

Abstract
Nuclear factor (NF)-κB is a transcription factor implicated in the pathogenesis of autoimmune disorders such as rheumatoid arthritis (RA). Here we have examined the effect of intra-articular administration of the IKK inhibitor, NEMO-binding domain peptide (NBD), on the severity of collagen-induced arthritis (CIA). NBD peptides were injected intra-articularly into the knee joints of DBA/1J mice after the onset of disease. Collagen-injected mice given a scrambled peptide served as controls. Arthritis severity was determined by visual examination of paws. Intra-articular NBD injection reduced the arthritis score and ameliorated morphological signs of bone destruction compared to the controls. Serum levels of type-II collagen-specific immunoglobulin (Ig)G2a antibodies were lower in NBD-treated mice versus the control mice, whereas the levels of type-II collagen-specific IgG1 antibodies were increased by NBD treatment. NBD treatment diminished the proinflammatory cytokines interleukin (IL)-17 and interferon (IFN)-γ in serum, but increased the regulatory cytokine IL-10. NBD-treated CIA mice exhibited significantly higher percentages and numbers of forkhead box protein 3 (FoxP3+)CD4+CD25+ regulatory T cells than controls. Immunofluorescence analysis of NBD-treated mice revealed that FoxP3 and Ym1, a marker of alternatively activated macrophages, were juxtaposed to each other within draining inguinal lymph nodes. Intra-articular administration of NBD peptide is effective as an experimental therapy in a murine model of RA. Nevertheless, the intra-articular treatment modality is still associated with systemic effects on the immune system.
Keywords: alternatively activated macrophages, autoimmunity, collagen-induced arthritis, NBD peptide, regulatory T cells
Introduction
Collagen-induced arthritis (CIA) is a well-established animal model of human rheumatoid arthritis (RA). In rodents, CIA can be induced by immunization with type II collagen (CII) in Freund's complete adjuvant (CFA), which leads to CII-specific antibody production 1,2. Phenotypically, CIA animals show uncontrolled proliferation of synoviocytes, joint infiltration by leucocytes and high local expression of nuclear factor (NF)-κB. Enhanced expression of NF-κB is a key pathophysiological feature of CIA because pharmacological inhibition of NF-κB is known to correlate with a diminished inflammatory response in RA 3. NF-κB is involved mechanistically in regulating both innate and adaptive immunity, and substantial evidence suggests a positive correlation between NF-κB expression and the severity of inflammation 4. Conversely, NF-κB can also limit inflammation in metabolic diseases via NF-κB-dependent generation of CD4+CD25+ regulatory T cells (Treg) by M2 macrophages 5. Thus, therapeutic modulation of the NF-κB pathway must be approached carefully.
NF-κB/Rel becomes activated following phosphorylation of the inhibitor of κB (IκBα) protein by the IκB kinase (IKK) protein complex. This causes NF-κB to be released from IκBα and allows it to translocate to the nucleus. In a non-inflammatory setting, the IKK complex acts as a containment mechanism for NF-κB, preventing its inappropriate activation. Production of proinflammatory cytokines and ligation of Toll-like receptors causes IκBα to be released from NF-κB either by phosphorylation or polyubiquitination-mediated degradation. The functions of NF-κB in inflammation are complex, as IKK-β, one of the two homologous catalytic kinase subunits of the IKK complex, is essential for NF-κB activation in the presence of proinflammatory molecules. This event releases NF-κB dimers from the NF-κB–IκBα complex, leading to the translocation of NF-κB dimers to the nucleus with further binding to the κB enhancer region of target genes 4. Thus, IKK is a key convergence point for many different stimuli responsible for NF-κB activation.
NF-κB essential modulator (NEMO) is a regulatory subunit of the IKK complex that might be a promising target for IKK-centred intervention 6. Thus, we determined to evaluate the therapeutic potential of NF-κB blockade, and decided to target the NEMO pathway. NEMO-binding domain peptide (NBD) acts as an IκB kinase (IKK) inhibitor, and previous work has shown that it could sufficiently block autoimmune and inflammatory actions of NF-κB 7,8. However, these approaches require repeated treatments and have a high probability of triggering unfavourable systemic side effects 9. Considering that NF-κB serves significant homeostatic roles in several inflammatory diseases, including RA, systemic inhibition of NF-κB may not be an optimal treatment option for RA patients. In order to side-step the undesirable systemic side effects we explored the use of localized IKK inhibition as a treatment for mice with CIA. We posited that local intra-articular administration of NBD would target IKK in inflamed joints, thereby diminishing the adverse side effects associated with systemic NF-κB inhibition.
Materials and methods
Animals
Six to 8-week-old male DBA/1J mice (The Jackson Laboratory, Bar Harbor, ME, USA) were maintained in groups of two to four animals in polycarbonate cages in a specific pathogen-free environment and were fed standard chow (Ralston Purina, St Louis, MO, USA) and water ad libitum. All experimental procedures were examined and approved by the Animal Research Ethics Committee at the UT Southwestern Medical Center.
Induction and evaluation of arthritis
CIA induction was conducted as described previously 10. Bovine CII was dissolved in 0·05 N acetic acid at a concentration of 2 mg/ml and emulsified (1:1 ratio) with complete Freund's adjuvant. As a primary immunization, a 0·1 ml emulsion containing 100 μg CII was injected into the tail. Two weeks later (D14), a booster injection of 100 μg CII dissolved similarly and emulsified 1:1 with incomplete Freund's adjuvant was administered to the hind leg. Starting 18 days after the primary immunization, three independent observers examined the severity of arthritis three times a week for up to 6 weeks. The severity of arthritis was recorded as the mean arthritic index on a 0–4 scale according to the following criteria: 0 = no oedema or swelling; 1 = slight oedema and erythema limited to the foot or ankle; 2 = slight oedema and erythema from the ankle to the tarsal bone; 3 = moderate oedema and erythema from the ankle to the tarsal bone; and 4 = oedema and erythema from the ankle to the entire leg 11. The final score was an average value of the three independent joint evaluations.
NBD peptide injections into knee joints
NEMO binding domain for IKK complex formation, a cell-permeable IKK complex inhibitor NBD peptide (DRQIKIWFQNRRM KWKKTALDWSWLQTE), and a scrambled control peptide (DRQIKIWFQNRRMKW KKQLRRPADRELAE), were synthesized at the UT Southwestern Medical Center Protein Chemistry Technology Center. Mice were anaesthetized using a ketamine cocktail. Injections were performed using a 31-gauge insulin syringe (BD, Franklin Lakes, NJ, USA) through the medial meniscus and infrapatellar fat pad between the medial condyle of the tibia and the medial epicondyle of the femur. One week after the booster injection (on D21), NBD peptide (150 μg to each joint) or scrambled control peptide (150 μg to each joint) was administered six times over the course of 2 weeks into alternate knees.
Histology of joint tissues
Hind paws were obtained from each mouse, trimmed of skin, fixed in 10% phosphate-buffered formalin for 1 day, decalcified in 15% ethylenediamine tetraacetic acid (EDTA) for 3 weeks, then embedded in paraffin. Tissue sections (7 μm) were prepared and stained with haematoxylin and eosin (H&E). Inflammation and joint damage were investigated by scoring five parameters. Animals were scored on a scale of 0–5, as described 12,13, using the following criteria: (0) normal; (1) infiltration of inflammatory cells; (2) synovial hyperplasia; (3) pannus formation; (4) bone erosion; and (5) bone destruction. Two independent examiners evaluated the histological slides.
Measurement of autoantibodies
Blood was collected from the orbital sinus of NBD- and scrambled control peptide-treated mice. Serum specimens were stored at −20°C until use. Total immunoglobulin (Ig)G1 and IgG2a antibodies were measured using the mouse IgG1/IgG2a enzyme-linked immunosorbent assay (ELISA) quantitation kit (Bethyl Lab Co., Montgomery, TX, USA). For the measurement of anti-CII antibodies, microplates were coated with 4 μg/ml CII overnight, blocked with 1% bovine serum albumin (Sigma-Aldrich, St Louis, MO, USA), and incubated subsequently with sera at a dilution of 1:5000. Bound total or CII-specific IgG1 or IgG2a were detected by incubation with horseradish peroxidase (HRP)-conjugated anti-mouse Ig isotype-specific antibodies (Bethyl Lab Co.) at 37°C for 1 h. Plates were then washed with phosphate-buffered saline–Tween 20 (PBST) and developed with 3,3′,5,5′-tetramethylbenzidine (TMB) substrate. The reaction was terminated with 4·5 N H2SO4. Optical density (OD) values were measured at 450 nm using an Automatic Microplate Reader (BLx808; Bio-Tek, Winooski, VT, USA). The OD measured in the standard serum, diluted serially, and the arbitrary units each exhibited good linear correlation at all determinations (R = 0·99; data not shown).
[3H]-thymidine incorporation in mouse spleen and draining lymph nodes
Draining inguinal lymph nodes (dLNs) and spleens were excised and homogenized, and cell suspensions cultured in RPMI-1640 containing fetal calf serum (10% vol/vol), 2-mercaptoethanol (20 μM), L-glutamine (1% wt/vol), penicillin (100 U/ml) and streptomycin (100 μg/ml) in the presence or absence of type II collagen. During the last 16–18 h of the 3-day assay, cells were pulsed with 1 uCi of [3H]-thymidine (GE Healthcare, Little Chalfont, UK) per well. The incorporation of [3H]-thymidine was determined using a Betaplate scintillation counter (Perkin-Elmer, Wellesley, MA, USA).
Isolation of cells from joints, spleen and lymph nodes
Joint cells were dissociated enzymatically in 0·125% Dispase II (Roche Diagnostics, Indianapolis, IN, USA) and 0·2% collagenase II (Sigma-Aldrich) in Hanks's balanced salt solution (HBSS) for 1 h at 37°C and strained through a 70-μm cell strainer to generate a single-cell suspension. Spleen and draining lymph nodes were minced directly through a cell strainer. Red blood cells (RBCs) were lysed by incubating the preparations in 0·83% NH4Cl and live cells were counted in a haemocytometer using trypan blue exclusion.
Intracellular staining for forkhead box protein 3 (FoxP3) and interleukin (IL)-17
Single-cell suspensions were isolated from draining lymph nodes (dLNs) and joint cells. For flow cytometric analysis, cells were surface-stained with anti-CD4 and anti-CD25 antibodies. Intracellular staining for FoxP3 was performed according to the appropriate eBioscience protocol. For intracellular IL-17 staining, cells were stimulated for 5 h with 25 ng/ml phorbol myristate acetate (PMA) (Sigma) and 250 ng/ml ionomycin (Sigma) and incubated with Golgiplug (BD PharMingen). Staining was performed using fixation/permeabilization buffer (eBioscience, San Diego, CA, USA) following the manufacturer's instructions. Directly conjugated isotype-matched rat anti-mouse antibodies (eBioscience) were used as controls for non-specific staining. Cytometric acquisition was performed using a FACSCalibur flow cytometer (BD Immunocytometry Systems, Franklin Lakes, NJ, USA).
All suppression assays were performed in 96-well round-bottomed plates (BD) in a final volume of 200 ul/well of complete RPMI-1640 media. Varying numbers of CD4+CD25+ T or CD4+CD25– T cells from dLNs of NBD-treated mice were cultured for 3 days in the presence of CII (40 μg/well) with CII-reactive CD4+ T cells (1 × 105 cells) and irradiated antigen-presenting cells (APCs) (1 × 105 cells) obtained from the spleens of CIA mice in triplicate. The cells were co-cultured at the following ratios: 1:100, 1:20, 1:10 and 1:2 (CD4+: CD4+CD25+ or CD4+: CD4+CD25–). Proliferative responses were measured as the amount of [3H]-thymidine incorporated during the last 18 h of incubation.
Fluorescence microscopy
Mouse dLNs were collected, embedded in octreotide (OCT) and snap-frozen in liquid nitrogen. Cryosections (6 um thick) were fixed with 4% paraformaldehyde, blocked with 10% horse serum for 30 min, and stained with various antibodies including anti-FoxP3-fluorescein isothiocyanate (FITC) (eBioscience) and rabbit polyclonal antibody against Ym1, followed by Alexa 555-labelled secondary antibodies (Invitrogen, Carlbad, CA, USA). Fluorescence images were acquired using an LSN510 confocal microscope (Carl Zeiss, Oberkochen, Germany).
Western blotting
Cells (3–4 × 106) were washed in phosphate-buffered saline (PBS) and lysed with a buffer composed of 20 mM Tris-HCl (pH 7·5), 150 mM NaCl, 1 mM Na2 EDTA, 1 μg/ml leupetin, 1% Triton X-100, 1 mM phenylmethylsulphonyl fluoride (PMSF) and 1 mM Na3VO4. The protein content of the samples was quantified by Bradford assay, and 10 μg was loaded per lane onto sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) gels. Samples were then transferred to nitrocellulose membranes (Millipore, Bedford, MA, USA), probed with the indicated antibodies, and developed with an enhanced chemiluminescence detection system (Amersham, Arlington Heights, IL, USA).
Detection of cytokine production by ELISA
Serially diluted serum specimens were assayed for interferon (IFN)-γ, IL-17 and IL-10 by ELISA using assay kits from R&D Systems (Minneapolis, MN, USA) following the manufacturer's instructions.
Statistical analysis
Data from in-vitro and ex-vivo experiments were analysed for statistical significance (GraphPad, InStat, version 2·02; GraphPad Software, Inc., San Diego, CA, USA), using Student's t-test or the Mann–Whitney U-test. A P-value < 0·05 was taken as the level of significance.
Results
Intra-articular NBD injection ameliorates CIA symptoms
We investigated the therapeutic potential of a specific IKK-β inhibitor, NBD, for local treatment of arthritis in the CIA animal model. For intra-articular treatment, the hind paws were injected either with NBD or a scrambled control peptide six times over the course of 2 weeks after primary immunization with collagen. The course of arthritis was monitored until week 6 after the primary immunization (Fig. 1a). As expected, the control peptide-injected mice developed severe arthritis, including marked swelling and erythema of the hind paws and the forepaws (Fig. 1b). In contrast, intra-articular injections with NBD peptide markedly reduced the clinical manifestations of fully developed CIA. This was evidenced by changes in the arthritic index and disease occurrence rate in DBA/1 mice injected with control peptide compared to NBD-treated mice (Fig. 1b). We assessed the histopathological changes in the inflamed joint by H&E staining, and found that these paralleled closely the clinical findings from individual mice. In the control peptide-injected mice, the joints revealed inflammatory cell infiltration, synovial hyperplasia, erosion of cartilage and bone destruction. Conversely, CIA mice receiving intra-articular NBD treatment showed relatively mild cartilage destruction in the injected ankle joints (Fig. 1c). The histopathological score was reduced significantly in NBD-treated mice versus control peptide-injected mice (2·96 ± 0·08 versus 1·43 ± 0·23, P < 0·01; Fig. 1d). Overall, these findings show that local injection of the small molecule NBD peptide ameliorates the arthritic disease process and associated bone destruction significantly.
Fig. 1.
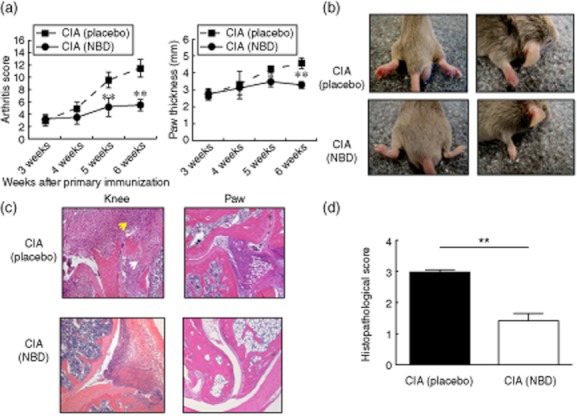
NEMO-binding domain peptide (NBD) peptide treatment ameliorates collagen-induced arthritis (CIA). (a) Severity of arthritis was assessed by clinical scoring (left) and paw thickness measurements (right) in CIA mice injected intra-articularly with either the control, scrambled peptide (placebo) or NBD peptide. Data are expressed as the mean arthritis score in each group, *P < 0·05; **P < 0·01; n = 11–12 mice per group, pooled from three independent experiments. (b) Representative examples of paw oedema in mice from the experiment shown in panel (a). (c) Histological sections of joint following control or NBD peptide injections; stained for haematoxylin and eosin (H&E) and examined microscopically. Lower arrows indicate areas of bone erosion and upper arrows indicate areas of infiltration by inflammatory cells. Data are representative of similar images from 11–12 mice per group. (d) The graph shows histological scores from the experiment in (c) (mean ± standard error of the mean (s.e.m.); **P < 0·001). Data reported are average ± s.e.m. Statistical analysis was by Student's t-test.
Effects of NBD treatment on serum levels of type-II-collagen-specific IgG subtypes
Among the IgG subtypes, IgG1 is reported to be associated with anti-inflammatory action, whereas IgG2a is known to be a promoter of inflammation with important roles in the pathogenesis of CIA 2. Total serum levels of IgG2a were reduced significantly in NBD-treated mice compared to control peptide-treated CIA mice (Fig. 2a). In contrast, IgG1 was elevated modestly in NBD-treated mice compared to CIA mice that received control peptide (Fig. 2a). To determine whether NBD treatment of CIA mice was associated with a change in the humoral immune response to type II collagen, the levels of IgG1 and IgG2a anti-type-II-collagen autoantibodies were measured by ELISA in serum samples 6 weeks after primary immunization. The serum concentrations of CII-specific IgG2a from NBD-treated mice were significantly lower than those of the control-treated mice (P < 0·001). In contrast, the levels of CII-specific IgG1 were increased significantly by NBD treatment (Fig. 2b). These data are consistent with the idea that IgG2a may have important roles in the pathogenesis of arthritis, as its reduction correlated with diminished arthritis in NBD-treated mice.
Fig. 2.

NEMO-binding domain peptide (NBD) treatment's effect on total serum immunoglobulin (Ig)G subtypes and type II collagen (CII)-specific antibodies in collagen-induced arthritis (CIA) mice. Six weeks after primary immunization, sera from CIA-induced mice (n = 11–12 mice per group, pooled from three independent experiments) were collected. Total IgG subtype and type II collagen-specific IgG1 and IgG2a were detected by enzyme-linked immunosorbent assay. The results were calculated in arbitrary units and are expressed as means ± standard deviation; *P < 0·05; **P < 0·01; ***P < 0·001. Placebo mice received a control, scrambled peptide.
NBD treatment decreases proliferative responses of lymphocytes to CII
It has been shown previously that CIA in mice is associated with significant abnormalities in CII-specific T cells 14. To investigate whether NBD-induced suppression of arthritis also impacted the clonal expansion of CII-specific T cell responses, splenocytes and draining lymph node (dLN) cells were isolated from all experimental groups at day 42 after primary immunization, and were cultured with bovine CII or anti-CD3 monoclonal antibody (mAb) for proliferation assays. As expected, draining lymph node cells in control peptide-injected mice showed a six- to sevenfold increase in [3H]-thymidine incorporation following CII stimulation, indicating that proliferation occurred in response to CII. In contrast, cells obtained from NBD-treated CIA mice exhibited a reduced proliferative response to CII (Fig. 3a) (control peptide versus NBD treated; 6·8 ± 0·8 versus 2·6 ± 0·4; P < 0·05). To assess spontaneous cell proliferation, we performed a carboxyfluorescein diacetate succinimidyl ester (CFSE)-based suppression assay with dLN cells. As shown in Fig. 3b, CFSE dilution was elevated greatly in the T cells from CIA mice but not in NBD-treated mice (upper panel, Fig. 3b). Similar changes were found in splenocytes (low panel, Fig. 3b). The effect of NBD treatment on the viability of dLN cells and splenocytes was determined by trypan blue exclusion. The number of cells in dLNs was reduced significantly in NBD-treated mice compared to control peptide-treated mice (Fig. 3c).
Fig. 3.

T cell proliferative response to antigenic stimulation with type II collagen (CII). (a) Cells from the draining inguinal lymph nodes (dLNs) and spleens of mice killed 6 weeks after primary immunization were cultured for 72 h in the presence of 40 μg/ml of CII. Panels show changes in the stimulation index (SI) of cells isolated from the draining lymph node (upper panel) and spleen (low panel). The data are representative of 11–12 mice per group pooled from three independent experiments; *P < 0·05; **P < 0·01. (b) Fluorescence of 5,6-carboxyfluorescein succinimidyl ester (CFSE)–labelled dLNs (upper panel) and spleens (low panel) obtained from each group of mice. Cells were cultured with type II collagen for 72 h, followed by flow cytometric analysis. (c) The total number of cells present in dLN cells and splenocytes in control (placebo) and NBD peptide-treated mice. Results are presented as the mean ± standard deviation of three independent experiments; *P < 0·05.
Intra-articular NBD treatment blocks IKK-β phosphorylation and NF-κB activation in arthritic joints
IKK-β activation is a critical mechanism by which proinflammatory stimuli induce NF-κB function 4,6. IKK-β expression is found in fibroblast-like synoviocytes and is vital to IL-1-β and tumour necrosis factor (TNF)-α-mediated NF-κB activation and subsequent inflammatory gene transcription in human monocytes 15. Thus, we examined whether inhibition by NBD would affect this pathway in our model of inflammation. NBD peptide blocked phosphorylation of IKK-β and thereby inhibited its activation in cellular extracts obtained from arthritic knee joints, as demonstrated by immunoblotting for active IKK molecules (Fig. 4a). As expected, these observations were accompanied by a dramatic inhibition of NF-κB activation in cells retrieved from arthritic joints (Fig. 4a). In contrast, phosphorylation of IKK-β and total NF-κB in control peptide-treated arthritic mice were increased markedly. Taken together, these data demonstrate that NBD peptide blocks IKK-β phosphorylation and NF-κB activity in cells from arthritic joints, resulting in reduced inflammation. Using densitometry, we found that the p-IKK-β/IKK-β ratio was reduced significantly in NBD-treated CIA mice compared with control peptide-treated mice (0·79 versus 0·12). NBD treatment also considerably reduced the total NF-κB/β-actin ratio relative to control peptide-treated mice (1·32 versus 0·44) (Fig. 4b).
Fig. 4.

NEMO-binding domain peptide (NBD) peptide inhibits nuclear factor (NF)-κB activation and modulates cell numbers in collagen-induced arthritis (CIA). (a) Immunoblotting using specific antibodies for p-IκB kinase (IKK)-β, IKK-β and NF-κB on the cellular extracts from the knee joints of CIA-induced mice. β-actin was used as a loading control. (b) Representative blots are shown in (a) with densitometric readings expressed as ratios in (b). ‘Placebo’ refers to the control peptide treated mice. Results are the mean ± standard deviation of three mice per group. **P < 0·01.
NBD peptide inhibits proinflammatory cytokine production
Inflammatory cytokines have been shown to be associated with pathogenic dysfunction in rheumatoid arthritis 16. Hence, intracellular staining was performed to determine if IL-17 was being produced by T cells in the draining lymph nodes (LN). Flow cytometric analysis illustrated that control peptide-treated CIA mice had a greater frequency and absolute number of IL-17-producing CD4+ T cells in the dLNs than did control peptide-treated CIA mice, after anti-CD3 mAb stimulation (Fig. 5a). In contrast, only 1·54% of dLN T cells obtained from NBD-treated CIA mice produced IL-17 after stimulation. We next elucidated the potential source of IL-17 in the arthritic joints. We observed that, in line with our findings in Fig. 5a, there was also markedly reduced IL-17 production by CD4+ T cells infiltrating the joint in the NBD-treated CIA mice compared with control peptide-treated CIA mice (Fig. 5c). Of note, CD4+ T cells from arthritic joints of NBD-treated mice produced low levels of IL-17. We also measured the production of the proinflammatory cytokines IFN-γ and IL-17, and the anti-inflammatory cytokine IL-10 in sera by ELISA. Serum levels of IFN-γ and IL-17 were reduced significantly in NBD-treated mice compared to control peptide-treated arthritic mice, whereas the production of IL-10 was increased in intra-articular NBD-treated mice (Fig. 5f).
Fig. 5.

Proinflammatory cytokine inhibition and expansion of regulatory T cells (Treg) cells in NEMO-binding domain peptide (NBD)-treated mice. NBD treatment decreased the percentage (upper panel) and absolute number (low panel) of CD4+interleukin (IL)-17+ T cells from draining inguinal lymph nodes (dLNs). Values are presented as the mean ± standard deviation (s.d.) of 11–12 mice per group, pooled from three independent experiments; **P < 0·01. (b) Frequency (upper panel) and calculated absolute numbers (low panel) of CD4+CD25+ forkhead box protein 3 (FoxP3+) Treg cells from NBD- or control peptide-treated mice. For these experiments, single-cell suspensions were prepared from the dLN cells of collagen-induced arthritis (CIA) mice 6 weeks after the primary immunization; ***P < 0·001. (c) Representative fluorescence activated cell sorter (FACS) plots showing intracellular staining for IL-17 in joint-derived CD4+ T cells of NBD- and control peptide-treated mice. (d) Representative flow cytometric analysis of CD4+FoxP3+ T cells in joint cells from NBD- and control peptide-treated mice. (e) CD4+CD25+ and CD4+CD25− T cells from the dLNs of NBD-peptide-treated mice were incubated in increasing ratios with a fixed number of type II collagen (CII)-reactive CD4+ T cells (1 × 105) and irradiated antigen-presenting cells (1 × 105) from the spleen of CIA mice in the presence of CII (40 μg/ml). After 72 h, cultures were pulsed for the last 18 h and [3H]-thymidine incorporation was assayed. Values are the mean ± s.d. from two independent experiments; *P < 0·05; **P < 0·001. cpm: counts per minute. (f) Serum concentrations of interferon (IFN)-γ, IL-10 and IL-17 (pg/ml) were measured 6 weeks after the primary immunization. Values are presented as the mean ± s.d., n = 8 mice per group, pooled from three independent experiments; *P < 0·05; **P < 0·01.
Involvement of Treg cells in the therapeutic effect of NBD in CIA
Treg expressing the transcription factor FoxP3 are known to prevent lethal autoimmunity. Treg cells also have protective effects during treatment of experimental CIA in mice, a T cell-mediated and organ-specific disease 17. Because NBD inhibited the severity of the inflammatory phase of CIA following the activation of antigen-specific CD4+ T helper type 1 (Th1) cells, we examined the possibility that NBD treatment induces Treg cells in CIA mice. NBD-treated CIA mice had significantly higher percentages and numbers of CD4+CD25+FoxP3+ cells in dLNs when compared to the control peptide-injected mice (Fig. 5b). These results indicate that NBD promotes the emergence of activated Treg cells during CIA. To determine the role of NBD peptide in modulating regulatory T cells response and joint inflammation in collagen-induced arthritis, FoxP3 expression on CD4+ T cells was examined by flow cytometry. During CIA induction, NBD treatment increased CD4+FoxP3+ cells considerably in the joint cells (Fig. 5d). After anti-CD3 stimulation, NBD-treated mice exhibited a 10-fold increase in CD4+FoxP3+ T cells compared with control peptide-treated mice (data not shown).
To examine further whether NBD-induced regulatory T cells are functional in suppressing the proliferation of CII-specific CD4+ T cells, we co-cultured CD4+CD25+ or CD4+CD25– T cells from NBD peptide-treated mice with CIA CD4+ effector cells from the spleen of CIA mice in a [3H]-thymidine-based suppression assay; 72 h after stimulation with 40 ug/ml of CII, proliferation was evaluated by [3H]-thymidine incorporation. Figure 5e shows that CD4+CD25+ T cells from the dLNs of NBD-treated mice inhibit significantly the proliferation of CII-specific CD4+ T cells in CIA mice. Suppression of cell proliferation was noted at a CD4+CD25+/CD4+ effector ratios of 1:20, 1:10 and 1:2. These results suggest that NBD treatment might ameliorate disease by inducing Treg activity and decreasing CD4+ effector cell proliferation in CIA.
Cross-talk between Ym1-expressing macrophages and Treg cells in NBD treated mice
Ym1 is a well-known marker of regulatory M2 macrophages and certain subsets of dendritic cells (DCs) 18. Therefore, we compared Ym1 expression on macrophages from control- and NBD-treated mice. Whereas Ym1 was barely detectable on CD11b+F4/80+ macrophages of dLNs from control peptide-treated mice [mean fluorescence intensity (MFI) = 11·2 ± 1·3], it was highly expressed in counterparts from NBD-treated mice (MFI = 27·5 ± 3·0; P < 0·01) as shown in Fig. 6a. We next investigated whether NBD treatment promoted a physical association between Ym1-expressing cells and Tregs in dLNs. Immunofluorescence analysis of NBD-treated mice revealed that FoxP3-expressing and Ym1-expressing cells were present and adjacent to each other in dLNs, unlike in control peptide-treated mice (Fig. 6b). Treg-mediated suppression is known to be a part of the response to chronic antigenic stimulation, originating from interaction with professional and non-professional APC 19. Given the proximity of Ym1+ macrophages with infiltrating FoxP3+ Treg cells, it is tempting to speculate that NF-κB blockade may promote the expansion of Treg cells and regulatory macrophages, possibly as a consequence of interaction with each other.
Fig. 6.

Identification of CD11b++F4/80++Ym1+ cells in NEMO-binding domain peptide (NBD)-treated mice. (a) Percentages of CD11b and F4/80 double-positive cells were determined in draining inguinal lymph nodes (dLNs) of NBD and control peptide-treated mice 6 weeks after the primary immunization. CD11b+/F4/80+-positive cells were gated, and expression of Ym1 was determined from dLN cells of these mice. Data are representative of three independent experiments. (b) Immunofluorescence analysis of dLNs from NBD or control peptide-treated mice, fixed and stained with antibodies specific for forkhead box protein 3 (FoxP3+) (green) and Ym1 (red). Original magnification, ×200. Photomicrographs shown are representative of two independent experiments.
Discussion
To test whether localized NF-κB inhibition can play a therapeutic role in the CIA murine model, we injected NBD peptide intra-articularly six times over 2 weeks after a booster injection of type II collagen. Our results show that NBD peptide reduced clinical signs of arthritis significantly 35 days after the primary immunization, even with few treatments. Histology revealed that despite the presence of some pathological changes, NBD-injected paws had noticeably less damaged cartilage and bone destruction when compared to the control peptide-injected paws. This indicates that early, short, local therapeutic blockade of NF-κB at the time of symptom onset is protective in animals with chronic arthritis. Intra-articular NBD treatment reduced joint inflammation and impeded cartilage and bone deterioration, in accordance with previously reported results of systemic treatment in CIA 20.
We also demonstrated that intra-articular injection of NBD had a marked effect on the humoral response to bovine type II collagen, with a reduction of anti-CII specific IgG2a antibodies. Anti-CII IgG2a is an extremely important determinant in the pathogenesis of CIA 21. IgG aggregation and complement fixation are known to promote the development of arthritis 20. In our study, we found a reduction in anti-CII IgG2a titres 42 days after disease onset. This decrease in the amount of anti-CII IgG2a also modified the ratio of IgG2a to IgG1, indicating an overall Th2-type immune response. These serological differences may have contributed in part to the observed therapeutic benefit of NBD peptide treatment.
NF-κB involvement has been demonstrated in diverse cellular processes, ranging from apoptosis to proliferation 22. Many autoimmune disorders exhibit characteristically ineffective control of T cell proliferation, facilitating a self-reactive immune response 23. Activation of T cells and macrophages is a well-established major pathogenic determinant in CIA 24. Specifically, antigen-activated CD4+ T cells are involved critically in RA pathogenesis 14. Once activated, CD4+ T cells enhance B cell production of rheumatoid factor and receptor activator of nuclear factor kappa-B ligand (RANKL) reciprocally, which potentiate osteoclastogenesis, leading eventually to bone erosion 25. Here we observed that NF-κB is responsible for the proliferation of such cells, as suppression of NF-κB inhibited the disease-eliciting proliferation of dLN cells and splenocytes in CIA.
We found that NF-κB blockade also inhibited the production of the proinflammatory cytokines IL-17 and IFN-γ. IL-17 is a T cell-derived cytokine involved in the pathogenesis of human RA as well as several other models of autoimmunity 26. IL-17 interaction with other proinflammatory cytokines, such as IL-1 and TNF-α, induces inflammation and leads to cartilage destruction and bone resorption in arthritis 16 26. IFN-γ is another cytokine known to contribute to chronic inflammation and progressive tissue destruction in CIA 16. Regulation of these cytokines has been shown to be dependent upon NF-κB 27–29. In our study, production of the proinflammatory cytokines IFN-γ and IL-17 was reduced significantly in NBD-treated mice compared to control peptide-treated mice. Furthermore, production of the anti-inflammatory, protective cytokine IL-10 was increased significantly by NBD treatment (Fig. 5f). These findings suggest that NBD is a mediator that modulates both NF-κB-responsive proinflammatory and anti-inflammatory cytokines beneficially.
Immunoblotting of arthritic joints revealed reduced NF-κB activity in NBD-treated mice significantly compared with control peptide-treated mice (Fig. 4a). Further, our results show that DBA/1J mice treated with NBD peptide were consistently resistant to CIA and experienced alleviation of arthritic symptoms, including paw and joint swelling (Fig. 1c). NBD peptides have been investigated in previous studies both in vivo and in vitro to block NF-κB-mediated inflammation 7,8, cell proliferation 30 and/or tumour cell infiltration 31. Jimi et al. have reported that NBD peptides ameliorate CIA when administered intraperitoneally (i.p.) daily to rats 32. Our study confirms these results using short-term, local administration of NBD, and advances Tregs and APCs as potential protective mechanisms.
Generation of CD4+CD25+ Treg cells is controlled by FoxP3, which is also known to play a significant role in assembling the Rel family transcription factors such as nuclear factor of activated T cells (NFAT) and NF-κB 33. FoxP3 has also been shown to suppress the effector function of pathogenic, autoreactive T cells that produce such cytokines as IL-2, IL-4 and IFN-γ 34. Further, FoxP3 knock-out mice have an increased susceptibility for autoimmune disease development and demonstrate abnormally elevated NFAT and NF-κB transcriptional activities 33,35. Interestingly, we found that the percentages and absolute numbers of CD4+CD25+FoxP3+ Treg cells are increased considerably in NBD-treated mice compared with control peptide-treated mice, and that these cells suppress the proliferation of CII-specific effector T cells in CIA mice. It is tempting to conclude that the induction of Treg cells by intra-articular injection of NBD may have been responsible for the increase observed in alternatively activated macrophages (M2). Such an assumption is supported by the findings of this study, as well as several previous studies that demonstrated an association between CD4+CD25+FoxP3+ Treg cells and M2 macrophages. It has been shown that CD4+CD25+FoxP3+ Treg cells can interact with APCs such as B cells, DCs, neutrophils and monocytes 36–38. Moreover, Tiemessen et al. reported that CD4+CD25+FoxP3+ Treg cells co-operate with human macrophages/monocytes, thereby generating alternatively activated macrophages 5.
M2 macrophages are sometimes divided further into subtypes. M2a and M2c macrophages express mannose receptors, Arginase I, FIZZ-1 and Ym1 39,40, and their role appears to be anti-inflammatory in nature. Differentiation of M2 macrophages is tied closely to NF-κB signalling, an overabundance of which can lead to a predominance of the M1 phenotype 41. M2a cells appear after stimulation with IL-4 and IL-13, whereas M2c differentiation is controlled by IL-10, transforming growth factor (TGF)-β and glucocorticoid hormones 5,36. Recent experimental studies have shown that Treg cells may cause phenotypic alteration from M1 to M2 phenotype-like macrophages and vice versa 5,42. The percentages of CD11b+F4/80+ macrophages in dLNs were enhanced considerably following NBD treatment in our studies, compared to the controls. Strikingly, the expression of Ym1 on CD11b+F4/80+ macrophages was also increased in NBD-treated mice, juxtaposed to the Treg cells. Of note, Liu et al. have demonstrated that CD4+CD25+ Treg cells and CD4+CD25– T effector cells control the activation of M1 and M2 macrophages, respectively 5,36. This cross-talk between Th cells (effector or regulatory cells) and macrophages (M1 or M2) is essential for sustaining proper immune responses and homeostasis via reciprocal mechanism(s). This idea is consistent with a recent observation showing that human CD4+CD25+ Treg cells can induce an M2 phenotype in vitro during macrophage differentiation from monocytes 5. To our knowledge, this is the first report to describe the interaction of Treg cells and M2 macrophages induced by NF-κB inhibition.
The findings of this study suggest that NF-κB blockade using NBD may be used as a potential therapeutic agent in the management of chronic inflammatory processes and alleviation of subsequent joint destruction in arthritis. However, in contrast to our initial hypothesis, even intra-articular blockade of NF-κB is associated with prominent systemic effects, including the modulation of humoral autoimmunity and the genesis of regulatory T and myeloid cells in the immune system.
Acknowledgments
This work is supported in part by grant from NIH (R01 AR050812). S. M. was supported in part by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2005-0048480).
Disclosure
None.
References
- 1.Bevaart L, Vervoordeldonk MJ, Tak PP. Collagen-induced arthritis in mice. Methods Mol Biol. 2010;602:181–192. doi: 10.1007/978-1-60761-058-8_11. [DOI] [PubMed] [Google Scholar]
- 2.Brand DD, Marion TN, Myers LK, et al. Autoantibodies to murine type II collagen in collagen-induced arthritis: a comparison of susceptible and nonsusceptible strains. J Immunol. 1996;157:5178–5184. [PubMed] [Google Scholar]
- 3.Aya K, Alhawagri M, Hagen-Stapleton A, Kitaura H, Kanagawa O, Novack DV. NF-(kappa)B-inducing kinase controls lymphocyte and osteoclast activities in inflammatory arthritis. J Clin Invest. 2005;115:1848–1854. doi: 10.1172/JCI23763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Q, Verma IM. NF-kappaB regulation in the immune system. Nat Rev Immunol. 2002;2:725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 5.Tiemessen MM, Jagger AL, Evans HG, van Herwijnen MJ, John S, Taams LS. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc Natl Acad Sci USA. 2007;104:19446–19451. doi: 10.1073/pnas.0706832104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karin M, Delhase M. The I kappa B kinase (IKK) and NF-kappa B: key elements of proinflammatory signalling. Semin Immunol. 2000;12:85–98. doi: 10.1006/smim.2000.0210. [DOI] [PubMed] [Google Scholar]
- 7.Dai S, Hirayama T, Abbas S, Abu-Amer Y. The IkappaB kinase (IKK) inhibitor, NEMO-binding domain peptide, blocks osteoclastogenesis and bone erosion in inflammatory arthritis. J Biol Chem. 2004;279:37219–37222. doi: 10.1074/jbc.C400258200. [DOI] [PubMed] [Google Scholar]
- 8.May MJ, D'Acquisto F, Madge LA, Glockner J, Pober JS, Ghosh S. Selective inhibition of NF-kappaB activation by a peptide that blocks the interaction of NEMO with the IkappaB kinase complex. Science. 2000;289:1550–1554. doi: 10.1126/science.289.5484.1550. [DOI] [PubMed] [Google Scholar]
- 9.Furuta H, Osawa K, Shin M, et al. Selective inhibition of NF-kappaB suppresses bone invasion by oral squamous cell carcinoma in vivo. Int J Cancer. 2012;131:E625–635. doi: 10.1002/ijc.27435. [DOI] [PubMed] [Google Scholar]
- 10.Park MJ, Min SY, Park KS, et al. Indoleamine 2,3-dioxygenase-expressing dendritic cells are involved in the generation of CD4+CD25+ regulatory T cells in Peyer's patches in an orally tolerized, collagen-induced arthritis mouse model. Arthritis Res Ther. 2008;10:R11. doi: 10.1186/ar2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosloniec EF, Cremer M, Kang AH, Myers LK, Brand DD. Collagen-induced arthritis. Curr Protoc Immunol. 2010;Chapter 15:1–25. doi: 10.1002/0471142735.im1505s89. Unit 15 5. [DOI] [PubMed] [Google Scholar]
- 12.Tao J, Kamanaka M, Hao J, et al. IL-10 signaling in CD4+ T cells is critical for the pathogenesis of collagen-induced arthritis. Arthritis Res Ther. 2011;13:R212. doi: 10.1186/ar3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baharav E, Bar-Yehuda S, Madi L, et al. Antiinflammatory effect of A3 adenosine receptor agonists in murine autoimmune arthritis models. J Rheumatol. 2005;32:469–476. [PubMed] [Google Scholar]
- 14.Brand DD, Myers LK, Terato K, et al. Characterization of the T cell determinants in the induction of autoimmune arthritis by bovine alpha 1(II)-CB11 in H-2q mice. J Immunol. 1994;152:3088–3097. [PubMed] [Google Scholar]
- 15.Aupperle KR, Bennett BL, Boyle DL, Tak PP, Manning AM, Firestein GS. NF-kappaB regulation by I kappa B kinase in primary fibroblast-like synoviocytes. J Immunol. 1999;163:427–433. [PubMed] [Google Scholar]
- 16.McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 2007;7:429–442. doi: 10.1038/nri2094. [DOI] [PubMed] [Google Scholar]
- 17.Morgan ME, Flierman R, van Duivenvoorde LM, et al. Effective treatment of collagen-induced arthritis by adoptive transfer of CD25+ regulatory T cells. Arthritis Rheum. 2005;52:2212–2221. doi: 10.1002/art.21195. [DOI] [PubMed] [Google Scholar]
- 18.Bellon T, Martinez V, Lucendo B, et al. Alternative activation of macrophages in human peritoneum: implications for peritoneal fibrosis. Nephrol Dial Transplant. 2011;26:2995–3005. doi: 10.1093/ndt/gfq771. [DOI] [PubMed] [Google Scholar]
- 19.Vlad G, Cortesini R, Suciu-Foca N. License to heal: bidirectional interaction of antigen-specific regulatory T cells and tolerogenic APC. J Immunol. 2005;174:5907–5914. doi: 10.4049/jimmunol.174.10.5907. [DOI] [PubMed] [Google Scholar]
- 20.Stuart JM, Tomoda K, Yoo TJ, Townes AS, Kang AH. Serum transfer of collagen-induced arthritis. II. Identification and localization of autoantibody to type II collagen in donor and recipient rats. Arthritis Rheum. 1983;26:1237–1244. doi: 10.1002/art.1780261011. [DOI] [PubMed] [Google Scholar]
- 21.Min SY, Hwang SY, Park KS, et al. Induction of IL-10-producing CD4+CD25+ T cells in animal model of collagen-induced arthritis by oral administration of type II collagen. Arthritis Res Ther. 2004;6:R213–219. doi: 10.1186/ar1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baldwin AS., Jr The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 23.Weyand CM, Goronzy JJ. T-cell-targeted therapies in rheumatoid arthritis. Nat Clin Pract Rheumatol. 2006;2:201–210. doi: 10.1038/ncprheum0142. [DOI] [PubMed] [Google Scholar]
- 24.Myers LK, Rosloniec EF, Cremer MA, Kang AH. Collagen-induced arthritis, an animal model of autoimmunity. Life Sci. 1997;61:1861–1878. doi: 10.1016/s0024-3205(97)00480-3. [DOI] [PubMed] [Google Scholar]
- 25.Kong YY, Feige U, Sarosi I, et al. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999;402:304–309. doi: 10.1038/46303. [DOI] [PubMed] [Google Scholar]
- 26.Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol. 2003;171:6173–6177. doi: 10.4049/jimmunol.171.11.6173. [DOI] [PubMed] [Google Scholar]
- 27.Lentsch AB, Shanley TP, Sarma V, Ward PA. In vivo suppression of NF-kappa B and preservation of I kappa B alpha by interleukin-10 and interleukin-13. J Clin Invest. 1997;100:2443–2448. doi: 10.1172/JCI119786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cho YG, Cho ML, Min SY, Kim HY. Type II collagen autoimmunity in a mouse model of human rheumatoid arthritis. Autoimmun Rev. 2007;7:65–70. doi: 10.1016/j.autrev.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 29.Weyand CM, Kaiser M, Yang H, Younge B, Goronzy JJ. Therapeutic effects of acetylsalicylic acid in giant cell arteritis. Arthritis Rheum. 2002;46:457–466. doi: 10.1002/art.10071. [DOI] [PubMed] [Google Scholar]
- 30.Ianaro A, Tersigni M, Belardo G, et al. NEMO-binding domain peptide inhibits proliferation of human melanoma cells. Cancer Lett. 2009;274:331–336. doi: 10.1016/j.canlet.2008.09.038. [DOI] [PubMed] [Google Scholar]
- 31.Hayakawa Y, Maeda S, Nakagawa H, et al. Effectiveness of IkappaB kinase inhibitors in murine colitis-associated tumorigenesis. J Gastroenterol. 2009;44:935–943. doi: 10.1007/s00535-009-0098-7. [DOI] [PubMed] [Google Scholar]
- 32.Jimi E, Aoki K, Saito H, D'Acquisto F, et al. Selective inhibition of NF-kappa B blocks osteoclastogenesis and prevents inflammatory bone destruction in vivo. Nat Med. 2004;10:617–624. doi: 10.1038/nm1054. [DOI] [PubMed] [Google Scholar]
- 33.Le Bras S, Geha RS. IPEX and the role of Foxp3 in the development and function of human Tregs. J Clin Invest. 2006;116:1473–1475. doi: 10.1172/JCI28880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bettelli E, Dastrange M, Oukka M. Foxp3 interacts with nuclear factor of activated T cells and NF-kappa B to repress cytokine gene expression and effector functions of T helper cells. Proc Natl Acad Sci USA. 2005;102:5138–5143. doi: 10.1073/pnas.0501675102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ulmanen I, Halonen M, Ilmarinen T, Peltonen L. Monogenic autoimmune diseases – lessons of self-tolerance. Curr Opin Immunol. 2005;17:609–615. doi: 10.1016/j.coi.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 36.Liu G, Ma H, Qiu L, et al. Phenotypic and functional switch of macrophages induced by regulatory CD4+CD25+ T cells in mice. Immunol Cell Biol. 2011;89:130–142. doi: 10.1038/icb.2010.70. [DOI] [PubMed] [Google Scholar]
- 37.Ralainirina N, Poli A, Michel T, et al. Control of NK cell functions by CD4+CD25+ regulatory T cells. J Leukoc Biol. 2007;81:144–153. doi: 10.1189/jlb.0606409. [DOI] [PubMed] [Google Scholar]
- 38.Tarbell KV, Yamazaki S, Steinman RM. The interactions of dendritic cells with antigen-specific, regulatory T cells that suppress autoimmunity. Semin Immunol. 2006;18:93–102. doi: 10.1016/j.smim.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 39.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 40.Raes G, De Baetselier P, Noel W, Beschin A, Brombacher F, Hassanzadeh Gh G. Differential expression of FIZZ1 and Ym1 in alternatively versus classically activated macrophages. J Leukoc Biol. 2002;71:597–602. [PubMed] [Google Scholar]
- 41.Porta C, Rimoldi M, Raes G, et al. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor κB. Proc Natl Acad Sci USA. 2009;106:14978–14983. doi: 10.1073/pnas.0809784106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117:175–184. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]