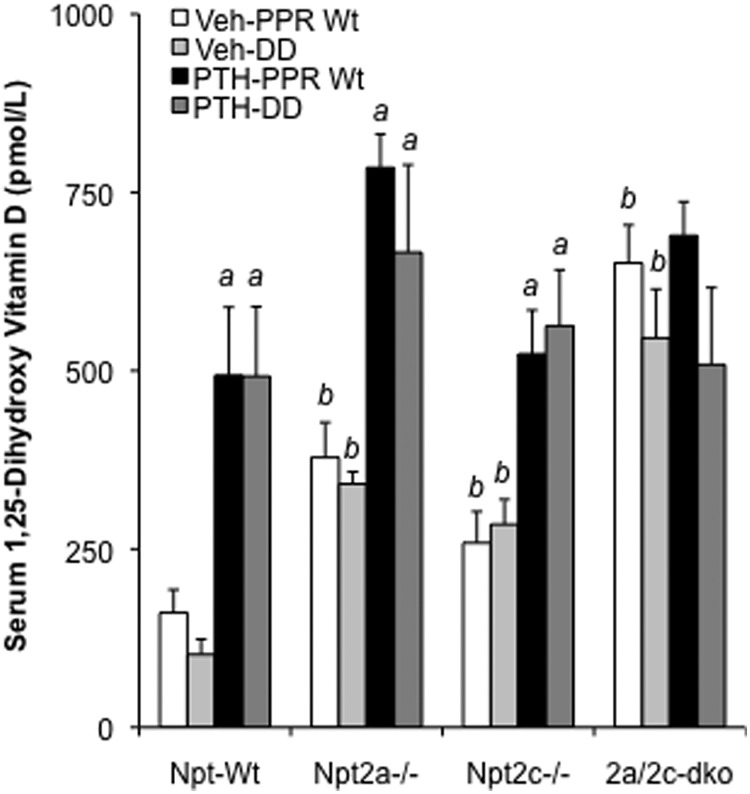
Effect of continuous PTH on serum 1,25(OH)2D3. All the groups of mice received a continuous infusion of PTH or vehicle for 8 days, as described in Figure 1. Serum 1,25(OH)2D3 of the infused mice was determined on day 8 after infusion. Data were expressed as mean ± SEM (n = 6). a, P < .05 (compared with vehicle infused mice with the same genotype); b, P < .05 (compared with Npt-Wt mice with PPR Wt or DD).
Continuous PTH had no impact on serum creatinine levels
To determine whether continuous infusion of PTH led to renal failure in the infused mice with persistent hypercalcemia, we next measured serum creatinine levels. No increase in serum levels of creatinine was observed in any groups of mice with continuous PTH infusion (Supplemental Figure 2), indicating that continuous PTH infusion applied here did not lead to renal failure.
Effect of continuous PTH on renal Npt2a and Npt2c expression
To explore the molecular mechanisms underlying the abnormal phosphate homeostasis in the DD mice with continuous infusion of PTH, we next examined the renal levels of mRNAs encoding Npt2a and Npt2c in the continuously infused mice. Continuous PTH infusion caused a significant decrease in the expression of Npt2a mRNA by approximately 50% in the Wt mice but not in the DD mice (Figure 6A), and a similar reduction of Npt2a mRNAs was also observed in the PPR Wt/Npt2c−/− mice but not in the DD mice deficient for Npt2c (Figure 6B). No effect of continuous PTH on Npt2c mRNA expression was observed in either Wt or DD mice (Figure 6A). Interestingly, continuous PTH increased the levels of mRNAs encoding vitamin D 1α-hydroxylase and 24α-hydroxylase in both PPR Wt and DD mice with or without ablation of Npt2c (Figure 6, A and B). No significant alteration of mRNAs encoding PPR was induced by a continuous PTH infusion in either PPR Wt or DD mice.
Figure 6.
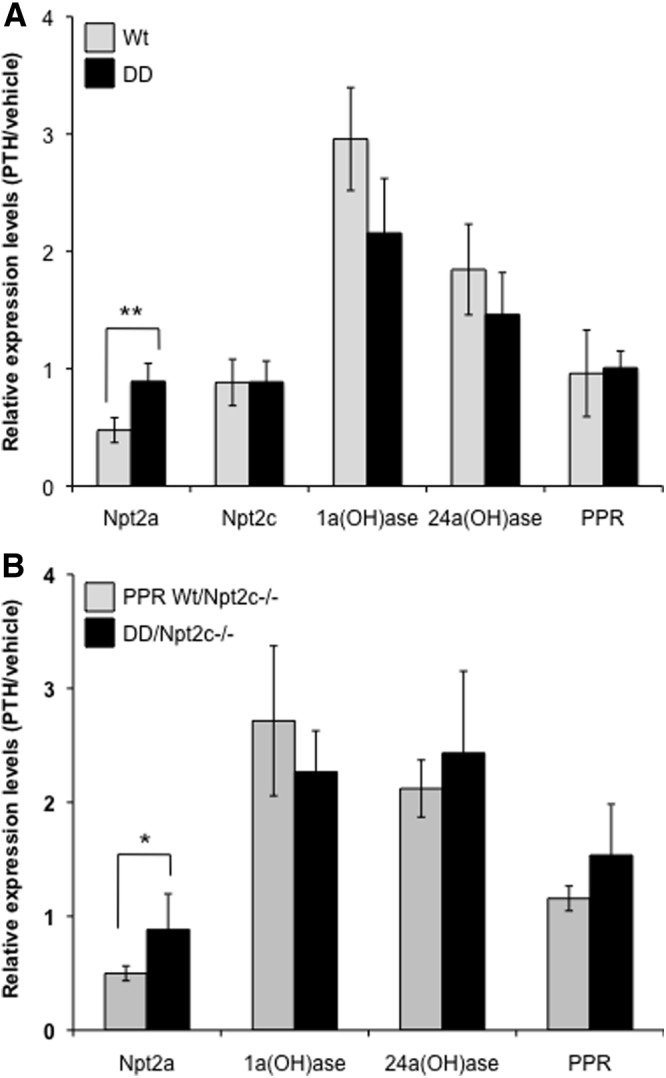
Real-time PCR analysis of the PTH-regulated genes in the kidney. Real-time PCR analysis of Npt2a, Npt2c, 1α-hydroxylase [1α(OH)ase)], 24α-hydroxylase ([24α(OH)ase)] and PPR as well as Gapdh (used as internal control for normalization) was performed with total RNA isolated from the kidneys of PPR Wt and DD mice (A) or from Npt2c knockouts with PPR Wt or DD (B) 8 days after a continuous infusion of PTH or vehicle. Data were expressed as mean ± SEM (n = 6) of fold changes in PTH infusion/vehicle infusion of the same group of mice after normalization with Gapdh. *P < .05; **P < .01.
Discussion
PTH, one of the most potent phosphaturic factors, binds to and activates the PPR, which reduces the expression of the two predominant sodium-dependent phosphate cotransporters in the kidney, namely Npt2a and Npt2c. Both transporters are expressed in the brush border membrane of the proximal tubules, and both contribute to phosphate reabsorption and thus the maintenance of phosphate homeostasis. The effects of PTH on the two cotransporters have been extensively studied, mainly ex vivo and in vitro, and the relative contribution of Npt2a and Npt2c to PTH-induced phosphaturia has been estimated (5, 17, 20, 24, 25, 33–39). The acute in vivo regulation of phosphate homeostasis through PTH appears to involve predominantly the cAMP/PKA pathway at the PPR, which is consistent with the diminished or absent response to exogenous PTH observed in patients with pseudohypoparathyroidism type Ia, who are carriers of maternally inherited inactivating Gsα mutations. However, our recent findings in mice expressing a mutant PPR that is unable to efficiently stimulate the PI-PLC/IP3 signaling pathway in response to PTH strongly indicated that the latter pathway is importantly involved in the PTH-dependent control of urinary phosphate excretion through the regulation of Npt2a and Npt2c (31). Because we have not carefully examined the effect of the DSEL mutation on other possible pathways downstream of the PPR, such as Gi, G12, G13, and arrestin, we cannot eliminate possible roles for these pathways in the effects of the DSEL mutation. Here we used multiple genetically manipulated mouse models to explore the relative contribution of Npt2a and Npt2c to the effect of infused PTH on phosphaturia and the role of PI-PLC signaling downstream of the activated PPR in the Npt2a/2c-mediated actions of PTH in phosphate balance. Our data indicated that PI-PLC signaling via the PPR is required for the sustained but not the acute hypophosphatemic response to continuous PTH. We also showed that both Npt2a and Npt2c are mediators of the phosphaturic response to PTH.
Normally an increase in serum PTH is associated with renal phosphate-wasting and hypophosphatemia, as observed here in the wild-type mice continuously infused with PTH. Surprisingly, the PTH-infused DD mice, expressing the PI-PLC-defective PPR that activates AC normally, were only transiently hypophosphatemic during the first 2 days of PTH infusion, in contrast to the PTH-infused Wt mice that exhibited sustained hypophosphatemia and urinary phosphate excretion. Although a previous study documented that the acute action of PTH on phosphate balance is mediated by the AC/PKA pathway activated by the PPR (20), our current findings indicate that the AC/PKA signaling of the activated PPR is not sufficient for PTH to efficiently induce its prolonged hyperphosphatemic effect. This strongly suggests that the long-term in vivo effect of elevated levels of PTH on serum phosphate homeostasis requires PI-PLC signaling via the PPR, although there may be some contribution of AC/PKA signaling.
In mammals, the kidney contributes importantly to phosphate homeostasis with approximately 70% of the phosphate filtered in the glomerulus being reabsorbed in the proximal tubule. To evaluate renal phosphate handling in the DD mice, we estimated the fractional excretion index of phosphate and observed a significant waning of the increase in the fractional excretion index of phosphate in the mutant animal. This indicates that the increase in renal phosphate excretion was not maintained in the mutant mice and suggests that the attenuated hypophosphatemia in the PPR-mutant mice resulted from abnormal phosphate handling by the kidney.
PTH regulates three distinct sodium-dependent phosphate transporters, Npt2a, Npt2c, and type III sodium-dependent phosphate transporter 2 (PiT2), which are expressed in the brush border membrane of the proximal tubules (6, 24, 34). In the proximal tubules, these transporters function to control the reabsorption of phosphate from the tubule back into blood, thus increasing serum phosphate concentration. Of the three transporters, Npt2a mediates most of phosphate reabsorption in the kidney and is responsible for more than 70% of the phosphate reabsorbed, whereas Npt2c handles some of the remaining 30% (40). PTH, by binding to the PPR, activates both AC/PKA and PC-PLC/PKC signaling pathways and results in the suppression of Npt2a protein expression in the proximal tubules (20, 30, 35, 38).
Although the effect of PTH on the regulation of Npt2a has been extensively investigated, the relative contribution of the PI-PLC signaling pathway downstream of the activated PPR in regulating Npt2a-mediated phosphate reabsorption in vivo remains incompletely understood. Continuous PTH persistently caused a dramatic decrease in serum phosphate and an increase in urine phosphate excretion in Wt mice, whereas the PTH-infused DD mice developed hypophosphatemia and phosphaturia only transiently during the first 2 days after starting the infusion. Continuous PTH infusion induced a less dramatic hypophosphatemia and phosphaturia in the Npt2a-null animal compared with that seen in the Wt mice; this is consistent with the previous demonstration that regulation of Npt2a mediates most of the PTH-induced hypophosphatemia and phosphaturia in mice (41). Moreover, continuous infusion of PTH for 8 days significantly decreased the levels of mRNA encoding Npt2a in PPR Wt but not DD mice. This decrease in mRNA may contribute to the persistent effect of PTH in the Wt but not the DD mice. The present data indicate that the long-term hypophosphatemic and phosphaturic effect of PTH require PI-PLC signaling at the PPR and affects mostly Npt2a expression. This long-term, PI-PLC-mediated PTH effect is distinct from the PC-PLC/PKC-dependent effect. PI-PLC/IP3-independent PKC activation can be mimicked by PTH(3–34), a PTH analog that cannot activate AC/PKA (19), appears to involve only apical PTH receptors, and requires Na+/H+ exchanger regulatory factor-1 for efficient activation of PKC signaling (17, 18, 42).
In contrast to the actions of continuous PTH in Npt2a-null mice, PTH infusion caused only a modest attenuation in PTH-induced hypophosphatemia and phosphaturia in animals lacking Npt2c. Our current data furthermore confirm that Npt2a plays, in comparison with Npt2c, a much more prominent role in the PTH-dependent regulation of phosphate homeostasis. Ablation of both Npt2a and Npt2c caused no further response to continuous PTH in either serum phosphate or urine phosphate excretion, indicating that PTH uses only Npt2a and Npt2c to control urinary phosphate excretion and to maintain serum phosphate balance. Moreover, attenuation of PTH-mediated hypophosphatemia and phosphaturia was less dramatic in the DD mice with ablation of Npt2c than in DD mice with intact cotransporters. Our observations thus suggest that Npt2c may also play a role in the long-term effect of PTH on phosphate balance through PI-PLC signaling, although we failed to observe the down-regulation of Npt2c mRNAs by continuous PTH infusion, even in Wt mice.
We further determined whether the PI-PLC-dependent long-term effect of PTH on phosphate balance was partly due to indirect actions mediated by abnormal levels of 1,25(OH)2D3 or FGF23 in the PTH-infused DD mice. Elevated PTH is known to enhance renal 1α-hydroxylase and increase serum 1,25(OH)2D3. This biologically active vitamin D analog not only increases gastrointestinal phosphate absorption but also stimulates osteocyte production of FGF23 (43), the centrally important hormonal regulator of phosphate homeostasis (44–46). Thus, alteration in serum 1,25(OH)2D3 levels would potentially impact serum phosphate concentration through FGF23-dependent urine phosphate excretion. Continuous PTH infusion increased serum 1,25(OH)2D3 similarly in both Wt and DD mice, even with the ablation of either Npt2a or Npt2c. Moreover, the stimulatory effect of continuous PTH on the levels of renal 1α-hydroxylase mRNAs also was comparable for Wt and DD mice. Thus, continuous PTH elevated serum 1,25(OH)2D3 equally in both Wt and DD mice, likely due to the increased expression of 1α-hydroxylase, although the PTH-stimulated increase in 24α-hydroxylase mRNAs may have prevented a further 1,25(OH)2D3 increase in both mice. Because serum FGF23 increased equivalently in both Wt and DD mice, PI-PLC signaling downstream of the activated PPR does not appear to contribute to 1,25(OH)2D3-dependent or -independent FGF23 production. PTH infusion for 24 hours failed to increase serum FGF23, even in Wt mice but continuous PTH infusion for 8 days significantly increased serum FGF23 in both Wt and DD mice. These in vivo findings imply that the long-term PTH effect on phosphate homeostasis that are mediated through PPR-activated PI-PLC signaling occur independent of changes in serum FGF23 levels. Thus, neither changes in vitamin D metabolism or in FGF23 levels can explain the waning of the phosphaturic response to PTH after 2 days of PTH infusion, making it necessary to explore in future studies the proximal tubular mechanisms downstream of the PPR that contribute to phosphate homeostasis.
Administration of PTH is known to increase serum calcium both in humans and in animals. It is not clear yet whether PI-PLC signaling through the PPR contributes to the PTH-dependent control of serum calcium homeostasis in vivo; in fact, current data suggest that cAMP/PKA-dependent mechanisms downstream of the PPR are sufficient for enhancing calcium reabsorption by the distal renal tubules (13, 47). Here we examined the blood calcium response in DD mice expressing the PI-PLC-defective PPR that activates AC/PKA normally and showed that continuous PTH infusion dramatically increased blood ionized calcium levels in both Wt and DD mice with or without ablations of Npt2a and/or Npt2c. In all groups of mice, PTH administration induced persistent elevation of ionized calcium, regardless of baseline levels. Consistent with previous findings by others (13, 47), our data thus indicate that PI-PLC signaling downstream of the activated PPR is not required for regulating serum calcium levels, which is in contrast to the PI-PLC-dependent long-term effect of PTH on serum phosphate homeostasis.
In summary, our studies here of chronic elevation of PTH levels suggest that PI-PLC activation by the PPR appear to contribute importantly to the normal regulation of phosphate but not of calcium homeostasis. Selective activation of this signaling pathway may improve urinary phosphate excretion in patients with chronic renal disease, if it causes no major increase in blood calcium levels, and may limit the degree of hyperphosphatemia in patients with pseudohypoparathyroidism. It is also possible that PLC activation by the PPR contributes also to the physiological regulation of phosphate homeostasis, but the normal serum phosphate levels in the DD mouse at baseline argue that this role is modest.
Acknowledgments
This work was supported by National Institutes of Health Grant PO1 DK11794.
Present address for L.S.: Department of Endocrinology, Tongji Hospital, Tongji University School of Medicine, Shanghai 200065, China.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AC
- adenylyl cyclase
- 2a/2c-dko
- absence of both Npt2a−/− and Npt2c−/− phosphate transporters
- DD
- genetically manipulated knock-in mice
- FEIPi
- fractional excretion index for Pi
- FGF23
- fibroblast growth factor 23
- hPTH
- human PTH
- IP3
- inositol trisphosphate
- Npt2a
- NaPi-IIa
- Npt2c
- NaPi-IIc
- 1,25(OH)2D3
- 1,25 dihydroxyvitamin D3
- PC-PLC
- phosphatidylcholine-dependent phospholipase C
- PI-PLC
- phosphatidylinositol-specific phospholipase C
- PKA
- protein kinase A
- PKC
- protein kinase C
- PLC
- phospholipase C
- PPR
- PTH/PTHrP receptor
- Wt
- wild type.
References
- 1. Datta NS, Abou-Samra AB. PTH and PTHrP signaling in osteoblasts. Cell Signal. 2009;21:1245–1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pfister MF, Ruf I, Stange G, et al. Parathyroid hormone leads to the lysosomal degradation of the renal type II Na/Pi cotransporter. Proc Natl Acad Sci USA. 1998;95:1909–1914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Traebert M, Roth J, Biber J, Murer H, Kaissling B. Internalization of proximal tubular type II Na-P(i) cotransporter by PTH: immunogold electron microscopy. Am J Physiol Renal Physiol. 2000;278:F148–F154 [DOI] [PubMed] [Google Scholar]
- 4. Bacic D, Lehir M, Biber J, Kaissling B, Murer H, Wagner CA. The renal Na+/phosphate cotransporter NaPi-IIa is internalized via the receptor-mediated endocytic route in response to parathyroid hormone. Kidney Int. 2006;69:495–503 [DOI] [PubMed] [Google Scholar]
- 5. Segawa H, Onitsuka A, Furutani J, et al. Npt2a and Npt2c in mice play distinct and synergistic roles in inorganic phosphate metabolism and skeletal development. Am J Physiol Renal Physiol. 2009;297:F671–F678 [DOI] [PubMed] [Google Scholar]
- 6. Biber J, Hernando N, Forster I, Murer H. Regulation of phosphate transport in proximal tubules. Pflugers Arch. 2009;458:39–52 [DOI] [PubMed] [Google Scholar]
- 7. Beck L, Karaplis AC, Amizuka N, Hewson AS, Ozawa H, Tenenhouse HS. Targeted inactivation of Npt2 in mice leads to severe renal phosphate wasting, hypercalciuria, and skeletal abnormalities. Proc Natl Acad Sci USA. 1998;95:5372–5377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tomoe Y, Segawa H, Shiozawa K, et al. Phosphaturic action of fibroblast growth factor 23 in Npt2 null mice. Am J Physiol Renal Physiol. 2010;298:F1341–F1350 [DOI] [PubMed] [Google Scholar]
- 9. Bergwitz C, Roslin NM, Tieder M, et al. SLC34A3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria predict a key role for the sodium-phosphate cotransporter NaPi-IIc in maintaining phosphate homeostasis. Am J Hum Genet. 2006;78:179–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lorenz-Depiereux B, Benet-Pages A, Eckstein G, et al. Hereditary hypophosphatemic rickets with hypercalciuria is caused by mutations in the sodium-phosphate cotransporter gene SLC34A3. Am J Hum Genet. 2006;78:193–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hoenderop JG, van Leeuwen JP, van der Eerden BC, et al. Renal Ca2+ wasting, hyperabsorption, and reduced bone thickness in mice lacking TRPV5. J Clin Invest. 2003;112:1906–1914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. van Abel M, Hoenderop JG, van der Kemp AW, Friedlaender MM, van Leeuwen JP, Bindels RJ. Coordinated control of renal Ca(2+) transport proteins by parathyroid hormone. Kidney Int. 2005;68:1708–1721 [DOI] [PubMed] [Google Scholar]
- 13. de Groot T, Lee K, Langeslag M, et al. Parathyroid hormone activates TRPV5 via PKA-dependent phosphorylation. J Am Soc Nephrol. 2009;20:1693–1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Amizuka N, Lee HS, Kwan MY, et al. Cell-specific expression of the parathyroid hormone (PTH)/PTH-related peptide receptor gene in kidney from kidney-specific and ubiquitous promoters. Endocrinology. 1997;138:469–481 [DOI] [PubMed] [Google Scholar]
- 15. Ba J, Brown D, Friedman PA. Calcium-sensing receptor regulation of PTH-inhibitable proximal tubule phosphate transport. Am J Physiol Renal Physiol. 2003;285:F1233–F1243 [DOI] [PubMed] [Google Scholar]
- 16. Lupp A, Klenk C, Rocken C, Evert M, Mawrin C, Schulz S. Immunohistochemical identification of the PTHR1 parathyroid hormone receptor in normal and neoplastic human tissues. Eur J Endocrinol. 2010;162:979–986 [DOI] [PubMed] [Google Scholar]
- 17. Capuano P, Bacic D, Roos M, et al. Defective coupling of apical PTH receptors to phospholipase C prevents internalization of the Na+-phosphate cotransporter NaPi-IIa in NHERF1-deficient mice. Am J Physiol Cell Physiol. 2007;292:C927–C934 [DOI] [PubMed] [Google Scholar]
- 18. Traebert M, Volkl H, Biber J, Murer H, Kaissling B. Luminal and contraluminal action of 1–34 and 3–34 PTH peptides on renal type IIa Na-P(i) cotransporter. Am J Physiol Renal Physiol. 2000;278:F792F798. [DOI] [PubMed] [Google Scholar]
- 19. Takasu H, Guo J, Bringhurst FR. Dual signaling and ligand selectivity of the human PTH/PTHrP receptor. J Bone Miner Res. 1999;14:11–20 [DOI] [PubMed] [Google Scholar]
- 20. Nagai S, Okazaki M, Segawa H, et al. Acute down-regulation of sodium-dependent phosphate transporter NPT2a involves predominantly the cAMP/PKA pathway as revealed by signaling-selective parathyroid hormone analogs. J Biol Chem. 2011;286:1618–1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jan de Beur SM, Ding CL, LaBuda MC, Usdin TB, Levine MA. Pseudohypoparathyroidism 1b: exclusion of parathyroid hormone and its receptors as candidate disease genes. J Clin Endocrinol Metab. 2000;85:2239–2246 [DOI] [PubMed] [Google Scholar]
- 22. Wu WI, Schwindinger WF, Aparicio LF, Levine MA. Selective resistance to parathyroid hormone caused by a novel uncoupling mutation in the carboxyl terminus of Gα(s). A cause of pseudohypoparathyroidism type Ib. J Biol Chem. 2001;276:165–171 [DOI] [PubMed] [Google Scholar]
- 23. Bastepe M, Altug-Teber O, Agarwal C, Oberfield SE, Bonin M, Jüppner H. Paternal uniparental isodisomy of the entire chromosome 20 as a molecular cause of pseudohypoparathyroidism type Ib (PHP-Ib). Bone. 2010;48:659–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Segawa H, Yamanaka S, Onitsuka A, et al. Parathyroid hormone-dependent endocytosis of renal type IIc Na-Pi cotransporter. Am J Physiol Renal Physiol. 2007;292:F395–F403 [DOI] [PubMed] [Google Scholar]
- 25. Matsumoto N, Hemmi A, Yamato H, et al. Immunohistochemical analyses of parathyroid hormone-dependent downregulation of renal type II Na-Pi cotransporters by cryobiopsy. J Med Invest. 2010;57:138–145 [DOI] [PubMed] [Google Scholar]
- 26. Lanzano L, Lei T, Okamura K, et al. Differential modulation of the molecular dynamics of the type IIa and IIc sodium phosphate cotransporters by parathyroid hormone. Am J Physiol Cell Physiol. 2011;301:C850–C861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lotscher M, Scarpetta Y, Levi M, et al. Rapid downregulation of rat renal Na/P(i) cotransporter in response to parathyroid hormone involves microtubule rearrangement. J Clin Invest. 1999;104:483–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pfister MF, Lederer E, Forgo J, et al. Parathyroid hormone-dependent degradation of type II Na+/Pi cotransporters. J Biol Chem. 1997;272:20125–20130 [DOI] [PubMed] [Google Scholar]
- 29. Iida-Klein A, Guo J, Takemura M, et al. Mutations in the second cytoplasmic loop of the rat parathyroid hormone (PTH)/PTH-related protein receptor result in selective loss of PTH-stimulated phospholipase C activity. J Biol Chem. 1997;272:6882–6889 [DOI] [PubMed] [Google Scholar]
- 30. Guo J, Chung UI, Kondo H, Bringhurst FR, Kronenberg HM. The PTH/PTHrP receptor can delay chondrocyte hypertrophy in vivo without activating phospholipase C. Dev Cell. 2002;3:183–194 [DOI] [PubMed] [Google Scholar]
- 31. Guo J, Liu M, Yang D, et al. Phospholipase C signaling via the parathyroid hormone (PTH)/PTH-related peptide receptor is essential for normal bone responses to PTH. Endocrinology. 2010;151:3502–3513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guo J, Chung UI, Yang D, Karsenty G, Bringhurst FR, Kronenberg HM. PTH/PTHrP receptor delays chondrocyte hypertrophy via both Runx2-dependent and -independent pathways. Dev Biol. 2006;292:116–128 [DOI] [PubMed] [Google Scholar]
- 33. Hernando N, Forgo J, Biber J, Murer H. PTH-Induced downregulation of the type IIa Na/P(i)-cotransporter is independent of known endocytic motifs. J Am Soc Nephrol. 2000;11:1961–1968 [DOI] [PubMed] [Google Scholar]
- 34. Picard N, Capuano P, Stange G, et al. Acute parathyroid hormone differentially regulates renal brush border membrane phosphate cotransporters. Pflugers Arch. 2010;460:677–687 [DOI] [PubMed] [Google Scholar]
- 35. Weinman EJ, Biswas RS, Peng Q, et al. Parathyroid hormone inhibits renal phosphate transport by phosphorylation of serine 77 of sodium-hydrogen exchanger regulatory factor-1. J Clin Invest. 2007;117:3412–3420 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36. Weinman EJ, Steplock D, Cha B, et al. PTH transiently increases the percent mobile fraction of Npt2a in OK cells as determined by FRAP. Am J Physiol Renal Physiol. 2009;297:F1560–F1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Weinman EJ, Steplock D, Shenolikar S, Blanpied TA. Dynamics of PTH-induced disassembly of Npt2a/NHERF-1 complexes in living OK cells. Am J Physiol Renal Physiol. 2011;300:F231–F235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cunningham R, Biswas R, Brazie M, Steplock D, Shenolikar S, Weinman EJ. Signaling pathways utilized by PTH and dopamine to inhibit phosphate transport in mouse renal proximal tubule cells. Am J Physiol Renal Physiol. 2009;296:F355–F361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang LE, Maunsbach AB, Leong PK, McDonough AA. Differential traffic of proximal tubule Na+ transporters during hypertension or PTH: NHE3 to base of microvilli vs. NaPi2 to endosomes. Am J Physiol Renal Physiol. 2004;287:F896–F906 [DOI] [PubMed] [Google Scholar]
- 40. Tenenhouse HS, Martel J, Gauthier C, Segawa H, Miyamoto K. Differential effects of Npt2a gene ablation and X-linked Hyp mutation on renal expression of Npt2c. Am J Physiol Renal Physiol. 2003;285:F1271–F1278 [DOI] [PubMed] [Google Scholar]
- 41. Zhao N, Tenenhouse HS. Npt2 gene disruption confers resistance to the inhibitory action of parathyroid hormone on renal sodium-phosphate cotransport. Endocrinology. 2000;141:2159–2165 [DOI] [PubMed] [Google Scholar]
- 42. Muff R, Fischer JA, Biber J, Murer H. Parathyroid hormone receptors in control of proximal tubule function. Annu Rev Physiol. 1992;54:67–79 [DOI] [PubMed] [Google Scholar]
- 43. Liu S, Tang W, Zhou J, et al. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. J Am Soc Nephrol. 2006;17:1305–1315 [DOI] [PubMed] [Google Scholar]
- 44. Quarles LD. Endocrine functions of bone in mineral metabolism regulation. J Clin Invest. 2008;118:3820–3828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gattineni J, Bates C, Twombley K, et al. FGF23 decreases renal NaPi-2a and NaPi-2c expression and induces hypophosphatemia in vivo predominantly via FGF receptor 1. Am J Physiol Renal Physiol. 2009;297:F282–F291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bergwitz C, Jüppner H. Regulation of phosphate homeostasis by PTH, vitamin D, and FGF23. Annu Rev Med. 2010;61:91–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. de Groot T, Kovalevskaya NV, Verkaart S, et al. Molecular mechanisms of calmodulin action on TRPV5 and modulation by parathyroid hormone. Mol Cell Biol. 2011;31:2845–2853 [DOI] [PMC free article] [PubMed] [Google Scholar]