

Abstract
Asthma imposes considerable patient and economic burdens, with the most severe cases causing the greatest affliction. Identifying stimuli that worsen asthma severity is an essential step to controlling both disease morbidity and the lessening economic impact. This study provides the first mechanistic investigation into how acute ethanol exposure will increase asthma severity in a murine model of mild cockroach allergen (CRA)-induced asthma. Outbred mice were sensitized to induce mild allergic asthma, with intratracheal CRA exposures on days 0 and 14. On day 21 mice were gavaged with water or 32% ethanol, and the third allergen exposure was given 30 min post-gavage. Asthmatic responses were measured at several time-points up to 42 h after the third allergen challenge. Ethanol-gavaged mice showed increased asthma severity within 90 min post-allergen challenge, with exacerbations lasting for 24 h. Ethanol caused greater airways obstruction, including an eightfold increase in epithelial cell mucin and increased mucus plugs, resulting in a 50% reduction in bronchiole patency. Ethanol gavage also induced significant increases in airways hyperreactivity. While T helper type 1 (Th1) and Th2 cytokines were not altered by ethanol gavage, pulmonary neutrophil and eosinophil recruitment were augmented. This increase was associated with increased chemokine production. Administration 2 h prior to ethanol gavage of a neutralizing antibody cocktail to keratinocyte-derived chemokine, macrophage inflammatory protein-2, eotaxin-1 and eotaxin-2 prevented ethanol-induced eosinophil recruitment and airways hyperreactivity. These data provide evidence that acute alcohol exposure immediately prior to a mild allergen-triggered asthmatic episode will exacerbate asthma severity mediated by increased production of chemokines.
Keywords: alcohol, cytokines, eosinophils, histology, pulmonary
Introduction
Asthma was first described accurately more than 1800 years ago 1. Despite millennia of opportunity, a continually deeper understanding of disease aetiology and recent advances in personalized medicine, there is still no cure. Instead, asthma has developed into one of the top chronic conditions in the nation, afflicting upwards of 10% of adults and 30% of children in the western world 2. Most research and clinical efforts focus on controlling the disease through the development of tailored therapies; for example, allergen-specific and cytokine-directed immunotherapy 3,4. Despite these newer treatment options, asthma continues to burden patient quality of life and health-care resources 5,6, costing the United States alone more than $15 billion in annual asthma-related expenses 2. Instead of focusing research efforts on therapeutic options, it could be argued that time and resources would be better spent developing asthma prevention strategies through the identification and control of factors that exacerbate symptoms 7. More meaningful approaches may result by unearthing modifiable lifestyle components that affect the development, persistence and magnitude of the disease either directly or indirectly. With a thorough understanding of these factors, asthma morbidity may be better controlled through these lifestyle changes to avoid triggers, potentially minimizing the need for therapeutic intervention. Prevention strategies will undoubtedly improve patient quality of life and lessen the economic burden.
Several clinical studies have cited that acute alcohol consumption triggers asthmatic symptoms 8–14. It has been reported that 40% of asthmatic individuals indicate that drinking alcohol worsens their asthma symptoms 15, and a direct challenge with alcohol will trigger an asthmatic attack 16. This ability of alcohol to induce asthma is not restricted to Asian populations, who have a high incidence of a deficiency of alcohol dehydrogenase 14,15. A 2010 study demonstrated that Caucasian populations, with a genetic variation resulting in the fast metabolism of alcohol, have a nearly twofold increased risk of alcohol-induced hypersensitivity reactions 17. Despite this clinical information, public awareness on this interaction is minimal, and alcohol is not included in common lists of asthma triggers 2,18. This may be a direct result from the lack of basic research addressing the alcohol–asthma relationship, suggesting the need for studies focusing on how alcohol consumption exacerbates asthma.
We recently published that acute alcohol exposure in a cockroach allergen-sensitized mouse will trigger asthma-like pulmonary inflammation 19.The current study utilizes a unique murine model to investigate the basic mechanisms of how ethanol ingestion exacerbates asthma-like pulmonary inflammation. Through these investigations, specific asthmatic responses exacerbated by ethanol were identified, including several hallmarks of asthma – mucin overproduction, respiratory obstruction, lung granulocyte infiltration and airways hyperreactivity. These new insights include a mechanistic approach that identifies chemokines as key molecules involved in orchestrating the exacerbations.
Materials and methods
Mice
Female 18–20 g outbred HSD-ICR (Institute for Cancer Research) mice (Harlan Sprague Inc., Frederick, MD, USA) were used for all experiments. Outbred mice were used for these studies to render the data more relevant to patient populations. Previous work has demonstrated that these mice develop robust asthma-like pulmonary inflammation similar to inbred mice 20. Mice were housed in an approved temperature-controlled facility with a 12-h light/dark cycle. All animal experiments were performed under the approval of Boston University's Institutional Animal Care and Use Committee (IACUC).
Experimental design
Cockroach allergen (CRA; Greer Laboratories, Lenoir, NC, USA) was administered by intratracheal (i.t.) administration of CRA diluted in sterile Hanks's balanced salt solution (HBSS) in two 25-μl aliquots on days 0 and 14, as described previously 20,21. For CRA dose–response experiments, mice received a third i.t. CRA exposure on day 21, underwent whole-body plethysmography (WBP) as described previously 22 3 h post-CRA challenge, and were killed 2 h post-WBP. Thirty min after ethanol (EtOH) or water gavage (described below), mice received a third and final i.t. CRA exposure. Respiratory physiology was assessed by unrestrained WBP (Buxco Research Systems, Wilmington, NC, USA) at 1·5–40 h post-CRA challenge. The results from WBP were confirmed by forced ventilation (FlexiVent; SCIREQ, Montreal, Canada) at 14 h post-CRA challenge, as described in a previous publication 23. Animals used for WBP were killed for collection of plasma, bronchoalveolar (BAL) fluid and lungs 2 h post-WBP at 3·5, 7·5, 16, 24 and 42 h post-CRA challenge. The sensitized group of mice were exposed to CRA on days 0 and 14, then killed on day 21, prior to gavage or third CRA challenge.
Ethanol exposure
Mice were sensitized to CRA as described above and on day 21 mice underwent a 2-h food deprivation to allow gastric emptying. Ethanol was administered by oral gavage of 32% EtOH (300 μl per mouse; approx. 3 g/kg) under light isoflurane anaesthesia using our previously published method 19. Blood ethanol levels peaked at 1 h post-gavage and ranged from 32 to 42 mM. This method of ethanol administration did not result in pulmonary aspiration of EtOH 19. Water was given by gavage using the same protocol.
Euthanasia procedure and collection of BAL, lungs, plasma
Mice were anaesthetized by intraperitoneal (i.p.) ketamine/xylazine injection (87 and 13 μg/g body weight, respectively), blood collected into ethylenediamine tetraacetic acid (EDTA)-containing tube by retro-orbital exsanguination and killed by cervical dislocation. BAL fluid, cells and lungs were collected and processed as described previously 21,24. BAL was performed using 250 μl lavage aliquots of 37°C sterile HBSS for a total of 2 ml. BAL and blood samples were spun at 600 g and plasma and BAL supernatant or BAL cells were isolated. Lungs and heart were perfused with sterile saline, a single-lobe (left) lung fixed in ethanol for periodic acid-Schiff (PAS)-staining/histology processing, and multi-lobed (right) lung homogenized in protease inhibitor solution to obtain a lung homogenate (LH). Total cell counts and differentials were performed as described previously 24.
Measurements and analysis
BAL and LH chemokines and cytokines were measured by sandwich enzyme-linked immunosorbent assay (ELISA) using matched antibody pairs (R&D Systems, Minneapolis, MN, USA) 25, with BAL diluted 1:2 and the LH diluted 1:10. Lung eosinophil peroxidase (EPO) and neutrophil myeloperoxidase (MPO) were assayed as described 21,23 and expressed as percentage over blank [sample optical density (OD)/blank OD]. CRA-specific immunoglobulin (Ig)E was measured by coating a 96-well plate with CRA, incubating 1:10 diluted plasma samples, and detecting CRA-bound IgE with goat-anti-mouse IgE-horseradish peroxidase (HRP) (Bethyl Laboratories, Montgomery, TX, USA) and the results expressed as the OD (OD465–OD590) 26. Mucin quantification of PAS-stained histology was performed through morphometric analysis of digital images using ImageJ freeware (http://rsbweb.nih.gov/ij/) 22. Bronchiole patency was determined by two measurements. First, the area of the entire bronchiole cross-section was determined from the basement membrane inwards, including epithelial cell layer, mucin and open space. Secondly, the area of the open space was determined, excluding bronchoepithelial cells, goblet cells and mucin. The area of open space was divided by the total bronchiole area to determine the percentage patency of the airways. This was performed for each large airway, identified by the presence of epithelial cells per lung slice, and an average patency determined per mouse.
Flow cytometry
After 1·5 and 16 h post CRA-challenge, BAL cells were collected and red blood cells (RBCs) lysed with ammonium chloride-potassium (ACK) lysis buffer (Lonza, Allendale, NJ, USA). Cells were resuspended in fluorescence activated cell sorter (FACS) buffer [0·5% bovine serum albumin (BSA) in ×1 phosphate-buffered saline (PBS)] and incubated with anti-CD16/CD32 (Fc-Block) for 20 min at room temperature prior to staining with fluorescein isothiocyanate (FITC)-Ly6G and AlexaFluor-CD193 or isotype control antibodies (BD Pharmingen, San Diego, CA, USA) for 45 min at 4°C. Cells were washed twice, then fixed in 2% formaldehyde prior to analysis using a FACSCalibur (BD Biosciences, San Diego, CA, USA). Eosinophils were identified as Ly6Glow, CD193+, and neutrophils were identified as Ly6Ghigh, CD193– using FlowJo software (Treestar Inc., Ashland, OR, USA).
Administration of neutralizing antibodies
Neutralizing antibodies to murine keratinocyte-derived chemokine (KC), macrophage inflammatory protein (MIP)-2, eotaxin-1 and eotaxin-2 were diluted in sterile HBSS and given i.t. as two 25 μl aliquots 2 h prior to ethanol gavage following a previously successful protocol 23. Each antibody administration included a cocktail of 10 μg of each specific antibody for a total of 40 μg total antibody per mouse. All chemokine-neutralizing antibodies were monoclonal rat IgG2a or IgG2b (R&D Systems). For the antibody control group, ChromPure Rat IgG (Jackson ImmunoResearch, West Grove, PA, USA) was diluted in sterile HBSS and given i.t. as 40 μg total in two 25 μl aliquots.
Statistical analysis
Data are presented as mean ± standard error of the mean (s.e.m.). For CRA dose–response experiments in Fig. 1, all comparisons were performed by one-way analysis of variance (anova) with Dunnett's post-hoc analysis comparing the groups to normal mice. For time–course kinetics experiments, all comparisons were first analysed with a two-way anova to determine if there was a significant interaction between time and gavage group (water or ethanol). All the analyses indicated a significant effect of time on the measured parameters. Comparisons between three groups (sensitized, water gavage and ethanol gavage) were performed with one-way anova and Dunnett's post-hoc test. For Fig. 8c, eosinophil recruitment control rat IgG was compared to anti-chemokine by Fisher's exact test using the highest number of BAL eosinophils as the threshold level. All statistical analyses were performed using Graphpad Prism version 5·02 (La Jolla, CA, USA). Sample size and number of times each experiment was performed are noted in each figure legend.
Fig. 1.

Model of mild atopic asthma. Female outbred mice were sensitized and challenged with cockroach allergens for three exposures (days 0, 14 and 21), respiratory physiology measured 3 h post-cockroach allergen (CRA)-challenge and mice killed 5 h post-CRA challenge. Four doses of CRA were tested, each mouse receiving the same dose for each of the three allergen exposures. The x-axis numbers refer to the dilution of the CRA (actual concentrations are found in Table 1). (a) Cockroach allergen-specific immunoglobulin (Ig)E. Allergen-specific IgE displayed a dose-dependent decrease with decreasing allergen concentration. (b) Total bronchoalveolar lavage (BAL) cells. (c) BAL eosinophils and lung eosinophil peroxidase (EPO) activity. (d) BAL neutrophils and lung myeloperoxidase (MPO) activity all display dose-dependent decreases with decreasing allergen concentration. (e) Lung histology depicting inflammatory cell infiltrate ‘i’ and mucin ‘m’. F–H: Pulmonary physiology by whole-body plethysmography (WBP), given for measurements without methacholine. (f) Higher CRA corresponded to lengthened times of expiration, Te. (g) Breathing frequency was depressed significantly with higher CRA concentrations. (h) Airways hyperreactivity represented by enhanced pause (PenH) was exacerbated with higher doses of CRA at baseline and after exposure to 25 mg/ml methacholine (McH). *P < 0·05 versus normal; n = 2–6 per group for one experiment.
Fig. 8.
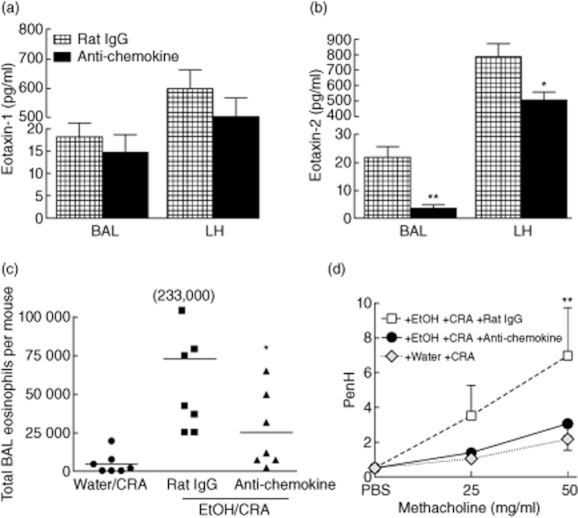
Chemokine-neutralizing antibodies prevent ethanol-induced eosinophil recruitment and airways hyperreactivity (AHR). Ethanol-gavaged and cockroach allergen (CRA)-challenged mice were treated with a cocktail of anti-chemokine antibodies or control rat immunoglobulin (Ig)G 2 h prior to CRA challenge. Mice were killed at 1·5 h post-CRA challenge at the height of eosinophil recruitment or 14 h for AHR measurement. (a) Eotaxin-1 was reduced slightly in both lung compartments, although not significantly. (b) Eotaxin-2 was significantly lower in the bronchoalveolar lavage (BAL) and lung homogenate (LH) of mice receiving chemokine-neutralizing antibodies. (c) BAL eosinophils. Administration of chemokine neutralizing antibodies decreased BAL eosinophil infiltrate. Each symbol represents an individual mouse and the horizontal bar is the mean. The 233 000 is a single mouse whose value did not fit onto the scale. (d) AHR. Development of AHR was prevented in mice receiving chemokine-neutralizing antibodies. Data are mean ± standard deviation of the mean for three to eight mice from two separate experiments. *P < 0·05, **P < 0·01 anti-chemokine compared to control rat IgG.
Results
Inducing mild atopic asthma in mice
CRA-induced murine asthma has been shown previously as a reproducible animal model to study atopic asthma 20–22,26,27. In order to allow for ethanol-induced exacerbations with increases in inflammatory responses, a submaximal asthmatic model was developed. Groups of mice were sensitized on day 0 and challenged on days 14 and 21 with 1:2, 1:10, 1:40 or 1:80 dilution of the CRA stock, corresponding allergen and lipopolysaccharide (LPS) concentrations in Table 1. Following the CRA exposure on day 21, plasma levels of CRA-specific IgE displayed a dose-dependent response and all CRA dilutions evoked significant elevations in allergen-specific IgE over normal mice (Fig. 1a). Bronchoalveolar lavage (BAL) fluid was collected and there was a dose-dependent increase in recovered inflammatory cells (Fig. 1b). Differentials of the BAL cells showed that eosinophils (Fig. 1c) and neutrophils (Fig. 1d) follow a similar dose-dependent response (grey hatched bars) as well as eosinophil and neutrophil peroxidases in the lung tissue (black bars). The BAL eosinophil numbers mirrored the EPO activity within the lung, and the myeloperoxidase mirrored the neutrophil numbers.
Table 1.
Cockroach allergen (CRA) dilutions and corresponding allergen and lipopolysaccharide (LPS) concentrations. Cockroach allergens Bla g1/2 and LPS were measured and are given as dose, per mouse, per 50 μl intratracheal exposure
| CRA dilution | Bla g1/2 (ng/dose) | LPS (ng/dose) |
|---|---|---|
| 1:2 | 2000 | 600 |
| 1:10 | 400 | 120 |
| 1:40 | 100 | 30 |
| 1:80 | 50 | 15 |
Histological processing and PAS staining of ethanol-fixed lungs show severe inflammation and massive mucin production in mice receiving 1:2 CRA, and the severity of inflammation and magnitude of mucin staining decreased with decreasing allergen concentration (Fig. 1e). Obstruction of airways often leads to worsened respiratory physiology; for example, lengthened expiratory time 22,28. Respiration was analysed by WBP and showed increases in the time of expiration prior to methacholine challenge with higher allergen concentration exposure (Fig. 1f), signifying increasing intrapleural obstructions in these mice. In addition, we have shown previously that breathing frequency (F) drops with increasing asthma morbidity 22, and this parameter also decreased in a dose-dependent manner (Fig. 1g). Lastly, airways hyperreactivity was measured, as represented by enhanced pause (PenH) (Fig. 1h), and showed that mice receiving higher concentrations of allergen displayed higher airways hyperreactivity with or without exposure to the bronchoconstrictor methacholine. Because we wished to develop a mild asthma model, the 1:40 CRA dose was chosen for all subsequent studies.
Ethanol accelerates acute mucin production and airways obstruction in CRA-challenged mice
One of the major contributors to asthma morbidity is mucus overproduction in the lungs 29, often leading to considerable occlusion of the airways and in severe cases plug formation and complete occlusion 30. As previous work shows that ethanol gavage does not induce mucin in non-allergen sensitized mice, a group of ethanol-alone mice was not studied 19. The effect of concurrent ethanol exposure with CRA challenge on mucin production was investigated. Airway epithelial cell mucin accumulation was measured by morphometric analysis of PAS-stained sections of lung from CRA-sensitized mice gavaged with either water or 32% ethanol, challenged with cockroach allergens 30 min post-gavage, and killed at multiple time-points post-CRA challenge (Fig. 2a). Mucin in lungs of sensitized, non-gavaged, non-CRA-challenged mice was also measured (S). Ethanol gavage increased mucin production acutely nearly threefold over water-gavaged controls within 1·5 h post-CRA challenge, which was a significant eightfold elevation over the levels found in sensitized mice. The airway mucin levels in the ethanol gavaged mice and water-gavaged group continued to increase and airway mucin in both groups reached maximal levels at 42 h post-CRA challenge. Representative histology from PAS-stained lungs of sensitized mice and mice killed at 1·5 h post-CRA challenge is shown in Fig. 2b. Sensitized mice have little to no mucin accumulation in the lungs prior to gavage or CRA challenge (Fig. 2b, Sensitized panel), and water gavage and CRA-challenged mice showed small amounts of mucin accumulation in the bronchoepithelial cells (Fig. 2b, +Water +CRA panel). Sensitized mice with ethanol gavage and CRA challenge show massive mucin production at this time (Fig. 2b, +EtOH +CRA panel).
Fig. 2.

Ethanol administration augments acute mucin production resulting in significant airway obstruction. (a) Bronchiole airway mucin was measured by computer-aided morphometry of digital images at the indicated times after cockroach allergen (CRA) challenge. Ethanol caused a significant elevation in mucin production at 1·5 h post-CRA challenge. (b) Representative ×40 magnification of periodic acid Schiff (PAS)-stained histology from mouse lungs depicting bronchial epithelial cell mucin production in sensitized mice (Sensitized) and mice gavaged with water, then challenged with CRA (+Water +CRA) or 32% ethanol and challenged with CRA (+EtOH +CRA). Mice were killed 1·5 h post-CRA challenge. (c) Representative ×4 magnification of histology from B, depicting area of total bronchiole A1, area of bronchiole opening A2 and occluded (O) and closed (C) bronchioles. (d) Ethanol-induced airways obstruction was noted by a significant decrease in bronchiole patency. (e) A significant increase in mucus plugs was observed in ethanol-gavaged, CRA-challenged mice. (f) Time of expiration (Te), a whole-body plethysmography (WBP) parameter used to assess intrapleural airways obstruction, was increased in ethanol-gavaged mice at 1·5 h post-CRA challenge; 25 mg/ml methacholine. Data are mean ± standard deviation of the mean for two separate experiments; n = 3–8 mice. *P < 0·05 +EtOH+CRA compared to both Sensitized (S) and +Water +CRA. #P < 0·05 compared to sensitized.
To investigate whether ethanol-induced mucin overproduction at 1·5 h post-CRA challenge was leading to significant obstruction of the airways, histological airway measurements were taken of PAS-stained lungs from this time-point. Representative histology of lung bronchioles is shown in Fig. 2c and digital images of these slides were used to measure bronchiole patency (Fig. 2d) and the extent of mucin plugging (Fig. 2e). Percentage patency of the bronchioles was determined by measuring the area of the bronchiole lumen (Fig. 2c, area A2, dotted black line) divided by the total bronchiole diameter, including the epithelial cell layer (Fig. 2c, area A1, solid black line). Sensitized mice prior to gavage or third CRA challenge display approximately 55% airway patency (Fig. 2d). Water gavage and third CRA challenge caused a decrease in patency to 40%, although this was not significant. Ethanol gavage with the third CRA challenge caused a significant decrease in airway patency to 27%, approximately half the bronchiole luminal space of their sensitized mice counterparts.
As shown in the right panel of Fig. 2c, mucin overproduction can lead to the accumulation of extracellular mucin plugs, which can result in closing of the airways (C) or complete occlusion (O). The total number of mucus plugs resulting in either airways closing or occlusion was counted per mouse (Fig. 2e). Sensitized mice did not show mucin plug formation. Water-gavaged CRA-challenged mice showed a small amount of mucin plug formation. In comparison, ethanol-gavaged CRA-challenged mice display a significant eightfold increase in mucin plug formation compared to water-gavaged counterparts. WBP was used to investigate whether the histological findings of airways obstruction led to respiratory impairment (Fig. 2f). Expiratory time, a WBP parameter, is a breathing parameter linked closely to a worsened breathing ability and intrapleural airways obstruction 28,31. WBP was used for these measurements rather than forced ventilation as the study measured spontaneous time of expiration, rather than airways hyperreactivity. Concurrent water and CRA exposures led to a modest lengthening of expiratory time; however, the effects of ethanol were greater, leading to a significant twofold increase in expiratory time when compared to sensitized mice and a significant elevation over water-gavaged CRA-challenged controls. Together with the histological findings, these data show that concurrent ethanol gavage with CRA exposure leads to acute, significant mucin overproduction and respiratory obstruction. In comparison, concurrent water gavage and allergen exposure did not exacerbate either significantly at the early time-point.
Ethanol has limited impact on Th1/Th2 cytokine profiles
Cytokines are critical mediators of asthma and are often used in classifying the degree of severity 32. To determine whether ethanol would alter Th1 and Th2 cytokine expression in this model of CRA-induced allergic asthma, BAL Th1 (Fig. 3a) and Th2 (Fig. 3b) cytokines were measured. We have reported previously a prompt increase in BAL tumour necrosis factor (TNF) levels after the third allergen exposure 33. Figure 3a shows that the third exposure to CRA increases BAL levels of Th1 cytokines including TNF, interleukin (IL)-12 and interferon (IFN)-γ, as anticipated. Ethanol gavage increased BAL TNF levels significantly compared to the sensitized mice (i.e. time 0 mice) within 1·5 h after CRA challenge. TNF levels after CRA exposure in the water-gavaged mice did not increase significantly until 3·5 h. While the ethanol-gavaged mice had greater BAL TNF levels, these were not increased statistically significantly compared to water gavage. Th2 cytokines were also increased in the BAL by allergen exposure but, again, ethanol never resulted in significant increases compared to water (Fig. 3b). These data demonstrate that the ethanol gavage did not globally alter all inflammatory responses.
Fig. 3.
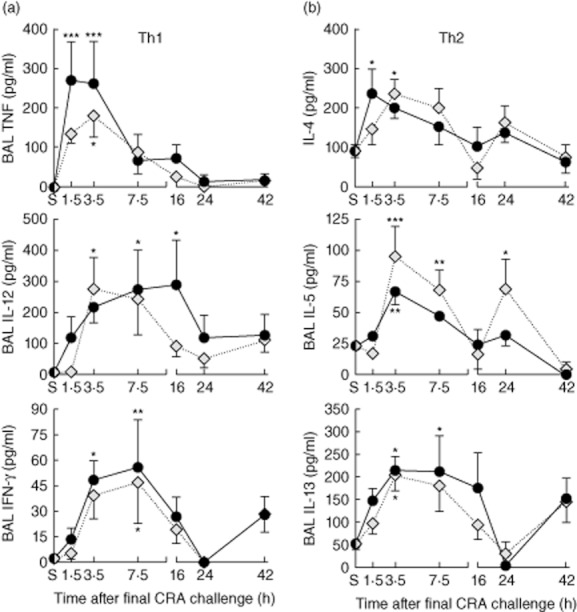
Bronchoalveolar lavage (BAL) T helper type 1 (Th1)/Th2 cytokine production. Mice were gavaged with ethanol or water and challenged 30 min later with cockroach allergen (CRA). BAL was collected at the indicated times. (a) BAL Th1 cytokines including tumour necrosis factor (TNF), interleukin (IL)-12 and interferon (IFN-γ). (b) BAL Th2 cytokines including IL-4, IL-5 and IL-13. Statistical analysis showed that time had a significant impact on cytokine production, but ethanol did not increase the BAL cytokine levels compared to water gavage; *P < 0·05, **0·01, ***0·001 versus sensitized (S).
Ethanol exposure prior to allergen challenge increases BAL neutrophil recruitment
As shown in Fig. 2, ethanol gavage, together with allergen challenge, resulted in significant airflow obstructions, suggesting an ethanol-induced increase in asthma severity. Neutrophil infiltration into the lung is another predictor of increased asthma severity, and is found often in conjunction with airflow obstruction 34,35. Furthermore, rapid chemokine production has been documented in the CRA asthma model 24. Given this background, the effect of ethanol exposure prior to CRA challenge on BAL neutrophil infiltration was investigated (Fig. 4). Several protein mediators play a significant role in the migration of neutrophils to an inflammatory site, including the CXC chemokines MIP-2 and KC 36. BAL MIP-2 (Fig. 4a) and KC (Fig. 4b) were measured from sensitized mice (S) and sensitized mice gavaged with either water or ethanol followed by CRA challenge. Both CXC chemokines exhibited similar kinetics, increasing sharply to peak levels within 1·5 h and resolving to near baseline by 7·5 h post-CRA challenge. Water-gavaged, CRA-challenged mice showed modest CXC production which was not elevated significantly over sensitized levels, while ethanol-gavaged CRA-challenged mice showed significant CXC elevations. Next, BAL neutrophil infiltration was quantified (Fig. 4c). Allergen challenge caused neutrophil recruitment to the lungs in both the water- and ethanol-gavaged groups, although the increase in BAL neutrophils was not evident until 3·5 h post-CRA challenge. While the water-gavaged CRA-challenged mice displayed increased BAL neutrophils, no time–point from this group was elevated significantly over sensitized levels. However, the ethanol-gavaged mice maintained significant BAL neutrophil elevations between 3·5 and 16 h post-CRA challenge compared to sensitized levels.
Fig. 4.
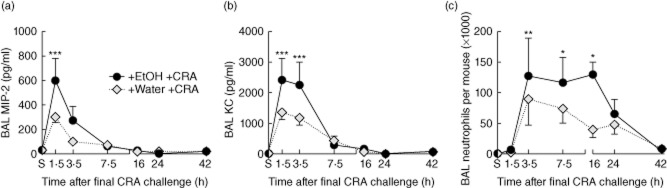
Ethanol gavage enhances CXC chemokine production and neutrophil recruitment in the lung. Bronchoalveolar lavage (BAL) macrophage inflammatory protein (MIP)-2 (a) and keratinocyte-derived chemokine (KC) (b) were elevated post-cockroach allergen (CRA) challenge, peaking at 1·5 h, with greater increases in ethanol-gavaged mice. Only ethanol-gavaged CRA-challenged mice exhibit significant CXC chemokine increases over non-gavaged sensitized (S) mice. (C) BAL neutrophils. Ethanol gavage induced significant increases in BAL neutrophils between 3·5 and 16 h post-CRA challenge, while water-gavaged CRA-challenged mice also show increased neutrophils, although more modest and not significantly elevated from sensitized mice. Data are mean ± standard deviation of the mean for four to eight mice from two separate experiments; *P < 0·05, **0·01, ***0·001 versus sensitized.
Ethanol exposure prior to allergen challenge increases BAL eosinophil recruitment
Another hallmark of asthma is the recruitment of eosinophils to the lung. The presence of these cells is correlated strongly with worsening disease and impaired respiratory function 34, as they act as effector cells through secretion of toxic and proinflammatory granule products 37,38. Recruitment of eosinophils into the bronchoalveolar space, an important step in asthma pathogenesis 39, is orchestrated by a number of proteins including the CC chemokines eotaxin-1 and eotaxin-2. Eotaxin-1 has been shown to be a critical mediator in CRA-induced asthma 25. BAL fluid was analysed for the presence of eotaxins and eosinophils post-CRA challenge, as well as in sensitized mice prior to gavage and third allergen challenge. BAL eotaxin-1 (Fig. 5a) was elevated acutely upon CRA exposure in both groups, although the increase occurred more quickly in the ethanol-gavaged group compared to the water-gavaged group. BAL eotaxin-2 was also measured (Fig. 5b) and showed higher concentrations than eotaxin 1, with levels in ethanol-gavaged mice increased significantly over sensitized mice at 16 h post-CRA challenge.
Fig. 5.

Ethanol enhances pulmonary eotaxin production and eosinophil recruitment. (a) Bronchoalveolar lavage (BAL) eotaxin-1 is elevated significantly above baseline levels in both gavage groups, with higher concentrations found in mice gavaged with ethanol. (b) BAL eotaxin-2 is elevated significantly only in ethanol-gavaged mice. (c) BAL eosinophils were quantified. Significant eosinophil recruitment over sensitized levels was observed in ethanol-gavaged mice at all time-points measured post-cockroach allergen (CRA) challenge. Water-gavaged mice showed more modest eosinophil recruitment. (d) Representative cytospin pictographs 1·5 h post-CRA challenge, highlighting ethanol-induced BAL eosinophil recruitment at this early time. Data are mean ± standard deviation of the mean for four to eight mice, representative of two separate experiments; *P < 0·05, **0·01, ***0·001 compared to the sensitized group; ##P < 0·001 compared to both sensitized and water-gavaged group.
BAL eosinophils for each time-point above were quantified (Fig. 5c). As anticipated, allergen challenge induced the recruitment of eosinophils into the lung. Interestingly, prior exposure to ethanol caused a significant increase in recruitment of pulmonary eosinophils compared to water at the first measured time-point, i.e. within 90 min of allergen challenge. Representative BAL cytospin photomicrographs from 1·5 h post-CRA challenge are shown in Fig. 5d. BAL cells from sensitized mice were primarily macrophages (Fig. 5d, left panel), and the BAL cells from water-gavaged CRA-challenged mice were also primarily macrophages (Fig. 5d, centre panel). Sensitized mice gavaged with ethanol and CRA-challenged have much greater eosinophil recruitment (Fig. 5d, right panel). To the best of our knowledge, this is the first publication to show that acute ethanol exposure aggravates eosinophilic inflammation after exposure to an allergen.
Neutrophils are typical first responders to sites of inflammation, followed usually at a later stage by eosinophil recruitment 40–42. The CRA preparation used for these studies is the same material used for skin testing in patients and contains a number of Toll-like receptor (TLR) agonists such as LPS. Because LPS is present, the pulmonary recruitment of neutrophils was not unexpected 43. However, the findings that prior ethanol exposure results in eosinophil recruitment preceding that of neutrophils after allergen exposure was not anticipated. Figure 6a shows the relative percentages of eosinophils and neutrophils in the BAL cells at 1·5 and 16 h post-CRA challenge in ethanol-gavaged mice, as quantified by differential counting of cytospin slides. At the 1·5-h time-point, eosinophils represent 40% of total BAL cells, while neutrophils represent fewer than 3%. At the 16-h time-point both eosinophils and neutrophils are present in approximately equal numbers. To verify that the earlier responders were indeed eosinophils, flow cytometry was used to assess cells recovered from the BAL in the ethanol-gavaged mice at 1·5 h (Fig. 6b). At this early time-point, flow cytometry confirmed low neutrophil numbers, which represented fewer than 1% of the recruited cells. By 16 h post-CRA challenge the neutrophils and eosinophils are present in equal proportions (Fig. 6c).
Fig. 6.
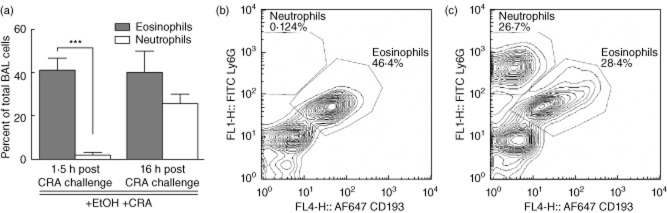
Eosinophil and neutrophil recruitment confirmed by flow cytometry. (a) Percentages of bronchoalveolar lavage (BAL) eosinophils and neutrophils at 1·5 and 16 h post-cockroach allergen (CRA) challenge, as determined by light microscopy. Identification of eosinophils (CD193+ Ly6Glow) and neutrophils (CD193-Ly6Ghigh) was performed. Relative cell percentages at (b) 1·5 h and (c) 16 h post-CRA challenge. Flow cytometry confirms that the early BAL cell recruitment was composed primarily of eosinophils with almost no neutrophils at this time. By 16 h both neutrophils and eosinophils represent a significant percentage of BAL cell recruitment. Data for (a) are mean ± standard deviation of the mean for seven to eight mice from two separate experiments; ***P < 0·001 eosinophils versus neutrophils. Flow cytometry pictographs representative of four mice per time-point.
Airways hyperreactivity exacerbated upon alcohol exposure
Figures 5 and 6 demonstrated that concurrent alcohol and allergen exposures led to exacerbated eosinophilic inflammation in the lung. Airway eosinophils have been correlated strongly with increased respiratory distress 44 and are thought to contribute to pulmonary dysfunction through degranulation and the production of mediators, including proinflammatory cytokines, cytotoxic proteases and cysteinyl leukotrienes 45,46. These mediators can lead to exacerbated bronchoconstriction and airways hyperreactivity (AHR), another hallmark of asthma 47. AHR was investigated in our mice by analysis of the WBP PenH in sensitized mice prior to gavage or CRA challenge (S), and in mice gavaged with water or ethanol and analysed post-CRA challenge (Fig. 7a). WBP was used for these studies to screen experimental groups quickly using multiple measurements. In this mild model of asthma, water gavage prior to CRA challenge did not increase PenH, consistent with the data in Fig. 1. However, ethanol-gavaged, CRA-challenged mice displayed increased PenH, which was elevated compared to baseline sensitized mice within 1·5 h. Given controversy with the methodology, WBP was confirmed by forced ventilation measurements of pulmonary resistance (R; FlexiVent) (Fig. 7b) 14 h post-CRA challenge, a time chosen to reduce the possibility of mucin differences as a confounding factor. Water-gavaged mice challenged with CRA showed increased R at this time; however, similar to PenH results, the response to methacholine was not elevated significantly over baseline responses. Resistance of ethanol-gavaged CRA-challenged mice was greater than that of the water-gavaged control group, and responses to methacholine were elevated significantly over baseline responses. These data indicate that ethanol exposure prior to allergen challenge evokes more severe AHR exacerbations.
Fig. 7.
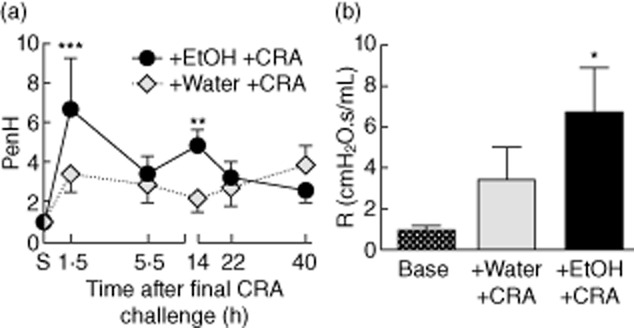
Ethanol exacerbates airways hyperreactivity (AHR). (a) AHR was measured by whole-body plethysmography (WBP) by assessing enhanced pause (PenH) at the indicated time-points. PenH was exacerbated at both 1·5 and 14 h post-cockroach allergen (CRA) challenge in mice receiving ethanol, while it was not elevated significantly in the water-gavaged group. (b) AHR was confirmed by measuring airways resistance (R) 14 h post-CRA challenge at baseline and after a 10-min exposure to 25 mg/ml methacholine. Only ethanol-gavaged CRA-challenged mice had significantly elevated resistance over baseline measurements. WBP responses are to 50 mg/ml methacholine for six to eight mice, representative of two separate experiments. Airways resistance responses were in response to 25 mg/ml methacholine, three mice per group. All data represent mean ± standard deviation of the mean; *P < 0·05, **0·01, ***0·001 compared to the sensitized group.
Chemokine-neutralizing antibodies prevent eosinophil recruitment and AHR
Ethanol exacerbates the pulmonary responses to the third CRA challenge, as shown by the significant increases compared to baseline that occurred only in the ethanol plus allergen group. The presence of greater numbers of eosinophils and neutrophils in the lung has been correlated closely with propagating asthmatic responses such as AHR 36,48. To investigate the mechanism of ethanol-induced granulocyte recruitment and the potential role of these cells in respiratory exacerbations, neutralizing antibodies were administered as a cocktail targeting the eosinophil and neutrophil chemokines eotaxin-1 and -2, and MIP-2 and KC, respectively. Neutralizing antibodies were given intratracheally 2 h prior to ethanol gavage, then BAL and LH chemokine levels and granulocyte inflammation were measured at1·5 h post-CRA challenge. In separate groups of mice, AHR was measured at 14 h.
Interestingly, chemokine-neutralizing antibodies did not decrease pulmonary neutrophil chemokines or cell recruitment (data not shown), using an antibody neutralizing protocol which has proved effective in the asthma model 23. Additionally, the anti-chemokine-treated mice did not show a reduction of eotaxin-1 in BAL and LH (Fig. 8a). However, eotaxin-2 was reduced significantly in anti-chemokine-treated mice in both the BAL and lung homogenates (Fig. 8b). Importantly, this chemokine neutralization protocol blocked ethanol-induced eosinophil BAL infiltrate significantly, reducing eosinophil numbers almost threefold from 72 000 in the control antibody group to 24 000 in the chemokine-neutralizing antibody group (Fig. 8c).
The literature has shown that eosinophil accumulation in the airspaces represents a major component responsible for the development of AHR 49,50–52. To test whether the decreases in eosinophils observed in mice receiving anti-chemokine treatment may also result in decreased AHR, PenH was measured by WBP (Fig. 8d). Mice receiving control antibodies prior to ethanol gavage and CRA challenge had a significant increase in AHR, consistent with Fig. 7. In contrast, mice receiving chemokine-neutralizing antibodies prior to ethanol gavage and CRA challenge were prevented from developing ethanol-induced exacerbations in AHR, with responses comparable to those of water-gavaged CRA-challenged mice at this time.
These data show that a single intratracheal administration of chemokine-neutralizing antibodies reduced CC chemokine levels successfully in the lung and was sufficient to prevent the ethanol-enhanced acute infiltration of eosinophils and the development of AHR. Altogether, these results suggest that chemokines play a substantial role in mediating ethanol-induced asthmatic exacerbations, and shed light on the mechanisms through which the exacerbations occur.
Discussion
The data provided here show that acute alcohol exposure exacerbates key allergen-induced asthmatic responses. A remarkable finding was the alcohol-induced augmentation of BAL eosinophil recruitment, which is orchestrated most probably through alcohol-specific increases in CC chemokines, as we have shown that administration of chemokine-neutralizing antibodies prior to alcohol and allergen exposures will prevent this early eosinophil response. These data are unorthodox for two reasons. Neutrophils are cited conventionally as the first cells recruited to the site of allergic inflammation, followed by later eosinophil recruitment 40–42. Further, previous studies have shown that acute ethanol exposure actually decreases CC chemokine production and eosinophilic inflammation 53,54. In this study we have provided a novel example where acute alcohol exposure not only increases CC chemokines and eosinophil inflammation, but also alters the kinetics of cell recruitment, so that eosinophil influx precedes that of neutrophils.
These early BAL eosinophils are probable candidates for orchestrating the time–course of ethanol-induced asthmatic exacerbations. Administration of chemokine-neutralizing antibodies reduced an important mediator involved in eosinophil migration, blocked early eosinophil infiltration into the lung and prevented the development of AHR. A possible explanation for improvements in respiratory physiology could be the importance of chemokines, as eotaxins have been shown to be essential in mediating eosinophil-induced asthmatic inflammation and exacerbations in AHR 41,44,50,55. Our model suggests that chemokine-neutralizing antibodies may prevent ethanol-induced exacerbations in AHR through the reduced recruitment of inflammatory cells and protein mediators contributing to AHR. Future work needs to be conducted to investigate the roles of these cells and molecules in mediating ethanol-exacerbations in airways responses.
Interestingly, in historical times, when less was understood about asthma triggers and inflammatory responses, alcohol was used as asthma therapy 56. During that same time in history many reported asthma-related deaths failed to demonstrate pathological changes associated with the severe, chronic inflammation in asthma deaths reported today 57,58, suggesting that acute responses to inappropriate therapies may have been the cause of these deaths. In light of the data from our study presented here, one would not think to suggest alcohol as a current therapeutic option for asthma. A better understanding of asthma triggers is important not only for asthma prevention, but is also crucial for the development of effective, safe therapeutics.
Perhaps most noteworthy are the comparisons between the magnitude of responses between the water-gavaged and ethanol-gavaged CRA-challenged mice. In our mild asthma mice, water gavage and the third CRA challenge induced only weak asthmatic responses; most parameters investigated were not elevated significantly from pre-gavage sensitized levels at any time-point. In contrast, mice receiving oral alcohol prior to a third allergen exposure displayed significant, marked increases in almost all asthmatic responses measured. In effect, the insult of oral ethanol exposure coupled with the allergen exposure pushed sensitized mice with otherwise mild asthmatic reactions into having more significant, severe reactions.
By showing that ethanol exposure caused more severe asthmatic responses, these data lend support to the proposition that multiple inflammatory stimuli are necessary for the development of severe airway disease 59. CRA-induced asthma is a clinically relevant model that provides an excellent framework to study multiple-stimuli-induced asthma – CRA is heterogeneous and already provides inflammatory provocation from several pathways, including innate immune stimulation with proteases, LPS and chitin, and adaptive immunity via allergen exposures 60,61. LPS has been reported to impact neutrophil activation directly 62, providing a potential explanation as to why the anti-chemokine intervention did not reduce neutrophil recruitment. Additional inflammatory ‘hits’ in this model in the human context of disease are highly likely, especially with epidemiology showing that the populations most probably affected by CRA-induced asthma are younger subjects living in inner-city and poverty conditions, which are consequently exposed to asthma triggers including diesel particulates, cigarette smoke and mould, and are likely to engage in social drinking 63–65. Our studies use a model of a sensitized individual who becomes acutely intoxicated, replicating a young person who engages in binge drinking. Binge drinking has been identified as a significant issue by the US Centers for Disease Control 66.
IgE is an important component of the allergic response to allergens, and clinical studies have also demonstrated that patients who consume alcohol have increased serum IgE levels 67. Another study demonstrated that self-reported alcohol exposure in humans is associated strongly with increased IgE levels 68. This increase in IgE occurs to allergens such as peanuts, even when the patients are asymptomatic 69. Ethanol administration to mice will also increase IgE levels 70.
Previous investigators have published that alcohol will depress asthmatic responses in experimental animals, findings which conflict directly with the current data. It has been reported that acute ethanol exposure depresses AHR, but those studies were not performed in allergen-sensitized mice 71. Oldenburg et al. published findings that contradict our results directly; specifically, that oral alcohol will depress AHR and pulmonary eosinophil recruitment 72. There were substantial differences in the experimental models. For example, our study utilized the outbred HSD-ICR mice as opposed to inbred strains. Additionally, the prior group chronically provided alcohol in the drinking water for 6 weeks, then exposed the mice to allergens. This would reproduce the clinical scenario of a chronic alcoholic, whose first allergen exposure occurs after chronic alcohol consumption has already been initiated. In contrast, our model mimics a different clinical scenario where an allergen-sensitized individual acutely consumes alcohol. In this setting, ethanol will exacerbate the asthmatic responses. It is quite possible that when mildly asthmatic, relatively asymptomatic CRA-sensitized individuals consume alcohol during a time of allergen exposure, asthmatic responses are strengthened.
Acknowledgments
The authors wish to thank Jessica Allen for the computer-aided morphometry in Fig. 2. This work was supported in part by NIH grant ES 013528.
Disclosure
The authors declare no conflicts of interest.
References
- 1.Karamanou M, Androutsos G. Aretaeus of Cappadocia and the first clinical description of asthma. Am J Respir Crit Care Med. 2011;184:1420–1421. doi: 10.1164/ajrccm.184.12.1420b. [DOI] [PubMed] [Google Scholar]
- 2.Jackson DJ, Sykes A, Mallia P, Johnston SL. Asthma exacerbations: origin, effect, and prevention. J Allergy Clin Immunol. 2011;128:1165–1174. doi: 10.1016/j.jaci.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walsh GM. An update on emerging drugs for asthma. Expert Opin Emerg Drugs. 2012;17:37–42. doi: 10.1517/14728214.2012.657625. [DOI] [PubMed] [Google Scholar]
- 4.Calderon MA, Casale TB, Togias A, Bousquet J, Durham SR, Demoly P. Allergen-specific immunotherapy for respiratory allergies: from meta-analysis to registration and beyond. J Allergy Clin Immunol. 2011;127:30–38. doi: 10.1016/j.jaci.2010.08.024. [DOI] [PubMed] [Google Scholar]
- 5.Akinbami LJ, Moorman JE, Liu X. Asthma prevalence, health care use, and mortality: United States, 2005–2009. Natl Health Stat Rep. 2011:1–14. [PubMed] [Google Scholar]
- 6.Meltzer EO, Blaiss MS, Nathan RA, Doherty DE, Murphy KR, Stoloff SW. Asthma burden in the United States: results of the 2009 Asthma Insight and Management survey. Allergy Asthma Proc. 2012;33:36–46. doi: 10.2500/aap.2011.32.3519. [DOI] [PubMed] [Google Scholar]
- 7.Crocker DD, Kinyota S, Dumitru GG, et al. Effectiveness of home-based, multi-trigger, multicomponent interventions with an environmental focus for reducing asthma morbidity: a community guide systematic review. Am J Prev Med. 2011;41:S5–32. doi: 10.1016/j.amepre.2011.05.012. [DOI] [PubMed] [Google Scholar]
- 8.Ayres JG. Alcohol-induced bronchial asthma. J Allergy Clin Immunol. 1997;99:860. doi: 10.1016/s0091-6749(97)80026-7. [DOI] [PubMed] [Google Scholar]
- 9.Ayres JG, Clark TJ. Alcoholic drinks and asthma: a survey. Br J Dis Chest. 1983;77:370–375. [PubMed] [Google Scholar]
- 10.Kurosawa M, Kobayashi S. Investigation of the effect of ketotifen on alcohol-induced asthma: a case study. J Int Med Res. 1990;18:435–439. doi: 10.1177/030006059001800513. [DOI] [PubMed] [Google Scholar]
- 11.Shimoda T, Kohno S, Takao A, et al. Investigation of the mechanism of alcohol-induced bronchial asthma. J Allergy Clin Immunol. 1996;97:74–84. doi: 10.1016/s0091-6749(96)70285-3. [DOI] [PubMed] [Google Scholar]
- 12.Sisson JH. Alcohol and airways function in health and disease. Alcohol. 2007;41:293–307. doi: 10.1016/j.alcohol.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vally H, de Klerk N, Thompson PJ. Alcoholic drinks: important triggers for asthma. J Allergy Clin Immunol. 2000;105:462–467. doi: 10.1067/mai.2000.104548. [DOI] [PubMed] [Google Scholar]
- 14.Zellweger JP. Alcohol-induced asthma: not only in Asians. J Allergy Clin Immunol. 1997;99:860. doi: 10.1016/s0091-6749(97)80027-9. [DOI] [PubMed] [Google Scholar]
- 15.Vally H, Thompson PJ. Allergic and asthmatic reactions to alcoholic drinks. Addict Biol. 2003;8:3–11. doi: 10.1080/1355621031000069828. [DOI] [PubMed] [Google Scholar]
- 16.Dahl R, Henriksen JM, Harving H. Red wine asthma: a controlled challenge study. J Allergy Clin Immunol. 1986;78:1126–1129. doi: 10.1016/0091-6749(86)90261-7. [DOI] [PubMed] [Google Scholar]
- 17.Linneberg A, Gonzalez-Quintela A, Vidal C, et al. Genetic determinants of both ethanol and acetaldehyde metabolism influence alcohol hypersensitivity and drinking behaviour among Scandinavians. Clin Exp Allergy. 2010;40:123–130. doi: 10.1111/j.1365-2222.2009.03398.x. [DOI] [PubMed] [Google Scholar]
- 18.Young C. Patient education. Avoiding asthma triggers: a primer for patients. J Am Osteopath Assoc. 2011;111:S30–32. [PubMed] [Google Scholar]
- 19.Bouchard JC, Kim J, Beal DR, Vaickus LJ, Craciun FL, Remick DG. Acute oral ethanol exposure triggers asthma in cockroach allergen-sensitized mice. Am J Pathol. 2012;181:845–857. doi: 10.1016/j.ajpath.2012.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vaickus LJ, Bouchard J, Kim J, Natarajan S, Remick DG. Inbred and outbred mice have equivalent variability in a cockroach allergen-induced model of asthma. Comp Med. 2010;60:420–426. [PMC free article] [PubMed] [Google Scholar]
- 21.Vaickus LJ, Bouchard J, Kim J, Natarajan S, Remick DG. Oral tolerance inhibits pulmonary eosinophilia in a cockroach allergen induced model of asthma: a randomized laboratory study. Respir Res. 2010;11:160–171. doi: 10.1186/1465-9921-11-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vaickus LJ, Bouchard J, Kim J, Natarajan S, Remick DG. Assessing pulmonary pathology by detailed examination of respiratory function. Am J Pathol. 2010;177:1861–1869. doi: 10.2353/ajpath.2010.100053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim J, Natarajan S, Vaickus LJ, et al. Diesel exhaust particulates exacerbate asthma-like inflammation by increasing CXC chemokines. Am J Pathol. 2011;179:2730–2739. doi: 10.1016/j.ajpath.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKinley L, Kim J, Bolgos GL, Siddiqui J, Remick DG. CXC chemokines modulate IgE secretion and pulmonary inflammation in a model of allergic asthma. Cytokine. 2005;32:178–185. doi: 10.1016/j.cyto.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 25.Kim J, Merry AC, Nemzek JA, Bolgos GL, Siddiqui J, Remick DG. Eotaxin represents the principal eosinophil chemoattractant in a novel murine asthma model induced by house dust containing cockroach allergens. J Immunol. 2001;167:2808–2815. doi: 10.4049/jimmunol.167.5.2808. [DOI] [PubMed] [Google Scholar]
- 26.Natarajan S, Kim J, Bouchard J, Cruikshank W, Remick DG. Reducing LPS content in cockroach allergens increases pulmonary cytokine production without increasing inflammation: a randomized laboratory study. BMC Pulm Med. 2011;11:12–25. doi: 10.1186/1471-2466-11-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Natarajan S, Kim J, Bouchard J, Cruikshank W, Remick DG. Pulmonary endotoxin tolerance protects against cockroach allergen-induced asthma-like inflammation in a mouse model. Int Arch Allergy Appl Immunol. 2012;158:120–130. doi: 10.1159/000330896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tantucci C, Ellaffi M, Duguet A, et al. Dynamic hyperinflation and flow limitation during methacholine-induced bronchoconstriction in asthma. Eur Respir J. 1999;14:295–301. doi: 10.1183/09031936.99.142. [DOI] [PubMed] [Google Scholar]
- 29.Lai HY, Rogers DF. Mucus hypersecretion in asthma: intracellular signalling pathways as targets for pharmacotherapy. Curr Opin Allergy Clin Immunol. 2010;10:67–76. doi: 10.1097/ACI.0b013e328334643a. [DOI] [PubMed] [Google Scholar]
- 30.Rogers DF. Airway mucus hypersecretion in asthma: an undervalued pathology? Curr Opin Pharmacol. 2004;4:241–250. doi: 10.1016/j.coph.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 31.Baydur A, Wilkinson L, Mehdian R, Bains B, Milic-Emili J. Extrathoracic expiratory flow limitation in obesity and obstructive and restrictive disorders: effects of increasing negative expiratory pressure. Chest. 2004;125:98–105. doi: 10.1378/chest.125.1.98. [DOI] [PubMed] [Google Scholar]
- 32.Williams CM, Rahman S, Hubeau C, Ma HL. Cytokine pathways in allergic disease. Toxicol Pathol. 2012;40:205–215. doi: 10.1177/0192623311430694. [DOI] [PubMed] [Google Scholar]
- 33.Kim J, McKinley L, Natarajan S, et al. Anti-tumor necrosis factor-alpha antibody treatment reduces pulmonary inflammation and methacholine hyper-responsiveness in a murine asthma model induced by house dust. Clin Exp Allergy. 2006;36:122–132. doi: 10.1111/j.1365-2222.2005.02407.x. [DOI] [PubMed] [Google Scholar]
- 34.Fahy JV. Eosinophilic and neutrophilic inflammation in asthma: insights from clinical studies. Proc Am Thorac Soc. 2009;6:256–259. doi: 10.1513/pats.200808-087RM. [DOI] [PubMed] [Google Scholar]
- 35.Orihara K, Matsuda A. Pathophysiological roles of microvascular alterations in pulmonary inflammatory diseases: possible implications of tumor necrosis factor-alpha and CXC chemokines. Int J Chron Obstruct Pulmon Dis. 2008;3:619–627. doi: 10.2147/copd.s3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Turato G, Baraldo S, Zuin R, Saetta M. The laws of attraction: chemokines, neutrophils and eosinophils in severe exacerbations of asthma. Thorax. 2007;62:465–466. doi: 10.1136/thx.2006.070656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sampson AP. The role of eosinophils and neutrophils in inflammation. Clin Exp Allergy. 2000;30(Suppl. 1):22–27. doi: 10.1046/j.1365-2222.2000.00092.x. [DOI] [PubMed] [Google Scholar]
- 38.Monteseirin J. Neutrophils and asthma. J Invest Allergol Clin Immunol. 2009;19:340–354. [PubMed] [Google Scholar]
- 39.Uhm TG, Kim BS, Chung IY. Eosinophil development, regulation of eosinophil-specific genes, and role of eosinophils in the pathogenesis of asthma. Allergy Asthma Immunol Res. 2012;4:68–79. doi: 10.4168/aair.2012.4.2.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gauvreau GM, Watson RM, O'Byrne PM. Kinetics of allergen-induced airway eosinophilic cytokine production and airway inflammation. Am J Respir Crit Care Med. 1999;160:640–647. doi: 10.1164/ajrccm.160.2.9809130. [DOI] [PubMed] [Google Scholar]
- 41.Lukacs NW, Strieter RM, Chensue SW, Widmer M, Kunkel SL. TNF-alpha mediates recruitment of neutrophils and eosinophils during airway inflammation. J Immunol. 1995;154:5411–5417. [PubMed] [Google Scholar]
- 42.Carroll N, Carello S, Cooke C, James A. Airway structure and inflammatory cells in fatal attacks of asthma. Eur Respir J. 1996;9:709–715. doi: 10.1183/09031936.96.09040709. [DOI] [PubMed] [Google Scholar]
- 43.Natarajan S, Kim J, Remick DG. Acute pulmonary lipopolysaccharide tolerance decreases TNF-alpha without reducing neutrophil recruitment. J Immunol. 2008;181:8402–8408. doi: 10.4049/jimmunol.181.12.8402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blease K, Lukacs NW, Hogaboam CM, Kunkel SL. Chemokines and their role in airway hyper-reactivity. Respir Res. 2000;1:54–61. doi: 10.1186/rr13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bystrom J, Patel SY, Amin K, Bishop-Bailey D. Dissecting the role of eosinophil cationic protein in upper airway disease. Curr Opin Allergy Clin Immunol. 2012;12:18–23. doi: 10.1097/ACI.0b013e32834eccaf. [DOI] [PubMed] [Google Scholar]
- 46.Jacobsen EA, Ochkur SI, Lee NA, Lee JJ. Eosinophils and asthma. Curr Allergy Asthma Rep. 2007;7:18–26. doi: 10.1007/s11882-007-0026-y. [DOI] [PubMed] [Google Scholar]
- 47.Meurs H, Gosens R, Zaagsma J. Airway hyperresponsiveness in asthma: lessons from in vitro model systems and animal models. Eur Respir J. 2008;32:487–502. doi: 10.1183/09031936.00023608. [DOI] [PubMed] [Google Scholar]
- 48.Watt AP, Schock BC, Ennis M. Neutrophils and eosinophils: clinical implications of their appearance, presence and disappearance in asthma and COPD. Curr Drug Targets Inflamm Allergy. 2005;4:415–423. doi: 10.2174/1568010054526313. [DOI] [PubMed] [Google Scholar]
- 49.Kraneveld AD, Folkerts G, Van Oosterhout AJ, Nijkamp FP. Airway hyperresponsiveness: first eosinophils and then neuropeptides. Int J Immunopharmacol. 1997;19:517–527. doi: 10.1016/s0192-0561(97)00085-4. [DOI] [PubMed] [Google Scholar]
- 50.Nakae S, Lunderius C, Ho LH, Schafer B, Tsai M, Galli SJ. TNF can contribute to multiple features of ovalbumin-induced allergic inflammation of the airways in mice. J Allergy Clin Immunol. 2007;119:680–686. doi: 10.1016/j.jaci.2006.11.701. [DOI] [PubMed] [Google Scholar]
- 51.Amrani Y. Airway smooth muscle modulation and airway hyper-responsiveness in asthma: new cellular and molecular paradigms. Expert Rev Clin Immunol. 2006;2:353–364. doi: 10.1586/1744666X.2.3.353. [DOI] [PubMed] [Google Scholar]
- 52.Anticevich SZ, Hughes JM, Black JL, Armour CL. Induction of human airway hyperresponsiveness by tumour necrosis factor-alpha. Eur J Pharmacol. 1995;284:221–225. doi: 10.1016/0014-2999(95)00463-u. [DOI] [PubMed] [Google Scholar]
- 53.Bukara M, Bautista AP. Acute alcohol intoxication and gadolinium chloride attenuate endotoxin-induced release of CC chemokines in the rat. Alcohol. 2000;20:193–203. doi: 10.1016/s0741-8329(99)00100-7. [DOI] [PubMed] [Google Scholar]
- 54.Vanha-Perttula TP. The influence of vitamin C on eosinophil response to acute alcohol intoxication in rats. Acta Endocrinol (Copenh) 1960;35:585–593. doi: 10.1530/acta.0.xxxv0585. [DOI] [PubMed] [Google Scholar]
- 55.Radinger M, Johansson AK, Sitkauskiene B, Sjostrand M, Lotvall J. Eotaxin-2 regulates newly produced and CD34 airway eosinophils after allergen exposure. J Allergy Clin Immunol. 2004;113:1109–1116. doi: 10.1016/j.jaci.2004.03.022. [DOI] [PubMed] [Google Scholar]
- 56.Ayres JG. The history of the use of alcohol in the treatment of respiratory diseases. Br J Dis Chest. 1987;81:80–86. doi: 10.1016/0007-0971(87)90112-4. [DOI] [PubMed] [Google Scholar]
- 57.Siegel SC. History of asthma deaths from antiquity. J Allergy Clin Immunol. 1987;80:458–462. doi: 10.1016/0091-6749(87)90075-3. [DOI] [PubMed] [Google Scholar]
- 58.Weitzman JB, Kanarek NF, Smialek JE. Medical examiner asthma death autopsies: a distinct subgroup of asthma deaths with implications for public health preventive strategies. Arch Pathol Lab Med. 1998;122:691–699. [PubMed] [Google Scholar]
- 59.Pavord ID, Birring SS, Berry M, Green RH, Brightling CE, Wardlaw AJ. Multiple inflammatory hits and the pathogenesis of severe airway disease. Eur Respir J. 2006;27:884–888. doi: 10.1183/09031936.06.00128105. [DOI] [PubMed] [Google Scholar]
- 60.Gao P. Sensitization to cockroach allergen: immune regulation and genetic determinants. Clin Dev Immunol. 2012;2012:563760. doi: 10.1155/2012/563760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rosenstreich DL, Eggleston P, Kattan M, et al. The role of cockroach allergy and exposure to cockroach allergen in causing morbidity among inner-city children with asthma. N Engl J Med. 1997;336:1356–1363. doi: 10.1056/NEJM199705083361904. [DOI] [PubMed] [Google Scholar]
- 62.Fittschen C, Sandhaus RA, Worthen GS, Henson PM. Bacterial lipopolysaccharide enhances chemoattractant-induced elastase secretion by human neutrophils. J Leukoc Biol. 1988;43:547–556. doi: 10.1002/jlb.43.6.547. [DOI] [PubMed] [Google Scholar]
- 63.Balmes JR, Earnest G, Katz PP, et al. Exposure to traffic: lung function and health status in adults with asthma. J Allergy Clin Immunol. 2009;123:626–631. doi: 10.1016/j.jaci.2008.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.D'Amato G, Liccardi G, D'Amato M, Holgate S. Environmental risk factors and allergic bronchial asthma. Clin Exp Allergy. 2005;35:1113–1124. doi: 10.1111/j.1365-2222.2005.02328.x. [DOI] [PubMed] [Google Scholar]
- 65.Brunialti MK, Martins PS, Barbosa de Carvalho H, Machado FR, Barbosa LM, Salomao R. TLR2, TLR4, CD14, CD11B, and CD11C expressions on monocytes surface and cytokine production in patients with sepsis, severe sepsis, and septic shock. Shock. 2006;25:351–357. doi: 10.1097/01.shk.0000217815.57727.29. [DOI] [PubMed] [Google Scholar]
- 66.Li C, Balluz LS, Okoro CA, et al. Surveillance of certain health behaviors and conditions among states and selected local areas – Behavioral Risk Factor Surveillance System, United States, 2009. MMWR Surveill Summ. 2011;60:1–250. [PubMed] [Google Scholar]
- 67.Coutinho V, Vidal C, Vizcaino L, Gonzalez-Quintela A. Effect of alcohol consumption and cessation on serum total immunoglobulin E concentrations. J Invest Allergol Clin Immunol. 2011;21:327–329. [PubMed] [Google Scholar]
- 68.Friedrich N, Husemoen LL, Petersmann A, Nauck M, Volzke H, Linneberg A. The association between alcohol consumption and biomarkers of alcohol exposure with total serum immunoglobulin E levels. Alcohol Clin Exp Res. 2008;32:983–990. doi: 10.1111/j.1530-0277.2008.00655.x. [DOI] [PubMed] [Google Scholar]
- 69.Vidal C, Vizcaino L, Diaz-Peromingo JA, et al. Immunoglobulin-E reactivity to a glycosylated food allergen (peanuts) due to interference with cross-reactive carbohydrate determinants in heavy drinkers. Alcohol Clin Exp Res. 2009;33:1322–1328. doi: 10.1111/j.1530-0277.2009.00961.x. [DOI] [PubMed] [Google Scholar]
- 70.Alonso M, Gomez-Rial J, Gude F, Vidal C, Gonzalez-Quintela A. Influence of experimental alcohol administration on serum immunoglobulin levels: contrasting effects on IgE and other immunoglobulin classes. Int J Immunopathol Pharmacol. 2012;25:645–655. doi: 10.1177/039463201202500311. [DOI] [PubMed] [Google Scholar]
- 71.Oldenburg PJ, Wyatt TA, Factor PH, Sisson JH. Alcohol feeding blocks methacholine-induced airway responsiveness in mice. Am J Physiol Lung Cell Mol Physiol. 2009;296:L109–114. doi: 10.1152/ajplung.00487.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Oldenburg PJ, Poole JA, Sisson JH. Alcohol reduces airway hyperresponsiveness (AHR) and allergic airway inflammation in mice. Am J Physiol Lung Cell Mol Physiol. 2012;302:L308–315. doi: 10.1152/ajplung.00077.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]