

Abstract
Although diabetes due to insulin deficiency or insulin resistance is a major cause of heart disease, the pathogenesis of cardiac dysfunction during the development of diabetic cardiomyopathy is not fully understood. Varying degrees of defects in subcellular organelles, such as sarcolemma, mitochondria, sarcoplasmic reticulum, myofibrils and extracellular matrix have been observed in the diabetic heart. These subcellular abnormalities in chronic diabetes become evident with the occurrence of hormonal imbalance, metabolic defects, oxidative stress and intracellular Ca2+ overload. During the initial stages of diabetes, hormonal imbalances, including elevated plasma levels of catecholamines and angiotensin II, as well as metabolic defects, appear to favour the development of oxidative stress; these changes lead to subcellular defects in the myocardium. Reductions in sarcoplasmic reticular Ca2+ pump and Ca2+ release channel function are associated with cardiac dysfunction, whereas alterations in sarcolemmal Na+/Ca2+ exchanger and Na+/K+ ATPase activities contribute to intracellular Ca2+ overload at late stages of diabetes. The continued accumulation of Ca2+ in mitochondria produces Ca2+ overload in these organelles, and this change induces impairment of energy production and depletion of energy stores as well as further promotion of oxidative stress in chronic diabetes. Generation of oxyradicals due to impaired electron transport results in the opening of mitochondrial pores, leakage of toxic proteins and myocardial cell damage in diabetes. These observations support the view that alterations in sarcoplasmic reticular and mitochondrial functions produce intracellular Ca2+ overload and depletion of energy stores and, thus, play an important role in the development of cardiac dysfunction in diabetic cardiomyopathy.
Keywords: Diabetic cardiomyopathy, Mitochondria, Sarcoplasmic reticulum, Subcellular remodelling
Diabetes is one of the major metabolic diseases and affects a large number of people. It has been estimated that the prevalence of diabetes will increase from 2.8% in 2000 to 4.4% in 2030 (1). Diabetes is caused either by insulin deficiency, due to a lack of insulin production, or insulin resistance, in which the target cells no longer respond to insulin. These insulin-related abnormalities cause membrane defects that are associated with changes in cation permeability, especially for Ca2+. It is now well accepted that altered Ca2+ permeability results in defects in smooth muscle, endothelial cells, cardiac muscle and neuronal cells, which can lead to microangiopathy, atherosclerosis, cardiomyopathy and neuropathy, respectively (2). Ultimately, these processes induce local and generalized ischemia, contractile dysfunction, loss of sympathetic influences and, eventually, impaired cardiac performance (2–4). In epidemiological studies, diabetes is associated with an increased incidence of heart failure even after adjusting for the presence of hypertension and coronary artery disease (5). Although the exact reasons for cardiac dysfunction in diabetic cardiomyopathy are not clear, metabolic derangements with respect to excessive use of free fatty acids (FFAs) and the reduced use of glucose, as well as hormonal imbalances and subcellular defects due to insulin deficiency or insulin resistance have been suggested to account for structural defects in the diabetic heart (2,3). In addition, marked alterations in biochemical and functional activities of different subcellular organelles such as extracellular matrix, sarcolemma (SL), sarcoplasmic reticulum (SR), mitochondria and myofibrils have been identified in the heart under chronic diabetic conditions (2). Because of the role of the SR in Ca2+ handling and the role of mitochondria in energy production (3,6), the present review intends to focus on the mechanisms, as well as the significance, of SR and mitochondrial defects in diabetic cardiomyopathy. Because Ca2+ handling defects in cardiac membranes are invariably associated with the occurrence of intracellular Ca2+ overload (3,4,6), the involvement of this mechanism in the pathogenesis of diabetic cardiomyopathy will also be discussed briefly. The contribution of changes in SL, which maintains Ca2+ homeostasis, myofibrils, which serve as contractile machinery, and extracellular matrix, which controls the permeability of cations, during the development of diabetic cardiomyopathy have been described previously (3,7) and, thus, it is not our intention to deemphasize their role in diabetes-induced cardiac dysfunction.
CARDIAC SR DYSFUNCTION IN DIABETES
By virtue of its ability to release and accumulate Ca2+, SR are considered to play a major role in the process of excitation-contraction coupling in the myocardium. Voltage-gated L-type Ca2+ channels in the SL membrane are activated on depolarization of cardiomyocytes and permit the entry of Ca2+, which releases more Ca2+ from the SR Ca2+ stores through the ryanodine receptor (6). This Ca2+ reaches myofibrils, binds to troponin C, releases the inhibition of actomyosin by troponin I and triggers the sliding of thick and thin filaments resulting in cardiac contraction. The increased level of cytosolic Ca2+ is then lowered by the combined actions of the SR Ca2+ pump ATPase (SERCA2a), the SL Na+/Ca2+ exchanger and the SL Ca2+-stimulated ATPase, as well as the mitochondrial uniporter (6). It has been shown that SERCA2a is the main mediator for lowering the cytoplasmic concentration of Ca2+ and is regulated by a SR protein, phospholamban. Although Ca2+ is bound to calsequestrin in the lumen of SR, the formation of a quaternary complex of SR proteins (junction, triadin, calsequestrin and the ryanodine receptor) is believed to release Ca2+ from the SR (8). Thus, different SR proteins are involved in the accumulation, binding and release of Ca2+ from the SR tubules, and changes in their contents and activities appear to disturb Ca2+ homeostasis. In fact, a depression in the SR Ca2+-release activity would depress cardiac contraction, whereas a decrease in the SR Ca2+-uptake activity would impair cardiac relaxation (6).
A wide variety of alterations in the SR Ca2+-transport activities have been observed in hearts during the development of chronic diabetes induced by streptozotocin (2,3). Ganguly et al (9) reported that a decrease in Ca2+-uptake activity in SR vesicles was associated with a depression in SERCA2a activity; these changes in diabetic animals were prevented by insulin administration. Similar defects in the cardiac SR Ca2+-transport activity due to diabetes were shown by other investigators (10,11). Abnormalities in SR Ca2+release were also identified in the diabetic heart (12,13). By using an experimental model of alloxan-induced diabetes in rats, Golfman et al (14) showed that SR Ca2+-uptake and Ca2+-release activities were depressed in the diabetic heart. Furthermore, impaired Ca2+-uptake and Ca2+-release activities were seen in noninsulin-dependent diabetic hearts (15,16). A depression of SR Ca2+ release channels in diabetes was also evident from experiments using the rapid cooling myocardial contracture technique as well as ryanodine receptor binding studies (17). These observations suggest that SR function in the diabetic heart may be defective as well as contributory to depressed cardiac performance in chronic diabetes. Depression of the SR Ca2+-uptake activity for a prolonged period in chronic diabetes can also produce intracellular Ca2+ overload.
MECHANISMS OF SR DEFECTS
The decreased SR-transport activities in the diabetic heart may be related to alterations in protein content as well as regulatory mechanisms in the SR membrane. In this regard, decreases in Ca2+ uptake and Ca2+release were associated with depressions in the levels of SERCA2a, ryanodine receptor and phospholamban proteins in diabetic hearts (12,18,19), except in one study where phospholamban protien content was increased (12). While some investigators have also reported reductions in SR ryanodine receptor (20,21) and phospholamban protein content (20), others have shown no change in SERCA2a protein content (20–22) in the diabetic heart. The SERCA2a messenger RNA levels were either unaltered (21,22) or decreased (20), and messenger RNA levels for the ryanodine receptor were depressed (20,21), whereas those for phospholamban were either unaltered (20) or increased (21). The conflicting results with respect to gene and protein expressions for SERCA2a and phospholamban may be due to differences in the duration and severity of diabetes in these various studies. Differential changes in SR activities and protein content during the development of diabetes have also been described previously (14,23,24). Nonetheless, these observations support the view that alterations in SR function and SR remodelling occur in the diabetic heart (3). Remodelling of other subcellular organelles, including SL and myofibrils, has also been reported during the development of diabetic cardiomyopathy (3,4).
Impaired SR function in the diabetic heart is not only associated with depressions in the contents of Ca2+-cycling proteins but may also be due to a decrease in the phosphorylation of phospholamban (18,19,25). Although the activities of cyclic AMP-dependent protein kinase and Ca2+ dependent protein kinase were increased, the reduction in phospholamban phosphorylation was attributed to an increase in SR-associated protein phosphotase activity (25). The decrease in SR function in the diabetic heart has also been suggested to be due to an increase in the formation of advanced glycation end products (26). Because NADPH oxidase activation (27) and increased phosphotidyl inositol turnover (28) have been observed in diabetic hearts, it is likely that the depression in SR function may be due to oxidative modification of SR proteins and/or alterations in the phospholipid composition of the SR membrane, respectively. Furthermore, in view of the increase in the activities of proteolytic enzymes due to hyperglycemia and diabetes (7,29), alterations in SR function may be a consequence of proteolysis of membrane proteins. The functional significance of changes in SR proteins is suggested from observations indicating that depressed levels of SERCA2a are associated with reduced cardiac performance in the diabetic heart (30,31). In addition, overexpression of SERCA2a in transgenic mice was found to improve cardiac function in diabetic cardiomyopathy (32).
DEVELOPMENT OF INTRACELLULAR Ca2+ OVERLOAD
Several investigators have examined diabetes-induced changes in the intracellular concentration of free Ca2+ ([Ca2+]i) in cardiomyocytes as well as total Ca2+ content in the myocardium. The basal (Ca2+)i was observed to be either unaltered (33), increased (34) or decreased (35–37) in cardiomyocytes from diabetic animals. Elevated levels of (Ca2+)i in the diabetic heart appear to be due to depression of the SR Ca2+-uptake activity (9–11). On the other hand, the inability to detect the elevated levels of (Ca2+)i in diabetic cardiomyocytes may be due to accumulation of Ca2+ in mitochondria because these organelles are known to serve as Ca2+ sinks in pathological conditions (6,38). Because (Ca2+)i in cardiomyocytes is maintained by Ca2+ influx and Ca2+ efflux at the SL level as well as Ca2+ release and Ca2+ uptake by both SR and mitochondria (38), it is possible that the observed differences in the results for (Ca2+)i in the diabetic heart from different laboratories may be due to differential changes in Ca2+-transport activities in these subcellular organelles. Such differences in the observed changes in (Ca2+)i may also be attributable to differences in the stage and severity of diabetes (3). It should be noted that depressed Ca2+ efflux (32,39) and SL Ca2+ pump ATPase activity (40,41) as well as increased Ca2+influx (28,42) and increased SL Ca2+/Mg2+ ecto-ATPase activity (3,43) in the diabetic heart would also raise (Ca2+)i and, thus, contribute to the occurrence of intracellular Ca2+ overload.
Increased Ca2+ permeability due to changes in the composition of SL membranes has been suggested to raise (Ca2+)i in diabetic myocardium (44). Furthermore, marked depressions in SL Na+/K+ ATPase activity (45–47) and SL Na+/Ca2+ exchanger activity (41,47–49) appear to play a major role in the development of intracellular Ca2+ overload in the diabetic heart. In this regard, a depression in SL Na+/K+ ATPase activity would increase the intracellular concentration of free Na+([Na+]i) and this is exactly what was reported in diabetic cardiac muscle (50). Because SL Na+/Ca2+ exchangers are considered to be involved in Ca2+ efflux under normal conditions (3,6), the observed depression of its activity in diabetic heart would decrease Ca2+ efflux and raise (Ca2+)i in cardiomyocytes. On the other hand, an increase in (Na+)i in diabetes (50) can be seen to enhance the entry of Ca2+ in cardiomyocytes through the stimulation of SL Na+/Ca2+ exchangers in reverse mode (3,6,38). In fact, a net gain of Ca2+ or increased level of total tissue Ca2+ has been demonstrated in the diabetic myocardium (15,16,51,52). It is noteworthy that interventions such as SL Ca2+ channel blockers and angiotensin receptor antagonists, which are known to attenuate the occurrence of intra-cellular Ca2+ overload, have been shown to partially prevent subcellular defects, cardiac dysfunction and ultrastructural damage in diabetic cardiomyopathy (13,53–55). These observations suggest that in addition to SR defects, alterations in SL Ca2+ cycling proteins may also participate in raising (Ca2+)i and inducing intracellular Ca2+ overload in the diabetic heart.
CARDIAC MITOCHONDRIAL DYSFUNCTION IN DIABETES
While defects in SR function may play a critical role in the development of impaired cardiac performance in chronic diabetes, abnormalities in other subcellular organelles, including mitochondria, have been identified to occur in the diabetic heart (2–4). It should be noted that the main function of mitochondria in the heart is to produce energy in the form of ATP, which is required for cardiac contractile activity. These organelles are known to be intimately involved in the process of glucose and FFA metabolism by cardiomyocytes, and their oxidative phosphorylation activity is generally impaired in different types of failing hearts (56–58). Defect in energy production are invariably associated with impairment of the electron transport system and the increased formation of oxyradicals in the mitochondria. Such events promote the development of oxidative stress, opening of mitochondrial pores, leakage of different mitochondrial proteins and the development of cardiomyocyte damage (2–4,16). In addition, mitochondria are known to serve as Ca2+ sinks in the cell and, when cardiomyocytes are faced with conditions of intracellular Ca2+ overload, it can result in Ca2+overload in the mitochondria, defecencies in the process of oxidation of phosphorylation and enhanced production of oxyradicals (4,6,38). Thus, it is essential to discuss the role of mitochondria in the process of substrate utilization and energy production, regulation of intracellular Ca2+ and the development of oxidative stress in the diabetic heart.
ENERGY PRODUCTION AND OXIDATION OF FFA
Although plasma levels of both glucose and FFA are elevated in diabetic subjects, glucose metabolism is markedly reduced due to impaired glucose transport in cardiomyocytes (2,16). Glucose uptake in the diabetic myocardium is also reduced due to the elevated levels of FFA (57–59). On the other hand, FFA uptake is increased in the diabetic heart and FFAs are either incorporated into triglycerides or catabolized in the mitochondria (58). The increase in fatty acid oxidation in the diabetic heart (60–62) has been demonstrated to be due to the upregulation of perioxisome proliferator activated receptor α (PPARα) (63–65). Thus, it is evident that energy production in the diabetic myocardium is primarily dependent on the oxidation of FFA by mitochondria; however, increased oxidation of FFAs for a prolonged period can impair the electron transport chain and mitochondrial oxidative phosphorylation activity. In fact, the respiratory and oxidative phosphorylation activities of cardiac mitochondria have been observed to be depressed in chronic diabetes (66–68). These changes in mitochondrial function were found to be associated with the depletion of high energy phosphate stores as well as depression in the performance of diabetic hearts (67,68).
Excessive lipid uptake and increased mitochondrial fatty acid oxidation are now well known to be associated with the accumulation of lipid droplets in the myocardium and the development of diabetic cardiomyopathy (2,16). In this regard, the upregulation of PPARα has been reported to play a critical role in mediating diabetes-induced lipotoxicity and pathological alterations in the myocardium (69–72). High levels of FFAs in the diabetic myocardium have been suggested to be intimately involved in the pathogenesis of cardiac cell damage as well as cardiac dysfunction (2,73). Such observations are further supported by the fact that palmitate and long-chain saturated fatty acids were found to induce apoptosis and cell death in cardiomyocytes (74,75). Accordingly, a shift in myocardial metabolism with respect to increased uptake, utilization and oxidation of FFAs may play an important role in the development of diabetic cardiomyopathy.
OXIDATIVE STRESS AND MITOCHONDRIAL ALTERATIONS
It is becoming clear that oxidative stress generated by different sources, including mitochondria, is a major factor in the pathogenesis of diabetic cardiomyopathy (3,4,76–80). Elevated levels of plasma glucose and advanced protein glycation end products have been shown to be involved in contributing toward generating oxidative stress in the diabetic heart (81–85). Increased levels of plasma hormones, such as angiotensin II, catecholamines and endothelins, have also been considered to promote oxidative stress in diabetic cardiomyopathy (3,86). Cardiac mitochondria have been reported to generate reactive oxygen species including superoxide radicals, hydroxyl radicals and hydrogen peroxide due to microangiopathy and subsequent hypoxia in chronic diabetes (3,82,87). Impaired insulin signalling as well as auto-oxidation of glucose has also been demonstrated to affect mitochondria and to promote the development of oxidative stress in the diabetic heart (88,89). Hyperglycemia-induced apoptosis in the diabetic heart has been shown to involve mitochondrial cytochrome C-activated caspase-3, as well as depression in the mitochondrial reduced glutathione content (90,91). In fact, normalizing mitochondrial superoxide production as well as overexpression have been observed to prevent hyperglycemia-induced cell damage (88,92). Furthermore, diabetic cardiomyopathy and cardiac dysfunction have been prevented by different antioxidants such as vitamin E, catalase and metallothionein (3,93,94). Taken together, the oxidative stress generated through the participation of mitochondria, as a consequence of hyperglycemia, excessive utilization of FFAs, impaired electron transport and oxidative phosphorylation processes, seems to be a crucial factor for the genesis of diabetic cardiomyopathy.
Ca2+ HANDLING BY MITOCHONDRIA
In view of the ability of mitochondria to accumulate large amounts of Ca2+, these organelles are known to prevent and/or delay the occurrence of intracellular Ca2+ overload in cardiomyocytes under different pathological conditions (6,38). During the development of cardiac dysfunction and intracellular Ca2+ overload in chronic diabetes, mitochondria are believed to continue accumulating Ca2+, serving as a protective mechanism (3). However, these organelles become overloaded with Ca2+ with time and, thus, their respiratory and oxidative phosphorylation activities are impaired in the diabetic heart (3). Different investigators have reported a depression in the mitochondrial Ca2+ uptake activity in the diabetic myocardium under chronic conditions (14,67,68). Such a defect in mitochondrial Ca2+ uptake has been reported to occur following the loss of SR Ca2+-pump activity in diabetic cardiomyopathy (24) and has been proposed to be due to oxidative stress (3,4). Mitochondrial abnormalities in the process of energy production in the diabetic heart have also been reported to increase the intracellular concentration of H+, which is considered to promote the occurrence of (Na+)i overload and cardiomyocyte swelling (2,16). Although mitochondria are being established as a source of cellular Ca2+ signalling (95,96), the exact contribution of changes in these mechanisms in diabetic cardiomyopathy remains to be investigated. Nevertheless, these observations regarding the participation of mitochondria in the regulation of intracellular Ca2+ at early and late stages of diabetic cardiomyopathy are consistent with the view that mitochondria play an important role in health and disease (97).
CONCLUSIONS
From the foregoing discussion, it is clear that insulin deficiency or insulin resistance in diabetes is associated with a decrease in glucose utilization and an increase in the use of FFAs for the production of energy in cardiomyocytes. This metabolic shift, along with the elevated levels of different hormones, including angiotensin II and catecholamines, as well as hyperglycemia result in the occurrence of oxidative stress. Such mechanisms promote the development of intra-cellular Ca2+ overload due to alterations in the SR and SL Ca2+-transport systems and, thus, lead to cardiac dysfunction. These events during the development of diabetic cardiomyopathy are depicted in Figure 1. It is known that excessive oxidation of FFAs for a prolonged period of time, as well as intracellular Ca2+ overload due to the loss of SR Ca2+-pump activity impair the electron transport system, generate oxyradicals and contribute to further promoting oxidative stress in the diabetic heart. Defects in mitochondrial function in diabetes also result in the occurrence of (Na+)i overload and cardiomyocyte swelling. In addition, mitochondrial abnormalities lead to a depression in oxidative phosphorylation and depletion of high energy stress as well as release of mitochondrial cytochrome C and the induction of cellular death. A scheme depicting all of these mitochondrial alterations is shown in Figure 2. The present article has attempted to emphasize the role of changes in both SR and mitochondria in the pathogenesis of cardiac dysfunction in diabetic cardiomyopathy.
Figure 1).
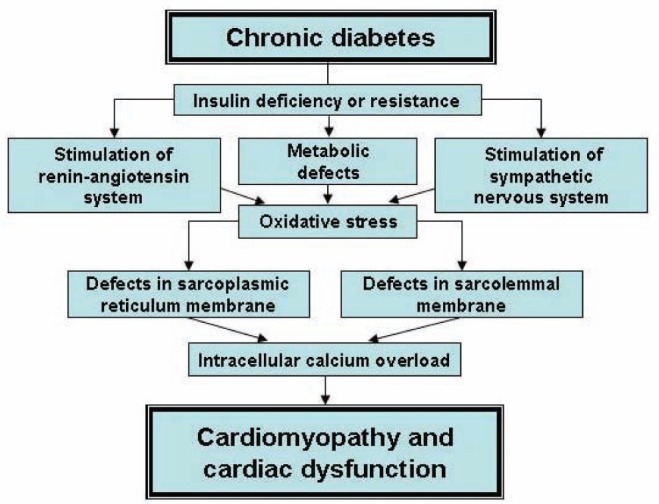
Role of oxidative stress in sarcolemma and sarcoplasmic reticulum defects in the development of cardiomyopathy and cardiac dysfunction in chronic diabetes
Figure 2).
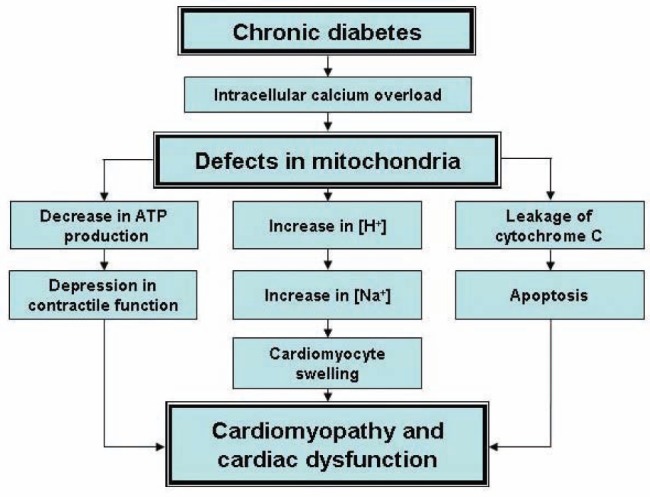
Role of mitochondrial defects in the development of cardiomyopathy and cardiac dysfunction in chronic diabetes
Acknowledgments
The research reported in this article was supported by a grant from the Canadian Institutes of Health Research. The infrastructure for this project was provided by the St Boniface Hospital Research Foundation (Winnipeg, Manitoba).
REFERENCES
- 1.Wild S, Roglic G, Green A, et al. Global prevalence of diabetes: Estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047–53. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- 2.Dhalla NS, Pierce GN, Innes IR, et al. Pathogenesis of cardiac dysfunction in diabetes mellitus. Can J Cardiol. 1985;1:263–81. [PubMed] [Google Scholar]
- 3.Dhalla NS, Liu X, Panagia V, et al. Subcellular remodeling and heart dysfunction in chronic diabetes. Cardiovasc Res. 1998;40:239–47. doi: 10.1016/s0008-6363(98)00186-2. [DOI] [PubMed] [Google Scholar]
- 4.Dhalla NS, Temsah RM, Netticadan T. Role of oxidative stress in cardiovascular diseases. J Hypertens. 2000;18:655–73. doi: 10.1097/00004872-200018060-00002. [DOI] [PubMed] [Google Scholar]
- 5.Boudina S, Albel ED. Diabetic cardiomyopathy revisited. Circulation. 2007;115:3213–23. doi: 10.1161/CIRCULATIONAHA.106.679597. [DOI] [PubMed] [Google Scholar]
- 6.Dhalla NS, Pierce GN, Panagia V, et al. Calcium movements in relation to heart function. Basic Res Cardiol. 1982;77:117–39. doi: 10.1007/BF01908167. [DOI] [PubMed] [Google Scholar]
- 7.Müller AL, Dhalla NS. Role of various proteases in cardiac remodeling and progression of heart failure. Heart Fail Rev. 2012;17:395–409. doi: 10.1007/s10741-011-9269-8. [DOI] [PubMed] [Google Scholar]
- 8.Zhang L, Kelley J, Schmeisser G, et al. Complex formation between junctin, triadin, calsequestrin, and the ryanodine receptor proteins of the cardiac junctional sarcoplamic reticulum membrane. J Bio Chem. 1997;272:23389–97. doi: 10.1074/jbc.272.37.23389. [DOI] [PubMed] [Google Scholar]
- 9.Ganguly PK, Pierce GN, Dhalla KS, et al. Defective sarcoplasmic reticular calcium transport in diabetic cardiomyopathy. Am J Physiol Endocrinol Metab. 1983;244:E528–35. doi: 10.1152/ajpendo.1983.244.6.E528. [DOI] [PubMed] [Google Scholar]
- 10.Penpargkul S, Fein F, Sonnenblick EH, et al. Depressed cardiac sarcoplasmic reticular function from diabetic rats. J Mol Cell Cardiol. 1981;13:303–9. doi: 10.1016/0022-2828(81)90318-7. [DOI] [PubMed] [Google Scholar]
- 11.Lopaschuk GD, Tahiliani AG, Vadlamudi RV, et al. Cardiac sarcoplasmic reticulum function in insulin- or carnitine-treated diabetic rats. Am J Physiol Heart Circ Physiol. 1983;245:H969–76. doi: 10.1152/ajpheart.1983.245.6.H969. [DOI] [PubMed] [Google Scholar]
- 12.Choi KM, Zhong Y, Hoit BD, et al. Defective intracellular Ca2+ signalling contributes to cardiomyopathy in type 1 diabetic rats. Am J Physiol Heart Circ Physiol. 2002;283:H1398–408. doi: 10.1152/ajpheart.00313.2002. [DOI] [PubMed] [Google Scholar]
- 13.Yaras N, Bilginoglu A, Vassort G, et al. Restoration of diabetes-induced abnormal local Ca2+ release in cardiomyocytes by angiotensin II receptor blockade. Am J Physiol Heart Circ Physiol. 2007;292:H912–20. doi: 10.1152/ajpheart.00824.2006. [DOI] [PubMed] [Google Scholar]
- 14.Golfman LS, Takeda N, Dhalla NS. Cardiac membrane Ca2+ -transport in alloxan-induced diabetes in rats. Diabetes Res Clin Pract. 1996;31:S73–7. doi: 10.1016/0168-8227(96)01233-8. [DOI] [PubMed] [Google Scholar]
- 15.Schaffer SW, Mozaffari MS, Artman M, et al. Basis for myocardial mechanical defects associated with non-insulin dependent diabetes. Am J Physiol Endocrinol Metab. 1989;256:E25–30. doi: 10.1152/ajpendo.1989.256.1.E25. [DOI] [PubMed] [Google Scholar]
- 16.Schaffer SW. Cardiomyopathy associated with non-insulin dependent diabetes. Mol Cell Biochem. 1991;107:1–20. doi: 10.1007/BF02424571. [DOI] [PubMed] [Google Scholar]
- 17.Yu Z, Tibbits GF, McNeill JH. Cellular functions of diabetic cardiomyocytes: Contractility, rapid cooling contracture and ryanodine binding. Am J Physiol Heart Circ Physiol. 1994;266:H2082–9. doi: 10.1152/ajpheart.1994.266.5.H2082. [DOI] [PubMed] [Google Scholar]
- 18.Netticadan T, Temsah RM, Kent A, et al. Depressed levels of Ca2+ cycling proteins may underlie sarcoplamic reticulum dysfunction in the diabetic heart. Diabetes. 2001;50:2133–8. doi: 10.2337/diabetes.50.9.2133. [DOI] [PubMed] [Google Scholar]
- 19.Zhong Y, Ahmed S, Grupp IL, et al. Altered SR protein expression associated with contractile dysfunction in diabetic rat hearts. Am J Physiol Heart Circ Physiol. 2001;281:H1137–47. doi: 10.1152/ajpheart.2001.281.3.H1137. [DOI] [PubMed] [Google Scholar]
- 20.Xu YJ, Elimban V, Takeda S, et al. Cardiac sarcoplasmic reticulum function and gene expression in chronic diabetes. Cardiovasc Pathobiol. 1996;1:89–96. [Google Scholar]
- 21.Zhou BQ, Hu SJ, Wang GB. The analysis of ultrastructure and gene expression of sarco/endoplasmic reticulum calcium handling proteins in alloxan-induced diabetic rat myocardium. Acta Cardiol. 2006;61:21–7. doi: 10.2143/AC.61.1.2005136. [DOI] [PubMed] [Google Scholar]
- 22.Zarain-Herzberg A, Yano K, Elimban V, et al. Cardiac sarcoplamic reticulum Ca2+ATPase expression in streptozotocin-induced diabetic rat heart. Biochem Biophys Res Commun. 1994;203:113–20. doi: 10.1006/bbrc.1994.2156. [DOI] [PubMed] [Google Scholar]
- 23.Golfman L, Dixon IM, Takeda N, et al. Differential changes in cardiac myofibrillar and sarcoplamic reticular gene expression in alloxan-induced diabetes. Mol Cell Biochem. 1999;200:15–25. doi: 10.1023/a:1006950218597. [DOI] [PubMed] [Google Scholar]
- 24.Takeda N, Dixon IM, Hata T, et al. Sequence of alterations in subcellular organelles during the development of heart dysfunction in diabetes. Diabetes Res Clin Pract. 1996;30:S113–22. doi: 10.1016/s0168-8227(96)80047-7. [DOI] [PubMed] [Google Scholar]
- 25.Vasanji Z, Dhalla NS, Netticadan T. Increased inhibition of SERCA2 by phospholamban in the type 1 diabetic heart. Mol Cell Biochem. 2004;261:245–9. doi: 10.1023/b:mcbi.0000028762.97754.26. [DOI] [PubMed] [Google Scholar]
- 26.Bidasee KR, Zhang Y, Shao CH, et al. Diabetes increases formation of advanced glycation end products on Sarco(endo)plasmic reticulum Ca2+ATPase. Diabetes. 2004;53:463–73. doi: 10.2337/diabetes.53.2.463. [DOI] [PubMed] [Google Scholar]
- 27.Li SY, Yang X, Ceylan-Isik AF, et al. Cardiac contractile dysfunction in Lep/Lep obesity is accompanied by NADPH oxidase activation, oxidative modification of sarco(endo)plasmic reticulum Ca2+-ATPase and myosin heavy chain isozyme switch. Diabetologia. 2006;49:1434–46. doi: 10.1007/s00125-006-0229-0. [DOI] [PubMed] [Google Scholar]
- 28.Bergh CH, Hjalmarson A, Sjogren KG, et al. The effect of diabetes on phosphatidylinositol turnover and calcium influx in myocardium. Horm Metab Res. 1988;20:381–6. doi: 10.1055/s-2007-1010842. [DOI] [PubMed] [Google Scholar]
- 29.Li Y, Li Y, Feng Q, et al. Calpain activation contributes to hyperglycemia-induced apoptosis in cardiomyocytes. Cardiovasc Res. 2009;84:100–10. doi: 10.1093/cvr/cvp189. [DOI] [PubMed] [Google Scholar]
- 30.Belke DD, Swanson EA, Dillman WH. Decreased sarcoplasmic reticulum activity and contractility in diabetic db/db mouse heart. Diabetes. 2004;53:3201–8. doi: 10.2337/diabetes.53.12.3201. [DOI] [PubMed] [Google Scholar]
- 31.Zhao XY, Hu SJ, Li J, et al. Decreased cardiac sarcoplasmic reticular reticulum Ca2+-ATPase activity contributes to cardiac dysfunction in streptozotocin-induced diabetic rats. J Physiol Biochem. 2006;62:1–8. doi: 10.1007/BF03165800. [DOI] [PubMed] [Google Scholar]
- 32.Suarez J, Scott B, Dillmann WH. Conditional increase in SERCA2a protein is able to reverse contractile dysfunction and abnormal calcium flux in established diabetic cardiomyopathy. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1439–45. doi: 10.1152/ajpregu.00736.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu Z, Quamme GA, McNeill JH. Depressed responses to (Ca2+)i isoproterenol and cAMP in isolated cardiomyocytes from experimental diabetic rats. Am J Physiol Heart Circ Physiol. 1994;266:H2334–42. doi: 10.1152/ajpheart.1994.266.6.H2334. [DOI] [PubMed] [Google Scholar]
- 34.Yaras N, Ugur M, Ozdemir S, et al. Effects of diabetes on ryanodine receptor Ca release channel (RyR2) and Ca2+ homeostasis in rat heart. Diabetes. 2005;54:3082–8. doi: 10.2337/diabetes.54.11.3082. [DOI] [PubMed] [Google Scholar]
- 35.Noda N, Hayashi H, Miyata H, et al. Cytosolic Ca2+ concentration and pH of diabetic rat myocytes during metabolic inhibition. J Mol Cell Cardiol. 1992;24:435–46. doi: 10.1016/0022-2828(92)93197-r. [DOI] [PubMed] [Google Scholar]
- 36.Lagadic-Gossmann D, Buckler KJ, Prigent KL, et al. Altered Ca2+ handling in ventricular myocytes isolated from diabetic rats. Am J Physiol Heart Circ Physiol. 1996;270:H1529–37. doi: 10.1152/ajpheart.1996.270.5.H1529. [DOI] [PubMed] [Google Scholar]
- 37.Horackova M, Murphy MG. Effects of chronic diabetes mellitus on electrical contractile activities, 45Ca2+ transport, fatty acid, profiles and ultrastructure of isolated rat ventricular myocytes. Pflugers Arch. 1988;411:564–72. doi: 10.1007/BF00582379. [DOI] [PubMed] [Google Scholar]
- 38.Dhalla NS, Wang X, Beamish RE. Intracellular calcium handling in normal and failing hearts. Exp Clin Cardiol. 1996;1:7–20. [Google Scholar]
- 39.Pereira L, Matthes J, Schuster I, et al. Mechanisms of (Ca2+)i transient in cardiomyopathy of db/db type 2 diabetic mice. Diabetes. 2006;55:608–15. doi: 10.2337/diabetes.55.03.06.db05-1284. [DOI] [PubMed] [Google Scholar]
- 40.Heyliger CE, Prakash A, McNeill JH. Alterations in cardiac sarcolemmal Ca2+ pump activity during diabetes mellitus. Am J Physiol Heart Circ Physiol. 1987;252:H540–4. doi: 10.1152/ajpheart.1987.252.3.H540. [DOI] [PubMed] [Google Scholar]
- 41.Makino N, Dhalla KS, Elimban V, et al. Sarcolemmal Ca2+ transport in streptozotocin-induced diabetic cardiomyopathy in rats. Am J Physiol Endocrinol Metab. 1987;253:E202–7. doi: 10.1152/ajpendo.1987.253.2.E202. [DOI] [PubMed] [Google Scholar]
- 42.Sauviat MP, Feuvray D. Electrophysiological analysis of the sensitivity to calcium in ventricular muscle from alloxan diabetic rats. Basic Res Cardiol. 1986;81:489–96. doi: 10.1007/BF01907755. [DOI] [PubMed] [Google Scholar]
- 43.Borda E, Pascual J, Wald M, et al. Hypersensitivity to calcium associated with an increased sarcolemmal Ca2+ATPase activity in diabetic rat heart. Can J Cardiol. 1988;4:97–101. [PubMed] [Google Scholar]
- 44.Pierce GN, Kutryk MJ, Dhalla NS. Alterations in Ca2+ binding by and composition of the cardiac sarcolemmal membrane in chronic diabetes. Proc Natl Acad Sci USA. 1983;80:5412–6. doi: 10.1073/pnas.80.17.5412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pierce GN, Dhalla NS. Sarcolemmal Na+-K+ ATPase activity in diabetic rat heart. Am J Physiol Cell Physiol. 1983;245:C241–7. doi: 10.1152/ajpcell.1983.245.3.C241. [DOI] [PubMed] [Google Scholar]
- 46.Kjeldsen K, Braendgaard H, Sidenius P, et al. Diabetes decreases Na+-K+ pump concentration in skeletal muscles, heart ventricular muscle, and peripheral nerves of rat. Diabetes. 1987;36:842–8. doi: 10.2337/diab.36.7.842. [DOI] [PubMed] [Google Scholar]
- 47.Golfman L, Dixon IM, Takeda N, et al. Cardiac sarcolemmal Na+-Ca2+exchange and Na+/K+ ATPase activities and gene expression in alloxan-induced diabetes in rats. Mol Cell Biochem. 1998;188:91–101. [PubMed] [Google Scholar]
- 48.Kashihara H, Shi ZQ, Yu JZ, et al. Effects of diabetes and hypertension on myocardial Na+-Ca2+ exchange. Can J Physiol Pharmacol. 2000;78:7812–19. doi: 10.1139/cjpp-78-1-12. [DOI] [PubMed] [Google Scholar]
- 49.Hattori Y, Mastuda N, Kimura J, et al. Diminished function and expression of Na+-Ca2+ exchanger in diabetic rats: Implications in Ca2+ overload. J Physiol. 2000;527:85–94. doi: 10.1111/j.1469-7793.2000.00085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lagadic-Gossmann D, Feuvray D. Intracellular sodium activity in papillary muscle from diabetic rat hearts. Exp Physiol. 1991;76:147–9. doi: 10.1113/expphysiol.1991.sp003478. [DOI] [PubMed] [Google Scholar]
- 51.Nagase N, Saijo Y, Nitta H, et al. Myocardial disorders caused by magnesium deficiency in diabetic KK mice. Magnesium. 1989;8:307–15. [PubMed] [Google Scholar]
- 52.Regan TJ, Wu CF, Yeh CK, et al. Myocardial composition and function in diabetes. The effects of chronic insulin use. Circ Res. 1981;49:1268–77. doi: 10.1161/01.res.49.6.1268. [DOI] [PubMed] [Google Scholar]
- 53.Afzal N, Pierce GN, Elimban V, et al. Influence of verapamil on some subcellular defects in diabetic cardiomyopathy. Am J Physiol Endocrinol Metab. 1989;256:E453–8. doi: 10.1152/ajpendo.1989.256.4.E453. [DOI] [PubMed] [Google Scholar]
- 54.Afzal N, Ganguly PK, Dhalla KS, et al. Beneficial effects of verapamil in diabetic cardiomyopathy. Diabetes. 1988;37:936–42. doi: 10.2337/diab.37.7.936. [DOI] [PubMed] [Google Scholar]
- 55.Liu X, Suzuki H, Sethi R, et al. Blockade of the renin-angiotensin system attenuates sarcolemma and sarcoplasmic reticulum remodeling in chronic diabetes. Ann N Y Acad Sci. 2006;1084:141–54. doi: 10.1196/annals.1372.003. [DOI] [PubMed] [Google Scholar]
- 56.Dhalla NS, Das PK, Sharma GP. Subcellular basis of cardiac contractile failure. J Mol Cell Cardiol. 1978;10:363–85. doi: 10.1016/0022-2828(78)90384-x. [DOI] [PubMed] [Google Scholar]
- 57.Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Phys Rev. 2005;85:1093–129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- 58.Rodrigues B, Cam MC, McNeill JH. Myocardial substrate metabolism: Implications in diabetic cardiomyopathy. J Mol Cell Cardiol. 1995;27:169–79. doi: 10.1016/s0022-2828(08)80016-8. [DOI] [PubMed] [Google Scholar]
- 59.Randle PJ, Garland PB, Hales CN, et al. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;1:785–9. doi: 10.1016/s0140-6736(63)91500-9. [DOI] [PubMed] [Google Scholar]
- 60.Mazumder PK, O’Neill BT, Roberts MW, et al. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes. 2004;53:2366–74. doi: 10.2337/diabetes.53.9.2366. [DOI] [PubMed] [Google Scholar]
- 61.Carley AN, Severson DL. Fatty acid metabolism is enhanced in type 2 diabetic hearts. Biochim Biophys Acta. 2005;1734:112–126. doi: 10.1016/j.bbalip.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 62.Herrero P, Peterson LR, McGill JB, et al. Increased myocardial fatty acid metabolism in patients with type 1 diabetes mellitus. J Am Coll Cardiol. 2006;47:598–604. doi: 10.1016/j.jacc.2005.09.030. [DOI] [PubMed] [Google Scholar]
- 63.Finck BN, Kelly DP. Peroxisome proliferator-activated receptor alpha (PPARalpha) signalling in the gene regulatory control of energy metabolism in the normal and diseased heart. J Mol Cell Cardiol. 2002;34:1249–57. doi: 10.1006/jmcc.2002.2061. [DOI] [PubMed] [Google Scholar]
- 64.Banke NH, Wende AR, Leone TC, et al. Preferential oxidation of triacylglyceride-derived fatty acids in heart is augmented by the nuclear receptor PPAR alpha. Circ Res. 2010;107:233–41. doi: 10.1161/CIRCRESAHA.110.221713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Duncan JG, Fong JL, Medeiros DM, et al. Insulin-resistant heart exhibits a mitochondrial biogenic response driven by the peroxisome proliferator-activated receptor-alpha/PGC-1alpha gene regulatory pathway. Circulation. 2007;115:909–17. doi: 10.1161/CIRCULATIONAHA.106.662296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kuo TH, Moore KH, Giacomelli F, et al. Defective oxidative metabolism of heart mitochondria from genetically diabetic mice. Diabetes. 1983;32:781–7. doi: 10.2337/diab.32.9.781. [DOI] [PubMed] [Google Scholar]
- 67.Pierce GN, Dhalla NS. Heart mitochondrial function in chronic experimental diabetes in rats. Can J Cardiol. 1985;1:48–54. [PubMed] [Google Scholar]
- 68.Flarsheim CE, Grupp IL, Matlib MA. Mitochondrial dysfunction accompanies diastolic dysfunction in diabetic rat heart. Am J Physiol Heart Circ Physiol. 1996;271:H192–202. doi: 10.1152/ajpheart.1996.271.1.H192. [DOI] [PubMed] [Google Scholar]
- 69.Finck BN, Lehman JJ, Leone TC, et al. The cardiac phenotype induced by PPARalpha over expression mimics that caused by diabetes mellitus. J Clin Invest. 2002;109:121–30. doi: 10.1172/JCI14080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Finck BN, Han X, Courtois M, et al. A critical role for PPARalpha-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: Modulation by dietary fat content. Proc Natl Acad Sci USA. 2003;100:1226–31. doi: 10.1073/pnas.0336724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Finck BN. The role of the peroxisome proliferator-activated receptor alpha pathway in pathological remodelling of the diabetic heart. Curr Opin Clin Nutr Metab Care. 2004;7:391–6. doi: 10.1097/01.mco.0000134371.70815.32. [DOI] [PubMed] [Google Scholar]
- 72.Cheng L, Ding G, Qin Q, et al. Cardiomyocyte-restricted peroxisome proliferator-activated receptor-delta deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat Med. 2004;10:1245–50. doi: 10.1038/nm1116. [DOI] [PubMed] [Google Scholar]
- 73.Yagyu H, Chen G, Yokoyama M, et al. Lipoprotein lipase (LpL) on the surface of cardiomyocytes increases lipid uptake and produces a cardiomyopathy. J Clin Invest. 2003;111:419–26. doi: 10.1172/JCI16751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Listenberger LL, Ory DS, Schaffer JE. Palmitate-induced apoptosis can occur through a ceramide-independent pathway. J Biol Chem. 2001;276:14890–5. doi: 10.1074/jbc.M010286200. [DOI] [PubMed] [Google Scholar]
- 75.Ostrander DB, Sparagna GC, Amoscato AA, et al. Decreased cardiolipin synthesis corresponds with cytochrome C release in palmitate-induced cardiomyocyte apoptosis. J Biol Chem. 2001;276:38061–7. doi: 10.1074/jbc.M107067200. [DOI] [PubMed] [Google Scholar]
- 76.Baynes JW, Thorpe SR. Role of oxidative stress in diabetic complications: A new perspective on an old paradigm. Diabetes. 1999;48:1–9. doi: 10.2337/diabetes.48.1.1. [DOI] [PubMed] [Google Scholar]
- 77.Giardino I, Fard AK, Hatchell DL, et al. Aminoguanidine inhibits reactive oxygen species formation, lipid peroxidation, and oxidant-induced apoptosis. Diabetes. 1998;47:1114–20. doi: 10.2337/diabetes.47.7.1114. [DOI] [PubMed] [Google Scholar]
- 78.Ohuwa T, Sato Y, Naoi M. Hydroxyl radical formation in diabetic rats induced by streptozotocin. Life Sci. 1995;56:1789–98. doi: 10.1016/0024-3205(95)00150-5. [DOI] [PubMed] [Google Scholar]
- 79.Boudina S, Abel ED. Diabetic cardiomyopathy, causes and effects. Rev Endocr Metab Disord. 2010;11:31–9. doi: 10.1007/s11154-010-9131-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kowluru RA, Engerman RL, Kern TS. Diabetes-induced metabolic abnormalities in myocardium: Effect of antioxidant therapy. Free Radic Res. 2000;32:67–74. doi: 10.1080/10715760000300071. [DOI] [PubMed] [Google Scholar]
- 81.Brownlee M. Advanced protein glycosylation in diabetes and aging. Annu Rev Med. 1995;46:223–34. doi: 10.1146/annurev.med.46.1.223. [DOI] [PubMed] [Google Scholar]
- 82.Taniguchi N, Kaneto H, Asahi M, et al. Involvement of glycation and oxidative stress in diabetic macroangiopathy. Diabetes. 1996;45:S81–3. doi: 10.2337/diab.45.3.s81. [DOI] [PubMed] [Google Scholar]
- 83.Wolff SP, Jiang ZY, Hunt JV. Protein glycation and oxidative stress in diabetes mellitus and ageing. Free Radic Biol Med. 1991;10:339–59. doi: 10.1016/0891-5849(91)90040-a. [DOI] [PubMed] [Google Scholar]
- 84.Yan SD, Schmidt AM, Anderson GM, et al. Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J Biol Chem. 1994;269:788–91. [PubMed] [Google Scholar]
- 85.Peiro C, Lafuente N, Matesanz N, et al. High glucose induces cell death of cultured human aortic smooth muscle cells through the formation of hydrogen peroxide. Br J Pharmacol. 2001;133:967–74. doi: 10.1038/sj.bjp.0704184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kajstura J, Fiordaliso F, Andreoli AM, et al. IGF-1 overexpression inhibits the development of diabetic cardiomyopathy and angiotensin II-mediated oxidative stress. Diabetes. 2001;50:1414–24. doi: 10.2337/diabetes.50.6.1414. [DOI] [PubMed] [Google Scholar]
- 87.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–44. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nishikawa T, Edelstein D, Du XL, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–90. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 89.Boudina S, Bugger H, Sena S, et al. Contribution of impaired myocardial insulin signalling to mitochondrial dysfunction and oxidative stress in the heart. Circulation. 2009;119:1272–83. doi: 10.1161/CIRCULATIONAHA.108.792101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cai L, Li W, Wang G, et al. Hyperglycemia-induced apoptosis in mouse myocardium: mitochondrial cytochrome C-mediated caspase-3 activation pathway. Diabetes. 2002;51:1938–48. doi: 10.2337/diabetes.51.6.1938. [DOI] [PubMed] [Google Scholar]
- 91.Ghosh S, Pulinilkunnil T, Yuen G, et al. Cardiomyocyte apoptosis induced by short-term diabetes requires mitochondrial GSH depletion. Am J Physiol Heart Circ Physiol. 2005;289:H768–76. doi: 10.1152/ajpheart.00038.2005. [DOI] [PubMed] [Google Scholar]
- 92.Shen X, Zheng S, Metreveli NS, et al. Protection of cardiac mitochondria by over expression of MnSOD reduces diabetic cardiomyopathy. Diabetes. 2006;55:798–805. doi: 10.2337/diabetes.55.03.06.db05-1039. [DOI] [PubMed] [Google Scholar]
- 93.Ye G, Metreveli NS, Donthi RV, et al. Catalase protects cardiomyocyte function in models of type 1 and type2 diabetes. Diabetes. 2004;53:1336–43. doi: 10.2337/diabetes.53.5.1336. [DOI] [PubMed] [Google Scholar]
- 94.Cai L, Wang Y, Zhou G, et al. Attenuation by metallothionein of early cardiac cell death via suppression of mitochondrial oxidative stress results in a prevention of diabetic cardiomyopathy. J Am Coll Cardiol. 2006;48:1688–97. doi: 10.1016/j.jacc.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 95.Szabadkai G, Duchen MR. Mitochondria: the hub of cellular Ca2+ signalling. Physiology. 2008;23:84–94. doi: 10.1152/physiol.00046.2007. [DOI] [PubMed] [Google Scholar]
- 96.Ryu SY, Beutner G, Dirksen RT, et al. Mitochondrial ryanodine receptors and other mitochondrial Ca2+ permeable channels. FEBS Lett. 2010;584:1948–55. doi: 10.1016/j.febslet.2010.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Duchen MR. Roles of mitochondria in health and disease. Diabetes. 2004;53:S96–102. doi: 10.2337/diabetes.53.2007.s96. [DOI] [PubMed] [Google Scholar]