

Abstract
Sympathetic stimulation regulates cardiac excitation-contraction coupling in hearts but can also trigger ventricular arrhythmias caused by early afterdepolarizations (EADs) in pathological conditions. Isoproterenol (ISO) stimulation can transiently cause EADs which could result from differential kinetics of L type calcium (Ca) current (ICaL) vs. delayed rectifier potassium current (IKs) effects, but multiple PKA targets complicate mechanistic analysis. Utilizing a biophysically detailed model integrating Ca and β-adrenergic signaling, we investigate how different phos-phorylation kinetics and targets influence β-adrenergic-induced transient EADs. We found that: 1) The faster time course of ICaL vs. IKs increases recapitulated experimentally observed ISO-induced transient EADs (which are due to ICaL reactivation). These EADs disappear at steady state ISO and do not occur during more gradual ISO application. 2) This ICaL vs. IKs kinetic mismatch with ISO can also induce transient EADs due to spontaneous sarcoplasmic reticulum (SR) Ca release and Na/Ca exchange current. The increased ICaL, SR Ca uptake and action potential duration (APD) raise SR Ca to cause spontaneous SR Ca release, but eventual IKs activation and APD shortening abolish these EADs. 3) Phospholemman (PLM) phosphorylation decreases both types of EADs by increasing outward Na/K-ATPase current (INaK) for ICaL-mediated EADs, and reducing intracellular Na and Ca loading for SR Ca-release-mediated EADs. Slowing PLM phosphorylation kinetics abolishes this protective effect. 4) Blocking phos-pholamban (PLB) phosphorylation has little effect on ICaL-mediated transient EADs, but abolishes SR Ca-release-mediated transient EADs by limiting SR Ca loading. 5) RyR phosphorylation has little effect on either transient EAD type. Our study emphasizes the importance of understanding non-steady state kinetics of several systems in mediating β-adrenergic-induced EADs and arrhythmias.
Keywords: β-adrenergic signaling, transient early afterdepolarization, isoproterenol, spontaneous Ca release, phosphorylation kinetics
Introduction
β-adrenergic receptors (β-AR) critically modulate ventricular excitation-contraction coupling (ECC) through signaling cascades (Fig. 1A) that lead to PKA-dependent phosphorylation of several proteins to orchestrate the positive inotropic and lusitropic cell response [1]. Briefly, βAR agonists, such as isoprotenorol (ISO) or norepinephrine (NE), activate stimulatory G protein causing adenylate cyclase (AC) to enhance cAMP production, which in turn activates protein kinase A (PKA) [2, 3]. In ventricular myocytes, PKA phosphorylates multiple targets that regulate L-type calcium (Ca) current (ICaL), ryanodine receptor (RyR) gating, phospholamban (PLB)-dependent sarcoplasmic reticulum (SR) Ca-ATPase (SERCA), phospholemman (PLM)-dependent Na-K ATPase (NKA), slowly-activating delayed rectifier potassium current (IKs), cystic fibrosis transmembrane regulator current (ICFTR), and myofilament sensitivity.
Figure 1. β-AR model and kinetics of phosphorylation.

A. Schematic of β-AR signaling network and ECC. β-AR agonists bind to βAR and cause PKA-phosphorylation of numerous cellular targets. (PDE, phosphodiesterase; PKI, protein kinase inhibitor; PP1, protein phosphatase-1; PP2A, protein phosphatase-2A; I1, inhibitor-1.) B. Normalized phosphorylation time course of ICaL and IKs following 1 μM ISO application in the original SS model (solid) [15] and in experimental data (dashed lines) from Lui et al. [19]. C. Our modified IKs kinetics (solid lines) recapitulate experiment data of IKs vs. ICaL increase upon ISO (as in B). Shaded are is where EADs are observed in Fig. 2.
β-AR stimulation can also induce ventricular arrhythmias caused by early afterdepolarizations (EADs) [4]. EADs have been widely investigated both in experiments [5–7] and simulations [8–12], and key dynamical mechanisms of EAD formation have been established [13, 14], but most studies focused on events occurring at steady state. Detailed information about the time courses and kinetics of PKA-dependent modulation of its numerous targets is incomplete, although phosphorylation of sarcolemmal ion channels seems to be faster than that of cytosolic targets [15]. Differential kinetics of modulation of cellular targets may cause transient instabilities which are especially arrhythmogenic. This picture is further complicated by effects of the ISO-induced elevation of intracellular [Ca] ([Ca]i) on ICaL (via Ca dependent inactivation, CDI), Na/Ca exchanger (NCX) current (INCX), Ca-activated Cl− current (ICl(Ca)), and IKs, all of which can influence AP configuration. Computer models of cardiac myocyte electro-physiology integrating Ca and β-AR signaling are especially useful to analyze these complex interactions. A recent theoretical study has shown that a time delay between the PLM- and PLB-mediated effects on NKA and SERCA (a slower decrease in [Na]i caused by the former vs. a more rapid [Ca]i elevation in [Ca]i by the latter) leads to adaptation of the Ca transient (CaT), causing a temporarily larger CaT followed by a smaller steady state CaT [16]. Transient prolongation of action potential duration (APD) was also found experimentally upon ISO application [17, 18]. Recent experiments also found that ICaL increases much faster than IKs upon ISO application [19], suggesting that PKA-dependent regulation of these channels may have different kinetics, which were assumed similar in previous models [15, 20].
Here, we utilize a computational framework to assess whether differential kinetics of PKA phosphorylation of ICaL and IKs are sufficient to induce transient EADs following βAR stimulation, as found experimentally [19]. We also analyze the impact of altered kinetics of the signaling cascade. Also, we test whether and how this transient EAD formation is affected by changes in [Ca]i due to PKA-dependent phosphorylation of RyR and PLB. Further, we incorporate PKA phosphorylation of PLM, which by modulating INaK influences both membrane voltage and [Na]i, and thus [Ca]i via INCX. All these effects might impact EAD generation. In addition to demonstrating EADs due to ICaL recovery (referred as ICaL-mediated EADs), the differential kinetics of ICaL and IKs regulation also induce transient EADs due to spontaneous Ca releases (Ca-release-mediated EADs). Understanding the mechanism underlying transient EADs could facilitate the development of anti-arrhythmic drugs and therapeutic strategies.
Methods
We use the β-adrenergic signaling branch of the Soltis-Saucerman (SS) rabbit myocyte model [15], which integrated the Shannon-Bers ECC model [21] with the β-adrenergic and CaMKII signaling networks (Fig. 1A). In the SS model, the time constant of IKs phosphorylation (IKsp) is similar to that of L-type Ca channel phosphorylation (LCCp) (Fig. 1B, solid, τIcaL=7.7s, τIKsp=7.5s). However, recent experimental data [19] shows that upon ISO application IKs grows much more slowly than ICaL (Fig. 1B and C red dotted). Therefore, we slowed down the rate constants of IKs phosphorylation and dephosphorylation from 84 and 8.52 s−1 to 1.87 and 0.19 s−1, respectively (τIKsp=39.7s), to recapitulate the measured kinetics, but without perturbing the steady state effect (Fig. 1C solid lines and Fig S1, at α=45). Since the rise is better fit by a sigmoid function (vs. exponential), we used a Gompertz sigmoid approximation to the data for time constant τ.
A previous study used time constants for ICaL and IKs changes in relatively simple simulations [19], but here we use a detailed β-adrenergic signaling model to alter PKA and phosphatase activity. This also allows us to consider the effect of other PKA targets and investigate Ca-release-mediated EADs. This is also advantageous in providing a mechanistic signaling framework for devising further novel experimental tests. For example, both LTCCs and IKs channels have associated AKAPs that localize PKA to the channel [22, 23], but the rate of PKA activation could differ because: a) the relative extent of local PKA activation, b) the proximity of the AKAP-bound PKA and phosphatase to the target site (which influences the effective local [PKA] and [phosphatase]), c) the proximity and coupling efficiency of β-AR/Gs/adenylate cyclase to the AKAP (which for LTCC are in the same complex). These and other factors could cause the differential ICa,L and IKs activation kinetics. The formulation for PLM phosphorylation by PKA and NKA modulation was that in Yang and Saucerman [24].
Transient EADs caused by ICaL recovery (Type 1 EAD, EAD1) and spontaneous SR Ca releases (Type 2 EAD, EAD2) are studied separately. Table 1 shows parameter changes (in %) for EAD1 and EAD2 compared to the original SS model [15]. For EAD2, the only changes were reducing GKr by 40% to prolong APD and enhance EAD occurrence, and a slight (15%) increase in ICaL maximal conductance, GCaL (to match what we used for EAD1 below). Using the original SS ICa,L produced the same qualitative results (just slightly fewer total EAD2s; not shown).
Table 1.
Parameter modifications in the baseline models (comparing with the original model in %)
For EAD1, we prevent spontaneous SR Ca releases by increasing RyR EC50 for intra-SR [Ca] ([Ca]SR) by 78% and increasing GCaL by 15% to ensure ECC fidelity with the desensitized RyR2. To maintain cell Ca balance and limit SR Ca loading we also increased the maximal conductance of INCX (GNCX) by 25%. In addition, the maximal conductance of rapid-activating delayed rectifier potassium current (IKr) was strongly inhibited (GKr -95%) as in the experimental data [19], which prolongs APD, and enhances EAD propensity.
For both cell models we used a pacing cycle length (PCL) of 4 s, to explicitly match direct experimental data regarding EAD induction in rabbit myocytes [19] (faster PCL was also explored). At this PCL, the 40% GKr inhibition in the EAD2 case prolongs baseline APD from 409 to 588 ms. The Ca handling changes in the EAD1 model prolong baseline APD to 573 ms. With the additional block of GKr APD lengthens further to 1148 ms. Similar long APDs have been reported with IKr block [25, 26] or ICaL inactivation mutation [27] at slow rate (PCL>2s).
No EADs were observed in either basal state. Maximal ISO-induced increase of ICaL was increased (by 70%) to allow roughly doubling of ICaL upon 1μM ISO application in both models, consistent with experiments [28]. Unless specified, 1 μM ISO is abruptly applied to the myocyte at time zero. The model was implemented in C++, and simulations run with a 1 μs integration time step using the forward Euler method.
While blocking IKr (as in genetic or acquired LQT2) prolongs APD in a relevant manner, we explored other methods, such as increasing late Na current (INaL), to test whether the results were IKr-dependent. Likewise, we explored alternative mechanisms to prevent spontaneous SR Ca release (other than shifting RyR2 EC50). None of these influenced the mechanistic conclusions of our study (see below).
Results
ICaL-mediated transient EADs
In the original SS model, the time constants of IKs and ICaL increase in response to ISO are almost identical (Fig 1B), and in this case no EADs were observed during abrupt ISO application (Fig. 2A, top), and APD smoothly shortened after the first beat (grey in Fig. 2B). However, with more realistic differences of these time courses (Fig 1C) prominent EADs appeared transiently upon ISO application (Fig. 2A, bottom). APD increased progressively (black in Fig. 2B), until EADs occur from the 4th to 10th beat, where the PKA-dependent increase of ICaL is further advanced than that of IKs (red shaded area in Fig. 1C). The point of ICaL reactivation precedes EAD takeoff (marked by the dashed line in Fig. 2C), which is followed by the SR Ca release. This indicates that the secondary Ca release is not spontaneous but triggered by Ca entry via ICaL. EADs persist for 7 beats although APD decreases progressively due to increasing IKs. As IKs enhancement overcomes the larger ICaL at the 11th beat, EADs are suppressed, and at steady state the APD is much shorter (Fig 2B). This simulation demonstrates that transient EADs occurring during ISO application are caused by ICaL reactivation in a time window where a large enough mismatch exists between LCCp and IKsp. Fig. 2D shows that as the time constant for IKs (τIKs) is made slower than 14.2s, EADs start appearing as the kinetic mismatch with ICaL grows (τLCCp =7.7s; Fig. S1D). The larger the difference, the higher number of transient EADs.
Figure 2. Abrupt ISO application induces ICaL-mediated transient EADs due to differential PKA phosphorylation kinetics of ICaL vs. IKs.

A. Vm traces for the original SS model and after modifying IKs activation kinetics upon ISO application (* transient EADs). B. APD transiently prolongs (* EADs) and then shortens at steady state. C. EADs are caused by reactivation of ICaL (dashed vertical line), which occurs before EAD takeoff and causes SR Ca release. D. Number of total EADs (black) and maximum transient APD (grey) upon ISO application.
We also explored whether this kinetic regulatory mismatch between ICaL and IKs would persist during a more gradual rise in ISO concentration ([ISO]), whereby [ISO] increases gradually with time from 0 μM to 1 μM (Fig. 3A). For example, Fig. 3 shows that even a stepwise increase in [ISO] fails to generate EADs. Here [ISO] was increased to 5, 10 and 1000 nM for 2 min each (Fig. 3A). During this protocol, peak ICaL and IKs changes are shown in Fig. 4B. At the beginning of each [ISO] step, APD is first prolonged and then shortened without EADs (Fig. 3C). In this gradual application, the transient increase of both ICaL and IKs are attenuated (Fig. 3B). Fig. 4 shows APD, ICaL and IKs for three stages of the abrupt (left) vs. gradual (right) ISO application. With the progressive [ISO] increase maximal ICaL occurs later (c vs. b) at a time when there is already substantial IKs activation, which limits ICaL-induced APD prolongation and EAD opportunity. Therefore, we can extrapolate that slowing the ISO-induced ICaL enhancement (at any upstream step in the signaling cascade) might abolish transient EADs.
Figure 3. Gradual ISO application fails to induce EADs.
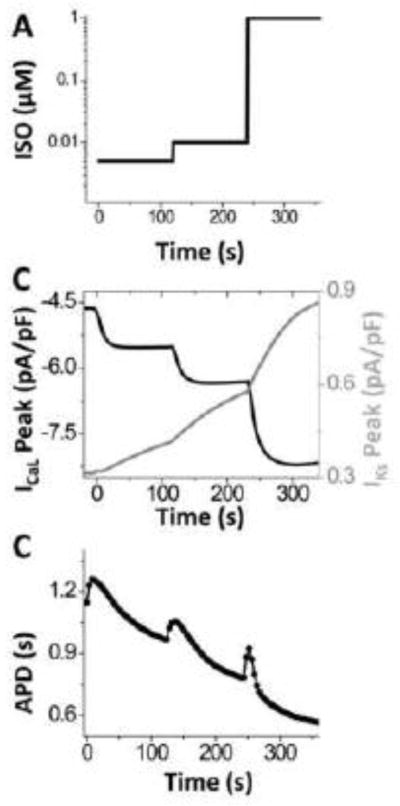
A. Protocol of gradual application of ISO. B. Time courses of peak ICaL and IKs during gradual ISO application. C. APD changes during gradual ISO application. While APD prolongs, no EADs occur.
Figure 4. Comparison between sudden and gradual ISO application.

AP (top), ICaL (middle) and IKs (bottom) before (black) and after (dotted) ISO application at steady state, and during the 1st EAD (grey) with abrupt (A) and gradual (B, as in Fig. 3A) ISO application. Inset in B (top) shows APs before and after gradual ISO application (occurring at time 0). Removal of time delay between ICaL and IKs phosphorylation by gradual ISO application abolishes EADs.
Effects of phosphorylation of PLB, RyR, and PLM on ICaL-mediated transient EADs
As ICaL recovery can be affected by [Ca]i, we also test how phosphorylation of the other PKA targets might influence transient EADs. Experimental data [29, 30] have shown that the time constants of PLB phosphorylation and PKA-dependent activation of INaK (due to PLM phos-phorylation) are ~10–15 s. Consistent with these observations, PLB and PLM phosphorylation kinetics are intermediate between ICaL and IKs in our model (Fig. S2D). Fig. 5A shows that preventing PLB phosphorylation slightly prolongs APD and increases EAD incidence (right panel). Without PLB phosphorylation SERCA function will not increase, which limits [Ca]SR and the rise in peak [Ca]i (green in Fig. S3F and B). This smaller [Ca]i contributes to less Ca-dependent ICaL inactivation, very slightly higher peak ICaL (green in Fig. S3A), which causes prolonged transient and steady state APDs vs. control (Fig. 5A, left). The greater integrated inward ICaL appears to dominate the reduced inward INCX (green in Fig S3E) with respect to APD.
Figure 5. Effects of PLB, PLM and RyR phosphorylation on ICaL-mediated transient EADs.

A. Inhibition of PLB phosphorylation prolongs APD (left) and increases EAD occurrence (* right). B. PLM phosphorylation reduces ISO effect of prolonging APD and decreases EAD occurrence. Only 1 EAD occurs at 6th beat after ISO application. C. Inhibition of RyR phosphorylation has little effect on APD prolongation and EAD formation.
Inclusion of PLM phosphorylation decreases APD and transient EADs (vs. control, Fig. 5B) due to a rapid increase of INaK (Fig. S3D, red). Although ICaL is temporarily larger (Fig. S3A, red), due to slightly lower [Ca]i (Fig. S3B, red) caused by activation of INCX following [Na]i decrease (Fig. S3C, red), the increase of INaK adds repolarizing current and helps limit APD while IKs is still growing. Slowing PLM phosphorylation (Fig. S4B) slows INaK enhancement and fails to suppress transient EADs. [Na]i changes more slowly than does INaK, but that leads to a progressive increase in INCX (Fig. S3E, red) and a secondary decrease in INaK which approaches a new steady state in parallel to the slower [Na]i changes. RyR phosphorylation in the cleft is fast, similar to ICaL (Fig. S2D), but inhibition of RyR phosphorylation has little effect on [Ca]i (Fig. S3B blue). Consequently it has almost no effect on ICaL or EAD formation (Fig. 5C). In this condition, the elevation in [Ca]SR is balanced by the reduced sensitivity of RyR (due to inhibition of RyR phosphorylation), leading to little effect in the Ca transient amplitude.
Influence of the heart rate acceleration by ISO
β-adrenergic stimulation also influences heart rate by effects on sino-atrial node cells (SANC). We used experimental results by Tanaka et al [31] on SANC firing in response to abrupt 10 μM ISO application (Fig. 6A inset), to control PCL in our ventricular myocyte model (Fig. 6A). This is justified by the fact that dose-dependency of heart rate acceleration by ISO saturates at 1 μM [32, 33], which is the [ISO] simulated here. In the experiment by Tanaka et al., ISO increased heart rate by 36.5% with a half time of 9.5 s at physiological rates [31]. We simulated both 36.5% (black) and 100% (red) acceleration on heart rate and found that even when heart rate is accelerated concomitant with the other PKA effects, the induction of potentially arrhythmogenic transient EADs was unaltered (Figs. 6B and C). As we reduced PCL from 4 to 1.5 s (Fig. 6D), there is relative large PCL window where transient EADs occur (Fig. 6D).
Figure 6. Transient EADs still occur at faster heart rate.

A. Simulated acceleration of heart rate (PCL reduction) by 1 μM ISO mimics experiment data (inset, dots are experimental data[31]; solid line is sigmoidal fit). B–C. The total number of EADs and the corresponding maximal APD are not appreciably affected by acceleration in PCL by 36.5% (black) and 100% (red). D. This reveals a wide PCL window that is permissive of EAD formation.
SR Ca-release-mediated transient EADs
When we restored the normal SS model (normal RyR2 gating) and only blocked IKr by 40% to prolong APD (and retain the small enhancement of ICa as above) there were no EADs present prior to ISO addition. That was also true if ICaL and IKs activation kinetics were similar (as in the SS model; Fig. 7A, top and 7B, gray). However, when using the realistic kinetic difference between ICaL and IKs phosphorylation, ISO addition again led to a train of transient EADs (Fig. 7A, bottom and Fig. 7B, black). In this case the EADs were due to SR Ca release because the EAD takeoff (dotted line in Fig. 7D) occurs after SR Ca release and before the slowing of ICaL decline. No net reactivation of ICaL was observed during EADs, indicating that these transient EADs are due to spontaneous SR Ca release and INCX. The increase in local [Ca]i and more positive Em should both accelerate ICaL decline, so the slowing of ICaL decline may reflect a synergy between INCX and ICaL to produce a larger EAD than would the small INCX at this voltage.
Figure 7. Induction of SR Ca-release-mediated EADs upon ISO application.

A. APs during ISO application for the SS model (top) and when IKs activation is slowed (bottom). B. APD for simulation in A, EADs occur at all APs between lines. C. Number of total EADs (black) and maximum transient APD (grey) upon ISO application as IKs activation is slowed. D. EADs are caused by SR Ca release (which starts before the dashed vertical line), which occurs before EAD takeoff and activates inward INCX.
The mismatch between ICaL and IKs phosphorylation kinetics allows a transient prolongation of APD upon abrupt ISO application. This allows the rapid increase in Ca influx (via ICaL) and SR Ca uptake to increase cellular Ca gain and [Ca]SR (Fig. S5B and F black). The ability of NCX to compete with SERCA is also limited at more positive Em during the long APD and this exacerbates the cellular and SR Ca gain. As [Ca]SR reaches the threshold for spontaneous Ca release, spontaneous Ca releases occur from the 3rd beat after ISO application (bar in Fig. S5B). This Ca release and INCX is not large enough to produce net depolarization at the plateau Em at this beat, but further prolongs APD, favoring further Ca gain. EADs start to occur from the 5th beat, where the accumulated [Ca]SR and release-induced INCX is large enough to produce net depolarization during the plateau (Fig. 7D). EADs continue until the 24th beat as enhanced IKs causes APD shortening (Fig. 7B black), and spontaneous Ca release stops when [Ca]i falls below the level supporting that release. Without the kinetic mismatch of PKA mediated increase of ICaL vs. IKs, the APD smoothly shortens after the 1st beat (Fig. 7B grey) and Ca loading is limited such that [Ca]SR remains below the spontaneous release threshold. Thus, no spontaneous SR Ca releases or EADs occur. The larger the kinetic mismatch is for PKA dependent increase of ICaL vs. IKs, the more transient EADs occurs (Fig. 7C).
Unlike ICaL-mediated transient EADs, SR Ca-release-mediated EADs can be significantly modulated by PLB or PLM. Slowing of PLB phosphorylation decreases transient EAD incidence (Fig. S7A). Complete dephosphorylation of PLB abolishes EADs (Fig. S6A) as [Ca]SR does not rise as much and no spontaneous Ca releases occur (Fig. S5F, green). The inclusion of PLM phosphorylation reduces EAD incidence (Fig. S6B) as it removes Na and Ca from the cell and thus reduces [Ca]SR (Fig. S5 red). Accelerating PLM phosphorylation significantly decreases EAD incidence (Fig. S7B), but does not abolish them, presumably because the decline in [Na]i is much slower than the rise in [Ca]i and [Ca]SR (Fig. S5C vs. B red). RyR dephosphorylation reduces the incidence of transient EADs (Fig. S6C) because the threshold [Ca]SR for these events is increased.
Discussion
EADs are recognized as a cellular cause of arrhythmias and sudden cardiac death [34, 35]. Theoretical studies have helped to understand the dynamic mechanisms underlying EADs [13, 14], but they have mainly focused on the steady state. Here we focus on the transient state, upon acute perturbation with β-AR activation which is known to be a trigger for arrhythmias. This area has been largely overlooked, in part because computational models with realistic kinetics for signaling cascades are limited. Here we use such a model [15, 24] and show how acute βAR stimulation can cause transient AP prolongation and EADs (as observed experimentally [17–19]) that are potentially arrhythmogenic, even though these EADs can be eliminated at steady state. We examine how kinetic differences among several key PKA targets may make this acute transition especially dangerous, and indeed EADs via both of the known mechanisms may be involved (ICaL reactivation and SR Ca release).
Our key findings are: 1) Incorporating measured differential kinetics of ICaL and IKs phos-phorylation into the SS rabbit ventricular myocyte model recapitulated experimentally observed transient EADs following ISO administration (in conditions of prolonged repolarization)[19]. 2) EAD occurrence was prevented by more gradual β-AR activation. 3) This kinetic mismatch was key for transient EADs mediated by both ICaL reactivation and SR Ca release. 4) ICaL-mediated transient EADs were enabled by AP prolongation, but not much influenced by preventing PLB or RyR phosphorylation. However, a rapid increase in outward INaK due to PLM phosphorylation was protective with respect to transient EADs. 5) SR Ca-release-mediated transient EADs occur due to progressive SR Ca loading by rapid increases in ICaL and SR Ca uptake during long APs, before the increase in IKs limits APD. These SR Ca-release-mediated EADs can be blunted by PLB and RyR dephosphorylation and by PLM phosphorylation (due mainly to reduced [Na]i and [Ca]SR). Thus, while the steady state co-activation of ICaL, IKs, SERCA, RyR and NKA by PKA can stabilize the inotropic state and APD shape induced by β-AR activation, kinetic mismatches create unique windows of vulnerability during acute activation.
Two types of EADs induced by β-AR activation
The importance of β-AR signaling in hearts has been well documented [1, 12, 36], but the signaling network is complex and includes feedback mechanisms, including that PKA contributes to β-AR desensitization [2] but also enhances cAMP levels by effects of Inhibitor-1 to reduce phosphatase 1 activity [37], as included in the model. Acute PKA activation can rapidly enhance Ca transients (as critical for rapid inotropy and lusitropy), but more slowly via PLM phosphorylation PKA limits the rise in [Na]i that occurs as an integral part of the fight-or-flight response with β-AR [38]. That NKA activation was shown to limit arrhythmogenic events in the steady state, but the slower changes in [Na]i could again leave this vulnerable window during acute sympathetic activation. Our results here agree with this notion.
EADs tend to occur at slow pacing rate, where repolarization is prolonged [35]. We use slow pacing and IKr block, as done in some experiments to understand EAD formation [19]. This may relate to bradycardias, such as, sick sinus node or atrial-ventricular node disorder [39]. EADs occur over a large range of PCLs in both experimental and theoretical studies [11, 18, 40].
EADs have been shown to be induced by 1) ICaL reactivation (EAD1) or 2) SR Ca release and activation of INCX (EAD2) [18, 41, 42]. EAD2s are thus mechanistically the same as for delayed afterdepolarizations (DADs) except that for DADs the inward INCX current for a given SR Ca release is larger because the INCX driving force is much stronger at more negative Em (vs. EADs during the plateau) [1]. EAD1s occur at slow pacing rates and long APD (e.g. in genetic or drug-induced LQT), while EAD2s are more likely when cell and SR Ca load are increased, typically at faster heart rates and with β-AR activation [43]. Notably, long APDs prolong Ca entry via ICaL and limit Ca extrusion via NCX both during the AP and in the shortened time at diastolic Vm, all of which favor SR Ca loading. This degree of Ca loading may not cause spontaneous SR Ca release. However, ISO rapidly increases Ca influx and SR Ca uptake and greatly enhances the likelihood of spontaneous SR Ca release events (EAD2s) during these long APs, and that can be exacerbated by RyR sensitization. We show that acute (but not gradual) β-AR activation can induce transient appearance of both EAD1s and EAD2s under the conditions studied. If we had not examined differential target activation kinetics, these effects would not have been seen.
We used different baseline conditions to demonstrate the transient appearance of EAD1s and EAD2s separately upon β-AR activation to investigate the underlying mechanisms. Whereas the EAD1s may be more likely to occur in bradycardia and in conditions of prolonged repolarization (e.g., Long QT Syndrome), the latter could be more relevant to faster heart rates (e.g., Catecholaminergic Polymorphic Ventricular Tachycardia), although they may coexist when the SR is overloaded [18]. As one may imagine, making the conditions favoring EAD1s or EAD2s stronger (e.g. longer APD) makes the transient EADs continue for more beats or become chronic. So in that sense the extent of GKr reduction in the two cases highlighted shows that this transient EAD occurrence happens under conditions where they would not occur in steady state. This may well explain why arrhythmias are sometimes transiently observed immediately after strong β-AR activation (e.g. [44]). These initiating events can also lead to life-threatening arrhythmias already in that vulnerable period.
Modeling of transient EAD1 vs. transient EAD2
For the EAD2 studies (Fig. 7) we used the baseline SS model with the only change being the 15% increase in ICaL at baseline (as in the EAD1 case; Fig. 2–6). If we reduce ICaL back to the control SS model, EAD2s were still transiently observed, although slightly fewer than the example in Fig 7. While the SS model demonstrates EAD2s, we limit our mechanistic analysis here because the SR Ca release events, which reflect Ca waves in myocytes, are not ideally modeled by common pool Ca models as used here. More detailed Ca wave induction and propagation will require spatially detailed intracellular models [45], because thousands of individual junctions are independently regulated and exert only local effects (unlike voltage gated channels which influence and are influenced by Vm cell-wide).
Studying the EAD1 mechanism separately during ISO exposure (Figs. 2–6) required: 1) preventing spontaneous SR Ca release events and 2) prolonging APD (to allow for ICaL recovery and reactivation). To prevent SR Ca release, we raised the Ca threshold for spontaneous SR Ca release. We also slightly increased basal ICaL to ensure realistic ECC efficacy with the desensitized RyR2, and INCX to maintain Ca flux balance and limiting SR Ca load. Notably, spontaneous Ca release can also be prevented by either doubling RyR passive leak or decreasing SR Ca uptake by 55%, as in the context of heart failure. Using either of those methods to limit SR Ca load still yielded transient EAD1s upon abrupt ISO application (not shown). The increased RyR passive leak also required INCX enhancement (250%) to limit diastolic [Ca]i rise. So, any method of preventing spontaneous SR Ca release suffices for requirement 1. To prolong APD and favor ICaL recovery at plateau Vm (requirement 2) [46, 47], we blocked IKr, as done pharmacologically or genetically in experimental EAD studies [19, 26]. Again, we still obtained transient EADs with abrupt ISO exposure with increased INaL as in LQT3 (not shown). However, enhanced INaL elevates diastolic [Na]i, [Ca]i and SR Ca load potentiating spontaneous Ca release, thereby complicating the separation between EAD1s and EAD2s. Since IKs activation kinetics is central to this study, we did not reduce that. So, this vulnerable period for β-AR-induced EADs is inherent to the differential ICa,L and IKs activation kinetics and not the details of the parameter settings. The model also allowed us to compare the impact of PKA kinetics at IKs, RyR, PLB and PLM on the appearance of EAD1s.
ISO induces Slower IKs vs. ICa activation and is modulated by other PKA targets
The faster ICa activation vs. IKs [19] suffices to drive the transient appearance of both EAD1s and EAD2s during ISO exposure. For EAD1s neither phosphorylation of RyR and PLB nor a wide range of phosphorylation kinetics had appreciable effects (Fig. 5 and S4). Without PLB phosphorylation the longest APD was higher, because the limited rise in SR Ca release caused less ICaL inactivation, but the number of EAD1s was not altered. In contrast, allowing PLM phos-phorylation suppressed EAD1s, and slowing PLM phosphorylation blocked this protective effect (Fig. 5 and S4). The majority of this protective effect was due to enhanced outward INaK and less APD prolongation, rather than effects on [Na]i or [Ca]i.
EAD2s depend upon increased SR Ca loading, and the fast increase of both ICaL and PLB phosphorylation contribute to this effect, because they load the SR (Fig. 7 and S6–7). Moreover, slowing PLB phosphorylation reduces the occurrence of transient EAD2s. Removing or slowing RyR phosphorylation had remarkably modest effects on EAD2s. Adding PLM phos-phorylation had little effect early (despite enhanced INaK), but could limit APD and EAD2s late, primarily because of lower [Na]i and consequently lower [Ca]SR (via NCX function). Thus the kinetics and extent of ICaL, IKs, PLB and PLM phosphorylation can all influence the occurrence of transient EADs, but the role of RyR phosphorylation seems limited.
Therefore, drugs or therapeutic strategies that tend to slow ICaL and/or speedup PLM phosphorylation by PKA might be therapeutically beneficial with respect to limiting EAD-related arrhythmias, and this might explain why some antiarrhythmic drugs have been ineffective [48]. IKs in the model is enhanced by elevated submembrane [Ca], but the sensitivity is high (Kd =38 nM) [49]. Thus, alterations in Ca transients do not appreciably alter IKs activation kinetics with ISO application.
Here we show that gradual ISO application, slowing β-AR activation upstream of PKA, abolishes transient EADs by reducing the kinetic mismatch between ICaL and IKs. Drugs or endo-genous proteins that speed up the upstream PKA activity will tend to increase the incidence of transient EADs, by amplifying the mismatch between ICaL and IKs. Conversely, agents that slow the activation of PKA could be protective against the type of transient EADs we describe here.
It is known that cAMP levels rise in distinct subcellular regions with β-AR stimulation [3, 36] and that target phosphorylation is also controlled by phosphatases, which are also in distinct microdomains [50]. While further refinement of the effective compartmentalization in the model [2, 15] could provide insight on cAMP compartmentalization, some details are controversial and it would require major model redesign and experimental validation. More importantly here, it would not alter our conclusions. The reason for that is that our model already reproduces the experimentally measured effects of (compartmentalized) β-AR activation on all of the ionic currents (e.g. IKs and ICaL), Ca-related aspects (e.g. SERCA and myofilament Ca buffering) and EADs, thereby functionally accounting for overall cAMP signaling.
Our focus here was on β-AR signaling, but the SS model includes CaMKII signaling (which we had disabled). Since CaMKII can also induce spontaneous Ca release-related arrhythmias when it is overexpressed [15], we tested whether including CaMKII effects would modify our conclusions. Figure S8 shows that a normal functional level of CaMKII slightly increased ICaL and APDs, and exacerbates the appearance of transient EADs (EAD1s and EAD2s) upon abrupt ISO application. However, CaMKII neither prevented EADs nor made them chronic under these conditions.
Fight-or-flight response
During the fight-or-flight response, norepinephrine is released locally within the myocardium and rapidly activates myocyte β-ARs. Under conditions of prolonged APD, such as LQT syndrome (genetic or drug-induced) or heart failure, we have shown that there is a sort of vulnerable period created by the temporal mismatch between ICa and IKs activation. Moreover, the APD shortening that is expected because of the concomitant increase in heart rate, does not prevent this vulnerable period for EAD induction (Fig. 6).
EADs are nonlinearly rate-dependent [11, 14, 40], but occur over a range of CLs [9, 11, 40]. Thus, EADs can still occur during heart rate acceleration. Indeed, the increase of ICaL may occur faster than the steady heart rate change upon adrenergic stimulation which takes several seconds [31, 51], creating a brief time window where EADs could also occur at long cycle lengths. Transient EADs are likely within a large CL window (Fig. 6D) as shown by both experimental and theoretical studies [9, 40]. Of course, as heart rate increases and when APD is short, this CL window for EADs shrinks and disappears. Therefore, this vulnerable period for EADs may not occur in the normal healthy heart during normal fight-or-flight responses, but may be a heretofore underappreciated problem in pathological conditions where acceleration of the (slow) heart rate alone by β-adrenergic stimulation fails to prevent EADs and arrhythmias [52].
The impact of cellular EADs like these at tissue level with respect to arrhythmogenesis has been previously examined [11, 53, 54]. All cells might respond as described here, which would create global EADs and QT prolongation. Additionally, heterogeneity (e.g. associated with local sympathetic activation) could create greater dispersion of repolarization and regional EAD islands where a critical number of cells would need to exhibit EADs synchronously to allow propagation to the whole tissue. Indeed, a local β-AR stimulation can induce PVC by synchronizing afterdepolarizations regionally and inducing whole heart arrhythmias [55].
In conclusion, our study provides a novel perspective for the mechanism of EAD formation via signaling pathways and may contribute to anti-arrhythmic drugs development. For example, targeting ion channels that have a slow time course is less likely to eliminate arrhythmias caused by transient EADs.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Research highlights.
We model differential PKA phosphorylation kinetics of ICaL vs IKs on transient EADs.
A mismatch between ISO-induced ICaL and IKs increase is key for transient EADs.
PLB dephosphorylation barely affects transient EAD1s, but reduces EAD2s.
PLM phosphorylation attenuates transient EAD1s and EAD2s by different mechanisms.
Acknowledgments
The authors thank Dr. CE Clancy for comments.
Sources of support:
Supported by National Institutes of Health grants K99-HL111334 (D.S.) and R37-HL30077 and P01-80101 (D.M.B.).
Footnotes
Disclosures: none declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 2.Saucerman JJ, Brunton LL, Michailova AP, McCulloch AD. Modeling beta-adrenergic control of cardiac myocyte contractility in silico. J Biol Chem. 2003;278:47997–8003. doi: 10.1074/jbc.M308362200. [DOI] [PubMed] [Google Scholar]
- 3.Saucerman JJ, Zhang J, Martin JC, Peng LX, Stenbit AE, Tsien RY, et al. Systems analysis of PKA-mediated phosphorylation gradients in live cardiac myocytes. Proc Natl Acad Sci U S A. 2006;103:12923–8. doi: 10.1073/pnas.0600137103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanich RF, Levine JH, Spear JF, Moore EN. Autonomic modulation of ventricular arrhythmia in cesium chloride-induced long QT syndrome. Circulation. 1988;77:1149–61. doi: 10.1161/01.cir.77.5.1149. [DOI] [PubMed] [Google Scholar]
- 5.Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440:463–9. doi: 10.1038/nature04710. [DOI] [PubMed] [Google Scholar]
- 6.Gilmour RF, Jr, Moïse S. Triggered activity as a mechanism for inherited ventricular arrhythmias in German shepherd dogs. Journal of the American College of Cardiology. 1996;27:1526–33. doi: 10.1016/0735-1097(95)00618-4. [DOI] [PubMed] [Google Scholar]
- 7.Song Y, Thedford S, Lerman BB, Belardinelli L. Adenosine-sensitive afterdepolarizations and triggered activity in guinea pig ventricular myocytes. Circulation Research. 1992;70:743–53. doi: 10.1161/01.res.70.4.743. [DOI] [PubMed] [Google Scholar]
- 8.Morotti S, Grandi E, Summa A, Ginsburg KS, Bers DM. Theoretical Study of L-type Ca2+ Current Inactivation Kinetics during Action Potential Repolarization and Early Afterdepolarizations. The Journal of Physiology. 2012 doi: 10.1113/jphysiol.2012.231886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tanskanen AJ, Greenstein JL, O’Rourke B, Winslow RL. The Role of Stochastic and Modal Gating of Cardiac L-Type Ca2+ Channels on Early After-Depolarizations. Biophysical Journal. 2005;88:85–95. doi: 10.1529/biophysj.104.051508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clancy CE, Rudy Y. Linking a genetic defect to its cellular phenotype in a cardiac arrhythmia. Nature. 1999;400:566–9. doi: 10.1038/23034. [DOI] [PubMed] [Google Scholar]
- 11.Sato D, Xie L-H, Sovari AA, Tran DX, Morita N, Xie F, et al. Synchronization of chaotic early afterdepolarizations in the genesis of cardiac arrhythmias. Proceedings of the National Academy of Sciences. 2009;106:2983–8. doi: 10.1073/pnas.0809148106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saucerman JJ, Healy SN, Belik ME, Puglisi JL, McCulloch AD. Proarrhythmic consequences of a KCNQ1 AKAP-binding domain mutation: computational models of whole cells and heterogeneous tissue. Circ Res. 2004;95:1216–24. doi: 10.1161/01.RES.0000150055.06226.4e. [DOI] [PubMed] [Google Scholar]
- 13.Luo CH, Rudy Y. A dynamic model of the cardiac ventricular action potential. II. Afterdepolarizations, triggered activity, and potentiation. Circulation Research. 1994;74:1097–113. doi: 10.1161/01.res.74.6.1097. [DOI] [PubMed] [Google Scholar]
- 14.Tran DX, Sato D, Yochelis A, Weiss JN, Garfinkel A, Qu Z. Bifurcation and Chaos in a Model of Cardiac Early Afterdepolarizations. Physical Review Letters. 2009;102:258103. doi: 10.1103/PhysRevLett.102.258103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soltis AR, Saucerman JJ. Synergy between CaMKII substrates and beta-adrenergic signaling in regulation of cardiac myocyte Ca2+ handling. Biophys J. 2010;99:2038–47. doi: 10.1016/j.bpj.2010.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang JH, Saucerman JJ. Computational models reduce complexity and accelerate insight into cardiac signaling networks. Circ Res. 2011;108:85–97. doi: 10.1161/CIRCRESAHA.110.223602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shimizu W, Antzelevitch C. Differential effects of beta-adrenergic agonists and antagonists in LQT1, LQT2 and LQT3 models of the long QT syndrome. Journal of the American College of Cardiology. 2000;35:778–86. doi: 10.1016/s0735-1097(99)00582-3. [DOI] [PubMed] [Google Scholar]
- 18.Volders PGA, Kulcsár A, Vos MA, Sipido KR, Wellens HJJ, Lazzara R, et al. Similarities between early and delayed afterdepolarizations induced by isoproterenol in canine ventricular myocytes. Cardiovascular Research. 1997;34:348–59. doi: 10.1016/s0008-6363(96)00270-2. [DOI] [PubMed] [Google Scholar]
- 19.Liu G-X, Choi B-R, Ziv O, Li W, de Lange E, Qu Z, et al. Differential conditions for early after-depolarizations and triggered activity in cardiomyocytes derived from transgenic LQT1 and LQT2 rabbits. The Journal of Physiology. 2012;590:1171–80. doi: 10.1113/jphysiol.2011.218164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saucerman JJ, McCulloch AD. Mechanistic systems models of cell signaling networks: a case study of myocyte adrenergic regulation. Progress in Biophysics and Molecular Biology. 2004;85:261–78. doi: 10.1016/j.pbiomolbio.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 21.Shannon TR, Wang F, Puglisi J, Weber C, Bers DM. A Mathematical Treatment of Integrated Ca Dynamics within the Ventricular Myocyte. Biophysical Journal. 2004;87:3351–71. doi: 10.1529/biophysj.104.047449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marx SO, Kurokawa J, Reiken S, Motoike H, D’Armiento J, Marks AR, et al. Requirement of a Macromolecular Signaling Complex for β Adrenergic Receptor Modulation of the KCNQ1-KCNE1 Potassium Channel. Science. 2002;295:496–9. doi: 10.1126/science.1066843. [DOI] [PubMed] [Google Scholar]
- 23.Hulme JT, Lin TW-C, Westenbroek RE, Scheuer T, Catterall WA. β-Adrenergic regulation requires direct anchoring of PKA to cardiac CaV1.2 channels via a leucine zipper interaction with A kinase-anchoring protein 15. Proceedings of the National Academy of Sciences. 2003;100:13093–8. doi: 10.1073/pnas.2135335100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang JH, Saucerman JJ. Phospholemman is a negative feed-forward regulator of Ca2+ in β-adrenergic signaling, accelerating β-adrenergic inotropy. Journal of Molecular and Cellular Cardiology. 2012;52:1048–55. doi: 10.1016/j.yjmcc.2011.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Antzelevitch C, Shimizu W, Yan G-X, Sicouri S, Weissenburger J, Nesterenko VV, et al. The M Cell. Journal of Cardiovascular Electrophysiology. 1999;10:1124–52. doi: 10.1111/j.1540-8167.1999.tb00287.x. [DOI] [PubMed] [Google Scholar]
- 26.Choi B-R, Burton F, Salama G. Cytosolic Ca2+ triggers early afterdepolarizations and torsade de pointes in rabbit hearts with type 2 long QT syndrome. The Journal of Physiology. 2002;543:615–31. doi: 10.1113/jphysiol.2002.024570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alseikhan BA, DeMaria CD, Colecraft HM, Yue DT. Engineered calmodulins reveal the unexpected eminence of Ca2+ channel inactivation in controlling heart excitation. Proceedings of the National Academy of Sciences. 2002;99:17185–90. doi: 10.1073/pnas.262372999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yuan W, Bers DM. Protein kinase inhibitor H-89 reverses forskolin stimulation of cardiac L-type calcium current. American Journal of Physiology - Cell Physiology. 1995;268:C651–C9. doi: 10.1152/ajpcell.1995.268.3.C651. [DOI] [PubMed] [Google Scholar]
- 29.Kockskämper J, Erlenkamp S, Glitsch HG. Activation of the cAMP protein–kinase A pathway facilitates Na+ translocation by the Na+–K+ pump in guinea-pig ventricular myocytes. The Journal of Physiology. 2000;523:561–74. doi: 10.1111/j.1469-7793.2000.t01-2-00561.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Valverde CA, Mundiña-Weilenmann C, Said M, Ferrero P, Vittone L, Salas M, et al. Frequency-dependent acceleration of relaxation in mammalian heart: a property not relying on phospholamban and SERCA2a phosphorylation. The Journal of Physiology. 2005;562:801–13. doi: 10.1113/jphysiol.2004.075432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tanaka H, Clark RB, Giles WR. Positive Chronotropic Responses of Rabbit Sino-Atrial Node Cells to Flash Photolysis of Caged Isoproterenol and Cyclic AMP. Proceedings of the Royal Society of London Series B: Biological Sciences. 1996;263:241–8. doi: 10.1098/rspb.1996.0038. [DOI] [PubMed] [Google Scholar]
- 32.Joung B, Tang L, Maruyama M, Han S, Chen Z, Stucky M, et al. Intracellular Calcium Dynamics and Acceleration of Sinus Rhythm by β-Adrenergic Stimulation. Circulation. 2009;119:788–96. doi: 10.1161/CIRCULATIONAHA.108.817379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park S, Park H, Hwang HJ, Shim J, Sung JH, Kim JY, et al. Heart Rate Acceleration of a Subsidiary Pacemaker by β-Adrenergic Stimulation. Korean Circ J. 2011;41:658–65. doi: 10.4070/kcj.2011.41.11.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maruyama M, Lin S-F, Xie Y, Chua S-K, Joung B, Han S, et al. Genesis of Phase 3 Early Afterdepolarizations and Triggered Activity in Acquired Long-QT Syndrome/Clinical Perspective. Circulation: Arrhythmia and Electrophysiology. 2011;4:103–11. doi: 10.1161/CIRCEP.110.959064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yan G-X, Wu Y, Liu T, Wang J, Marinchak RA, Kowey PR. Phase 2 Early Afterdepolarization as a Trigger of Polymorphic Ventricular Tachycardia in Acquired Long-QT Syndrome. Circulation. 2001;103:2851–6. doi: 10.1161/01.cir.103.23.2851. [DOI] [PubMed] [Google Scholar]
- 36.Heijman J, Volders PGA, Westra RL, Rudy Y. Local control of β-adrenergic stimulation: Effects on ventricular myocyte electrophysiology and Ca2+ transient. Journal of Molecular and Cellular Cardiology. 2011;50:863–71. doi: 10.1016/j.yjmcc.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nicolaou P, Hajjar RJ, Kranias EG. Role of protein phosphatase-1 inhibitor-1 in cardiac physiology and pathophysiology. Journal of Molecular and Cellular Cardiology. 2009;47:365–71. doi: 10.1016/j.yjmcc.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Despa S, Tucker AL, Bers DM. Phospholemman-Mediated Activation of Na/K-ATPase Limits [Na]i and Inotropic State During β-Adrenergic Stimulation in Mouse Ventricular Myocytes. Circulation. 2008;117:1849–55. doi: 10.1161/CIRCULATIONAHA.107.754051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aziz PF, Tanel RE, Zelster IJ, Pass RH, Wieand TS, Vetter VL, et al. Congenital long QT syndrome and 2:1 atrioventricular block: An optimistic outcome in the current era. Heart Rhythm. 2010;7:781–5. doi: 10.1016/j.hrthm.2010.02.035. [DOI] [PubMed] [Google Scholar]
- 40.Sato D, Xie L-H, Nguyen TP, Weiss JN, Qu Z. Irregularly Appearing Early Afterdepolarizations in Cardiac Myocytes: Random Fluctuations or Dynamical Chaos? Biophysical Journal. 2010;99:765–73. doi: 10.1016/j.bpj.2010.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Volders PGA, Vos MA, Szabo B, Sipido KR, de Groot SHM, Gorgels APM, et al. Progress in the understanding of cardiac early afterdepolarizations and torsades de pointes: time to revise current concepts. Cardiovascular Research. 2000;46:376–92. doi: 10.1016/s0008-6363(00)00022-5. [DOI] [PubMed] [Google Scholar]
- 42.Clusin WT. Calcium and Cardiac Arrhythmias: DADs, EADs, and Alternans. Critical Reviews in Clinical Laboratory Sciences. 2003;40:337–75. doi: 10.1080/713609356. [DOI] [PubMed] [Google Scholar]
- 43.Pogwizd SM, Bers DM. Cellular Basis of Triggered Arrhythmias in Heart Failure. Trends in Cardiovascular Medicine. 2004;14:61–6. doi: 10.1016/j.tcm.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 44.Myles RC, Wang L, Kang C, Bers DM, Ripplinger CM. Local β-Adrenergic Stimulation Overcomes Source-Sink Mismatch to Generate Focal Arrhythmia/Novelty and Significance. Circulation Research. 2012;110:1454–64. doi: 10.1161/CIRCRESAHA.111.262345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sato D, Bers DM. How does stochastic ryanodine receptor-mediated Ca leak fail to initiate a Ca spark? Biophys J. 2011;101:2370–9. doi: 10.1016/j.bpj.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu S, Melchert RB, Kennedy RH. Inhibition of L-Type Ca2+ Channel Current in Rat Ventricular Myocytes by Terfenadine. Circulation Research. 1997;81:202–10. doi: 10.1161/01.res.81.2.202. [DOI] [PubMed] [Google Scholar]
- 47.Mahajan A, Shiferaw Y, Sato D, Baher A, Olcese R, Xie L-H, et al. A Rabbit Ventricular Action Potential Model Replicating Cardiac Dynamics at Rapid Heart Rates. Biophysical Journal. 2008;94:392–410. doi: 10.1529/biophysj.106.98160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.A Comparison of Antiarrhythmic-Drug Therapy with Implantable Defibrillators in Patients Resuscitated from Near-Fatal Ventricular Arrhythmias. New England Journal of Medicine. 1997;337:1576–84. doi: 10.1056/NEJM199711273372202. [DOI] [PubMed] [Google Scholar]
- 49.Nitta J, Furukawa T, Marumo F, Sawanobori T, Hiraoka M. Subcellular mechanism for Ca2+-dependent enhancement of delayed rectifier K+ current in isolated membrane patches of guinea pig ventricular myocytes. Circulation Research. 1994;74:96–104. doi: 10.1161/01.res.74.1.96. [DOI] [PubMed] [Google Scholar]
- 50.Christ T, Boknik P, Wöhrl S, Wettwer E, Graf EM, Bosch RF, et al. L-Type Ca2+ Current Downregulation in Chronic Human Atrial Fibrillation Is Associated With Increased Activity of Protein Phosphatases. Circulation. 2004;110:2651–7. doi: 10.1161/01.CIR.0000145659.80212.6A. [DOI] [PubMed] [Google Scholar]
- 51.Khalsa SS, Rudrauf D, Sandesara C, Olshansky B, Tranel D. Bolus isoproterenol infusions provide a reliable method for assessing interoceptive awareness. International Journal of Psychophysiology. 2009;72:34–45. doi: 10.1016/j.ijpsycho.2008.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, et al. Genotype-Phenotype Correlation in the Long-QT Syndrome: Gene-Specific Triggers for Life-Threatening Arrhythmias. Circulation. 2001;103:89–95. doi: 10.1161/01.cir.103.1.89. [DOI] [PubMed] [Google Scholar]
- 53.de Lange E, Xie Y, Qu Z. Synchronization of Early Afterdepolarizations and Arrhythmogenesis in Heterogeneous Cardiac Tissue Models. Biophysical Journal. 2012;103:365–73. doi: 10.1016/j.bpj.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xie Y, Sato D, Garfinkel A, Qu Z, Weiss JN. So little source, so much sink: requirements for afterdepolarizations to propagate in tissue. Biophys J. 2010;99:1408–15. doi: 10.1016/j.bpj.2010.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Myles RC, Wang L, Kang C, Bers DM, Ripplinger CM. Local β-Adrenergic Stimulation Overcomes Source-Sink Mismatch to Generate Focal Arrhythmia. Circulation Research. 2012;110:1454–64. doi: 10.1161/CIRCRESAHA.111.262345. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.