

Abstract
Background
Very-long-chain acyl-CoA dehydrogenase (VLCAD) deficiency (VLCADD) is diagnosed in the US through newborn screening (NBS). NBS often unequivocally identifies affected individuals, but a growing number of variant patterns can represent mild disease or heterozygous carriers.
Aims
To evaluate the validity of standard diagnostic procedures for VLCADD by using functional in vitro tools.
Methods
We retrospectively investigated 13 patient samples referred to our laboratory because of a suspicion of VLCADD but with some uncertainty to the diagnosis. All 13 patients were suspected of having VLCADD either because of abnormal NBS or suggestive clinical findings. ACADVL genomic DNA sequencing data were available for twelve of them. Ten of the patients had an abnormal NBS suggestive of VLCADD, with three samples showing equivocal results. Three exhibited suggestive clinical findings and blood acylcarnitine profile (two of them had a normal NBS and the third one was unscreened). Assay of VLCAD activity and immunoblotting or immunohistologic staining for VLCAD were performed on fibroblasts. Prokaryotic mutagenesis and expression studies were performed for nine uncharacterized ACADVL missense mutations.
Results
VLCAD activity was abnormal in fibroblast cells from 9 patients (8 identified through abnormal NBS, 1 through clinical symptoms). For these 9 patients, immunoblotting/staining showed variable presence of VLCAD; all but one had two mutated alleles. Two patients with equivocal NBS results (and a heterozygous genotype) and the two patients with normal NBS exhibited normal VLCAD activity and normal VLCAD protein on immunoblotting/staining thus ruling out VLCAD deficiency. Nine pathogenic missense mutations were characterized with prokaryotic expression studies and showed a decrease in enzyme activity and variable stability of VLCAD antigen.
Conclusions
These results emphasize the importance of functional investigation of abnormal NBS or clinical testing suggestive but not diagnostic of VLCADD. A larger prospective study is necessary to better define the clinical and metabolic ramifications of the defects identified in such patients.
INTRODUCTION
Very-long-chain acyl-CoA dehydrogenase (VLCAD) deficiency (VLCADD) is a disorder of long chain mitochondrial fatty acid oxidation (FAO) [1, 2] with more than 400 reported patients [3]. VLCAD deficiency can present with a variety of clinical symptoms and a spectrum of severity that ranges from severe life threatening illness in the newborn period to relatively mild disease first developing late in childhood or early adulthood. Two major childhood phenotypes have been recognized [4–6]. The first consists of severe neonatal or early onset disease with recurrent episodes of hypoglycemia, acidosis, hepatic dysfunction, and cardiomyopathy. Patients who survive their initial presentation exhibit progressive cardiomyopathy and have a reported 75% mortality rate in the first few years of life. Children with later onset symptoms can have repeated episodes of hypoketotic hypoglycemia but are at low risk of developing cardiomyopathy, with a resultant lower mortality and better long term prognosis. Sequence analysis of the ACADVL gene has revealed some correlation of mutation genotype with phenotype however, this relationship is imperfect [5, 7]. Not surprisingly, patients with null mutations leading to complete absence of VLCAD tend to have more severe symptoms than those with some residual enzyme activity.
VLCADD is readily identified by newborn screening (NBS) of acylcarnitine profiles from blood spots with tandem mass spectrometry (often referred to as expanded newborn screening) in numerous countries, including the US, and has emerged as the second most common inborn error of fatty acid oxidation [6, 8–12]. The natural history of VLCADD has been radically modified by NBS due to pre-symptomatic treatment, with great improvement in short-term outcome [6, 13–15]. Implementation of such treatment requires that follow up procedures for confirmatory diagnosis for individuals flagged by NBS be as accurate as possible.
The acyl-CoA dehydrogenases (ACADs) are a family of enzymes that catalyze the α,β-dehydrogenation of acyl-CoA esters, transferring electrons to electron transferring flavoprotein (ETF) [16]. At least nine members of this enzyme family have been identified, each with a characteristic substrate specificity profile [17–23]. Very long, medium and short chain acyl-CoA dehydrogenases (VLCAD, MCAD, and SCAD respectively) catalyze the first step in the β-oxidation cycle with substrate optima of 16, 8 and 4 carbon chains respectively. ACAD9 is active against both saturated and unsaturated long chain substrates [24, 25]. The physiologic role of long chain acyl-CoA dehydrogenase (LCAD) remains unknown [26]. All ACADs are encoded in the nuclear genome as a precursor protein and function in the mitochondria [27–29]. While mature VLCAD and ACAD9 are homodimers, the other ACADs are homotetramers with each monomer containing a non-covalently bound flavin adenine dinucleotide molecule (FAD) as a prosthetic group [17, 30, 31]. VLCAD protein shares homology with other ACADs over much of its length, but has an additional 180 amino acids at its carboxy terminus which facilitates its interaction with the inner mitochondrial membrane and other fatty acid oxidation proteins [32–34].
In an attempt to evaluate the validity of the diagnostic procedures clinically available for VLCADD (i.e. blood acylcarnitine profile and molecular genetic testing of ACADVL), we used a combination of functional tests to retrospectively characterize enzymatic, biochemical, and molecular data of suspected VLCADD patients referred to our laboratory in the last three years with special emphasis on samples with some uncertainty raised by initial test results.
MATERIALS AND METHODS
Fibroblasts studies
Cell culture
Patient skin biopsies for fibroblast culture were performed on a clinical basis with written informed consent from patients and/or parents. Fibroblasts were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum and 100 μg/ml penicillin/streptomycin, 4.5 g/l glucose, 4 mM glutamine and 2 mM pyruvate. At 90% confluence in a T175 flask, cells were harvested by trypsinization, pelleted and stored at −80°C for further studies.
Western Blot
Cell pellets were resuspended in 250 μl ice-cold water and subjected to sonication on ice and centrifuged at 18,000 × g for 20 minutes at +4°C to remove cell debris. The supernatant was used for western blotting as described [33]. Briefly, 50 μg protein of an SDS treated sample was loaded onto the gel. Following electrophoresis, the gel was blotted onto a nylon membrane followed by visualization with a primary rabbit anti-VLCAD polyclonal antibody and secondary alkaline phosphatase goat anti-rabbit IgG antibody. Anti-VLCAD serum was produced as already reported [33].
Confocal imaging of fibroblasts
Fibroblasts from patients and healthy controls were seeded at a concentration of 5×104 cells/ml on tissue culture-treated glass cover slips and allowed to grow overnight at 37°C in a 5% CO2, 95% humidity incubator. Cells were then fixed in 4% paraformaldehyde for 10 min followed by 0.1% Triton X100 cell permeabilization and further blocked after brief washings in 5% donkey serum (Jackson ImmunoResearch, WestGrove, PA) for 1 hour on ice. This was followed by double primary antibody incubation with 1) anti-VLCAD antibody and 2) anti cytochrome c oxidase subunit 4 antibody (Abcam, Cambridge, MA) at 4°C overnight. After brief washing, cells were further incubated with donkey anti-rabbit secondary antibody Alexa fluor 488 (Invitrogen, Grand Island, NY) for VLCAD, and donkey anti-mouse secondary antibody Alexa Fluor 555 (Invitrogen) for cytochrome c oxidase subunit 4. Nuclei were counterstained with DAPI. The cover slips were then mounted using mounting media before imaging. All the images were taken using a Olympus Confocal FluoView FV1000 microscope at a magnification of 60X.
VLCAD activity measurement
Fibroblasts pellets were resuspended in 250 μl 50 mM Tris-Cl, pH 8.0, 10 mM EDTA, subjected to sonication on ice and centrifuged at 18,000 × g for 20 minutes at 4°C. The supernatant was assayed for VLCAD activity with the highly sensitive and specific electron transfer flavoprotein fluorescence reduction assay as described using 100 to 150 μg protein [35, 36].
Mutagenesis and expression studies
For prokaryotic mutagenesis studies, nine mutations (R162H, I189T, G289R, I420L, G439D, M443R, G514E, L540P and R567Q) were introduced into the VLCAD ΔEx3 pET-21a(+) expression construct using the QuickChange Site-Directed Mutagenesis Kit according to the manufacturer’s instructions (Stratagene, La Jolla, CA). Mutations were verified by sequencing and the plasmids were introduced into an E. coli expression strain (BL21), cultured at 37°C and induced for expression studies as previously described [33]. Enzyme activity and Western blotting were performed on cell-free extracts as above.
RESULTS
Patients and mutations
Thirteen patients with a suspicion of VLCADD were retrospectively studied (Table 1). Seven patients (PTs 1, 3, 4, 5, 6, 9 and 10) had been flagged by newborn screening as unequivocally suggestive of VLCADD due to a clearly elevated C14:1 and/or C14 acylcarnitine levels. For 3 patients (PTs 8, 12 and 13), the results of the NBS were equivocal for VLCADD (Table 1). For the other patients, NBS was normal in two (PTs 2 and 7) and not performed in one (PT 11), but all three exhibited clinical symptoms considered suggestive of VLCADD (Table 1). Blood acylcarnitine profiling was diagnostic of VLCADD in PT11 but only showed non-specific abnormalities in PTs 2 and 7. ACADVL genomic DNA sequencing data are shown in Table 1. Nine patients had two identified mutations, while three had only one mutated allele (PTs 8, 12, 13). Six new missense mutations (R162H, I189T, I420L, M443R, G514E, L540P), one previously reported [5, 37, 38] and functionally characterized [33] mutation (V283A), and 4 reported but uncharacterized missense mutations (L202P, G289R, G439D, R567Q) [38–40] were identified. Two splice donor site mutations were identified. The first mutation, previously reported, (c.1182+1G>A) induces exon 11 skipping [41], while the second (c.1182+3G>T) is new. Two small indels, insertion of 9 base pairs (c.1707_1716dup) and a 3 base-pair deletion (c.896_898del) are novel. The latter change induces a p.K299 deletion. This amino acid deletion and has previously been reported to be caused by a different mutation (c.895_897del) [42]. Patient 11, whose mutations have previously been reported [43], harbored 2 known mutations creating a frame shift: a deletion of 2 base pairs (c887_888del) [44] on one allele and a splice mutation on the other (c.1679-6G>A) [45]. Patient 2 harbored two variants likely to be non pathogenic (G43D, reported as a likely polymorphism [5], and L17F, reported as a non pathogenic variant: http://www.ncbi.nlm.nih.gov/sites/varvu?gene=37). For the patients with only one ACADVL exonic mutation, a macro or micro gene deletion/duplication was ruled out by prior clinical microarray analysis.
Table 1.
Clinical characteristics of the 13 patients.
| Patients | NBS1 C14:1 (μM)/C14 (μM) (normal values <0.6/<0.7) | Clinical signs at diagnosis (if NBS was negative) | Genotype | Last follow-up |
|---|---|---|---|---|
| 1 | 4.9/6.65 | Asymptomatic | c.T1619C (p.L540P) c.1707_1716dup |
6 years old: normal |
| 2 | N | 27 months: acute decompensation with dilated cardiomyopathy, rhabdomyolysis, hypoglycemia, hyperammonemia, lactic acidosis, renal failure ACP2 with non-diagnostic elevations of long chain species | c.G128A (p.G43D) c.C49T (p.L17F) |
Before 3 years of age: death from severe hypoglycemia |
| 3 | 3.29/4.71 | Asymptomatic | c.G1316A (p.G439D) c.T1328G (p.M443R) |
2 years: moderate CK3 increase during intercurrent infections |
| 4 | 1.2/0.93 | Asymptomatic | c.T848C (p.V283A) c.1182+3G>T |
1 year: normal |
| 5 | 2.83/3.49 | Asymptomatic | c.T566C (p.I189T) c.1182+1G>A |
19 months: moderate CK increase during intercurrent infections |
| 6 | 1.59/1.28 | Asymptomatic | c.T848C (p.V283A) c.G865A (p.G289R) |
6 months: normal |
| 7 | N | Unexpected sudden death before 6 months of age. Postmortem ACP with non-diagnostic elevations of long chain species | NA4 | |
| 8 | 0.585/0.656 | Asymptomatic | c.T605C (p.L202P) WT |
18 months: normal |
| 9 | 2.59/1.76 | Asymptomatic | c.T848C (p.V283A) c.A1258C (p.I420L) |
18 months: normal |
| 10 | 2.12/1.6 | Asymptomatic | c.G1541A (p.G514E) c.G1700A (p.R567Q) |
6 years of age: lost follow-up |
| 11 | NP7 | 3 months of age: severe hypertrophic cardiomyopathy enlarged liver, episodes of hypoglycemia. ACP diagnostic of VLCAD deficiency | c.887_888del c.1679-6G>A |
16 years 4/12: normal (under triheptanoin) |
| 12 | 0.74/0.74 | Asymptomatic | c.G485A (p.R162H) WT |
1 year: normal |
| 13 | 0.69/0.81 | Asymptomatic | c.896_898del (p.K299del) WT |
18 months: normal |
Newborn screening
Acylcarnitine profile
Creatine kinase
Not available
Flagged as borderline
Flagged as elevated
Not performed
Fibroblasts studies
-
Enzyme activity and immunoblotting (Fig. 1)
VLCAD activity in fibroblasts was deficient in all of the 7 patients with unambiguous abnormal NBS results, in one of the 2 patients with equivocal NBS results (PT 8), and in one of the 3 clinically suspected patients (PT 11), with activity ranging from not detectable to 38% of simultaneous controls (Fig. 1). VLCAD antigen was normal in one of these cell lines (PT 3), significantly reduced but present in 2 (PT 4 and 8), and absent in all of the others. Patients 2 and 7 (normal NBS), and 12 and 13 (equivocal NBS) had no abnormalities in VLCAD activity or antigen.
Immunofluorescence staining of patient and wild type fibroblasts for VLCAD revealed the variable presence of VLCAD antigen that most of the time, correlated well with the Western blotting results (Fig. 2) although immunostaining was more sensitive in detecting VLCAD antigen.
Figure 1.

Characterization of wild type (WT) and patient fibroblast samples for VLCAD activity and antigen. The top line shows mutations identified in patient cell lines. Western blot results using VLCAD antisera are shown in the next line. The bottom line gives the VLCAD activity from fibroblast extracts presented as a percentage of the concurrent wild type control (ND is non-detectable). All samples were also tested for MCAD activity and antigen as a control and gave normal levels of both (not shown).
Figure 2.


Immunofluorescent staining of wild type (A) and patient (B) fibroblasts. A. Wild type cells (WT). VLCAD antigen was visualized with green fluorescently tagged antibodies (top, middle panel) and mitochondrial cytochrome c oxidase (COX) was visualized with red fluorescently tagged antibodies (top, right panel). Nuclei were visualized with DAPI staining (blue; top, left panel). The merged image (bottom panel) shows colocalization of VLCAD and COX in mitochondria as yellow (white arrow). B. Patient fibroblasts. Each panel shows the merged only image of patients (PTs 1–6 and 8–11) with the corresponding Western blot results. The mutations identified in the cell line are shown at the top of each panel for convenience. Scale bar, 10.75 μm.
Prokaryotic mutagenesis and expression studies
All 9 mutations studied in an E. coli expression system had a clear impact on VLCAD activity and variable impact on the level of stable antigen (Fig. 3). Activities ranged between undetectable and 50% of that obtained with a wild type expression vector. Western blot of whole cell extracts with VLCAD antibody showed normal (G439D, M443R and G514E), decreased (G289R, I420L and R567Q) or absent (R162H, I189T and L540P) VLCAD antigen. Because enzyme activity and VLCAD antigen were normal in cells from patient 7, molecular testing was not further pursued.
Figure 3.
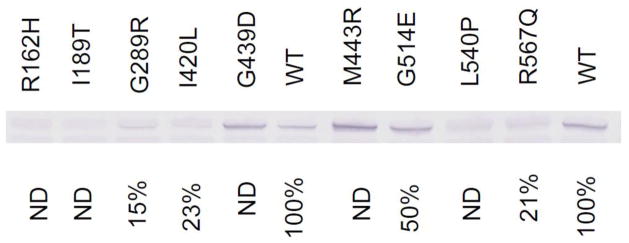
Prokaryotic mutagenesis and expression studies of 9 missense ACADVL mutations. Each mutation shown at the top of the figure was expressed in E. coli and the extract was analyzed by SDS-PAGE followed by western blotting with VLCAD antibodies (middle). Activity in cell-free extracts following prokaryotic expression is given on the bottom line. ND is non-detectable.
DISCUSSION
VLCAD deficiency presents with a broad spectrum of symptoms and patients identified through NBS are most often asymptomatic at birth. Our research laboratory routinely receives requests for functional testing for samples from individuals with equivocal NBS or diagnostic testing suggestive of VLCAD deficiency when variants of unknown significance are identified on molecular testing. Our aim in this study was to more completely characterize the utility of available functional tools to evaluate such patients. The menu of functional tools in our laboratory includes measurement of fibroblast VLCAD activity using ETF fluorescence reduction assay, PAGE followed by Western blotting, immunostaining of patient fibroblasts, and prokaryotic mutagenesis and expression studies. This combination of studies allowed us to unambiguously identify affected individuals with VLCADD from heterozygous carriers and unaffected patients. Of note, our goal was not to attempt to predict the clinical severity and outcome in individuals identified with VLCADD through NBS, though our results should ultimately feed back to allow this with further follow up of the patients. In this regard, clinical testing by acylcarnitine analysis of patient fibroblasts has shown some prognostic value for broad category of symptoms, but the correlation is imperfect. Comparing results of clinical fibroblast acylcarnitine profiling available to us (Fig. 4) confirms this imperfection. Indeed, patient 4 who is predicted to have a mild disease (residual VLCAD activity with stable antigen) had no abnormal acylcarnitine species, while, patients 5 and 6 (who had marginal elevations of C14 and C12 acylcarnitines, respectively) had greatly reduced VLCAD activity and antigen.
Figure 4.

Results from the fatty acid oxidation probe assay in fibroblasts for 10 of the 13 patients. Since the analyses were performed at two different clinical laboratories, abnormal C12, C14 and C16 acylcarnitines species are expressed as fold of increase relative to upper limit of control values of the testing lab rather than as actual values.
The three patients in this study not referred because of abnormal newborn screening (PTs 2, 7, 11) exhibited clinical symptoms compatible with VLCADD, specifically acute metabolic decompensation with cardiac and liver involvement (PT 2, 11) and sudden death (PT 7). The diagnosis was established in PT 11 by the presence of an appropriate pattern on acylcarnitine profile in blood at the time of symptoms but the pattern was nonspecific for patients 2 and 7. For those patients identified through newborn screening, VLCADD was suspected because of clearly (PTs 1, 3, 4, 5, 6, 9, 10) or equivocally (PTs 8, 12, 13) elevated levels of C14:1 and/or C14 acylcarnitines in blood spots. All patients in the unequivocal group proved to have absent or decreased VLCAD enzyme activity and/or protein in fibroblasts, while only one (PT 8) from the equivocal group appears to be affected. A second mutant allele could not be identified in this last patient who was heterozygous for the L202P mutation, making likely a promoter mutation or one leading to missplicing that would not be identified by standard sequencing of exonic regions along with intron-exon boundaries.
These data illustrate the value of the ETF reduction assay in confirming VLCADD [46]. Note that under our assay conditions, we typically find no or minimal enzyme activity reduction in obligate carrier parents (unpublished and as shown for PTs 12 and 13, Fig. 1). Both novel and known mutations in the ACADVL gene were described in this series of patients. Several of the mutations were easily predictable to be pathogenic including a multiple base pair duplication (c.1707_1716dup, PT1), two deletions (c.887_888del, PT 11 and c.896_898del, PT 13), and three mutations that likely affect splicing (c. 1182+3G>T, PT 4; c.1182+1G>A, PT 5; c.1679-6G>A, PT11). Experimentally, these mutations are confirmed as pathogenic by the lack of VLCAD antigen and/or activity in fibroblasts. Of the missense mutations, one (V283A) has previously been extensively studied and demonstrated to be deleterious [33]. Prokaryotic mutagenesis and expression studies confirm that all of the previously undescribed or uncharacterized mutations are clearly deleterious.
Molecular modeling of the identified mutations offers some insights into their pathologic mechanisms (Fig. 5). Mutations at G439 and M443 are likely to affect dimerization of the VLCAD subunits and/or FAD binding to the apoenzyme. Since the patient mutations at these positions lead to stable production of VLCAD protein in our prokaryotic expression system, dimerization is unlikely to be affected, thus implicating defective FAD binding as causing enzyme inactivity. In this event, the patients harboring these mutations might respond to some extent to riboflavin supplementation. Of note, enzyme activity in each of these 2 prokaryotic mutants was not restored by addition of 0.1 mM FAD (not shown). G514, L540, and R567 are all in the C-terminal region unique to VLCAD and ACAD9 among the ACADs. This domain mimics the structure provided by the tetrameric configuration of the other ACADs, and thus is critical to the stability of the homodimer. However, it also serves to help anchor the VLCAD dimer to the inner mitochondrial membrane. Mutations affecting the latter function have been reported to maintain stable enzyme activity when expressed in prokaryotes [33]. Indeed the G514E and R567Q mutations retain some stability and activity when expressed in E. coli and the fibroblast cell line with these mutations exhibits partial activity. The remainders of the mutations are scattered throughout the portion of the protein homologous to the other ACADs but distant to the active site. Thus, they are more likely to affect protein folding and/or stability rather than a direct effect on the enzyme mechanism. In fact, all of these mutations lead to unstable protein in the prokaryotic expression, though the effect was least with the G289R mutation located in a random coil motif on the surface of the protein.
Figure 5.

Molecular model of VLCAD crystal structure and mutation positions. The molecular structure of VLCAD from PDB file 2UXW was visualized with the MolView viewer (Accelerys, Inc, San Diego, CA). Subunit A is color coded according to structural motif: red, α-helix; blue, β-sheet; white, random coil, orange, C-terminal domain. The B subunit is colored purple. The mutations described in this manuscript are represented as yellow balls. The stick structures of FAD and palmitoyl-CoA are colored yellow and green, respectively.
Fibroblasts form patient 2 had normal enzyme activity and VLCAD antigen. The G43D variant identified in this cell line has previously been reported to be a polymorphism [5]. Both this variant, and a previously unreported L17F change are located within the mitochondrial targeting peptide of the VLCAD precursor. Neither is predicted to alter the amphipathic helix motif critical for this targeting, though introduction of a negatively charged aspartate residue near the amino terminus of the precursor may reduce the interaction of precursor VLCAD with the mitochondrial targeting and import machinery. Interestingly, the fibroblast cell line containing these alterations actually has excess VLCAD activity. Consistent with this finding, the acylcarnitine profile in this patient showed only nonspecific elevation of long chain species rather than a pattern suggestive of VLCADD specifically.
In summary, we found that eight out of ten patients identified through NBS appear to have true VLCADD while the remaining two are heterozygous carriers. Functional fibroblasts testing are valuable complimentary tools in order to unequivocally differentiate affected individuals from heterozygous carriers. These functional tests are also useful to confirm VLCADD in clinically suspected VLCADD patients. Albeit limited in number, no NBS negative samples proved to have deficiency. Finally, as already reported, our prokaryotic mutagenesis system is useful to identify pathogenic ACADVL mutations. Since VLCADD molecular heterogeneity makes it very difficult to predict the functional effects based on the genotype alone, we believe that such a multiple testing modalities approach seems suitable not only for identification of truly affected patients but also for the follow-up of equivocal NBS results. Implementation of such functional studies in clinical practice is impractical on a routine basis but could be considered in patients with unclear results from standard available tests.
Highlights.
VLCAD deficiency (VLCADD) is now diagnosed in the US through newborn screening
Numerous NBS variant patterns can represent mild disease or heterozygous carriers
13 patient samples suspected of VLCADD were retrospectively investigated
Prokaryotic expression studies were performed for nine missense ACADVL mutations
Functional investigation of equivocal NBS or clinical testing is crucial to diagnose disease
Acknowledgments
The authors are grateful to all the physicians who referred the samples and/or performed molecular analyses: Doctors Alice Basinger, Barbara Burton, Pranesh Chakraborty, Dimitar Gavrilov, Virginia Proud, Heidenreich Randall, Charles Roe, Alvaro Serrano-Russi, Lawrence Sweetman, and Arnold W. Strauss.
MS received grant supports from the Fulbright Scholar Program (Monahan Foundation), SFP (Société Française de Pédiatrie) and Institut Servier. JV was supported by PHS grant NIH R01 DK78775.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vockley J, Whiteman DA. Defects of mitochondrial beta-oxidation: a growing group of disorders. Neuromuscul Disord. 2002;12:235–246. doi: 10.1016/s0960-8966(01)00308-x. [DOI] [PubMed] [Google Scholar]
- 2.Houten SM, Wanders RJ. A general introduction to the biochemistry of mitochondrial fatty acid beta-oxidation. J Inherit Metab Dis. 2010;33:469–477. doi: 10.1007/s10545-010-9061-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McHugh DM, Cameron CA, Abdenur JE, Abdulrahman M, Adair O, Al Nuaimi SA, Ahlman H, Allen JJ, Antonozzi I, Archer S, Au S, Auray-Blais C, Baker M, Bamforth F, Beckmann K, Pino GB, Berberich SL, Binard R, Boemer F, Bonham J, Breen NN, Bryant SC, Caggana M, Caldwell SG, Camilot M, Campbell C, Carducci C, Cariappa R, Carlisle C, Caruso U, Cassanello M, Castilla AM, Ramos DE, Chakraborty P, Chandrasekar R, Ramos AC, Cheillan D, Chien YH, Childs TA, Chrastina P, Sica YC, de Juan JA, Colandre ME, Espinoza VC, Corso G, Currier R, Cyr D, Czuczy N, D’Apolito O, Davis T, de Sain-Van der Velden MG, Delgado Pecellin C, Di Gangi IM, Di Stefano CM, Dotsikas Y, Downing M, Downs SM, Dy B, Dymerski M, Rueda I, Elvers B, Eaton R, Eckerd BM, El Mougy F, Eroh S, Espada M, Evans C, Fawbush S, Fisher L, Franzson L, Frazier DM, Garcia LR, Bermejo MS, Gavrilov D, Gerace R, Giordano G, Irazabal YG, Greed LC, Grier R, Grycki E, Gu X, Gulamali-Majid F, Hagar AF, Han L, Hannon WH, Haslip C, Hassan FA, He M, Hietala A, Himstedt L, Hoffman GL, Hoffman W, Hoggatt P, Hopkins PV, Hougaard DM, Hughes K, Hunt PR, Hwu WL, Hynes J, Ibarra-Gonzalez I, Ingham CA, Ivanova M, Jacox WB, John C, Johnson JP, Jonsson JJ, Karg E, Kasper D, Klopper B, Katakouzinos D, Khneisser I, Knoll D, Kobayashi H, Koneski R, Kozich V, Kouapei R, Kohlmueller D, Kremensky I, la Marca G, Lavochkin M, Lee SY, Lehotay DC, Lemes A, Lepage J, Lesko B, Lewis B, Lim C, Linard S, Lindner M, Lloyd-Puryear MA, Lorey F, Loukas YL, Luedtke J, Maffitt N, Magee JF, Manning A, Manos S, Marie S, Hadachi SM, Marquardt G, Martin SJ, Matern D, Mayfield Gibson SK, Mayne P, McCallister TD, McCann M, McClure J, McGill JJ, McKeever CD, McNeilly B, Morrissey MA, Moutsatsou P, Mulcahy EA, Nikoloudis D, Norgaard-Pedersen B, Oglesbee D, Oltarzewski M, Ombrone D, Ojodu J, Papakonstantinou V, Reoyo SP, Park HD, Pasquali M, Pasquini E, Patel P, Pass KA, Peterson C, Pettersen RD, Pitt JJ, Poh S, Pollak A, Porter C, Poston PA, Price RW, Queijo C, Quesada J, Randell E, Ranieri E, Raymond K, Reddic JE, Reuben A, Ricciardi C, Rinaldo P, Rivera JD, Roberts A, Rocha H, Roche G, Greenberg CR, Mellado JM, Juan-Fita MJ, Ruiz C, Ruoppolo M, Rutledge SL, Ryu E, Saban C, Sahai I, Garcia-Blanco MI, Santiago-Borrero P, Schenone A, Schoos R, Schweitzer B, Scott P, Seashore MR, Seeterlin MA, Sesser DE, Sevier DW, Shone SM, Sinclair G, Skrinska VA, Stanley EL, Strovel ET, Jones AL, Sunny S, Takats Z, Tanyalcin T, Teofoli F, Thompson JR, Tomashitis K, Domingos MT, Torres J, Torres R, Tortorelli S, Turi S, Turner K, Tzanakos N, Valiente AG, Vallance H, Vela-Amieva M, Vilarinho L, von Dobeln U, Vincent MF, Vorster BC, Watson MS, Webster D, Weiss S, Wilcken B, Wiley V, Williams SK, Willis SA, Woontner M, Wright K, Yahyaoui R, Yamaguchi S, Yssel M, Zakowicz WM. Clinical validation of cutoff target ranges in newborn screening of metabolic disorders by tandem mass spectrometry: a worldwide collaborative project. Genet Med. 2011;13:230–254. doi: 10.1097/GIM.0b013e31820d5e67. [DOI] [PubMed] [Google Scholar]
- 4.Andresen BS, Bross P, Vianey-Saban C, Divry P, Zabot MT, Roe CR, Nada MA, Byskov A, Kruse TA, Neve S, Kristiansen K, Knudsen I, Corydon MJ, Gregersen N. Cloning and characterization of human very-long-chain acyl-CoA dehydrogenase cDNA, chromosomal assignment of the gene and identification in four patients of nine different mutations within the VLCAD gene. Hum Mol Genet. 1996;5:461–472. doi: 10.1093/hmg/5.4.461. [DOI] [PubMed] [Google Scholar]
- 5.Andresen BS, Olpin S, Poorthuis BJ, Scholte HR, Vianey-Saban C, Wanders R, Ijlst L, Morris A, Pourfarzam M, Bartlett K, Baumgartner ER, deKlerk JB, Schroeder LD, Corydon TJ, Lund H, Winter V, Bross P, Bolund L, Gregersen N. Clear correlation of genotype with disease phenotype in very-long-chain acyl-CoA dehydrogenase deficiency. Am J Hum Genet. 1999;64:479–494. doi: 10.1086/302261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spiekerkoetter U. Mitochondrial fatty acid oxidation disorders: clinical presentation of long-chain fatty acid oxidation defects before and after newborn screening. J Inherit Metab Dis. 2010;33:527–532. doi: 10.1007/s10545-010-9090-x. [DOI] [PubMed] [Google Scholar]
- 7.Andresen BS, Vianey-Saban C, Bross P, Divry P, Roe CR, Nada MA, Knudsen I, Gregersen N. The mutational spectrum in very long-chain acyl-CoA dehydrogenase deficiency. J Inherit Metab Dis. 1996;19:169–172. doi: 10.1007/BF01799421. [DOI] [PubMed] [Google Scholar]
- 8.Wright EL, Van Hove JL, Thomas J. Mountain States Genetics Regional Collaborative Center’s Metabolic Newborn Screening Long-term Follow-up Study: a collaborative multi-site approach to newborn screening outcomes research. Genet Med. 2010;12:S228–241. doi: 10.1097/GIM.0b013e3181fe5d50. [DOI] [PubMed] [Google Scholar]
- 9.Spiekerkoetter U, Haussmann U, Mueller M, ter Veld F, Stehn M, Santer R, Lukacs Z. Tandem mass spectrometry screening for very long-chain acyl-CoA dehydrogenase deficiency: the value of second-tier enzyme testing. J Pediatr. 2010;157:668–673. doi: 10.1016/j.jpeds.2010.04.063. [DOI] [PubMed] [Google Scholar]
- 10.Boneh A, Andresen BS, Gregersen N, Ibrahim M, Tzanakos N, Peters H, Yaplito-Lee J, Pitt JJ. VLCAD deficiency: pitfalls in newborn screening and confirmation of diagnosis by mutation analysis. Mol Genet Metab. 2006;88:166–170. doi: 10.1016/j.ymgme.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 11.Lindner M, Hoffmann GF, Matern D. Newborn screening for disorders of fatty-acid oxidation: experience and recommendations from an expert meeting. J Inherit Metab Dis. 2010;33:521–526. doi: 10.1007/s10545-010-9076-8. [DOI] [PubMed] [Google Scholar]
- 12.Arnold GL, Van Hove J, Freedenberg D, Strauss A, Longo N, Burton B, Garganta C, Ficicioglu C, Cederbaum S, Harding C, Boles RG, Matern D, Chakraborty P, Feigenbaum A. A Delphi clinical practice protocol for the management of very long chain acyl-CoA dehydrogenase deficiency. Mol Genet Metab. 2009;96:85–90. doi: 10.1016/j.ymgme.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilcken B. Fatty acid oxidation disorders: outcome and long-term prognosis. J Inherit Metab Dis. 2010;33:501–506. doi: 10.1007/s10545-009-9001-1. [DOI] [PubMed] [Google Scholar]
- 14.Baruteau J, Sachs P, Broue P, Brivet M, Abdoul H, Vianey-Saban C, Ogier de Baulny H. Clinical and biological features at diagnosis in mitochondrial fatty acid beta-oxidation defects: a French pediatric study of 187 patients. J Inherit Metab Dis. 2012 doi: 10.1007/s10545-012-9542-6. [DOI] [PubMed] [Google Scholar]
- 15.Hoffmann L, Haussmann U, Mueller M, Spiekerkoetter U. VLCAD enzyme activity determinations in newborns identified by screening: a valuable tool for risk assessment. J Inherit Metab Dis. 2012;35:269–277. doi: 10.1007/s10545-011-9391-8. [DOI] [PubMed] [Google Scholar]
- 16.Swigonova Z, Mohsen AW, Vockley J. Acyl-CoA dehydrogenases: Dynamic history of protein family evolution. J Mol Evol. 2009;69:176–193. doi: 10.1007/s00239-009-9263-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ikeda Y, Dabrowski C, Tanaka K. Separation and properties of five distinct acyl-CoA dehydrogenases from rat liver mitochondria. Identification of a new 2-methyl branched chain acyl-CoA dehydrogenase. J Biol Chem. 1983;258:1066–1076. [PubMed] [Google Scholar]
- 18.Ikeda Y, Okamura-Ikeda K, Tanaka K. Purification and characterization of short-chain, medium-chain, and long-chain acyl-CoA dehydrogenases from rat liver mitochondria. Isolation of the holo- and apoenzymes and conversion of the apoenzyme to the holoenzyme. J Biol Chem. 1985;260:1311–1325. [PubMed] [Google Scholar]
- 19.Ikeda Y, Tanaka K. Purification and characterization of 2-methyl-branched chain acyl coenzyme A dehydrogenase, an enzyme involved in the isoleucine and valine metabolism, from rat liver mitochondria. J Biol Chem. 1983;258:9477–9487. [PubMed] [Google Scholar]
- 20.Izai K, Uchida Y, Orii T, Yamamoto S, Hashimoto T. Novel fatty acid beta-oxidation enzymes in rat liver mitochondria. I. Purification and properties of very-long-chain acyl-coenzyme A dehydrogenase. J Biol Chem. 1992;267:1027–1033. [PubMed] [Google Scholar]
- 21.Rozen R, Vockley J, Zhou L, Milos R, Willard J, Fu K, Vicanek C, Low-Nang L, Torban E, Fournier B. Isolation and expression of a cDNA encoding the precursor for a novel member (ACADSB) of the acyl-CoA dehydrogenase gene family. Genomics. 1994;24:280–287. doi: 10.1006/geno.1994.1617. [DOI] [PubMed] [Google Scholar]
- 22.Willard J, Vicanek C, Battaile KP, Van Veldhoven PP, Fauq AH, Rozen R, Vockley J. Cloning of a cDNA for short/branched chain acyl-Coenzyme A dehydrogenase from rat and characterization of its tissue expression and substrate specificity. Arch Biochem Biophys. 1996;331:127–133. doi: 10.1006/abbi.1996.0290. [DOI] [PubMed] [Google Scholar]
- 23.Nguyen TV, Andresen BS, Corydon TJ, Ghisla S, Abd-El Razik N, Mohsen AW, Cederbaum SD, Roe DS, Roe CR, Lench NJ, Vockley J. Identification of isobutyryl-CoA dehydrogenase and its deficiency in humans. Mol Genet Metab. 2002;77:68–79. doi: 10.1016/s1096-7192(02)00152-x. [DOI] [PubMed] [Google Scholar]
- 24.Zhang J, Zhang W, Zou D, Chen G, Wan T, Zhang M, Cao X. Cloning and functional characterization of ACAD-9, a novel member of human acyl-CoA dehydrogenase family. Biochem Biophys Res Commun. 2002;297:1033–1042. doi: 10.1016/s0006-291x(02)02336-7. [DOI] [PubMed] [Google Scholar]
- 25.Ensenauer R, He M, Willard JM, Goetzman ES, Corydon TJ, Vandahl BB, Mohsen AW, Isaya G, Vockley J. Human acyl-CoA dehydrogenase-9 plays a novel role in the mitochondrial beta-oxidation of unsaturated fatty acids. J Biol Chem. 2005;280:32309–32316. doi: 10.1074/jbc.M504460200. [DOI] [PubMed] [Google Scholar]
- 26.He M, Pei Z, Mohsen AW, Watkins P, Murdoch G, Van Veldhoven PP, Ensenauer R, Vockley J. Identification and characterization of new long chain acyl-CoA dehydrogenases. Mol Genet Metab. 2011;102:418–429. doi: 10.1016/j.ymgme.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ikeda Y, Hale DE, Keese SM, Coates PM, Tanaka K. Biosynthesis of variant medium chain acyl-CoA dehydrogenase in cultured fibroblasts from patients with medium chain acyl-CoA dehydrogenase deficiency. Pediatr Res. 1986;20:843–847. doi: 10.1203/00006450-198609000-00007. [DOI] [PubMed] [Google Scholar]
- 28.Ikeda Y, Tanaka K. Immunoprecipitation and electrophoretic analysis of four human acyl-CoA dehydrogenases and electron transfer flavoprotein using antibodies raised against the corresponding rat enzymes. Biochem Med Metab Biol. 1987;37:329–334. doi: 10.1016/0885-4505(87)90044-2. [DOI] [PubMed] [Google Scholar]
- 29.Volchenboum SL, Vockley J. Mitochondrial import and processing of wild type and type III mutant isovaleryl-CoA dehydrogenase. J Biol Chem. 2000;275:7958–7963. doi: 10.1074/jbc.275.11.7958. [DOI] [PubMed] [Google Scholar]
- 30.Ikeda Y, Hine DG, Okamura-Ikeda K, Tanaka K. Mechanism of action of short-chain, medium-chain, and long-chain acyl-CoA dehydrogenases. Direct evidence for carbanion formation as an intermediate step using enzyme-catalyzed C-2 proton/deuteron exchange in the absence of C-3 exchange. J Biol Chem. 1985;260:1326–1337. [PubMed] [Google Scholar]
- 31.Ikeda Y, Okamura-Ikeda K, Tanaka K. Spectroscopic analysis of the interaction of rat liver short-chain, medium-chain, and long-chain acyl coenzyme A dehydrogenases with acyl coenzyme. A substrates Biochemistry. 1985;24:7192–7199. doi: 10.1021/bi00346a027. [DOI] [PubMed] [Google Scholar]
- 32.Souri M, Aoyama T, Yamaguchi S, Hashimoto T. Relationship between structure and substrate-chain-length specificity of mitochondrial very-long-chain acyl-coenzyme A dehydrogenase. Eur J Biochem. 1998;257:592–598. doi: 10.1046/j.1432-1327.1998.2570592.x. [DOI] [PubMed] [Google Scholar]
- 33.Goetzman ES, Wang Y, He M, Mohsen AW, Ninness BK, Vockley J. Expression and characterization of mutations in human very long-chain acyl-CoA dehydrogenase using a prokaryotic system. Mol Genet Metab. 2007;91:138–147. doi: 10.1016/j.ymgme.2007.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y, Mohsen AW, Mihalik SJ, Goetzman ES, Vockley J. Evidence for physical association of mitochondrial fatty acid oxidation and oxidative phosphorylation complexes. J Biol Chem. 2010;285:29834–29841. doi: 10.1074/jbc.M110.139493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frerman FE, Goodman SI. Fluorometric assay of acyl-CoA dehydrogenases in normal and mutant human fibroblasts. Biochem Med. 1985;33:38–44. doi: 10.1016/0006-2944(85)90124-3. [DOI] [PubMed] [Google Scholar]
- 36.Mohsen AW, Vockley J. High-level expression of an altered cDNA encoding human isovaleryl-CoA dehydrogenase in Escherichia coli. Gene. 1995;160:263–267. doi: 10.1016/0378-1119(95)00256-6. [DOI] [PubMed] [Google Scholar]
- 37.Djouadi F, Aubey F, Schlemmer D, Ruiter JP, Wanders RJ, Strauss AW, Bastin J. Bezafibrate increases very-long-chain acyl-CoA dehydrogenase protein and mRNA expression in deficient fibroblasts and is a potential therapy for fatty acid oxidation disorders. Hum Mol Genet. 2005;14:2695–2703. doi: 10.1093/hmg/ddi303. [DOI] [PubMed] [Google Scholar]
- 38.Gobin-Limballe S, Djouadi F, Aubey F, Olpin S, Andresen BS, Yamaguchi S, Mandel H, Fukao T, Ruiter JP, Wanders RJ, McAndrew R, Kim JJ, Bastin J. Genetic basis for correction of very-long-chain acyl-coenzyme A dehydrogenase deficiency by bezafibrate in patient fibroblasts: toward a genotype-based therapy. Am J Hum Genet. 2007;81:1133–1143. doi: 10.1086/522375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gobin-Limballe S, McAndrew RP, Djouadi F, Kim JJ, Bastin J. Compared effects of missense mutations in Very-Long-Chain Acyl-CoA Dehydrogenase deficiency: Combined analysis by structural, functional and pharmacological approaches. Biochim Biophys Acta. 2010;1802:478–484. doi: 10.1016/j.bbadis.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Olsen RK, Dobrowolski SF, Kjeldsen M, Hougaard D, Simonsen H, Gregersen N, Andresen BS. High-resolution melting analysis, a simple and effective method for reliable mutation scanning and frequency studies in the ACADVL gene. J Inherit Metab Dis. 2010;33:247–260. doi: 10.1007/s10545-010-9101-y. [DOI] [PubMed] [Google Scholar]
- 41.Strauss AW, Powell CK, Hale DE, Anderson MM, Ahuja A, Brackett JC, Sims HF. Molecular basis of human mitochondrial very-long-chain acyl-CoA dehydrogenase deficiency causing cardiomyopathy and sudden death in childhood. Proc Natl Acad Sci U S A. 1995;92:10496–10500. doi: 10.1073/pnas.92.23.10496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Souri M, Aoyama T, Orii K, Yamaguchi S, Hashimoto T. Mutation analysis of very-long-chain acyl-coenzyme A dehydrogenase (VLCAD) deficiency: identification and characterization of mutant VLCAD cDNAs from four patients. Am J Hum Genet. 1996;58:97–106. [PMC free article] [PubMed] [Google Scholar]
- 43.Roe CR, Sweetman L, Roe DS, David F, Brunengraber H. Treatment of cardiomyopathy and rhabdomyolysis in long-chain fat oxidation disorders using an anaplerotic odd-chain triglyceride. J Clin Invest. 2002;110:259–269. doi: 10.1172/JCI15311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mathur A, Sims HF, Gopalakrishnan D, Gibson B, Rinaldo P, Vockley J, Hug G, Strauss AW. Molecular heterogeneity in very-long-chain acyl-CoA dehydrogenase deficiency causing pediatric cardiomyopathy and sudden death. Circulation. 1999;99:1337–1343. doi: 10.1161/01.cir.99.10.1337. [DOI] [PubMed] [Google Scholar]
- 45.Cox GF, Souri M, Aoyama T, Rockenmacher S, Varvogli L, Rohr F, Hashimoto T, Korson MS. Reversal of severe hypertrophic cardiomyopathy and excellent neuropsychologic outcome in very-long-chain acyl-coenzyme A dehydrogenase deficiency. J Pediatr. 1998;133:247–253. doi: 10.1016/s0022-3476(98)70228-8. [DOI] [PubMed] [Google Scholar]
- 46.Vianey-Saban C, Divry P, Brivet M, Nada M, Zabot MT, Mathieu M, Roe C. Mitochondrial very-long-chain acyl-coenzyme A dehydrogenase deficiency: clinical characteristics and diagnostic considerations in 30 patients. Clin Chim Acta. 1998;269:43–62. doi: 10.1016/s0009-8981(97)00185-x. [DOI] [PubMed] [Google Scholar]