

Abstract
Doxorubicin (DOXO) is an efficient and low-cost chemotherapeutic agent. The use of DOXO is limited by its side effects, including cardiotoxicity, that may progress to cardiac failure as a result of multifactorial events that have not yet been fully elucidated. In the present study, the effects of DOXO at two different doses were analyzed to identify early functional and molecular markers of cardiac distress. One group of rats received 7.5 mg/kg of DOXO (low-dose group) and was followed for 20 weeks. A subset of these animals was then subjected to an additional cycle of DOXO treatment, generating a cumulative dose of 20 mg/kg (high-dose group). Physiological and biochemical parameters were assessed in both treatment groups and in a control group that received saline. Systolic dysfunction was observed only in the high-dose group. Mitochondrial function analysis showed a clear reduction in oxidative cellular respiration for animals in both DOXO treatment groups, with evidence of complex I damage being observed. Transcriptional analysis by quantitative polymerase chain reaction revealed an increase in atrial natriuretic peptide transcript in the high-dose group, which is consistent with cardiac failure. Analysis of transcription levels of key components of the cardiac ubiquitin-proteasome system found that the ubiquitin E3 ligase muscle ring finger 1 (MuRF1) was upregulated in both the low- and high-dose DOXO groups. MuRF2 and MuRF3 were also upregulated in the high-dose group but not in the low-dose group. This molecular profile may be useful as an early physiological and energetic cardiac failure indicator for testing therapeutic interventions in animal models.
Keywords: Cardiomyopathy, Doxorubicin, Echocardiography, Gene expression, Respirometry, Ubiquitin-proteasome system
Doxorubicin (DOXO) is one of the oldest and most widely used anthracyclines, and is often used to treat a broad spectrum of solid and hematopoietic tumours (1,2). These drugs, in association with innovations in surgical techniques and support measures, have led to improved survival in patients with cancer in recent decades (3). However, prolonged treatment with DOXO is hampered by undesired side effects, most notably the development of cardiotoxicity in a dose-dependent manner. This toxicity can present in two ways. In some cases, an acute and self-limited form of cardiomyopathy develops that can either cause death or resolve spontaneously. Alternatively, side effects can have a gradual onset and finally manifest as dilated cardiomyopathy with predominant left ventricular (LV) dysfunction. This can ultimately lead to death, despite the effective control of the treated cancer (1,2,4). The recommended cumulative dose is limited to 550 mg/m2. The dose that induces cardiac failure is notably variable (5,6), with some patients being able to tolerate higher DOXO doses that result in a better clinical outcome. The search for a better anthracycline that retains the same level of antitumour activity with lower cardiotoxicity has been unsuccessful to date. Therefore, strategies to improve cancer treatment with DOXO are still required.
The mechanism by which DOXO causes cardiac toxicity has yet to be fully elucidated and is probably distinct from the antitumour effects of DOXO (7,8). Although the latter relies on the blockade of cellular repair mechanisms, cardiac toxicity may occur through the generation of reactive oxygen species by mitochondrial NADH dehydrogenase (9). Oxygen radical generation results in a loss of mitochondrial function because of decreased complex I functionality. In addition, hypoactivity of glutathione peroxidase, the major detoxifying system of the heart, can contribute to cardiotoxicity (10). Recent studies have reported that decreased rates of state 3 and state 4 respiration in isolated mitochondria obtained from the hearts of animals that were treated with DOXO were associated with lower levels of uncoupling proteins 2 and 3 (11). Inhibition of components of the electron transport chain such as cytochrome c oxidase (which can be restored with the use of the antioxidant MitoQ) are also associated with DOXO-induced cardiotoxicity (12). The induction of the membrane permeability transition pore also contributes to mitochondrial function commitment (13), apoptosis (14) and changes in cardiac calcium metabolism (15). Disturbances of protein quality control or specific degradation of cardiac transcription factors mediated by the ubiquitin-proteasome sysem (UPS) have also been implicated (16–20). Therefore, understanding the clinically relevant cellular pathways that are involved in cardiac dysfunction should provide novel methods of suitable intervention.
Cardiomyocytes have high metabolic turnover rates and low levels of regeneration. Even under resting conditions, heart tissue is subjected to a stressful workload that can increase enormously in conditions that induce systolic or diastolic overload (21). In these circumstances, it is essential to maintain a proper balance between newly synthesized, correctly folded proteins and degraded proteins. These events are essential for cardiomyocyte homeostasis and are mediated by a complex system collectively known as protein quality control (19). This process is mediated by molecular chaperones and by the activity of the UPS. Molecular chaperones are essential for correct folding of nascent proteins, protection against protein unfolding and protein repair, whereas the UPS is necessary to terminally remove misfolded and unneeded cellular proteins, thereby preventing protein aggregation and cardiomyocyte dysfunction (22). This activity is critically important in postmitotic cells, such as neurons and cardiomyocytes, that cannot compensate for this damage through cell division (23). The strict regulatory function of the UPS is mediated by a biochemical enzyme cascade involving three classes of evolutionarily conserved proteins (E1, E2 and E3 enzymes) that direct the specific conjugation of ubiquitin moieties to cellular targets through isopeptide bonds (24). This process is repeated, leading to the polyubiquitination of target proteins that are then directed to the proteasome complex 26S and degraded into small peptides (24). The specificity of this system is determined by E3 ligases that, on signal activation, allow the recognition of cellular targets (25). At least nine E3 ligases have been described in the heart, including the Muscle Ring Finger protein family (MuRF1, MuRF2 and MuRF3) and muscle atrophy F-box (MAFbx)/atrogin-1 (26). These proteins are not only involved in cardiac development and maintenance of metabolism but also have a relevant function in cardiac pathology (27). The pathophysiological significance of UPS regulation in heart disease as a mediator of damage and/or as a consequence of injury is not yet clear, but strong evidence has emerged relating its function to cardiac hypertrophy (CH), dilated cardiomyopathy and ischemia (28–31). Recently, ubiquitin aggregates have been reported to be present in the hearts of patients with dilated cardiomyopathy (32,33), and several murine models have indicated that dysfunction of the UPS precedes cardiac failure (32).
Several articles have described UPS modulation by DOXO in heart tissue. Kumarapeli et al (17) described increased UPS-mediated proteolysis in mouse heart tissue and cultured cardiomyocytes following acute DOXO treatment. Liu et al (20) demonstrated an increase in proteasome activity when cardiomyocytes were exposed to low doses of DOXO. Interestingly, this effect was not reproduced at a higher dose, suggesting that the mechanisms underlying toxicity vary with the DOXO treatment protocol. In addition, overexpression of atrogin-1 has been observed after acute exposure to DOXO in rat cardiomyocytes in vitro and in vivo (33). Together, these findings suggest that with certain changes, hyperactivation of the UPS may also occur. To characterize the functional and molecular changes associated with cardiac dysfunction induced by DOXO, we developed a chronic model of cardiomyopathy in rats. The present study is, to the best of our knowledge, the first to address the subclinical and chronic effects of DOXO on mitochondrial function and E3 ligase expression. Our analysis of the temporal changes in cardiac function by serial echocardiography and by expression analysis of the E3 ligases of the UPS following exposure to different doses of DOXO demonstrates that cardiac changes in E3 ligase expression precede obvious LV dysfunction.
METHODS
Study design
Fifty-two female Wistar rats (Rattus norvegicus), 12 weeks of age, weighing approximately 250 g were housed in cages with a 12 h light/dark cycle at 25°C with ad libitum access to chow and water. All of the animals underwent echocardiographic analysis before drug administration. Animals were randomly assigned to receive saline at 1 mL/kg intraperitoneally (control group, n=15) or DOXO (Adriblastina RD, Pfizer Inc, USA) at 7.5 mg/kg intraperitoneally (low-dose group, n=37) that was administered in six doses of 1.25 mg/kg three times per week over two weeks to reduce local and systemic toxicity. The end of the first drug administration period was designated as week zero, as illustrated in Figure 1. The animals of each group were again subjected to echocardiography at two and four weeks following completion of the first cycle of DOXO administration. At four weeks, four animals from each group were euthanized, and their hearts were used for respirometry measurements and to quantify messenger RNA content.
Figure 1).
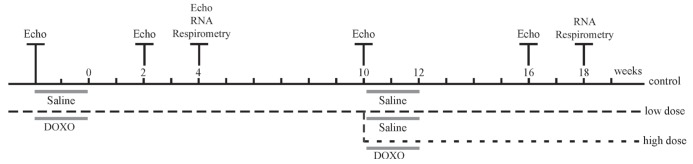
Timeline of study design. Initially, two experimental groups were established that included a control group that received saline (control; n=15) intraperitoneally and a low-dose group (low-dose, n=37) that received 7.5 mg/kg of doxorubicin (DOXO) intraperitoneally over two weeks, the end of which is indicated as time zero. Ten weeks after the end of the first cycle of DOXO, a subset of the animals in the low-dose group received an additional dose of 12.5 mg/kg DOXO (high-dose group, n=25), leaving 12 animals in the low-dose group at this point. The time of evaluation by echocardiogram (Echo), oxygen consumption tests (respirometry) and ventricular tissue extraction for messenger RNA expression analysis (RNA) are also indicated.
At week 10, echocardiography was performed to assess the LV systolic function for 11 control animals and 32 animals from the low-dose group (one animal from the low-dose group died). Twenty-five animals from the low-dose group received an additional DOXO dose of 12.5 mg/kg (2.5 mg/kg every other day over 10 days) for a total cumulative dose of 20 mg/kg. This new group was designated the high-dose group (n=25). At this timepoint, the low-dose group consisted of seven animals that received saline during the same period. At week 16, only nine animals from the high-dose group were still alive, and another functional analysis by echocardiography was performed for all of the groups. Two weeks later, four animals from each group were euthanized, and the heart mRNA content and respiratory rate were measured. At week 20, the remaining animals were euthanized as required by the ethics committee.
All of the procedures were performed within the guidelines established by the Ethics Committee for Caring of Experimental Animals, Instituto de Biofísica Carlos Chagas Filho, Universidade Federal do Rio de Janeiro. The experimental design, including interventions, is summarized in Figure 1.
Serial echocardiography
Echocardiographic analysis was performed before DOXO administration and at two, four, 10 and 16 weeks following DOXO administration. The anterior thorax of the animals was shaved following sedation with ketamine (50 mg/kg) and xylazine (5 mg/kg). Images were acquired using a 10 MHz annular phased-array electronic transducer connected to Megas (Esaote, Italy) ultrasonography equipment. The images were digitally captured, and the tests were recorded and subjected to posterior revision. Parasternal and apical views were investigated. The aorta-to-left atrial diameter ratio, the end-diastolic diameter of the LV and the end-systolic diameter of the LV, as well as the ejection fraction (EF) of the LV (LVEF) were measured. LV measurements were obtained from the short-axis views at the papillary muscle level. The left atrium and aorta diameters were measured at the base of the heart according to the Recommendations for Chamber Quantification (34). The LVEF was obtained by the Teichholz method (34), and the modified Simpson’s rule method (34,35) was used to calculate LV volumes. The stroke volume was obtained by subtracting the end-systolic volume from the end-diastolic volume, and cardiac output was estimated by multiplying the stroke volume by the heart rate/min. Only the results of the animals that were analyzed in all of the repetitions were included in the paired statistical analysis of echocardiographic measurements (control n=4, low-dose n=6 and high-dose n=8).
Mitochondrial respiratory activity in cardiac saponin-permeabilized fibres
The rate of mitochondrial respiration was assessed at different metabolic states in a high-resolution oxygraph (O2k, Oroboros Inc, Austria). The preparation of heart fibres was performed as described previously (36). The animals were euthanized by decapitation, and a small piece of the heart apex that was mainly composed of myofibrils was excised, weighed and immersed in ice-cold relaxing/preservation BIOPS solution (3 mM Ca2+-EGTA, 7 mM K+-EDTA, 6 mM Na+-ATP, 20 mM taurine, 15 mM phosphocreatine, 20 mM imidazol, 0.5 mM DTT and 50 mM MES, pH 7.1). The connecting tissue was removed, and individual fibre bundles were isolated and incubated under agitation in ice-cold BIOPS with 5 mg/mL saponin at 4°C for 30 min. The samples were subsequently transferred to ice-cold MIR05 mitochondrial respiration medium (0.5 mM EGTA, 3 mM MgCl2, 60 mM K+-lactobionate, 20 mM taurine, 10 mM KH2PO4, 1 mg/mL BSA and 20 mM HEPES, pH 7.1) under gentle agitation in an ice bath for 10 min. Mitochondrial respiratory activity was measured using Oxygraph-2k chambers containing air-saturated medium. The oxygen flow was normalized to the wet heart weight in all of the collected samples. Oxygen consumption was measured in the presence of the oxidizable complex I substrate mix of pyruvate/malate/glutamate (PMG) (5 mM, 2 mM and 10 mM, respectively) followed by the sequential addition of 1 mM ADP, an ATP synthesis substrate; 10 mM succinate, a complex II substrate; 0.5 μM carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP), an uncoupling drug; 1 μM rotenone, an inhibitor of complex I; and 20 mM Na+-azide. For all of the experiments, the electrode was calibrated between 0% and 100% saturation with atmospheric oxygen at 37°C, and the total reaction volume was 2.0 mL. The variability of the samples was low within each group studied (SEM <10%).
mRNA expression quantification
Ventricle fragments were removed from four animals from each experimental group immediately following euthanasia at four and 18 weeks after DOXO treatment. Tissues were homogenised with Ultra-Turrax (IKA, USA) in Trizol (Invitrogen, USA), and the total RNA was isolated following the manufacturer’s guide. The purified RNA was diluted in diethylpyrocarbonate-treated water and quantified spectrophotometrically at 260 nm. The integrity of the samples was confirmed by formaldehyde gel electrophoresis. Genomic DNA contamination was removed by incubation with RQ1 RNAse-free DNAse followed by treatment with RQ1 DNase stop solution (Promega, USA) to terminate the reaction. Total RNA (1 μg) from each animal was used to generate a pooled RNA template for first-strand synthesis of complementary DNA using random primers and Moloney murine leukemia virus reverse transcriptase (Promega, USA). Real-time reverse transcriptase-polymerase chain reaction (PCR) was performed on an ABI 7500 Fast Real-time PCR system with SYBR Green methodology (Applied Biosystems, USA) with 600 nM forward and reverse primers and complementary DNA in a 96-well array plate with a 25 μL final volume reaction. The primers were designed using Primer Express Software 3.0 (Applied Biosystems, USA) or Primer Blast (National Center for Biotechnology Information, USA). Melting curves and gel electrophoresis were performed to verify the specificity of all of the PCR products. The assay was run with the following cycling parameters: 50°C (2 min), 95°C (10 min) followed by 40 cycles at 95°C (15 s) and 60°C (1 min). All of the sample runs were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The samples were analyzed in triplicate and in a minimum of two independent experiments to ensure the experimental accuracy of the results. The relative gene expression was determined by the2∧(−ΔΔCt) method (37,38). The specific primer sequences and sizes of the PCR products are listed in Table 1.
TABLE 1.
Primer sequences
| Gene | Length of PCR product, bp | Forward | Reverse |
|---|---|---|---|
| GAPDH (NM_017008.3) | 85 | 5′−TGACTCTACCCACGGCAAGT | 5′−AGCATCACCCCATTTGATGT |
| Trim63 (MuRF1, NM_080903.1) | 351 | 5′−GAGCAATTGCATCTCCATGCTGGT | 5′−ACAATGCTCTTGATGAGCGGCTTG |
| Trim55 (MuRF2, NM_01012218.1) | 245 | 5′−ACATAGGGAAACCTGGTGGAA | 5′−TGTGTACACGCACCACACCTACA |
| Trim54 (MuRF3, NM_001013217.1) | 185 | 5′−ACTTCACGGTGGGTTTCAAG | 5′−TTGCCACAGAGGGTTAGAGG |
| Fbxo32 (MAFbx/atrogin1, NM_133521.1) | 368 | 5′−TCTACACTGCGAACAGCAGCTGAA | 5′−TCCAGGACAGAATGTGGCAGTGTT |
| Nppa (ANP, NM_012612.2) | 166 | 5′−TGAAAAGCAAACTGAGGGCT | 5′−GGATCTTTTGCGATCTGCTC |
ANP Atrial natriuretic peptide; GAPDH Glyceraldehyde-3-phosphate dehydrogenase; MAFbx Muscle atrophy F-box; MuRF Muscle ring finger; PCR Polymerase chain reaction
Statistical analysis
Comparisons between groups were performed using a two-tailed Student’s t test for unpaired samples. Values are plotted as the mean ± SEM. Differences were considered to be statistically significant at P<0.05.
RESULTS
Reduced survival rate after DOXO administration
Stress symptoms and survival were analyzed for 20 weeks to investigate the effects of DOXO on the animals. Animals in the control group experienced progressive weight gain and maintained appropriate levels of grooming with no observed deaths. One animal in the low-dose group died during the period of observation immediately following the first cycle of drug administration. This event could be a direct effect of the drug or the combined effects of the serial injection protocol that was used. Meanwhile, animals in the high-dose group became hypoactive, lost interest in grooming and experienced a high mortality rate. The survival curve is displayed in Figure 2A. This curve confirmed a clinically relevant effect of drug administration, although these deaths cannot be directly attributed to cardiac damage because DOXO is known to be associated with multisystem toxicity.
Figure 2).
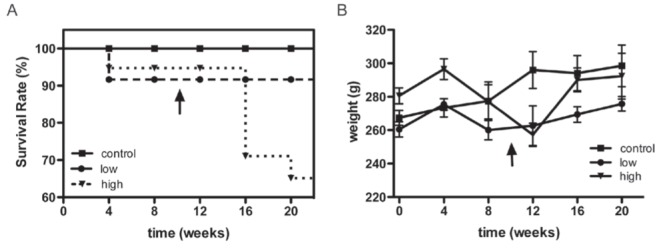
A Survival rate in weeks. Animals were monitored for 20 weeks immediately following the first cycle of doxorubicin (DOXO) administration (7.5 mg/kg). The vertical arrow indicates the second cycle of DOXO administration (12.5 mg/kg) to the high-dose group. No deaths occurred in the control group. One death occurred in the low-dose group shortly after drug administration. In contrast, several deaths occurred in the high-dose group after the additional dose of DOXO, indicating a dose-dependent pattern of mortality. B Weight measurement. The control group experienced gradual weight gain, while the low-dose group displayed a softened curve after the first four weeks. In addition to the initial weight loss beginning at four weeks, the high-dose group demonstrated intensified weight loss after the second DOXO cycle followed by subsequent weight gain that could be attributed to serositis (pleural and peritoneal effusions). The presence of serositis that had been detected in vivo during echocardiography of the high-dose animals was confirmed at the time of heart dissection.
The weight curve was also calculated (Figure 2B). Animals in the control group demonstrated gradual weight gain, while those in the low-dose group displayed a softened curve after four weeks. The high-dose group of animals lost weight after four weeks, and this weight loss was intensified after the second cycle of DOXO administration. In the weeks following the second DOXO cycle, weight loss reversal was observed that could be attributed to pleural and peritoneal effusions associated with serositis. The presence of serositis that had already been detected in vivo during echocardiography in all of the animals of the high-dose group was confirmed at heart dissection. These effusions were not present in animals from the other two groups.
DOXO-induced cardiomyopathy in rats
Serial echocardiograms were performed to analyze the effects of chronic DOXO treatment on cardiac function that lead to cardiomyopathy. The aorta-to-left atrial ratio was analyzed to identify evidence of LV dysfunction, because a reduction in this ratio is an indirect indicator of LV systolic dysfunction (Figure 3). Only the high-dose group developed a sharp decline compared with the control group at 16 weeks (0.85±0.16 versus 1±0, P<0.05; Figure 3). The LVEF was measured to survey the inotropic state of the myocardium directly (Figure 4A). No difference was observed between the groups during the first 10 weeks of treatment. After the second cycle of DOXO administration, the high-dose group displayed a significant reduction compared with the control group (64±5% versus 73±5%, P<0.05), whereas the low-dose group remained similar to the control group (76±5%).
Figure 3).
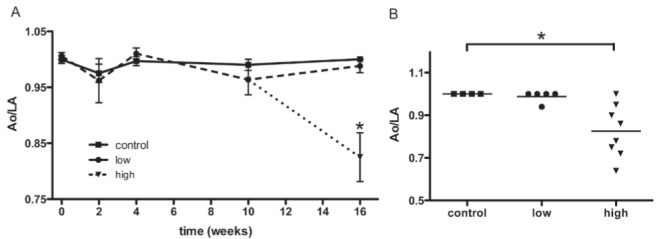
Echocardiographic measurements of the aorta-to-left atrial ratio (Ao/LA). The timecourse of left ventricular (LV) functional response to doxorubicin (DOXO) administration was indirectly inferred by the relationship between the transverse diameters of the aorta and the left atrium (control, n=4; low-dose, n=5; high-dose, n=8). A The Ao/LA is displayed over time, beginning after the completion of the first cycle of DOXO (7.5 mg/kg). There were no significant variations in the ratio between the control and the low-dose groups during the study. However, the high-dose group demonstrated a striking reduction after 16 weeks compared with the control group, indicating LV dysfunction. Data are expressed as mean ± SEM. B Individual results for each animal from tests performed at 16 weeks. Control, low- and high-dose groups are labelled. Control and low-dose groups showed similar results, while nearly all of the animals in the high-dose group exhibited a reduction in the ratio due to the atrial dilation that accompanies LV overload. *P<0.05
Figure 4).
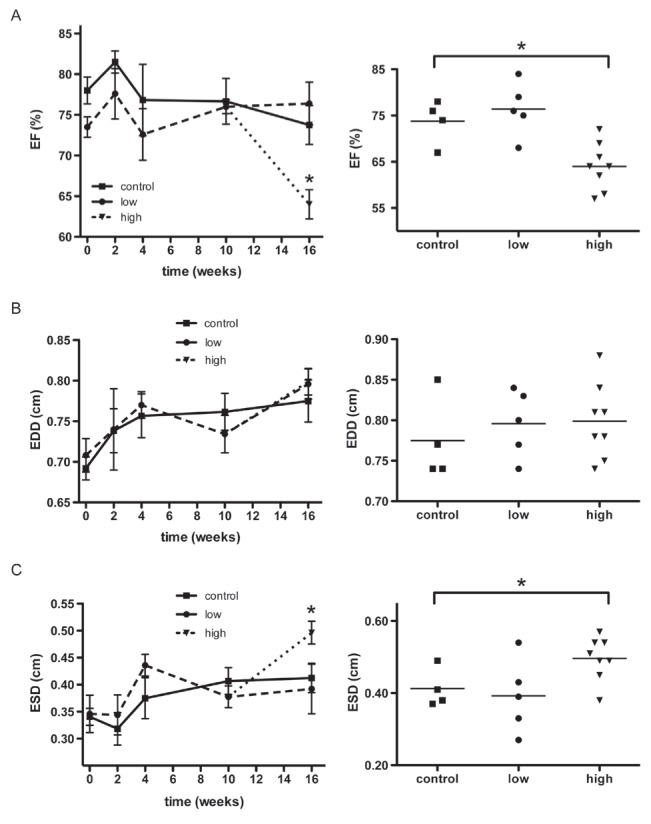
Echocardiographic analysis of left ventricle (LV) systolic performance. A Serial measurements were taken of the ejection fraction (EF) of the LV (left panel). Individual data for the animals from the three groups (control, n=4; low-dose, n=5; and high-dose, n=8) at week 16 is also provided (right panel). The EF of the high-dose group was significantly reduced at 16 weeks compared with the control group. B Serial measurements of the end-diastolic diameter (EDD) of the LV (left panel) and individual data at week 16 (right panel) are shown. There was no significant difference among the three groups. C Serial measurements of the end-systolic diameter (ESD) of the LV (left panel) and individual data at week 16 (right panel) are shown. The high-dose group displayed a significant increase in ESD compared with the control group at 16 weeks. The reduction of the EF in the high-dose group correlated with the increase in ESD in these animals, whereas the EDD did not differ from the control group. These results indicate that the high-dose group experienced LV dysfunction. Data are expressed as mean ± SEM. *P<0.05
All of the groups presented a progressive and similar increase in the end-diastolic diameter of the LV during the first four weeks (Figure 4B). The end-systolic diameters of the control and low-dose groups were notably similar until the end of the observation period at 16 weeks (0.41±0.05 cm versus 0.39±0.1 cm). The high-dose group developed a remarkable increase in the end-systolic diameter 16 weeks after the first cycle of administration compared with the control group (0.49±0.05 cm, P<0.05, Figure 4C). The high-dose group also displayed a striking reduction in cardiac output compared with the control group (Figure 5A), which was accompanied by a tendency toward a reduction in heart rate (Figure 5B). An M mode echocardiogram that is representative of one animal each from the control and high-dose groups is shown in Figure 5C. A discrete diastolic diameter change could be detected that was associated with a more distinct systolic diameter increase in the LV. In agreement with the echocardiographic evidence of LV systolic dysfunction, atrial natriuretic peptide mRNA expression increased by approximately eight-fold in the high-dose group compared with the control group (Figure 5D).
Figure 5).
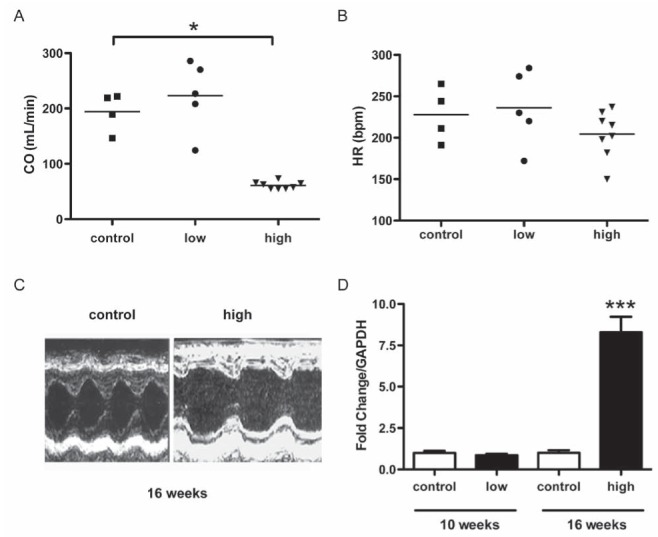
Functional analysis by echocardiography provides additional evidence for left ventricle dysfunction. Data are expressed as mean ± SEM. A The high-dose group exhibited a striking reduction in cardiac output at 16 weeks (60.95±6.7 mL/min) compared with the control group (194.17±35.09 mL/min). B There was no significant difference in heart rate between the three groups at 16 weeks (control 227±33 bpm, low-dose 236±45 bpm, high-dose 204±28 bpm) from baseline (data not shown). These results indicate that a reduction in stroke volume accounts for the decrease in high-dose cardiac output. C Representative M mode echocardiograms (transverse view) of the left ventricle of a rat from the control group (left) and from the high-dose group (right). The diastolic diameters are similar. D The left ventricle dysfunction was confirmed by a significant transcriptional increase in atrial natriuretic peptide in the high-dose group compared with the control group, determined by real-time PCR. *P<0.05; ***P<0.001
Molecular markers that could potentially identify subtle LV function alterations caused by the low DOXO dosages were also examined. Two parameters were investigated; mitochondrial activity in the experimentally treated rats using permeabilized heart fibres instead of isolated mitochondria and transcriptional levels of the E3 ligases of the UPS.
The mitochondrial oxygen consumption profile was consistent with DOXO-induced damage in the mitochondrial complex I respiratory system
The oxygen consumption rates of saponin-permeabilized cardiac fibres of experimental animals were measured in an oxygraph using high-resolution respirometry. Figure 6A presents a representative oxygen flux experiment using saponin-permeabilized cardiac tissue from one control animal to follow the reaction on the addition of substrates. In the oxygraph chamber, PMG was added to the medium as a substrate for complex I in the electron transport system (ETS). Next, 1 mM ADP, which enhances the respiration rate to produce ATP, was added to the reaction medium and was observed to increase the flux of oxygen consumption, as expected in well-coupled mitochondria. Further increases in the respiration rate were observed when 10 mM succinate, a substrate of complex II in the ETS, and FCCP, an uncoupler of oxidative phosphorylation that enhances the respiration rate, were added to the medium in sequence. Following these experiments, 1 μM rotenone produced a decrease in the rate of oxygen consumption, due to its inhibition of complex I in the ETS. NaN3 was added next, and a further decrease in the respiration rate – because of the inhibition of ETS complex IV – was observed, as expected.
Figure 6).
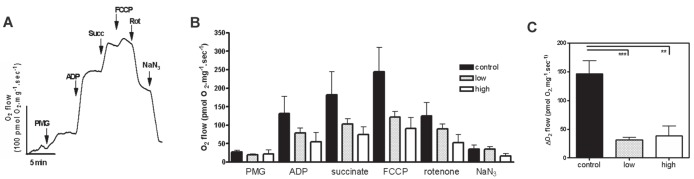
Doxorubicin (DOXO) treatment alters the mitochondrial oxygen consumption profile of rat heart tissue. A A representative oxygen flux experiment using saponin-permeabilized cardiac tissue from a control animal that was used to generate a normal response curve after administration of the substrates that increase the consumption of oxygen (pyruvate/malate/glutamate [PMG], ADP and succinate [Succ] and the uncoupling agent FCCP) followed by inhibitors that reduce oxygen consumption (rotenone [rot] and azide [NaN3]). B The bars represent the mean oxygen consumption that was measured in the control, low- and high-dose groups after each intervention. Data presented as mean ± SEM of three animals. Compared with controls, the low- and high-dose groups displayed a statistically insignificant reduction in the absolute values of oxygen consumption. C The graph depicts the change (Δ) in oxygen consumption at the rate of maximal respiration when using FCCP followed by rotenone inhibition of complex I. Both treatments induce a decrease in complex I activity in the DOXO groups compared with the control group. Fibres were used at 1 mg/mL to 5 mg/mL in the chamber. **P<0.01; ***P<0.005
Figure 6B shows the mean oxygen consumption rate that was measured in the cardiac fibres of experimental animals from the control, low- and high-dose DOXO treatment groups. Oxygen consumption in the presence of PMG was not significantly different between the DOXO treatments compared with control animals. The addition of ADP enhanced the respiration rate to produce ATP in all three groups without statistically significant differences among them. However, in low-dose and high-dose groups, this increase in the respiration rate was somewhat attenuated compared with the control group. The same trend was still observed after the addition of succinate and FCCP.
Surprisingly, when rotenone was added, the low- and high-dose DOXO groups did not respond with reduced oxygen consumption in the same range as the control group. This statistically significant difference demonstrated that animals from the control group were more sensitive to this drug than the low- or high-dose DOXO treatment groups. The further addition of NaN3 did not alter the rates of respiration.
The different sensitivity to rotenone between the control groups and the groups treated with DOXO is indicated in Figure 6C. This figure shows the inhibition of rotenone-sensitive oxygen consumption in animals from the control and DOXO treatment groups, after reaching the maximum respiration rate by the addition of the uncoupling drug FCCP. Tissue from control animals displayed the expected pattern of rotenone inhibition, with a 50% decrease in oxygen consumption using both complex I and II substrates (Figure 6C). However, tissues from both DOXO experimental groups displayed a lower inhibition response. The statistically significant difference between control and DOXO-treated animals observed after rotenone addition is consistent with DOXO-induced damage to the mitochondrial complex I respiratory system (Figure 6C).
DOXO cardiotoxicity is correlated with mRNA expression changes in the UPS
To determine whether cardiac dysfunction was correlated with changes in the UPS, gene expression of the major cardiac E3 ligases (MuRF1, MuRF2, MuRF3 and MAFbx) was analyzed after low-dose (7.5 mg/kg) DOXO administration but before evidence of LV systolic dysfunction onset. Gene expression was also analyzed 16 weeks after the first cycle of administration, following the development of LV systolic dysfunction in the hearts of the high-dose group. This evaluation was an attempt to detect early evidence of heart tissue dysfunction in the low-dose group and changes associated with chronic cardiac toxicity in the high-dose group.
The expression pattern of the E3 ligases exhibited remarkable changes corresponding to the dose of DOXO administration (Figure 7). MuRF1 displayed a dose-independent upregulation with a 1.6- to 1.8-fold change (P<0.001). For MuRF2 and MuRF3, an opposing pattern was observed, with downregulation in animals in the low-dose group (MuRF2 0.8-fold change; P<0.001 and MuRF3 0.6-fold change; P<0.001) and upregulation in animals in the high-dose group (1.4-fold change; P<0.01 for both). MAFbx mRNA levels exhibited a striking reduction at both doses (0.2- and 0.1-fold changes for the low- and high-dose groups, respectively; P<0.001).
Figure 7).
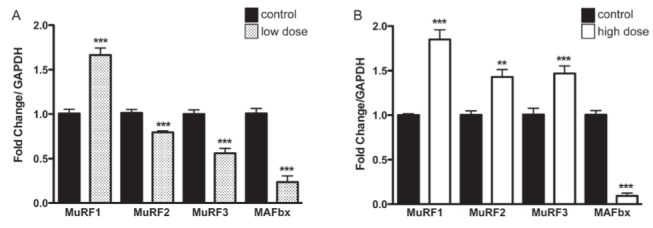
Messenger RNA expression profile of the E3 ligases in (A) the low-dose group, and (B) the high-dose group compared with the control group, determined by real-time polymerase chain reaction. The changes in muscle ring finger (MuRF)1 and MAFbx messenger RNA expression in the low-dose group were also observed in the high-dose group (MuRF1 induction and muscle atrophy F-box [MAFbx] repression). In contrast, the expression of MuRF2 and MuRF3 was suppressed in the low-dose group and induced in the high-dose group. **P<0.01; ***P<0.005
DISCUSSION
Cardiac remodelling after an injury involves a wide range of molecular rearrangements that differ from daily regulation in response to variations in hemodynamic load. These changes include variations in the mitochondrial respiration rate and the UPS, the alteration of which has been described in animal models and in patients with heart failure (HF).
Our approach used serial echocardiographic analysis of a rat model of chronic DOXO exposure that included two groups: a low-dose group that received a total cumulative dose of 7.5 mg/kg of DOXO; and a high-dose group that received a total cumulative dose of 20 mg/kg DOXO. Previous studies have also reproduced the cardiac injury caused by DOXO in small animals. The vast majority of these studies have focused on the EF and/or on fractional shortening (FS) measurements as an indicator of HF. This type of analysis has been performed to mirror the current assessment of human patients who receive DOXO to treat neoplasms. Based on the echocardiographical criteria, HF in animal models has been mainly characterized as a statistically significant reduction of EF or FS after DOXO administration. The current guidelines indicate a cutoff value of less than 55% for EF and less than 28% for FS to distinguish normal heart function from HF in humans. This approach, however, is limited by low detection sensitivity to the incipient and progressive worsening of heart function that can follow DOXO toxicity. Although it is a reliable indicator of HF, a clear reduction in EF/FS is a late-stage indicator of cardiotoxicity (39). Therefore, a search for subtle echocardiographic signs of heart damage that progress to HF is necessary.
The measurement of EF and FS were acquired from the systolic and diastolic diameters of the LV. Increases in both parameters have been described in several publications analyzing cardiac dysfunction following DOXO administration in rodents, primarily using doses varying from 15 mg/kg to 25 mg/kg (40–44). Only a few of these studies described a significant augmentation of the systolic LV diameter preceding diastolic augmentation (45–48). The results of the present study clearly demonstrate that animals in the high-dose group presented a significant EF reduction after 16 weeks of drug administration, and the reduction was caused by an isolated increase of the systolic diameter of the LV. These systolic diameter changes preceding diastolic enlargement were probably not detected in many previous studies because the measurements were acquired at later timepoints.
In contrast to the pattern of LV function findings detected by echocardiography, mitochondrial impairment was detected in the heart tissues of animals in the low-dose group as well as in the heart tissues of the high-dose animals. Moreover, the cardiomyocyte bioenergetics studies that were performed demonstrated noteworthy inhibition of NADH dehydrogenase complex activity.
Electron transport chain impairment has also been observed in previous studies using submitochondrial preparations from bovine heart tissue that was subjected to different concentrations of DOXO (49). In another group of experiments using rat heart tissue, it was observed that treatment with DOXO promoted a reduction in the respiratory control ratio, complex IV maximal respiration and cytochrome c oxidase activity (50). The authors suggested that the mitochondrial dysfunction induced by DOXO was likely caused by the formation of free radicals by the anthracycline in vivo at clinical concentrations (50). In fact, the exposure of rats to a cumulative dose of 15 mg/kg DOXO induced an early decrement in activity and protein levels of glutathione peroxidase and manganese superoxide dismutase in heart tissue (51). Both antioxidant enzymes are present in the mitochondrial matrix and protect mitochondrial components from oxidative damage. This could be related to the respiratory chain derangement observed even in the absence of evidence of functional LV damage in the low-dose group.
To preserve the stoichiometry of the internal membrane protein complexes, we studied respiration in permeabilized fibres instead of in isolated mitochondria, as proposed by Picard et al (52). Our experimental data are in agreement with previous observations, including a reduction of approximately 70% in rotenone-sensitive respiration in heart tissues from animals that were subjected to low and high doses of DOXO.
It has been reported that cardiac dysfunction is accompanied by a metabolic shift from fatty acid β-oxidation (FAO) to glucose oxidation (53,54). For the DOXO low-dose group that did not display systemic or echocardiographical evidence of toxicity, the increase in glycolytic flux should be able to compensate for the demand for ATP, as observed in the grey or hypoxic area surrounding infarcted heart tissue with mild to moderate ischemia that matches the similar rate of ATP production levels detected in healthy hearts under aerobic resting conditions (55,56). In this case, glycolysis should be one of the major upregulated energy-generating pathways to compensate for the energy deficit.
Another example of glycolytic cardiac adaptation comes from the observations of individuals who live in oxygen-deprived environments at high altitudes, such as the Quechuas in Peru. Glucose has been demonstrated to be the major fuel substrate supplying ATP demand in Quechuas’ hearts (57,58).
For animals that received higher doses of DOXO, this adaptation leads to a higher myocardial lipid accumulation that ultimately leads to cardiomyopathy, myocyte apoptosis and death. These characteristics were also shared by children with deficiencies in the enzymes involved in mitochondrial long-chain FAO (59) and also in mouse models in which the FAO enzyme very-long-chain acyl-CoA dehydrogenase has been disrupted (60,61). The authors speculated that the phosphocreatine/ATP ratio and absolute ATP levels were likely reduced in this situation, as observed in HF (53).
These results suggest that mitochondrial impairment of the heart represents the subacute effects of exposure to DOXO that precede the LV systolic commitment and may be a useful prognostic parameter. Interestingly, the different stages of heart function decline observed in the echocardiograms are accurately reflected by a differential transcriptional regulation profile for ubiquitin E3 ligases in low- and high-dose-treated animals.
Literature evidence suggests that the interplay between the UPS and heart pathophysiology is highly complex and specific to particular stress conditions or types of damage. MuRF1, MuRF2, MuRF3 and MAFbx interact with proteins of the contractile apparatus of cardiomyocytes and are clearly involved in cardiac development and remodelling. Although several lines of evidence indicate that suppression of the UPS and downregulation of E3 ligase expression can prevent cardiovascular disease progression, other studies suggest that UPS activation is essential for heart homeostasis (62). An example of this apparent paradox is that increased expression of MuRF1 has been associated with prevention of cardiac hypertrophy (63), but this increased expression is also an important factor that potentiates HF in a hypertrophic model in response to transaortic constriction (26). Therefore, the complex E3 ligase gene expression profile induced by DOXO, as demonstrated in the present study, may be a consequence of the intricate relationship between UPS activity and cardiac homeostasis. Moreover, differences in the E3 ligase profile observed in the present work may be interpreted as a consequence of distinct types of damage to cardiomyocytes that were caused by the DOXO treatment regimen.
The high-dose group, which exhibited clear cardiac functional alterations and mitochondrial dysfunction, also displayed upregulation of MuRF1, MuRF2 and MuRF3 E3 ligases. This massive activation of UPS components is consistent with a recent proteome analysis that was conducted using rabbits treated with an equivalent high-dose chronic daunorubicin treatment (3 mg/kg weekly for 10 weeks) (64). Differences in protein expression in LV cells from control and anthracycline-treated animals revealed an upregulation of proteasome subunits. Furthermore, UPS activation was confirmed by an increase in protein ubiquitination and trypsin-like activity. The same study also described the downregulation of several components of the respiratory pathway and upregulation of chaperones and proteins related to oxidative stress detoxification. These modifications suggest that the cardiomyocytes are under intense oxidative stress and that the UPS is acting with other components of protein homeostasis pathways in the degradation of misfolded, oxidized proteins (64). Conversely, the downregulation of MuRF2, MuRF3 and MAFbx suggests that a different scenario is occurring in the early stages of low-dose DOXO toxicity. An indication that low doses can induce a different cell response came from a study on mouse hearts that were perfused with 2 μM of DOXO for 2 h (65). This represents the lower limit of DOXO concentration found in the plasma of patients after bolus injection of the drug. The authors observed an overall decrease in gene expression, particularly in transcripts involved in cardiac stress response and stress signalling. In contrast, this experiment revealed upregulation of genes that are controlled by c-Myc, a transcription factor with activity that has been associated with the hypertrophic response in the heart. In this case, the downregulation of E3 ubiquitin ligases may be a consequence of this early hypertrophic response.
Interestingly, MuRF1 binds creatine kinase (CK), which mediates regeneration of ATP through the phosphotransferase pathway, thereby leading to its ubiquitination and possible degradation (66–69). In fact, CK downregulation was consistently reported in experiments involving single low-dose (65,70,71) or chronic anthracycline treatments (70), both at the transcriptional and protein level (64). Moreover, DOXO also perturbed the association of CK with the plasma membrane, which suggests that post-translational control is involved and could be mediated by ubiquitination (70).
We report that animals that received the low- or high-dose of DOXO exhibited an increase in MuRF1 mRNA expression that may result in downregulation of CK activity, consistent with reports that MuRF1 Tg+ mouse hearts exhibit a 20% to 25% decrease in total CK activity in vitro. These experiments also demonstrated that despite decreased CK activity, the level of total ATP did not significantly differ compared with wild-type animals.
In contrast, we found that animals in the low-dose group exhibited a reduction in MuRF2 and MuRF3 gene expression, while the high-dose group exhibited the inverse response, with an increase in MuRF2 and MuRF3 mRNA expression being observed.
We propose that in the low-dose animals, the imbalance in the CK shuttle provoked by an increase in MuRF1 is not enough to lower the amount of ATP necessary for the heart to work. The increase in MuRF2 and MuRF3, as detected in the high-dose DOXO group, could cause additional degradation of CK that may lead to a more pronounced decrease in ATP levels, provoking the cardiac damage that was observed by functional echocardiography analysis and consistent with having insufficient ATP levels for heart function.
We suggest that the specific profile observed for MuRF2 and MuRF3 transcripts could be useful as an early physiological and energetic cardiac failure indicator for testing therapeutic interventions in animal models. In the future, we will explore whether this difference can also be detected in skeletal muscle, where these proteins are also expressed, serving as a mirror of heart tissue regulation. It must be considered that DOXO is also able to alter proteasome activity by other mechanisms, independent of gene expression modulation. For example, direct binding of DOXO to the proteolysis apparatus has been demonstrated in mouse lymphocytic leukemia cells (72,73) and murine cardiomyocyte cultured cells (20).
The present study contributes to the understanding of how the UPS is associated with heart damage caused by DOXO administration. These changes demonstrate the dynamic aspect of the components of the UPS that have essential roles in heart homeostasis and are associated with the progression of cardiac dysfunction.
Acknowledgments
The present work was supported by grants from the Conselho Nacional de Desenvolvimento Científico e Tecnológico and the Fundação de Amparo a Pesquisa do Rio de Janeiro. The authors thank Dr Deivid Rodrigues and Luisa Hoffman (Universidade Federal do Rio de Janeiro, Instituto de Biofísica Carlos Chagas Filho) for help with the qPCR experiments and Reinaldo Santos for help with the oxymetry experiments (Universidade Federal do Rio de Janeiro, Instituto de Bioquímica Médica). The authors have no conflicts of interest to declare regarding the present study.
REFERENCES
- 1.Bonadonna G, Monfardini S, De Lena M, Fossati-Bellani F. Clinical evaluation of adriamycin, a new antitumour antibiotic. Br Med J. 1969;3:503–6. doi: 10.1136/bmj.3.5669.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carvalho C, Santos RX, Cardoso S, et al. Doxorubicin: The good, the bad and the ugly effect. Curr Med Chem. 2009;16:3267–85. doi: 10.2174/092986709788803312. [DOI] [PubMed] [Google Scholar]
- 3.Coleman MP, et al. Research commissioned by Cancer Research UK, 2010 < http://info.cancerresearchuk.org/cancerstats/survival/latestrates/index.htm> (Accessed September 6, 2010)
- 4.Bramwell VH, Anderson D, Charette ML. Doxorubicin-based chemotherapy for the palliative treatment of adult patients with locally advanced or metastatic soft-tissue sarcoma: A meta-analysis and clinical practice guideline. Sarcoma. 2000;4:103–12. doi: 10.1080/13577140020008066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Henderson IC, Allegra JC, Woodcock T, et al. Randomized clinical trial comparing mitoxantrone with doxorubicin in previously treated patients with metastatic breast cancer. J Clin Oncol. 1989;7:560–71. doi: 10.1200/JCO.1989.7.5.560. [DOI] [PubMed] [Google Scholar]
- 6.Bristow MR, Mason JW, Billingham ME, Daniels JR. Dose-effect and structure-function relationships in doxorubicin cardiomyopathy. Am Heart J. 1981;102:709–18. doi: 10.1016/0002-8703(81)90096-x. [DOI] [PubMed] [Google Scholar]
- 7.Takemura G, Fujiwara H. Doxorubicin-induced cardiomyopathy: From the cardiotoxic mechanisms to management. Prog Cardiovasc Dis. 2007;49:330–52. doi: 10.1016/j.pcad.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 8.Berthiaume JM, Wallace KB. Persistent alterations to the gene expression profile of the heart subsequent to chronic doxorubicin treatment. Cardiovasc Toxicol. 2007;7:178–91. doi: 10.1007/s12012-007-0026-0. [DOI] [PubMed] [Google Scholar]
- 9.Davies KJ, Doroshow JH, Hochstein P. Mitochondrial NADH dehydrogenase-catalyzed oxygen radical production by adriamycin, and the relative inactivity of 5-iminodaunorubicin. FEBS Lett. 1983;153:227–30. doi: 10.1016/0014-5793(83)80153-7. [DOI] [PubMed] [Google Scholar]
- 10.Doroshow JH, Locker GY, Myers CE. Enzymatic defenses of the mouse heart against reactive oxygen metabolites: Alterations produced by doxorubicin. J Clin Invest. 1980;65:128–35. doi: 10.1172/JCI109642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bugger H, Guzman C, Zechner C, Palmeri M, Russell KS, Russell RR., III Uncoupling protein downregulation in doxorubicin-induced heart failure improves mitochondrial coupling but increases reactive oxygen species generation. Cancer Chemother Pharmacol. 2011;67:1381–8. doi: 10.1007/s00280-010-1441-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chandran K, Aggarwal D, Migrino RQ, et al. Doxorubicin inactivates myocardial cytochrome c oxidase in rats: Cardioprotection by Mito-Q. Biophys J. 2009;96:1388–98. doi: 10.1016/j.bpj.2008.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wallace KB. Doxorubicin induced cardiac mitochondrionopathy. Pharmacol Toxicol. 2003;93:105–15. doi: 10.1034/j.1600-0773.2003.930301.x. [DOI] [PubMed] [Google Scholar]
- 14.Kiyomiya K, Kurebe M, Nakagawa H, Matsuo S. The role of the proteasome in apoptosis induced by anthracycline anticancer agents. Int J Oncol. 2002;20:1205–9. [PubMed] [Google Scholar]
- 15.Wallace KB. Adriamycin-induced interference with cardiac mitochondrial calcium homeostasis. Cardiovasc Toxicol. 2007;7:101–7. doi: 10.1007/s12012-007-0008-2. [DOI] [PubMed] [Google Scholar]
- 16.Poizat C, Sartorelli V, Chung G, Kloner RA, Kedes L. Proteasome-mediated degradation of the coactivator p300 impairs cardiac transcription. Mol Cell Biol. 2000;20:8643–54. doi: 10.1128/mcb.20.23.8643-8654.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumarapeli AR, Horak KM, Glasford JW, et al. A novel transgenic mouse models reveals deregulation of the ubiquitin-proteasome system in the heart by doxorubicin. FASEB J. 2005;19:2051–3. doi: 10.1096/fj.05-3973fje. [DOI] [PubMed] [Google Scholar]
- 18.Ito T, Fujio Y, Takahashi K, Azuma J. Degradation of NFAT5, a transcriptional regulator of osmotic stress-related genes, is a critical event for doxorubicin-induced cytotoxicity in cardiac myocytes. J Biol Chem. 2007;282:1152–60. doi: 10.1074/jbc.M609547200. [DOI] [PubMed] [Google Scholar]
- 19.Wang X, Su H, Ranek MJ. Protein quality control and degradation in cardiomyocytes. J Mol Cell Cardiol. 2008;45:11–27. doi: 10.1016/j.yjmcc.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu J, Zheng H, Tang M, Ryu YC, Wang X. A therapeutic dose of doxorubicin activates ubiquitin-proteasome system-mediated proteolysis by acting on both the ubiquitination apparatus and proteasome. Am J Physiol Heart Circ Physiol. 2008;295:H2541–50. doi: 10.1152/ajpheart.01052.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Su H, Wang X. The ubiquitin-proteasome system in cardiac proteinopathy: A quality control perspective. Cardiovasc Res. 2010;85:253–62. doi: 10.1093/cvr/cvp287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng Q, Wang X. Autophagy and the ubiquitin-proteasome system in cardiac dysfunction. Panminerva Med. 2010;52:9–25. [PMC free article] [PubMed] [Google Scholar]
- 23.Wang X, Robbins J. Heart failure and protein quality control. Circ Res. 2006;99:1315–28. doi: 10.1161/01.RES.0000252342.61447.a2. [DOI] [PubMed] [Google Scholar]
- 24.Zolk O, Schenke C, Sarikas A. The ubiquitin-proteasome system: Focus on the heart. Cardiovasc Res. 2006;70:410–21. doi: 10.1016/j.cardiores.2005.12.021. [DOI] [PubMed] [Google Scholar]
- 25.Mearini G, Schlossarek S, Willis MS, Carrier L. The ubiquitin-proteasome system in cardiac dysfunction. Biochim Biophys Acta. 2008;1782:749–63. doi: 10.1016/j.bbadis.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 26.Willis MS, Schisler JC, Li L, et al. Cardiac muscle ring finger-1 increases susceptibility to heart failure in vivo. Circ Res. 2009;105:80–8. doi: 10.1161/CIRCRESAHA.109.194928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rodriguez JE, Schisler JC, Patterson C, Willis MS. Seek and destroy: The ubiquitin-proteasome system in cardiac disease. Curr Hypertens Rep. 2009;11:396–405. doi: 10.1007/s11906-009-0069-7. [DOI] [PubMed] [Google Scholar]
- 28.Razeghi P, Baskin KK, Sharma S, et al. Atrophy, hypertrophy and hypoxemia induce transcriptional regulators of the ubiquitin proteasome system in the rat heart. Biochem Biophys Res Commun. 2006;342:361–4. doi: 10.1016/j.bbrc.2006.01.163. [DOI] [PubMed] [Google Scholar]
- 29.Depre C, Wang Q, Yan L, et al. Activation of the cardiac proteasome during pressure overload promotes ventricular hypertrophy. Circulation. 2006;114:1821–8. doi: 10.1161/CIRCULATIONAHA.106.637827. [DOI] [PubMed] [Google Scholar]
- 30.Meiners S, Dreger H, Fechner M, et al. Suppression of cardiomyocyte hypertrophy by inhibition of the ubiquitin-proteasome system. Hypertension. 2008;51:302–8. doi: 10.1161/HYPERTENSIONAHA.107.097816. [DOI] [PubMed] [Google Scholar]
- 31.Predmore JM, Wang P, Davis F, et al. Ubiquitin proteasome dysfunction in human hypertrophic and dilated cardiomyopathies. Circulation. 2010;121:997–1004. doi: 10.1161/CIRCULATIONAHA.109.904557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu J, Tang M, Mestril R, Wang X. Aberrant protein aggregation is essential for a mutant desmin to impair the proteolytic function of the ubiquitin-proteasome system in cardiomyocytes. J Mol Cell Cardiol. 2006;40:451–4. doi: 10.1016/j.yjmcc.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 33.Yamamoto Y, Hoshino Y, Ito T, et al. Atrogin-1 ubiquitin ligase is upregulated by doxorubicin via p38-MAP kinase in cardiac myocytes. Cardiovasc Res. 2008;79:89–96. doi: 10.1093/cvr/cvn076. [DOI] [PubMed] [Google Scholar]
- 34.Lang RM, Bierig M, Devereux RB, et al. American Society of Echocardiography’s Nomenclature and Standards Committee. Task Force on Chamber Quantification. American College of Cardiology Echocardiography Committee. American Heart Association. European Association of Echocardiography. European Society of Cardiology Recommendations for chamber quantification. Eur J Echocardiogr. 2006;7:79–108. doi: 10.1016/j.euje.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 35.Albin G, Rahko PS. Comparison of echocardiographic quantitation of left ventricular ejection fraction to radionuclide angiography in patients with regional wall motion abnormalities. Am J Cardiol. 1990;65:1031–2. doi: 10.1016/0002-9149(90)91009-u. [DOI] [PubMed] [Google Scholar]
- 36.Kuznetsov AV, Schneeberger S, Seiler R, et al. Mitochondrial defects and heterogeneous cytochrome c release after cardiac cold ischemia and reperfusion. Am J Physiol Heart Circ Physiol. 2004;286:H1633–41. doi: 10.1152/ajpheart.00701.2003. [DOI] [PubMed] [Google Scholar]
- 37.Livak K, Schimittgen T. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 38.Pfaffl MW. A new mathematical model for relative quantification in real time RT-PCR. Nucleics Acids Res. 2001;29:2002–7. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eidem BW. Identification of anthracycline cardiotoxicity: Left ventricular ejection fraction is not enough. J Am Soc Echocardiogr. 2008;21:1290–2. doi: 10.1016/j.echo.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 40.Karagoz B, Suleymanoglu S, Uzun G, et al. Hyperbaric oxygen therapy does not potentiate doxorubicin-induced cardiotoxicity in rats. Basic Clin Pharmacol Toxicol. 2008;102:287–92. doi: 10.1111/j.1742-7843.2007.00196.x. [DOI] [PubMed] [Google Scholar]
- 41.Das A, Durrant D, Mitchell C, et al. Sildenafil increases chemotherapeutic efficacy of doxorubicin in prostate cancer and ameliorates cardiac dysfunction. Proc Natl Acad Sci USA. 2010;107:18202–7. doi: 10.1073/pnas.1006965107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li L, Takemura G, Li Y, et al. Preventive effect of erythropoietin on cardiac dysfunction in doxorubicin-induced cardiomyopathy. Circulation. 2006;113:535–43. doi: 10.1161/CIRCULATIONAHA.105.568402. [DOI] [PubMed] [Google Scholar]
- 43.Weinstein DM, Mihm MJ, Bauer JA. Cardiac peroxynitrite formation and left ventricular dysfunction following doxorubicin treatment in mice. J Pharmacol Exp Ther. 2000;294:396–401. [PubMed] [Google Scholar]
- 44.Fujimura L, Matsudo Y, Kang M, Takamori Y, Tokuhisa T, Hatano M. Protective role of Nd1 in doxorubicin-induced cardiotoxicity. Cardiovasc Res. 2004;64:315–21. doi: 10.1016/j.cardiores.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 45.Teraoka KU, Hirano M, Yamaguchi K, Yamashina A. Progressive cardiac dysfunction in adriamycin-induced cardiomyopathy rats. Eur J Heart Fail. 2000;2:373–8. doi: 10.1016/s1388-9842(00)00111-2. [DOI] [PubMed] [Google Scholar]
- 46.Zhu W, Shou W, Payne MR, Caldwell R, Field LJ. A mouse model for juvenile doxorubicin-induced cardiac dysfunction. Pediatr Res. 2008;64:488–94. doi: 10.1203/PDR.0b013e318184d732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ohkura K, Lee JD, Shimizu H, et al. Mitochondrial complex I activity is reduced in latent adriamycin-induced cardiomyopathy of rat. Mol Cell Biochem. 2003;248:203–8. doi: 10.1023/a:1024161024231. [DOI] [PubMed] [Google Scholar]
- 48.Toko H, Oka T, Zou Y, et al. Angiotensin II type 1a receptor mediates doxorubicin-induced cardiomyopathy. Hypertens Res. 2002;25:597–603. doi: 10.1291/hypres.25.597. [DOI] [PubMed] [Google Scholar]
- 49.Marcillat O, Zhang Y, Davies KJA. Oxidative and non-oxidative mechanisms in the inactivation of cardiac mitochondrial electron transport chain components by doxorubicin. Biochem J. 1989;259:181–9. doi: 10.1042/bj2590181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Montaigne D, Marechal X, Baccouch R, et al. Stabilization of mitochondrial membrane potential prevents doxorubicin-induced cardiotoxicity in isolated rat heart. Toxicol Appl Pharmacol. 2010;244:300–7. doi: 10.1016/j.taap.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 51.Singal PK, Li T. Adriamycin-induced early changes in myocardial antioxidant enzymes and their modulation by probucol. Circulation. 2000;102:2105–10. doi: 10.1161/01.cir.102.17.2105. [DOI] [PubMed] [Google Scholar]
- 52.Picard M, Taivassalo T, Ritchie D, et al. Mitochondrial structure and function are disrupted by standard isolation methods. PLoS One. 2011;6:8–10. doi: 10.1371/journal.pone.0018317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huss JM, Kelly DP. Mitochondrial energy metabolism in heart failure: A question of balance. J Clin Invest. 2005;115:547–55. doi: 10.1172/JCI200524405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hansson A, Hance N, Dufour E, et al. A switch in metabolism precedes increased mitochondrial biogenesis in respiratory chain-deficient mouse hearts. PNAS. 2004;101:3136–41. doi: 10.1073/pnas.0308710100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Newsholme EA, Leech AR. Biochemistry for the medical sciences. Chichester: J. Wiley; 1983. pp. 232–3. [Google Scholar]
- 56.Pantely GA, Malone SA, Rhen WS, et al. Regeneration of myocardial phosphocreatine in pigs despite continued moderate ischemia. Circ Res. 1990;67:1481–93. doi: 10.1161/01.res.67.6.1481. [DOI] [PubMed] [Google Scholar]
- 57.Holden JE, Stone CK, Clark CM, et al. Enhanced cardiac metabolism of plasma glucose in high altitude natives: Adaptation against chronic hypoxia. J Appl Physiol. 1995;79:222–8. doi: 10.1152/jappl.1995.79.1.222. [DOI] [PubMed] [Google Scholar]
- 58.Rupert JL, Hochachka PW. The evidence for hereditary factors contributing to high altitude adaptation in Andean Natives: A review. High Alt Med Biol. 2001;2:235–56. doi: 10.1089/152702901750265332. [DOI] [PubMed] [Google Scholar]
- 59.Kelly DP, Strauss AW. Inherited cardiomyopathies. N Engl J Med. 1994;330:913–9. doi: 10.1056/NEJM199403313301308. [DOI] [PubMed] [Google Scholar]
- 60.Exil VJ, Roberts RL, Sims H, et al. Very-long-chain acyl-coenzyme a dehydrogenase deficiency in mice. Circ Res. 2003;93:448–55. doi: 10.1161/01.RES.0000088786.19197.E4. [DOI] [PubMed] [Google Scholar]
- 61.Kurtz DM, Rinaldo P, Rhead WJ, et al. Targeted disruption of mouse long-chain acyl-CoA dehydrogenase gene reveals crucial roles for fatty acid oxidation. Proc Natl Acad Sci USA. 1998;95:15592–7. doi: 10.1073/pnas.95.26.15592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li YF, Wang X. The role of the proteasome in heart disease. Biochim Biophys Acta. 2011;1809:141–9. doi: 10.1016/j.bbagrm.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Willis MS, Ike C, Li L, Wang DZ, Glass DJ, Patterson C. Muscle ring finger 1, but not muscle ring finger 2, regulates cardiac hypertrophy in vivo. Circ Res. 2007;100:456–9. doi: 10.1161/01.RES.0000259559.48597.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sterba M, Popelová O, Lenco J, et al. Proteomic insights into chronic anthracycline cardiotoxicity. J Mol Cell Cardiol. 2011;50:849–62. doi: 10.1016/j.yjmcc.2011.01.018. [DOI] [PubMed] [Google Scholar]
- 65.Tokarska-Schlattner M, Lucchinetti E, Zaugg M, et al. Early effects of doxorubicin in perfused heart: Transcriptional profiling reveals inhibition of cellular stress response genes. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1075–88. doi: 10.1152/ajpregu.00360.2009. [DOI] [PubMed] [Google Scholar]
- 66.Witt SH, Granzier H, Witt CC, Labeit S. MuRF1 and MuRF2 target a specific subset of myofibrilar proteins redundantly: Towards understanding MuRF-dependent muscle ubiquitination. J Mol Biol. 2005;350:713–22. doi: 10.1016/j.jmb.2005.05.021. [DOI] [PubMed] [Google Scholar]
- 67.Willis MS, Townley-Tilson D, Kang EY, Homeister JW, Patterson C. Sent to destroy: The ubiquitin proteasome system regulates cell signalling and protein quality control in cardiovascular development and disease. Circ Res. 2010;106:463–78. doi: 10.1161/CIRCRESAHA.109.208801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Willis MS, Zungu M, Patterson C. Cardiac muscle ring finger-1 – friend or foe? Trends Cardiovasc Med. 2010;20:12–6. doi: 10.1016/j.tcm.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhao TJ, Yan YB, Liu Y, Zhou HM. The generation of the oxidized form of creatine kinase is a negative regulation on muscle creatine kinase. J Biol Chem. 2007;282:12022–9. doi: 10.1074/jbc.M610363200. [DOI] [PubMed] [Google Scholar]
- 70.Tokarska-Schlattner M, Wallimann T, Schlattner U. Multiple interference of anthracyclines with mitochondrial creatine kinases: Preferential damage of the cardiac isoenzyme and its implications for drug cardiotoxicity. Mol Pharmacol. 2002;61:516–23. doi: 10.1124/mol.61.3.516. [DOI] [PubMed] [Google Scholar]
- 71.Tokarska-Schlattner M, Dolder M, Gerber I, Speer O, Wallimann T, Schlattner U. Reduced creatine-stimulated respiration in doxorubicin challenged mitochondria: Particular sensitivity of the heart. Biochim Biophys Acta. 2007;1767:1276–84. doi: 10.1016/j.bbabio.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 72.Kiyomiya K, Matsuo S, Kurebe M. Proteasome is a carrier to translocate doxorubicin from cytoplasm into nucleus. Life Sci. 1998;62:1853–60. doi: 10.1016/s0024-3205(98)00151-9. [DOI] [PubMed] [Google Scholar]
- 73.Kiyomiya K, Satoh J, Horie H, Kurebe M, Nakagawa H, Matsuo S. Correlation between nuclear action of anthracycline anticancer agents and their binding affinity to the proteasome. Int J Oncol. 2002;21:1081–5. [PubMed] [Google Scholar]