

Abstract
Shear stress stimulates nitric oxide (NO) release in endothelial cells. Stretch-activated ion channels (SACs) and the transient receptor potential vanilloid type 1 (TRPV1) receptor respond to mechanical stimulus and are permeable to Na+, Ca2+ and K+. The influence of SACs and the TRPV1 receptor on NO release on the heart and on the vascular reactivity of the thoracic aorta (TA) was studied. Experiments were performed in isolated perfused heart, cultured endothelial cells and TA rings from Wistar rats. Capsaicin (10 μM, 30 μM) was used as a NO release stimulator, capsazepine (6 μM, 10 μM) was used as a capsaicin antagonist and gadolinium (3 μM, 5 μM) was used as an inhibitor of SACs. NO was measured by the Kelm and Tenorio methods. Left ventricular pressure was recorded and coronary vascular resistance was calculated. Capsaicin increased NO release in the heart by 58% (395±8 pmol/mL to 627±23 pmol/mL). Capsazepine and gadolinium inhibited NO release by 74% and 82%, respectively. This tendency was similar in all experimental models. Capsaicin attenuated the effects of norepinephrine (10 M to 7 M) on TA and had no effect in the presence of Nω-nitro-L-arginine methyl ester. Therefore, the authors conclude that SACs and the TRPV1 receptor are both present in the coronary endothelium and that both participate in Ca2+-dependent NO release.
Keywords: Endothelium, Nitric oxide, Stretch-activated ion channels, TRPV1 receptors
Nociceptors are a group of neurons with afferent (sensorial) and efferent (motor) functions related to pain. These nociceptors participate in the regulation of vascular tone, cardiac functions, immunological processes and tisse growth, among other functions, in response to mechanical and thermal stimuli (1–4). Activation of nociceptors induces the release of neuropeptides, such as substance P, neurokinin-A, the calcitonin gene-related peptide (CGRP), somatostatin and others (3–5). CGRP is a cardiotonic agent dependent on nitric oxide (NO) production (5). NO is produced in almost all tissues and organs, exerting a variety of biological actions under both physiological and pathological conditions (5).
NO mediates vascular relaxation by increasing the levels of cyclic GMP and cyclic AMP in smooth muscle cells. NO release can be stimulated by sarcolemmal hyperpolarization and increased Ca2+ permeability (6) with the consequent activation of the endothelial NO synthase (NOS)-3 (7–10). In addition, during hypertension in humans and in animal models of salt-sensitive hypertension, CGRP exhibits important vasodilator actions, decreasing blood pressure through interactions with NO (11–16). Release of neuropeptides from nociceptors is also caused by activation of a member of the transient receptor potential (TRP) receptors subfamily, known as the vanilloid type 1 receptor (VR1) or the TRPV1 receptor, which is present in nociceptors (17–19). The TRPV1 receptor has also been described in endothelial cells of human pulmonary and cerebral arteries (18). The presence of the TRPV1 receptor has been suggested in other vascular regions as well (20–22). The TRPV1 receptor is sensitive to capsaicin (8-methyl-vannillyl-6-noneamide) which is a neurotoxin obtained from plants of the genus Capsicum (chili pepper) that has been used as an analgesic in diverse pain-related pathologies. This alkaloid, as well as its antagonist capsazepine, have been used as tools to study the structure and function of the TRPV1 receptor, which is an integral membrane protein (23–25). The TRPV1 receptor is also activated by cannabinoid substances, such as anandamide and 2-arachidonoyl-glycerol (26,27), which causes important vasodilator effects attributed to the actions of CGRP and NO (11–16,18,19).
The mechanisms modulating NO release in the endothelium are multifactorial. Shear stress generated by blood flow is well known to physiologically regulate NO release (28,29). Regulation of Ca2+ flux in the endothelium is mediated by at least one type of channel that is sensitive to mechanical stress, the stretch-activated ion channels (SACs) (30–32). SACs are functionally characterized; however, the molecular structure of SACs is not known. The TRPV1 receptor and SACs share functional characteristics including their sensitivity to mechanical stimuli and their selectivity to ions such as Na+, Ca2+ and K+ (24,30). SACs have been studied in the examination of Ca2+ currents in Xenopus cells, where it was found that the activity of this type of SAC can be blocked with the trivalent lanthanide gadolinium (Gd3+), thereby inhibiting the generation of NO dependent on NOS-3 activation (33–35).
The role of the TRPV1 receptor and SACs in NO release in the heart or in the aorta has only been partially explored. We hypothesized that there are functional similarities between SACs and the TRPV1 receptor. Therefore, we explored the functional similarities between the TRPV1 receptor and SACs on NO release in the heart, in the thoracic aorta (TA) and in endothelial cells in culture.
METHODS
Reagents
All reagents used in the present study were of analytical grade. Capsaicin, capsazepine, Gd3+, norepinephrine, acetylcholine, Nω-nitro-L-arginine methyl ester (L-NAME), human hemoglobin, dithionite, vanadium chloride (III), naphtylendiamine, sulfanilamide phosphoric acid, ethanol, collagenase type IV, trypsin, bovine serum albumin, M199 medium and Sephadex G-25 were obtained from Sigma Chemical Co (USA). Fetal bovine serum was obtained from HyClone (USA). Deionized water was made with the Simplicity system (Millipore SAS, France). Sodium pentobarbital was obtained from Pfizer Salud Animal, Pfizer México (Mexico).
Capsaicin was administered at 10 μM and 30 μM, capsazepine at 6 μM and 10 μM, Gd3+ at 3 μM and 5 μM, acetylcholine at cumulative doses of 2×10−9 M to 2×10−5 M, L-NAME at 300 μM and norepinephrine at cumulative doses of 2×10−9 M to 2×10−5 M. All drugs were administered to isolated hearts through continuous infusion (0.01 mL/min). For the TA rings and the endothelial cells in culture, drugs were administered through incubation. Sodium pentobarbital was administered at a surgical dose of 63 mg/kg.
Animals
Male Wistar rats, weighing 300 g to 350 g, were obtained from the animal facilities of the Instituto Nacional de Cardiología “Ignacio Chávez” (México, México). Rats were housed under standard temperature and light conditions (12 h light, 12 h dark) with a standard diet (LabDiet 5012, PMI Nutrition International, USA) and water ad libitum. All animals were handled according to the National Regulation for the Care and Handling of Experimental Animals (SAGARPA, NOM-062-ZOO-1999, Mexico) and the protocol was approved by the institutional Ethics Committee. All efforts were made throughout the entire experimental period to reduce the suffering of the animals to a minimum.
Isolated heart preparation according to Langendorff’s method
Rats were anesthetized with sodium pentobarbital. The heart was rapidly extracted and connected through the ascending aorta to a cannula attached to a nonrecirculating perfusion system. Krebs solution (KS) (7) was used as perfusant, maintained at 37±0.2 °C and a pH of 7.4±0.1, saturated with a mixture of 5% CO2 and 95% O2 (Aga, Mexico), during the entire experimental procedure. After cannulation, the heart was perfused for 30 min as an adaptation period. A latex balloon connected to a pressure transducer (SR25, Stalham Inc, Puerto Rico) was inserted into the left ventricle cavity and the maximal velocity of the left ventricular pressure (LVP, dp/dtmax) and the perfusion pressure (PP) were monitored and recorded on a computer system (Grass Instruments Co, USA). Coronary vascular resistance (CVR) was calculated from the PP data and the perfusion rate (PR) (CVR = PP/PR) (36). Heart rate was stabilized at 1 Hz by stimulating with a ventricular epicardial pacemaker (S5, Grass Instruments Co, USA) (37). The effect and potency of the drugs were measured following a study of gradual and sustained dose-response curves.
Experimental protocol
A group of 10 hearts was perfused at 10 mL/min (the approximate velocity of normal blood flow in the rat) (36), which resulted in an average of 50 mmHg of PP. Another group of 10 hearts was perfused at 5 mL/min, 10 mL/min, 15 mL/min, 20 mL/min, and 25 mL/min by means of a peristaltic pump (MasterFlex, Cole Palmer Instruments, USA) with the aim of increasing the shear stress on the coronary endothelium. Each heart served as its own control; hence, after a response curve to different flows in the absence of drugs, tests with 10 μM capsaicin, 6 μM capsazepine and 3 μM Gd3+ were performed. These substances were added to the perfusion solution at a rate of 0.01 mL/min using a continuous infusion pump (Hamilton Company, USA) via the cannula connected to the ascending aorta.
Culture of endothelial cells
Endothelial cells were obtained from coronary arteries of the rats as reported by Kan et al (38). Using Langendorff’s method, the heart was isolated and perfused with KS for 15 min. Thereafter, KS was substituted by a Ca2+-free KS solution at a pH of 7.2 to 7.4 until the mechanical activity of the heart ceased. Subsequently, a 15% collagenase solution in KS was used to perfuse the coronary arteries and to remove the endothelial layer. The vascular space was kept in contact with trypsin (0.25%) for 10 min. The heart was then perfused with 40 mL of KS. Cells were isolated from the perfusate by centrifugation and were subsequently resuspended in 10 mL of M199 medium enriched with 20% fetal bovine serum and plated onto tissue culture plates. Experiments were performed in the absence and presence of capsaicin (10 μM), capsazepine (6 μM), L-NAME (300 nM) and Gd3+ (5 μM).
Vascular reactivity in TA rings
Immediately after extracting the heart, the TA was removed and 2 mm TA rings, free of connective and fat tissue, with intact endothelium were obtained. TA rings were maintained in cold (2°C to 4°C) KS for 10 min. Subsequently, each ring was placed into a 5 mL isolated tissue chamber, fixed to the supports of a G-SRW46 (Grass Instruments Co, USA) tension transducer that was connected to a Grass 79 D polygraph (Grass Instruments Co, USA). TA rings were kept immersed in KS containing (mM): 118 NaCl, 6 KCl, 1.2 MgSO4, 1.2 KH2PO4, 25 NaHCO3, 1.6 CaCl2 and 11 D-glucose, at 37°C and pH 7.4, equilibrated with the 5% CO2 and 95% O2 mixture. Basal tension was 2 g and, after 1 h of adaptation, the TA rings were contracted using 2×10−7 M norepinephrine. Following this, acetylcholine was cumulatively added from 2×10−9 M to 2×10−5 M. The study was repeated varying the norepinephrine concentrations in each case from 2×10−9 M to 2×10−5 M.
To test the effects of NOS inhibition, aortic ring preparations were incubated for 10 min with L-NAME (300 μM). Subsequently, norepinephrine (2×10−7 M) was added and the maximal contraction was recorded. Relaxation of the TA rings was induced with cumulative concentrations of acetylcholine (range 2×10−9 M to 2×10−5 M).
Experiments were performed in the absence and presence of capsaicin (10 μM), capsazepine (6 μM), L-NAME (300 nM) and Gd3+ (5 μM).
Quantification of NO release in isolated hearts
NO release was measured in the perfusate by the modified Kelm and Schrader technique (39) under ultraviolet/visual spectrometry using an extinction coefficient of 411 nm to 401 nm at ambient temperature. The respective measurements were performed using a double-beam spectrometer (DW2000, SLM-Aminco, USA).
Quantification of NO in the culture medium of endothelial cells
NO was quantified in the cell culture medium as described by Tenorio et al (40), by the reduction of NO with vanadium (III) under ultraviolet/visual spectrometry using the extinction coefficient of 572 nm to 587 nm at ambient temperature. The respective measurements were performed with a double-beam spectrometer.
Expression of NOS-2 and NOS-3 in heart
To verify that capsaicin stimulates the release of NO, one group of 10 rats was treated with a daily intraperitoneal dose of capsaicin (5 mg/kg) during the four days before sampling until completing a final dose of 20 mg/kg (6) The cardiac tissue was prepared for the extraction of NOS and the protein concentration was determined using the method of Bradford (41). The tissue lysate was separated by electrophoresis on a 10% sodium dodecyl sulphate polyacrylamide gel (BioRad, USA) (42).
Statistical analysis
Results are expressed as mean ± SEM. Comparisons between means were analyzed by Student’s t tests or one-way ANOVAs followed by Bonferroni t tests, as appropriate. Differences were considered to be statistically significant at P<0.05 (43).
RESULTS
NO release from heart perfused at 10 mL/min
In the absence of drugs, NO released from hearts perfused at 10 mL/min was 395.12±8.25 pmol/mL, which decreased to 53.02±28.11 pmol/mL with the addition of Gd3+, and to 112.21±29.15 pmol/mL with capsazepine (Figure 1). In the presence of capsaicin, NO release increased to 627.52±23.35 pmol/mL (58%). This effect of capsaicin was reduced by capsazepine to 165.28±23.71 pmol/mL (26%) and by Gd3+ to 116.17±30.64 pmol/mL (13%).
Figure 1).
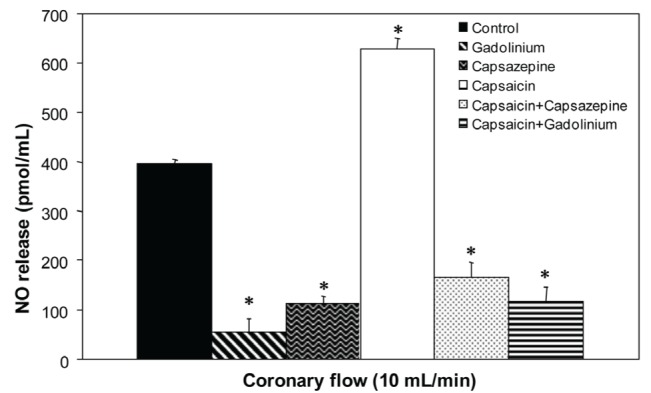
Effect of capsaicin, capsazepine and gadolinium on nitric oxide (NO) release in isolated rat hearts. Values represent mean ± SD; n=10. *P<0.05 by one-way ANOVA followed by Student’s t test
NO release from hearts perfused at different rates
Figure 2 shows the results of NO release from hearts subjected to variations in the PR in the absence or presence of different drugs. Under control conditions, the increase in NO release was dependent on the PR, and was increased by the action of capsaicin. This effect was blocked by capsazepine and, to a greater extent, by Gd3+. In the absence of drugs, the CVR remained constant (30×107 dyn·s·cm−5) even with the changes in pressure generated by the increase in the PR and NO release (Figure 3). Capsaicin, in this case, did not significantly alter the resistance. In contrast, capsazepine and Gd3+ promoted a constrictive effect at low flow rates, which reverted toward basal values with each increment in the PR.
Figure 2).
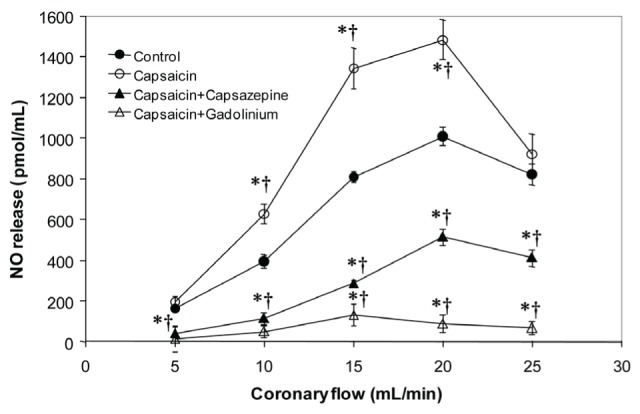
Effect of capsaicin, capsaicin+capsazepine and capsaicin+gadolinium on nitric oxide (NO) release in isolated rat hearts at different perfusion rates. Values represent mean ± SD; n=10. *P<0.05 by Student’s t test; †P<0.05 by one-way ANOVA followed by Student’s t test
Figure 3).
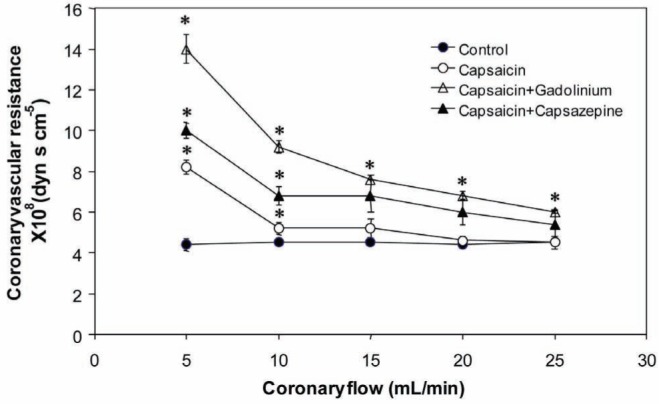
Effect of capsaicin, capsaicin+capsazepine and capsaicin+gadolinium on coronary vascular resistance in isolated rat hearts. Values represent mean ± SD; n=10. *P<0.05, one-way ANOVA followed by Student’s t test
Another parameter of cardiac function that was studied was the LVP (Figure 4), which, under control conditions, increased with increasing PR, an effect that was inhibited by capsaicin; this effect was accentuated by the action of capsazepine and Gd3+.
Figure 4).
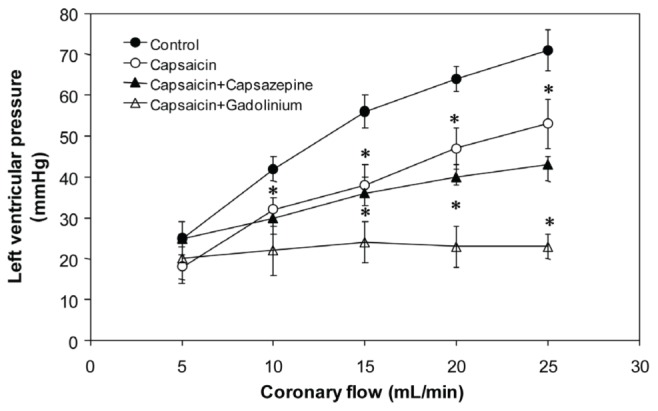
Effect of capsaicin, capsaicin+capsazepine and capsaicin+gadolinium on left ventricular pressure in isolated rat hearts. Values represent mean ± SD; n=10. *P<0.05, one-way ANOVA followed by Student’s t test
Coronary endothelial cells
Results for NO release in endothelial cells isolated from the coronary arteries are shown in Figure 5. The amount of NO released to the culture medium was 30.17 pmol/mL (n=3) in the absence of drugs. Acetylcholine (3 μM) was added to explore the functionality of the endothelial cells, and increased NO release to 130.26 pmol/mL, 433% higher than control levels. In contrast, Gd3+ (3 μM) reduced NO release to 20.36 pmol/mL. Capsaicin (10 μM) increased NO release to 80.30 pmol/mL. However, capsazepine (6 μM) did not change the control levels of NO release (34.09 pmol/mL). In the presence of Gd3+ (3 μM), acetylcholine (3 μM)-induced NO release was substantially reduced to 30.36 pmol/mL. Capsazepine (6 μM) blocked the effect of capsaicin (10 μM). In the presence of acetylcholine (3 μM) and capsaicin (10 μM), NO release was increased to 209.83 pmol/mL. The addition of capsazepine (6 μM) to acetylcholine (3 μM) had an antagonistic effect, which reduced NO release to 30.22 pmol/mL.
Figure 5).
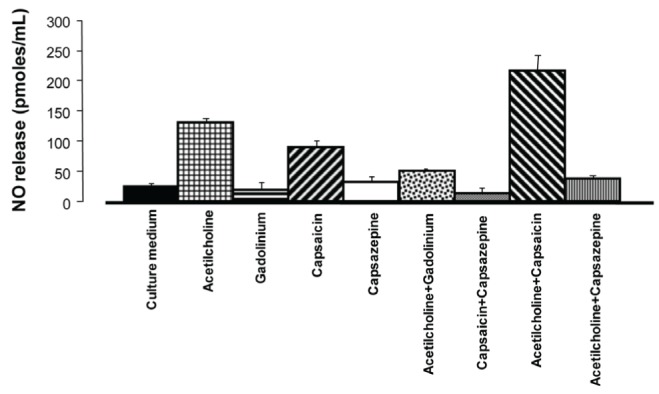
Nitric oxide release from cultured endothelial cells of rat coronary arteries after incubation with acetylcholine, gadolinium, capsaicin or capsazepine
Vascular reactivity
Measurements of vascular tone from the TA rings are shown in Figure 6. Vascular tone due to norepinephrine was not significantly modified by incubating with capsazepine (6 μM), Gd3+ (3 μM) or L-NAME (300 μM). The vascular tone was lower (46%) when incubating with capsaicin (10 μM), and this effect was blocked when capsazepine was added to the incubation medium. Similar results were observed when L-NAME was added.
Figure 6).
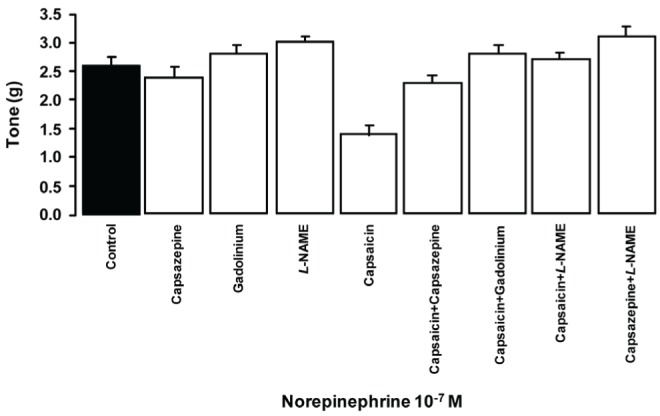
Effect of capsaicin, capsazepine, Nω-nitro-L-arginine methyl ester (L-NAME) and gadolinium on the tone of thoracic aorta rings induced by 10−7 M norepinephrine. P<0.05, one-way ANOVA followed by Student’s t test; n=8
Western blot
NOS-3 expression in heart tissue was substantially increased by the action of capsaicin and, to a lesser extent, was also stimulated by capsazepine, compared with control (Figure 7). NOS-2 protein expression was unchanged in the presence of capsaicin, while capsazepine caused an increased expression of the enzyme.
Figure 7).
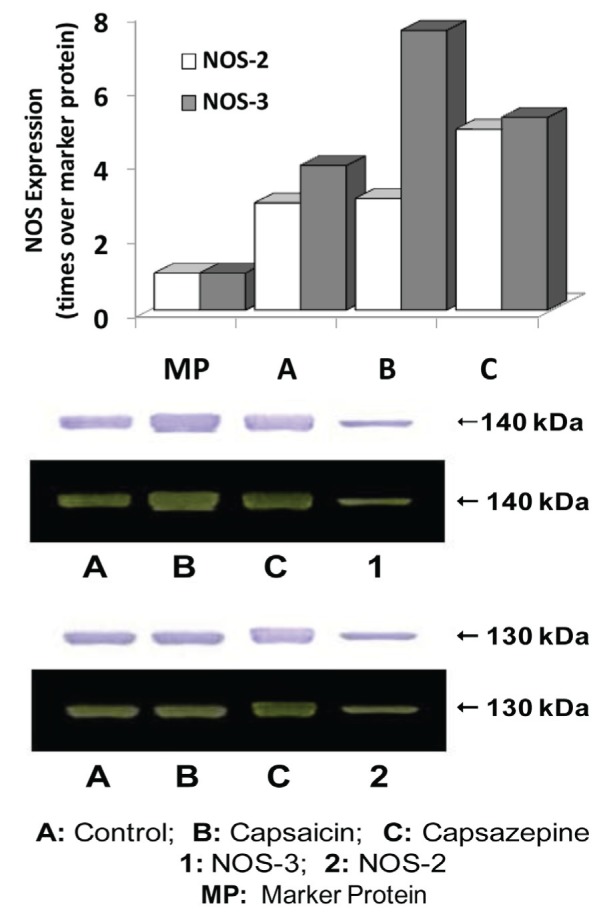
Expression of nitric oxide synthase (NOS)-3 and NOS-2 in cardiac tissue after four days of capsaicin and capsazepine treatment
DISCUSSION
The present study demonstrated that the TRPV1 receptor and SACs respond similarly to the mechanical stimuli generated by shear stress, increasing NO release in both the isolated heart and endothelial cells. There may be functional similarities between SACs and the TRPV1 receptor and it is possible that they result from structural similarity (44).
The inhibitory effect of Gd3+ on NO release in the heart due to physical stress generated by flow is attributed to the blocking of SACs (31), which prevents the increase in intracellular Ca2+ (31,32). Capsaicin induced a significant increase in NO release, which was inhibited similarly by Gd3+ and capsazepine. These results suggest that the inhibitory effect of capsazepine is also due to a decrease in intracellular Ca2+ concentrations in endothelial cells. It is possible that these effects are due to the presence of the TRPV1 receptor in the endothelial cells of coronary arteries. The TRPV1 receptor in endothelial cells may play an important role in the regulation of intracellular Ca2+ concentration, because NO generated by capsaicin’s action was also inhibited by Gd3+. To our knowledge, this finding has not been reported previously. It is possible that both effects are dependent on changes in Ca2+ kinetics or concentration (31,35).
The endothelial response to shear stress resulted in increased NO release when the PR was incrementally increased in isolated hearts. Compared with the control curve, capsaicin potentiated NO release. However, a saturation of the response system was observed at a PR of 25 mL/min, most likely due to the increase in perfusion flow producing mechanical deformations that generated a limit to the resistance of the coronary arteries, counteracting the endothelial response. Control and capsaicin-induced NO release were significantly inhibited by the antagonist capsazepine and by Gd3+, which allows us to infer that, due to the lack of Ca2+ to interact with NOS-3, NO production was decreased. Alterations in NO release produced by the drugs used and by the changes in PR were reflected in the CVR. Under control conditions, the level of resistance was not modified by increases in the PR, which confirms the participation of a self-regulating vascular tone system in the heart, with the participation of compensatory substances such as prostaglandins and prostacyclins (38).
On the other hand, the decrease in intracellular Ca2+ in the coronary endothelium due to the blocking of SACs by Gd3+was similar to the effect of the capsaicin antagonist capsazepine, which induced a constricting effect demonstrated by the initial increase in CVR. This constricting effect was decreased as the PR was increased. It is possible that this initial effect of Gd3+ occurred because of the decrease in NO release, which in turn stimulated the release of constricting substances such as endothelin-1 and constricting prostaglandins (4,45). Therefore, to counteract these adverse conditions, the endothelium releases vasodilator substances such as NO and adenosine (4). On the other hand, the LVP was decreased by the action of capsaicin, probably as a result of the increase in NO release. Capsazepine, as well as Gd3+, induce a negative ionotropic effect through a reduction in intracellular Ca2+ by blocking the TRPV1 receptor and SAC pathways. Furthermore, we also observed that capsaicin induced a negative inotropic effect, which was due to increased NO release (30).
NO release in cultured endothelial cells could be related to the action of capsaicin, as well as the antagonist action of capsazepine, which was exerted without the influence of nerves or tissues originating from outside of the endothelium, suggesting the presence of TRPV1 receptors in endothelial cells. Our results are similar to those recently published by Ching et al (44) while the present article was in revision.
Using an aortic ring model, we studied the effects of various drugs on vascular reactivity. We showed that the contractile response of the TA rings, precontracted with norepinephrine (2×10−7 M), was decreased in the presence of capsaicin; this decrement was blocked by the antagonistic effect of capsazepine and by NOS inhibition with L-NAME (25). These results suggest that the relaxing effect of capsaicin depends on NOS activation. This was confirmed in this model by results obtained by incubating TA rings with Gd3+. NOS-2 and NOS-3 expression in ventricular tissue in control conditions and after four days of capsaicin treatment demonstrates that the NO release system is stimulated by TRPV1 receptor activation. Our results show that activation of the TRPV1 receptor by capsaicin significantly stimulated NOS-3 protein expression.
NOS-2 was not stimulated by the action of the TRPV1 receptor, which may be because activation of this pathway is preferentially initiated by the action of Ca2+ entering the endothelial cell. An interesting explanation for the expression of NOS-3 stimulated by capsaicin was proposed by Ching et al (44); NOS-3 activation occurs due to the interaction of the TRPV1 receptor with the Ca2+-calmodulin complex, as well as an interaction with PI3K/Akt. These interactions are stimulated by TRPV1 receptor phosphorylation, but their mechanisms are not well understood. Controversially, Qiao et al (21), showed in PC12 cells that capsaicin at low concentrations (5 μM to 50 μM) stimulates the expression of NOS-2. Baylie and Brayden (22) showed that the relaxing effect produced by capsaicin is reduced by L-NAME, with NOS-3 being the isoenzyme most affected.
CONCLUSION
Our results demonstrated a role for SACs and the TRPV1 receptor systems in regulating NO release from the coronary arteries of the rat heart. In addition, we demonstrated, by using specific agonists and antagonists, that both systems activated NOS-3. Our data suggest that the NO-release pathway that triggers NOS-3 activation in both cases is Ca2+ dependent.
REFERENCES
- 1.Hoover BD, Chang Y, Hancock CJ, et al. Actions of tachykinins within the heart and their relevance to cardiovascular disease. Jpn J Pharmacol. 2000;84:367–73. doi: 10.1254/jjp.84.367. [DOI] [PubMed] [Google Scholar]
- 2.Szolcsanyi J, Oroszi G, Nemeth J, et al. Functional and biochemical evidence for capsaicin-induced neural endothelin release in isolated working rat heart. Eur J Pharm. 2001;419:215–21. doi: 10.1016/s0014-2999(01)00973-6. [DOI] [PubMed] [Google Scholar]
- 3.Zanesco A, Costa PKS, Riado RS, et al. Modulation of coronary flow and cardiomyocyte size by sensory fibers. Hypertension. 1999;34:790–4. doi: 10.1161/01.hyp.34.4.790. [DOI] [PubMed] [Google Scholar]
- 4.Ralevic V. Endothelial nitric oxide modulates perivascular sensory neurotransmission in the rat isolated mesenteric arterial bed. Br J Pharmacol. 2002;137:19–28. doi: 10.1038/sj.bjp.0704837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yeon D, Kwon S, Lee Y, et al. Capsaicin-induced relaxation in rabbit coronary artery. J Veter Medical Sci. 2001;63:499–503. doi: 10.1292/jvms.63.499. [DOI] [PubMed] [Google Scholar]
- 6.Gupta S, Lozano-Cuenca J, Villalón MC, et al. Pharmacological characterisation of capsaicin-induced relaxations in human and porcine isolated arteries. Naunyn-Schmiedeberg’s Arch Pharmacol. 2007;375:29–38. doi: 10.1007/s00210-007-0137-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou F-W, Li Y-J, Lu R, et al. Protection of calcitonin gene-related peptide-mediated preconditioning against coronary endothelial dysfunction induced by reperfusion in the isolated rat heart. Life Sci. 1999;64:1091–7. doi: 10.1016/s0024-3205(99)00037-5. [DOI] [PubMed] [Google Scholar]
- 8.Chang Y, Hoover BD, Hancock CJ. Endogenous tachykinins cause bradycardia by stimulating cholinergic neurons in the isolated guinea pig heart. Am J Physiol Reg Integ Comp Physiol. 2000;278:R1483–R9. doi: 10.1152/ajpregu.2000.278.6.R1483. [DOI] [PubMed] [Google Scholar]
- 9.Li Y-J, Peng J. The cardioprotection of calcitonin gene-related peptide-mediated preconditioning. Eur J Pharmacol. 2002;442:173–7. doi: 10.1016/s0014-2999(02)01538-8. [DOI] [PubMed] [Google Scholar]
- 10.Shen Y-T, Pittman JT, Buie SP, et al. Functional role of α-calcitonin gene-related peptide in the regulation of the cardiovascular system. J Pharmacol Exp Ther. 2001;298:551–8. [PubMed] [Google Scholar]
- 11.Csont T, Csonka C, Kovács P, et al. Capsaicin-sensitive sensory neurons regulate myocardial nitric oxide and cGMP signaling. Eur J Pharmacol. 2003;476:107–13. doi: 10.1016/s0014-2999(03)02117-4. [DOI] [PubMed] [Google Scholar]
- 12.Lo Y-Ch, Wu J-R, Wu S-N, et al. Glyceryl nonivamide: A capsaicin derivative with cardiac calcitonin gene-related peptide releasing, K+ channel opening and vasorelaxant properties. J Pharmacol Exp Ther. 1997;281:253–60. [PubMed] [Google Scholar]
- 13.Wang HD, Zhao Y. Increased salt sensitivity induced by impairment of sensory nerves: Is nephropathy the cause? J Hypertension. 2003;21:403–9. doi: 10.1097/00004872-200302000-00033. [DOI] [PubMed] [Google Scholar]
- 14.Wang HD. The vanilloid receptor and hypertension. Acta Pharmacol Sinica. 2005;26:286–94. doi: 10.1111/j.1745-7254.2005.00057.x. [DOI] [PubMed] [Google Scholar]
- 15.Okajima K, Harada N, Uchiba M, et al. Acivation of capsaicin-sensitive sensory neurons by carvedilol, a non-selective β-blocker, in spontaneous hypertensive rats. J Pharm Exp Ther. 2004;309:684–91. doi: 10.1124/jpet.103.061150. [DOI] [PubMed] [Google Scholar]
- 16.Watson RE, Supowit SC, Zhao H, et al. Role of sensory nervous system vasoactive peptides in hypertension. Braz J Medical Biol Res. 2002;35:1033–45. doi: 10.1590/s0100-879x2002000900004. [DOI] [PubMed] [Google Scholar]
- 17.Tominaga M, Tominaga T. Strucure and function of TRPV1. Eur J Physiol. 2005;451:143–50. doi: 10.1007/s00424-005-1457-8. [DOI] [PubMed] [Google Scholar]
- 18.Yao X, Garland JG. Recent developments in vascular endothelial cell transient receptor potential channels. Circ Res. 2005;97:853–63. doi: 10.1161/01.RES.0000187473.85419.3e. [DOI] [PubMed] [Google Scholar]
- 19.Akerman S, Kaube H, Goadsby PJ. Vanilloid type 1 receptors (VR1) on trigeminal sensory nerve fibres play a minor role in neurogenic dural vasodilatation, and are involved in capsaicin-induced dural dilation. Br J Pharmacol. 2003;140:718–24. doi: 10.1038/sj.bjp.0705486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poblete MI, Orliac LM, Briones R, et al. Anandamide elicits an acute release of nitric oxide through endothelial TRPV1 receptor activation in the rat arterial mesenteric bed. J Physiol. 2005;568:539–51. doi: 10.1113/jphysiol.2005.094292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qiao S, Li W, Tsubouchi R, et al. Role of vanilloid receptors in the capsaicin-mediated induction of iNOS in PC12 cells. Neurochem Res. 2004;29:687–93. doi: 10.1023/b:nere.0000018839.59457.5c. [DOI] [PubMed] [Google Scholar]
- 22.Baylie RL, Brayden JE. TRPV Channels and vascular function. Acta Physiol. 2011;203:99–116. doi: 10.1111/j.1748-1716.2010.02217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Szallasi A, Blumberg PM. Vanilloid (capsaicin) receptors and mechanisms. Pharmacol Rev. 1999;51:159–206. [PubMed] [Google Scholar]
- 24.Caterina JM, Schumacher AM, Tominaga M, et al. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature. 1997;389:816–24. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- 25.Geppetti P, Trevisani M. Activation and sensitisation of the vanilloid receptor: Role in gastrointestinal inflammation and function. British J Pharmacol. 2004;141:1313–20. doi: 10.1038/sj.bjp.0705768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mukhopadhyay S, Chapnick BM, Howlett CA. Anandamide-induced vasorelaxation in rabbit aortic rings has two components: G protein dependent and independent. Am J Physiol Heart Circ Physiol. 2002;282:H2046–54. doi: 10.1152/ajpheart.00497.2001. [DOI] [PubMed] [Google Scholar]
- 27.MacNaughton KW, Van Sickle DM, Keenan MC, et al. Distribution and function of the cannabinoid-1 receptor in the modulation of ion transport in the guinea pig ileum: Relationship to capsaicin-sensitive nerves. Am J Physiol Gastro Liv Physiol. 2003;286:G863–G71. doi: 10.1152/ajpgi.00482.2003. [DOI] [PubMed] [Google Scholar]
- 28.Boo CY, Sorescu G, Boyd N, et al. Shear stress stimulates phosphorylation of endothelial nitric-oxide synthase at Ser1179 by Akt-independent mechanisms. J Biol Chem. 2002;277:3388–96. doi: 10.1074/jbc.M108789200. [DOI] [PubMed] [Google Scholar]
- 29.Pyke EK, Dwyer ME, Tschakovsky EM. Impact of controlling shear rate on flow-mediated dilation responses in the brachial artery of humans. J Appl Physiol. 2004;97:499–508. doi: 10.1152/japplphysiol.01245.2003. [DOI] [PubMed] [Google Scholar]
- 30.Jacques-Fricke BT, Seow Y, Gottlieb AP, et al. Ca2+ influx through mechanosensitive channels inhibits neurite outgrowth in opposition to other influx pathways and release from intracellular stores. J Neuroscience. 2006;26:5656–64. doi: 10.1523/JNEUROSCI.0675-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nilus B, Droogmans G. Ion channels and their functional role in vascular endothelium. Physiol Rev. 2001;81:1415–59. doi: 10.1152/physrev.2001.81.4.1415. [DOI] [PubMed] [Google Scholar]
- 32.Munevar S, Wang Y-L, Dembo M. Regulation of mechanical interactions between fibroblasts and the substratum by stretch-activated Ca2+ entry. J Cell Sci. 2004;117:85–92. doi: 10.1242/jcs.00795. [DOI] [PubMed] [Google Scholar]
- 33.Xian-Cheng Y, Sachs F. Block of stretch-activated ion channels in Xenopus oocytes by gadolinium and calcium ions. Science. 1989;243:1068–71. doi: 10.1126/science.2466333. [DOI] [PubMed] [Google Scholar]
- 34.Li Y, Zheng J, Bird IM, et al. Mechanisms of shear stress-induced endothelial nitric-oxide synthase phosphorylation and expression in ovine fetoplacental artery endothelial cells. Biol Reprod. 2004;70:785–96. doi: 10.1095/biolreprod.103.022293. [DOI] [PubMed] [Google Scholar]
- 35.Zou H, Lifshitz LM, Tuft AR, et al. Visualization of Ca2+ entry through single stretch-activated cation channels. Proc Natl Acad Sci USA. 2002;99:6404–9. doi: 10.1073/pnas.092654999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Döring HJ. In: The isolated perfused warm-blooded heart acording to Langendorff. Dehnert H, editor. Friburgo de Brisgovia: Ed. Biomesstechnik-Verlag; 1988. pp. 1–131. [Google Scholar]
- 37.Flores ChLP, Infante VO, Sánchez TG, et al. Detección de signos vitales en ratas mediante métodos no invasivos. Vet Mex. 2002;33:179–87. [Google Scholar]
- 38.Kan H, Ruan Y, Malik UK. Signal transduction mechanism(s) involved in prostacyclin production elicited by acetylcholine in coronary endothelial cells of rabbit heart. J Pharm Exp Ther. 1997;282:113–22. [PubMed] [Google Scholar]
- 39.Kelm M, Schrader J. Nitric oxide release from the isolated guinea pig heart. Eur J Pharmacol. 1988;155:317–21. doi: 10.1016/0014-2999(88)90522-5. [DOI] [PubMed] [Google Scholar]
- 40.Tenorio LFA, Del Valle ML, Pastelín HG. Validación de un método analítico espectrofotométrico para la cuantificación de metabolitos estables de óxido nítrico en fluidos biológicos. Rev Mex Cienc Farmaceu. 2005;36:31–41. [Google Scholar]
- 41.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principe of protein dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 42.Towbin H, Stachelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Box G, Hunter W. Estadística para Investigadores: Introducción al Diseño de Experimentos, Análisis de Datos y Construcción de Modelos. 3a edn. México: Reverté; 1999. pp. 345–580. [Google Scholar]
- 44.Ching L-C, Kou YR, Shyue S-K, et al. Molecular mechanisms of activation of endothelial nitric oxide synthase mediated by transientreceptor potencial vanilloid type 1. Cardiovasc Res. 2011;91:492–501. doi: 10.1093/cvr/cvr104. [DOI] [PubMed] [Google Scholar]
- 45.Ramzy D, Rao V, Tumiati CL, et al. Elevated endothelin-1 levels impair nitric oxide homeostasis through a PKC-dependent pathway. Circulation. 2006;114:I319–26. doi: 10.1161/CIRCULATIONAHA.105.001503. [DOI] [PubMed] [Google Scholar]