

Abstract
Citrus, as one of the globally important fruit trees, has been an object of interest for understanding genetics and evolutionary process in fruit crops. Meta-analyses of 19 Citrus species, including 4 globally and economically important Citrus sinensis, Citrus clementina, Citrus reticulata, and 1 Citrus relative Poncirus trifoliata, were performed. We observed that codons ending with A- or T- at the wobble position were preferred in contrast to C- or G- ending codons, indicating a close association with AT richness of Citrus species and P. trifoliata. The present study postulates a large repertoire of a set of optimal codons for the Citrus genus and P. trifoliata and demonstrates that GCT and GGT are evolutionary conserved optimal codons. Our observation suggested that mutational bias is the dominating force in shaping the codon usage bias (CUB) in Citrus and P. trifoliata. Correspondence analysis (COA) revealed that the principal axis [axis 1; COA/relative synonymous codon usage (RSCU)] contributes only a minor portion (∼10.96%) of the recorded variance. In all analysed species, except P. trifoliata, Gravy and aromaticity played minor roles in resolving CUB. Compositional constraints were found to be strongly associated with the amino acid signatures in Citrus species and P. trifoliata. Our present analysis postulates compositional constraints in Citrus species and P. trifoliata and plausible role of the stress with GC3 and coevolution pattern of amino acid.
Keywords: Citrus, coevolution, mutational bias, Poncirus trifoliata, stress, GC3 biology
1. Introduction
Genome composition (such as GC- and AT-content) and subsequent balance of codon usage and eukaryotic translation machinery play an important role in the evolution of the nucleotides at the wobble position.1 Degeneracy in biased codon usage explains the concept behind the usage of same amino acids with multiple synonymous codons except methionine (Met) and tryptophan (Trp).2 This genome composition bias leads to the usage of some codons at a higher frequency as compared to the synonymous codons for encoding the particular amino acid. These differences in the usage of the synonymous codons have also been one of the factors for the evolution of proteome diversity and can help us to understand the evolution of those proteins that have structural differences in spite of being conserved at the sequence level.3–7
Two major paradigms of codon usage were proposed as plausible answers to clarify the non-randomness of codon usage at intra- and inter-species levels: (i) natural selection that is expected to yield a correlation with codon bias in highly expressed genes,8 rapidly regulated and variably expressed genes, and (ii) neutral processes, such as mutational biases (MBs), where some mutations occur more often than others across the genome of an organism because of local variations in the base composition. Several lines of evidence support the argument that the usage of synonymous codons with unequal frequencies in both prokaryotic and eukaryotic genes is the result of a complex balance between MB and/or natural selection and genetic drift.9–12 It has been suggested that the codon bias could be positively selected because of a more efficient and accurate translation, and favoured codons may correspond to the most highly expressed genes.2,13 In addition to neutral and selection processes, GC-biased gene conversion, which depends on the local recombination rate, is an important factor in shaping codon and amino acid usage.14
To date, wide variations have been observed in codon usage patterns in many organisms and have provided clues to understand the evolution of genes and gene families. The factors that potentially affect biased usage of codons are MB that correlates the codon usage bias (CUB) with the genomic GC content, Hill−Robertson effect explaining the interference of selection of one locus with another locus, translational selection, and a cumulative effect of replication and translational selection in shaping the codon usage across the genes of several bacterial species.15–19 It has been shown that CUB and protein functional conservation play a major role for the decelerated evolution of whole genome duplication in Saccharomyces cerevisiae.20 Several other factors that might affect the codon usage are amino acid conservation and hydrophobicity,21 gene expression,22 mRNA folding stability, codon−anticodon interaction, and gene length.22
Citrus is a diploid genus of the Rutaceae family, whose cultivated forms are important for human diet with more than 122 MT of annual world fruit production. We have systematically analysed codon usage patterns and genomic heterogeneity using a meta-analyses approach that involves multivariate tools and codon usage indices such as relative synonymous codon usage (RSCU) and effective number of codons (Nc) in the studied gene sets.23,24 In present study, we inferred global pattern of CUB, mutational pressures, and association of GC3 with potential stress events. We have collectively analysed horticulturally important Citrus species that includes recently sequenced double haploid Citrus sinensis genome25 and 18 additional Citrus species with expressed sequence tags (ESTs) counts of more than 1000 per species. To make this analysis comparative, we have also included Poncirus trifoliata that is considered to be a distant relative of Citrus genus. These species show wide variation in traits such as cold and drought tolerance and are of commercial importance. We have restricted our analyses to Citrus genera to develop resource information for Citrus species.
Our study demonstrated that CUB in Citrus species and P. trifoliata is biased towards AT richness, and a relatively higher occurrence of A- and T-ending codons was observed. We found several interesting and diverse patterns of optimal codons, and it was observed that two optimal codons coding for Alanine and Glycine were evolutionary conserved between the Citrus species and P. trifoliata. To the best of our knowledge, our analysis presents the first complete report on the identification of optimal codons across the entire Citrus genus and P. trifoliata, which could serve as the potential source for developing transgenic Citrus cultivars using codon optimization. GC3 and evolution were correlated using Hamming distance parameter to identify suggestive evolutionary pairs of genes. Co-orthologous genes matrix identified using the alignment ratio suggested close association of Citrus clementina, Citrus sinensis, and Citrus reticulata, as they belong to the same phylogenetic clade. We further demonstrated that nucleotide bias has a genome-wide influence on amino acid composition of Citrus species and P. trifoliata.
2. Materials and methodology
2.1. Sequence information and processing
Our dataset consists of genome-predicted coding regions and ESTs. All the genome-predicted coding sequences of C. sinensis were retrieved from recently sequenced Citrus genome.25 In case of other Citrus species, ESTs were downloaded from the National Center for Biotechnology Information (NCBI) EST repository (NCBI; http://www.ncbi.nlm.nih.gov). In addition, we downloaded putative unique transcripts for all studied species from the PlantGDB database (http://www.plantgdb.org). A detailed description of the data used is shown in Table 1. In case of ESTs, the ESTs were first clustered into contigs and singletons using CAP3 with default parameters.26 A minimum match percentage cutoff of 95% for 40 overlapping bases was used to assign 2 sequences to a cluster.
Table 1.
Genomic composition of coding region of Citrus species and P. trifoliata at GC, GC1, GC2, GC3, and GC3s
| No. | Citrus species | Genes/ESTa | Unigenes count | Genesb | GC | GC1 | GC2 | GC3 | GC3s |
|---|---|---|---|---|---|---|---|---|---|
| 1 | C. sinensis | 44 275 | – | 41362 | 43.92 | 50.4 | 40.33 | 41.03 | 38.8 |
| 2 | C. aurantifolia | 8219 | 7550 | 4715 | 47.78 | 52.96 | 43.87 | 46.52 | 44.73 |
| 3 | C. aurantium | 14 584 | 11 952 | 6787 | 46.93 | 52.47 | 42.59 | 45.72 | 43.94 |
| 4 | C. clementina | 118 365 | 51 591 | 33 765 | 47.04 | 51.84 | 42.51 | 46.76 | 44.99 |
| 5 | C. clementina × C. tangerina | 1843 | 1283 | 677 | 44.66 | 51.67 | 39.77 | 42.55 | 40.47 |
| 6 | C. jambhiri | 1017 | 858 | 701 | 45.86 | 51.79 | 41.13 | 44.66 | 42.67 |
| 7 | C. japonica var. margarita | 2924 | 1628 | 588 | 46.11 | 52.67 | 41.42 | 44.25 | 42.35 |
| 8 | C. limettioides | 8188 | 7933 | 3964 | 46.47 | 50.76 | 42.75 | 45.9 | 44.25 |
| 9 | C. limonia | 11 045 | 9761 | 4256 | 45.82 | 51.13 | 41.37 | 44.96 | 43.17 |
| 10 | C. medica | 1115 | 896 | 711 | 45.65 | 51.91 | 40.9 | 44.13 | 42.15 |
| 11 | C. reshni | 5768 | 3735 | 2644 | 45.2 | 51.51 | 40.57 | 43.52 | 41.5 |
| 12 | C. reticulata | 55 980 | 46 170 | 30 816 | 46.85 | 52.3 | 42.77 | 45.48 | 43.62 |
| 13 | C. reticulata × C. temple | 5823 | 3685 | 2253 | 46.48 | 52.41 | 41.64 | 45.41 | 43.41 |
| 14 | C. sinensis × P. trifoliata | 1837 | 1522 | 992 | 46.07 | 51.67 | 41.41 | 45.15 | 43.23 |
| 15 | C. sunki | 5216 | 4688 | 1746 | 47.58 | 51.51 | 43.58 | 47.64 | 46.17 |
| 16 | P. trifoliata | 62 695 | 35 740 | 19 445 | 46.78 | 52.33 | 42.67 | 45.35 | 43.52 |
| 17 | C. unshiu | 19 072 | 9289 | 4592 | 45.67 | 51.72 | 41.14 | 44.15 | 42.21 |
| 18 | C. paradisi | 8039 | 4621 | 2517 | 45.32 | 52.19 | 40.51 | 43.27 | 41.3 |
| 19 | C. paradisi × P. trifoliata | 7954 | 3335 | 2596 | 45.23 | 51.96 | 40.77 | 42.98 | 40.96 |
Means of GC% were calculated at the first, second, and third positions. GC3s represents the GC at the third synonymous position.
aIn C. sinensis, genome-predicted coding regions are used, whereas for the rest of the species, the number represents the EST count in the column (genes/EST).
bSelected genes above 300 bp threshold.
2.2. Frame correction of ESTs
All the UniGenes (contigs + singletons) were analysed for frame correction and prediction of protein-coding region using FrameDP.27,28 Briefly, the following pipeline was implemented using FrameDP to identify open reading frames (ORFs): firstly, each EST was compared against the TAIR database (Arabidopsis Information Resource; http://www.arabidopsis.org/) using BLASTX29 with E-value = 10−4, identity percent (%) = 40% over 100 amino acids.27 Secondly, the training dataset was generated from the BLASTX results, and, subsequently, the training matrix was calculated, which represents the coding style of the species. Thirdly, a collection of putative protein-coding sequences (CDSs) was generated for each Citrus species and P. trifoliata based on its homology with known protein dataset and on coding style recognition matrix.
2.3. Sequence filtering and GC variation
From the set of corrected sequences, we discarded proteins shorter than 100 amino acids to create a reliable dataset for all the studied Citrus species and P. trifoliata for further analysis.23 The final sequence dataset was subsequently analysed by tabulating the frequency of GC at the first, second, and third codon positions (GC1, GC2, GC3, and GC3s, respectively) using in-house written Perl and C++ scripts. GC3 is defined as the fraction of cytosines (C) and guanines (G) in the third position of the codon: GC3 = 3(C3 + G3)/L for the ORF of length L, whereas GC3S is defined as G + C base composition at the third synonymously degenerate position of codons. To define GC3-rich and GC3-poor groups, we have selected 5% of the genes with the highest and the lowest GC3 values. For the genes in the GC3-rich and-poor groups, we have computed two measures: (i) positional gradients of GC3 and (ii) CG3 skew that are defined as: CG3 skew is the difference in fraction of cytosines (C) and guanines (G) in the third position of the codon divided by the sum of C and G in the third position: CG3-skew = (C3−G3)/(C3 + G3) and positional gradient GC3 as a [G3 (x) + C3 (x)]/Nseq, where x is the distance measured as number of codons from the first ATG, and Nseq is the number of sequences.
2.4. Indices of codon usage and correspondence analysis
The effective number of codons (Nc) provides an independent measure of CUB, regardless of the gene length.23 The expected Nc values were computed according to the equation proposed by Wright, which assumes equal use of G and C (A and T) in degenerate codon groups.23
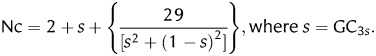 |
We have further analysed RSCU using multivariate analysis for all the 59 informative codons (excluding Met, Trp, and the three stop codons).30,31 As proposed earlier, if RSCU values are close to 1.0, it indicates that all the synonymous codons are used equally without any bias towards the usage of a particular codon in a gene of length L. In our study, axis 1 (COA/RSCU) and axis 2 (COA/RSCU) represent the first and second major axes of correspondence analysis. The Kyte–Doolittle scale was used to calculate the hydropathy score that is the arithmetic mean of the sum of hydropathic indices of each amino acid. This scale also provides information on transmembrane or surface regions.32
2.5. Identification of optimal codons
Optimal codons are believed to achieve faster translation rates and high accuracy, therefore the effect is more pronounced in highly expressed genes.33 For the identification of optimal codons, we used 10% of total genes from extreme ends of the principal axis of correspondence analysis (axis 1; COA/RSCU). Codon usage was then compared using χ2 contingency (χ2) of the two groups, and codons whose frequency of usage was significantly higher at three different levels of statistical precision (P-value < 0.5; P-value < 0.01; P-value < 0.001) in highly expressed genes when compared with lowly expressed genes were defined as the optimal codons. We also identified the genes related to the ribosomal proteins and observed the association of the ribosomal proteins with axis 1 (COA/RSCU) and axis 2 (COA/RSCU) of the correspondence analysis to optimize the identification of the optimal codons.
2.6. GO and stress-associated annotation, Hamming parameter, and coevolution of amino acid composition
Stress-related genes were identified using reciprocal best bidirectional hits (RBH) using NCBI BLASTP program with E-value 10−30, and gene ontology (GO) annotation of the model dicot plant Arabidopsis thaliana was then used by ‘guilt-by-association’ approach. To optimize our annotation pipeline, we selected GO annotations ‘involved in’ the category containing word ‘stress’ and/or ‘response’ and grouped together as ‘stress related’. The normalized Hamming distance was then evaluated according to the method described in Sablok et al.34 All the frame-corrected coding regions and the genome-predicted coding regions were parsed. Subsequent proteins were used to identify co-orthologous genes, and a suggestive phylogeny was drawn using the co-orthologous matrix as described in Lechner et al.35 To identify the effect of nucleotide bias on amino acid composition, we partitioned the codons according to GC-rich (the so-called GARP amino acids: Glycine, Alanine, Arginine, and Proline) and AT-rich, FYMINK amino acids (Phenylalanine, Tyrosine, Methionine, Isoleucine, Asparagine, and Lysine) amino acids.36,37 We have maintained the exclusion of Leucine and Arginine from the dataset as per Singer and Hickey.37
2.7. Statistical analysis
All the indices of codon usage were calculated using CodonW (http://codonw.sourceforge.net), and several in-house developed Perl, R, and C++ scripts were written to streamline the downstream analyses. Statistical analyses were performed using R in R studio (http://rstudio.org/). All the results were interpreted based on the non-parametric Spearman's rank correlation (ρ).
3. Results and discussion
3.1. Patterns of genomic content and codon usage variation across Citrus species and P. trifoliata
The recently sequenced nuclear genome of C. sinensis25 presents an opportunity to analyse the nucleotide compositional pressure and heterogenetic variation, which could possibly help us to understand the molecular adaptation of these horticulturally important fruit species. We have used meta-analyses approach using the predicted genomic coding regions of recently sequenced C. sinensis and frame-corrected ESTs of related species and P. trifoliata. Nucleotide composition analysis indicated that Citrus species are AT rich (∼46% GC, typical for many sequenced genomes of dicot species), and this bias towards the AT richness is dominant and is observed across all the studied species (Table 1).
Based on the above observation, we could hypothesize that these species are biased towards the A- and/or T-ending codons in the coding region across these two genera. Generally, most of the analysed dicot species prefer to use A- or T-ending codons at the third position; this observation is in accordance with a previous study in Citrus using a small dataset of 177 CDS regions.38 We found that average GC3 is significantly lower than GC1 and the observed GC (P-value < 0.05) which potentially explains that the genome composition is biased towards the dominating AT usage encoding genes at the third, ‘wobble’ position.
In eukaryotes, the intra-genomic heterogeneity is high, and interspecific variation of the average GC content is low.39,40 In Citrus species, GC usage varies by position, but with much greater variance, with higher usage in position 1 (∼51.8% GC) and lower GC usage in positions 2 and 3 (∼41.6 versus ∼44.7% and also at the third synonymous positions GC3s ∼ 42.8%). The observed results are consistent with the previous reports demonstrating preference for G is in position 1 and T/A in positions 2 and 341 and demonstrate the role of mutational pressure in the evolution of CUB across Citrus species and P. trifoliata. Furthermore, there is a significant wide variation in GC usage at the third synonymous position (GC3s ∼42.8%) (Table 1; Fig. 1). In our analysis, we estimated codon usage patterns of frame-corrected coding regions obtained from ESTs versus gene predictions derived from a fully assembled and annotated genome.
Figure 1.

The distribution of GC3 content in Citrus species, P. trifoliata and A. thaliana genes. The GC3 content showed unimodal distribution.
To look for biased association between the nucleotide content and codon usage, we plotted Nc against GC3s, also described as the Nc plot (Fig. 2) that has been widely demonstrated as an important parameter to evaluate codon usage variation among genes, as this codon usage index has a definite relationship with the GC3 (more compositionally biased DNA is expected to encode a smaller subset of codons).23,42 Wright23 argued that the comparison of actual distribution of genes, with expected distribution under no selection could be indicative, if CUB of genes has some other influences other than compositional constraints. A significant correlation was found between Nc and GC3s (0.466** C. sinensis; −0.025** Citrus aurantifolia; 0.070** Citrus aurantium; 0.125** C. clementina; 0.183** C. clementina × Citrus tangerina; 0.184** Citrus jambhiri; 0.179** Citrus japonica var. margarita; −0.048** Citrus limettioides; −0.031** Citrus limonia; 0.093** Citrus medica; 0.216** Citrus reshni; 0.061** C. reticulata; 0.101** C. reticulata × Citrus temple; 0.119** C. sinensis × P. trifoliata; −0.213** Citrus sunki; 0.061** P. trifoliata; 0.139** Citrus unshiu; 0.160** Citrus paradisi; 0.212** C. paradisi × P. trifoliata; **P-value < 0.01).
Figure 2.

Nc versus GC3s plot of Citrus species and P. trifoliata genes. The solid black line indicates the expected Nc value, if the codon bias is only due to GC3s. Species order: A = C. sinensis; B = C. aurantifolia; C = C. aurantium; D = C. clementina; E = C. clementina × C. tangerina; F = C. jambhiri; G = C. japonica var. margarita; H = C. limettioides; I = C. limonia; J = C. medica; K = C. reshni; L = C. reticulata; M = C. reticulata × C. temple; N = C. sinensis × P. trifoliata; O = C. sunki; P = P. trifoliata; Q = C. unshiu; R = C. paradisi; and S = C. paradisi × P. trifoliata.
We noted that most genes tend to lie below the standard trajectory path and are poised towards GC3s, which clearly demonstrates that MB is acting as a major factor for the wide variation in codon usage across the Citrus species and P. trifoliata (Fig. 2). However, there might be additional factors that might influence codon usage across these species. This dependence of codon usage on genome base composition (AT- or GC-richness) has been previously reported in several unicellular genomes.43,44 In a genome-wide analysis of eubacterial and archeal genomes, it has been suggested that genome-wide variance in codon usage is primarily due to MB as the GC content shows wide variation along the isochors.17 Simultaneously, alternative views associate GC variability with various factors such as transcriptional optimization, methylation, recombination, and horizontal gene transfer.45 It can be inferred that the cloning of orthologs and homologues conserved across the Citrus species and P. trifoliata that are AT rich and have low Nc values will require few degenerate primers. On the contrary, genes that are GC rich and having high GC values will require more degenerate primers for enhancement of cloning efficiency.
3.2. Correspondence analysis
It has been reported that there is a significant heterogeneity within the genes and genomes.33,46 A heat map was constructed using the observed RSCU values for all the 59 informative codons (excluding the Met, Trp, and the stop codons) using the average linkage clustering method (Fig. 3). Heat map and the supporting values (Supplementary Table 1) clearly define that the global codon usage is biased towards AT richness. We have partitioned the genes based on the high and low GC3s content to see the global pattern of deviation across the two principal axes displaying the major variation in accordance with the synonymous GC3s (Supplementary Fig. 1).
Figure 3.

Heat map of the average RSCU of the 59 degenerate codons in the Citrus species and P. trifoliata using Euclidean distance and average linkage clustering module.
Correspondence analysis showed that relative inertia tends to decrease along the axes, and in all the studies species, it was observed that axis 1 (COA/RSCU) contributed to the major portion of relative inertia indicating that the major trend of codon usage variation is associated with axis 1 (10.75 C. sinensis; 12.15 C. aurantifolia; 12.27 C. aurantium; 14.54 C. clementina; 6.65 C. clementina × C. tangrina; 11.62 C. jambhiri; 7.01 C. japonica var. margarita; 10.41 C. limettioides; 14.66 C. limonia; 11.24 C. medica; 10.67 C. reshni; 12.54 C. reticulata; 8.66 C. reticulata × C. temple; 10.85 C. sinensis × P. trifoliata; 13.61 C. sunki; 12.06 P. trifoliata; 9.11 C. unshiu; 8.72 C. paradisi; 10.75 C. paradisi × P. trifoliata). High significant correlation (+/−) was also observed between axis 1 and GC3s (0.860**, C. sinensis; 0.888**, C. aurantifolia; 0.887**, C. aurantium; 0.903**, −0.708** C. clementina; −0.836**, C. clementina × C. tangrina; −0.772**, C. jambhiri; −0.831**, C. japonica var. margarita; −0.803**, C. limettioides; 0.849**, C. limonia; 0.894**, C. medica; −0.855**, C. reshni; 0.894**, C. reticulata; 0.846**, C. reticulata × C. temple; −0.884**, C. sinensis × P. trifoliata; 0.806**, C. sunki; 0.879**, P. trifoliata; 0.834**, C. unshiu; −0.848**, C. paradisi; −0.879**, C. paradisi × P. trifoliata; **P-value < 0.01). The observed results indicate dominance of MB in the Citrus species and P. trifoliata and suggest that the variation in the usage of synonymous codons among the genes in Citrus species and P. trifoliata is largely a biased representation of nucleotide content of the genes.
3.3. Identification of optimal codons in Citrus species and P. trifoliata
Earlier and recent reports suggest that the usage of optimal codons and balanced codon usage enhances the efficiency of translation by increasing the translation rate of the preferred codons over the other synonymous codon choices.47,48 Genes using optimal codons have higher translation rate as compared to genes using non-optimal codons, which in turn increases the ribosome usage efficiency and potentially reduces the ribosome drop off.22,49,50 ESTs constitute partial transcriptome representation correlated with gene abundance and expression.51,52 In 14 Citrus species (C. aurantifolia; C. aurantium; C. clementina; C. jambhiri; C. limettioides; C. limonia; C. medica; C. reshni; C. reticulata; C. reticulata × C. temple; C. sinensis × P. trifoliata; C. unshiu; C. paradisi; C. paradisi × P. trifoliata) and P. trifoliata, we extracted the expression of ESTs count using the CAP3 assembly (.ace files) and compared the usage of codons in accordance with earlier reports.53–55 We also selected the ribosomal protein-encoding genes and quantified the association of the ribosomal encoding genes with axis 1 (COA/RSCU) and axis 2 (COA/RSCU) of correspondence analysis.
A star map showing optimal codons was constructed taking 10% of the genes from the extreme tails of multivariate analysis (Table 2), and several distinct trends of optimal codons were detected. In an earlier study,38 optimal codons in Citrus were estimated based on the correspondence analysis of codon usage, and the relative frequency of synonymous codon using 177 CDS and A- or T-ending optimal codons (TAA, GCT, GAT, CTT, AGG, AGA, and GTT) was observed. Overall, we observed that the trend of optimal codons usage was not conserved across all analysed species. However, in our analyses, we have observed that GCT (∼1.57; RSCU) and GGT (∼1.09; RSCU) representing Alanine and Glycine were found to be evolutionary conserved across all the species. Because both these optimal codons are T-ending codons, which is an indication of the dominant role of the MB in the conservation of the A- or T-ending codons, it potentially represents the biased genome composition. However, for the other observed optimal codons, pattern genomic composition was not able to explain the deviation. We further observed that for most amino acids with 2- to 6-fold degeneracy level, there has been a general preference for the usage of two or more codons as optimal codons. For example, in Glycine, two highly distributed optimal codons GGA and GGT were identified and they could be classified as the primary and secondary optimal codons preferentially based on the RSCU (GGA, ∼1.16 and GGT, ∼1.09). A curious trend observed among the Citrus species and P. trifoliata is that Leucine is frequently encoded optimally by a G-ending codon (TTG; ∼RSCU: 1.49) in four species (C. sinensis, C. clementina, C. reticulata, and P. trifoliata) rather than synonymous A- or T-ending codon. However, in other species, CTT (∼RSCU: 1.49) and CTC (∼RSCU: 0.89) (TTA, RSCU: 1.16; CTT RSCU: 1.51) were the optimal codons.
Table 2.
Star map showing optimal codon distribution in Citrus species and P. trifoliata
| Codon/AA | A | B | C | D | E | F | G | H | I | J | K | L | M | N | O | P |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TTT/Phe | * | * | * | * | ||||||||||||
| TTC/Phe | * | * | * | * | * | * | * | * | * | * | ||||||
| TTA/Leu | * | * | * | * | ||||||||||||
| TTG/Leu | * | * | * | * | ||||||||||||
| CTT/Leu | * | * | * | * | * | * | * | * | * | * | * | * | * | * | ||
| CTC/Leu | * | * | * | * | * | * | * | * | * | * | * | |||||
| CTA/Leu | * | * | * | * | * | |||||||||||
| CTG/Leu | * | * | * | |||||||||||||
| ATT/Ile | * | * | * | * | * | |||||||||||
| ATC/Ile | * | * | * | * | * | * | * | * | * | * | * | * | * | |||
| ATA/Ile | * | * | * | |||||||||||||
| GTT/Val | * | * | * | * | * | * | * | * | ||||||||
| GTC/Val | * | * | * | * | * | * | * | * | * | * | * | * | * | |||
| GTA/Val | * | * | * | * | ||||||||||||
| GTG/Val | * | * | ||||||||||||||
| TCT/Ser | * | * | * | * | * | * | * | * | * | * | * | * | * | * | ||
| TCC/Ser | * | * | * | * | * | * | * | * | * | |||||||
| TCA/Ser | * | * | * | * | * | * | * | * | * | * | * | |||||
| TCG/Ser | * | * | ||||||||||||||
| AGT/Ser | * | * | * | * | * | |||||||||||
| AGC/Ser | * | * | * | |||||||||||||
| CCT/Pro | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | |
| CCC/Pro | * | * | * | * | * | * | * | |||||||||
| CCA/Pro | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | |
| CCG/Pro | * | * | ||||||||||||||
| ACT/Thr | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | |
| ACC/Thr | * | * | * | * | * | * | * | * | * | * | * | |||||
| ACA/Thr | * | * | * | * | * | |||||||||||
| ACG/Thr | * | * | ||||||||||||||
| GCT/Ala | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * |
| GCC/Ala | * | * | * | * | * | * | * | * | * | * | * | |||||
| GCA/Ala | * | * | * | * | * | * | ||||||||||
| GCG/Ala | ||||||||||||||||
| TAT/Tyr | * | * | * | |||||||||||||
| TAC/Tyr | * | * | * | * | * | * | * | |||||||||
| CAT/His | * | * | * | * | * | * | ||||||||||
| CAC/His | * | * | ||||||||||||||
| CAA/Gln | * | * | * | * | ||||||||||||
| CAG/Gln | * | * | * | * | * | * | ||||||||||
| AAT/Asn | * | * | * | * | * | * | * | * | * | |||||||
| AAA/Lys | * | * | * | |||||||||||||
| AAG/Lys | * | * | * | * | * | * | * | * | * | * | * | * | ||||
| GAT/Asp | * | * | * | * | * | * | * | |||||||||
| GAA/Glu | * | * | * | * | ||||||||||||
| GAG/Glu | * | * | * | * | * | * | ||||||||||
| TGT/Cys | * | * | * | |||||||||||||
| TGC/Cys | * | * | * | * | * | * | * | * | * | * | ||||||
| CGT/Arg | * | * | * | * | * | * | * | * | * | * | * | * | * | |||
| CGC/Arg | * | * | * | * | * | * | * | * | * | * | * | * | ||||
| CGA/Arg | * | * | * | * | * | * | ||||||||||
| CGG/Arg | ||||||||||||||||
| AGA/Arg | * | * | * | * | * | * | * | |||||||||
| AGG/Arg | * | * | ||||||||||||||
| GGT/Gly | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * | * |
| GGC/Gly | * | * | * | * | * | |||||||||||
| GGA/Gly | * | * | * | * | * | * | * | * | * |
Species are in the following order: A = C. sinensis; B = C. aurantifolia; C = C. aurantium; D = C. clementina; E = C. jambhiri; F = C. limettioides; G = C. limonia; H = C. medica; I = C. reshni; J = C. reticulata; K = C. reticulata × C. temple; L = C. sinensis × P. trifoliata; M = P. trifoliata; N = C. unshiu; O = C. paradisi; and P = C. paradisi × P. trifoliata. AA represents amino acid.
We found deviations in the usage of optimal codons encoding Lysine. For example, Lysine is frequently encoded by AAA (∼RSCU: 0.98) as optimal codon in three species (C. sinensis, C. reticulata, and P. trifoliata), whereas AAG (∼RSCU: 1.01) was found to be the optimal codon for Lysine in rest of the Citrus species. In Arginine, we observed that in two species (C. aurantifolia and C. paradisi × P. trifoliata), AGG represents the potential optimal codon instead of the synonymous AGA. But based on the RSCU values, AGA (∼RSCU: 1.66) was assumed to be more dominant over the AGG codon (∼RSCU: 1.52) at the respective levels of significance P-value < 0.01 and P-value < 0.05. Using a mutation and selection model, Knight et al.56 demonstrated that ‘pairs of species with convergent GC content might also evolve convergent protein sequences, especially at functionally unconstrained positions’. For instance, the frequencies of both Lysine and Arginine are highly anti-correlated with GC content, and Lysine and Arginine can easily be substituted for one another in proteins. This observation explains the inclusion of Lysine and Arginine in our study and is well supported by an earlier study in nematodes.57 It has been proposed that Arginine and Leucine have a tendency to show different codon usage patterns because of the prevalence of the synonymous GC substitutions in the first and the third codon position.58 Recently, it has been postulated that codon optimization significantly enhanced MIR gene expression in Solanum lycopersicum cv. Microtom,59 which potentially depicts the importance and the usage of the optimal codons in gene expression and transgenics. To our knowledge, this is the first time large-scale identification of optimal codons in Citrus species and P. trifoliata, which could serve as a model repertoire to enhance the transformation efficiency.
3.4. Role of other selective constraints on codon bias in Citrus species and P. trifoliata
A recent study has revealed that MB deeply influences the folding stability of proteins, making proteins on the average less hydrophobic and, therefore, less stable with respect to unfolding and also less susceptible to misfolding and aggregation.60 To identify the potential effects of Gravy and aromaticity, we computed non-parametric correlation coefficients between axis 1 (COA/RSCU) and Gravy or aromaticity scores for all studied species. We observed that Gravy and aromaticity played a minor role in shaping the variation of codon usage across Citrus species and P. trifoliata. In the case of P. trifoliata, no significant correlation was observed for aromaticity, suggesting that aromaticity has no significant role in shaping the codon usage variation in this genus.
3.5. Variations in GC3 across Citrus species and P. trifoliata
Recent reports suggest that GC3 composition and GC gradient are acting as major factors along the orientation of transcription in monocots. It has been also suggested that GC composition is vital for understanding chromatin remodelling, gene expression, and recombination.45,61 Some studies failed to depict GC gradient along the genes of dicot plants using A. thaliana as a model.61 The reason for this failure is that high and low GC3 genes have opposite gradients along the genes, and when high and low GC3 genes are clumped together, the effect disappears. In Citrus species and P. trifoliata, unimodal bell-shaped distribution of GC3, centred at 0.39, is considered to be a typical mode of GC3 distribution for dicot species. We selected the top and bottom 5% of genes across each studied Citrus species and P. trifoliata, and strikingly distinct gradients of GC3 and CG3 skew for high and low GC3 genes were revealed. Genes with high GC3 showed higher level of GC3 codons in their middle coding regions than in terminal coding regions. In addition, high GC3 genes have a preference for C over G in the middle coding regions, and this preference was not found in the 3′-coding end ∼100 bp in size (Fig. 4; Supplementary Fig. 2).
Figure 4.

GC3 gradient across Citrus species and P. trifoliata. Species order: A = C. sinensis; B = C. aurantifolia; C = C. aurantium; D = C. clementina; E = C. clementina × C. tangerina; F = C. jambhiri; G = C. japonica var. margarita; H = C. limettioides; I = C. limonia; J = C. medica; K = C. reshni; L = C. reticulata; M = C. reticulata × C. temple; N = C. sinensis × P. trifoliata; O = C. sunki; P = P. trifoliata; Q = C. unshiu; R = C. paradisi; and S = C. paradisi × P. trifoliata. These plots show gradients of GC3 for 5% of GC3-rich and GC3-poor genes for all analysed genomes. Gradients from 5′ and 3′ ends of the CDS are shown in the same plot, separated by a vertical line. For all genomes, GC3-rich genes become more GC3 rich towards the middle of the CDS.
It was previously reported that stress-related genes in grasses are GC3-rich.45 We stratified all Citrus species and P. trifoliata genes into three groups by GC3: rich (top 5%), poor (bottom 5%), and medium (middle 90%). We observed that among the four major economically important Citrus species, in C. reticulata (1718), C. clementina (1522), and P. trifoliata (951), most of the GC3-rich genes were related to stress in comparison to the total number of observed stress-related genes, but in the case of C. sinensis, it was observed that a high number of medium GC3 genes (1857) were also abundant in stress-related genes. The relative abundance of the medium GC3 genes in C. sinensis may be due to the time-course adaptability of this species during the period of evolution, suggesting an adaptive evolution towards the stress.
A high abundance of stress-related genes in C. reticulata and P. trifoliata may be closely related to their high stress-tolerance trait and especially to P. trifoliata that has the highest cold-resistant character among Citrus species and its wild relatives. The occurrence of the high GC3 genes in all these species suggests an association between DNA methylation and GC3 as it has been previously suggested that high GC3 composition has been influenced by the GC mismatch-repair mechanism that is dominant in stress-associated genes as the absence of this positive bias may lead to the loss of the recombination repair mechanism and could be detrimental to the plant adaptation in the evolving stress conditions. Genomic regions under higher selective pressure are more frequently recombining, and as a result relative increase in GC3 content can be observed.45 As shown in Supplementary Table 2 and Fig. 5, in all Citrus species and P. trifoliata, the ratio of stress to non-stress-related genes in GC3-rich group was elevated in comparison to the GC3-medium group and depleted in the GC3-poor group. It is worth noting that GC3-poor group has fewer genes functionally described as stress-related when compared with the GC3-rich group.
Figure 5.

Distribution of stress-related genes in Citrus species and P. trifoliata according to the GO of Arabidopsis.
In all the studied species, GC3-rich and -poor genes have different trends from 5′ to 3′ end of the genes. GC3-rich genes become even more GC3 rich, and GC3-poor genes become more GC3 poor. Firstly, the GC3-rich group has more stress related genes than the GC3-poor group. Secondly, the GC3-rich group has a positive gradient of GC3 from 5′ and 3′ flanks to the middle portion of the CDS. The low-GC3 group has a negative gradient of GC3 from 5′ and 3′ flanks to the middle portion of the CDS. Thirdly, rich- and poor-GC3 groups showed different CG3 skew trends along the CDS: GC3-rich genes favour C3 over G3, and this preference is most pronounced in first 150 codons, whereas GC3-poor genes favour G3 over C3 (Supplementary Fig. 2). It was suggested that transcriptional and translational optimization of stress-related and tissue-specific genes is the major force responsible for maintaining high GC3 content.46 The replacement of the AT pair with the GC pair at the third codon position enhances the transcriptional activity that in turn enhances the array of ribosomal-binding proteins. Stayssman et al.62 provide a strong positive correlation between the methylation of internal unmethylated regions and expression of the host gene and postulated that the genes with high GC3 provide more targets for methylation. Genes involved in response to various stresses need to produce a protein with a faster rate as a response to external stimulus. This results in shortened length of transcript and preference for G and C (and especially, C) in the third position of the codon (to avoid abortive transcription and ribosome congestion). In addition, high GC3 genes have more methylation targets that allow for fine-tuning of transcriptional regulation.58
3.6. Evolution and coevolution of nucleotide and amino acid composition
We analysed the relationship between amino acid sequence divergence and variation in GC3, to test the effect of evolutionary divergence on codon usage. We observed that there is an overall positive correlation (0.43) between relative change in GC3 and normalized Hamming distance (Fig. 6). This was expected because more diverse amino acid sequences are likely to have more diverse nucleotide sequences. The trend is not uniform: for the genes with GC3 in the range between 0.5 and 0.6, the correlation is 0.596 and it drops to 0.32 for GC3 above 0.8 or below 0.4. There are some pairs of organisms that have negative correlation between GC3 and Hamming distance (C. jambhiri: C. limettioides, C. limonia: C. sinensis × P. trifoliata, C. aurantiifolia: C. jambhiri, C. jambhiri: C. paradisi, C. aurantiifolia: C. paradisi, C. jambhiri: P. trifoliata, C. medica: C. sinensis × P. trifoliata, C. jambhiri: C. unshiu, C. jambhiri: C. reshni, C. japonica var. margarita: C. paradisi, C. aurantiifolia: C. clementina × C. tangerina, C. aurantium: C. jambhiri, C. clementina × C. tangerina: C. jambhiri). Note that in the list of negative correlations, C. jambhiri occurs eight times.
Figure 6.

Hamming distance versus change in GC3 visualization across Citrus species and P. trifoliata. Relative difference GC3 composition between genes A and B, defined as GC3 (A)-GC3 (B)/[GC3 (A) + GC3 (B)] is positively correlated with the Hamming distance between corresponding protein sequences, calculated as 1–[I (AB) + I (BA)]/[I (AA) + I (BB)], where I (AB) is the number of identities in alignment of protein A to protein B. The resulting Hamming distance was rounded to 1 DP, and an average difference in GC3 was computed for each category.
Genes that have low GC3 content in C. jambhiri (<0.4) tend to have negative correlation between difference in GC3 and Hamming distance, and genes with high GC3 content (>0.6) are slightly positively correlated. This atypical behaviour can be explained by larger evolutionary distance between C. jambhiri and other species analysed in the present investigation. The highest correlation was observed between C. japonica var. margarita and C. limonia (ρ = 0.86, number of orthologous pairs N = 12, t-statistics t = 5.22, P-value = 0.0002). C. japonica var. margarita and C. limonia are evolutionary distant, and the number of orthologous pairs is only N = 12. Hence, a high value of correlation coefficient may be a result of an evolutionary pressure to conserve codon usage for a selected subset of conserved proteins. We observed that pairs of species having positive correlation between GC3 and Hamming distance are enriched in translation, transport, proteolysis, photorespiration, and genes involved in photosynthesis; pairs of species with negative correlation are enriched in stress-response genes.
Similar patterns of hierarchal clustering were depicted; when we clustered the species based on co-orthologous genes using genome-predicted and frame-corrected reconstructed proteins of the Citrus species and P. trifoliata. The phylogenetic clades were rerooted using P. trifoliata as an outgroup, and it was observed that C. sinensis, C. clementina, and C. reticulata all belong to the same clade, which are in accordance with the previous reports using major intrinsic proteins (XIP subfamily of aquaporins) and further support the conserved homology between these two species.63 These supportive views suggest that the identified frame-corrected coding regions are trustworthy and accurate to be used for the predictions in the species, with no genome information available till now (Supplementary Fig. 3A and B).
Because a nucleotide bias can lead to an overall bias in amino acid composition of proteins, it is possible that a genome with nucleotide bias may have introduced atypical amino acid substitutions in its proteome. Hence, AT-rich coding sequences would encode proteins rich in FYMINK amino acids (Phenylalanine, Tyrosine, Methionine, Isoleucine, Asparagine, and Lysine), whereas GC-rich coding sequences would produce proteins containing high levels of GARP amino acids (Glycine, Alanine, Arginine, and Proline). Because Citrus species and P. trifoliata are AT-rich, we found higher proportion of FYMINK [(FYMINK and CDSGC (y = 4.1431x + 22.341, R2 = 0.9099); GARP and CDSGC (y = 3.0687x + 45.414, R2 = 0.7803); P-value < 10−9)].
Because the third synonymous position does not influence the protein sequence, we computed the correlation coefficient between the FYMINK and GARP parameters and the GC3. This correlation is an important factor in describing nucleotide bias at synonymous and non-synonymous sites. We observed a high significant correlation between nucleotide composition and amino acid composition at the third synonymous position [(FYMINK and CDSGC3 (y = 10.862x + 142.4, R2 = 0.9296); GARP and CDSGC3 (y = 9.842x + 212.15, R2 = 0.7495)], suggesting that nucleotide bias has an influence on amino acid composition in Citrus species and P. trifoliata.
In summary, we found prevalence of A- or T-ending codons with an exception for Lysine and Arginine in P. trifoliata. We suggest that although the patterns of optimal codons were not conserved, two codons (GCT and GGT) were found to be conserved across all the Citrus species and P. trifoliata. We analysed GC3-rich and -poor genes and their association with stress, and our results provided novel insights into stress evolution of stress adaptation in Citrus species and P. trifoliata in accordance with the GC3 biology. This research is critical for all Citrus species, as it might facilitate understanding of genome dynamics and evolution in Citrus and P. trifoliata, transformation of interested target genes, designing multitargeting gene systems, and ultimately genetically improving these important fruit crops.
Supplementary data
Supplementary Data are available at www.dnaresearch.oxfordjournals.org.
Funding
This research was financially supported by the Ministry of Science and Technology of China (nos. 2011AA100205, 2011CB100606), the National NSF of China, and the Ministry of Agriculture of China (no. 200903044).
Supplementary Material
Acknowledgements
The authors thank Dr Kenta Nakai (the handling editor) and the two anonymous reviewers for helpful and constructive comments and Dr Yuepeng Han, Wuhan Botanic Garden (CAS) for critical reading of the manuscript. G.S. thanks the computational support provided from Research and Innovation Center, CRI-FEM, IASMA, Italy. T.V.T. thanks the Research Investment Scheme, University of Glamorgan, UK.
Footnotes
Edited by Prof. Kenta Nakai
References
- 1.Serres-Giardi L., Belkhir K., David J., Glémin S. Patterns and evolution of nucleotide landscapes in seed plants. Plant Cell. 2012;24:1379–97. doi: 10.1105/tpc.111.093674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hershberg R., Petrov D.A. General rules for optimal codon choice. PLoS Genet. 2009;5:e1000556. doi: 10.1371/journal.pgen.1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grantham R., Gautier C., Gouy M., Jacobzone M., Mercier R. Codon catalog usage is a genome strategy modulated for gene expressivity. Nucleic Acids Res. 1981;9:43–74. doi: 10.1093/nar/9.1.213-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Akashi H. Gene expression and molecular evolution. Curr. Opin. Genet. Dev. 2001;11:660–6. doi: 10.1016/s0959-437x(00)00250-1. [DOI] [PubMed] [Google Scholar]
- 5.Aragonès L., Guix S., Ribes E., Bosch A., Pintó R.M. Fine-tuning translation kinetics selection as the driving force of codon usage bias in the hepatitis A virus capsid. PLoS Pathol. 2010;6:e1000797. doi: 10.1371/journal.ppat.1000797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Medigue C., Rouxel T., Vigier P., Henaut A., Danchin A. Evidence for horizontal gene transfer in Escherichia coli speciation. J. Mol. Biol. 1991;222:851–6. doi: 10.1016/0022-2836(91)90575-q. [DOI] [PubMed] [Google Scholar]
- 7.Pascal G., M'edigue C., Danchin A. Universal biases in protein composition of model prokaryotes. Proteins. 2005;60:27–35. doi: 10.1002/prot.20475. [DOI] [PubMed] [Google Scholar]
- 8.Duret L. Evolution of synonymous codon usage in metazoans. Curr. Opin. Genet. Dev. 2002;12:640–9. doi: 10.1016/s0959-437x(02)00353-2. [DOI] [PubMed] [Google Scholar]
- 9.Bulmer M. The selection-mutation drift theory of synonymous codon usage. Genetics. 1991;129:897–907. doi: 10.1093/genetics/129.3.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharp P.M., Matassi G. Codon usage and genome evolution. Curr. Opin. Genet. Dev. 1994;4:851–60. doi: 10.1016/0959-437x(94)90070-1. [DOI] [PubMed] [Google Scholar]
- 11.Akashi H., Eyre-Walker A. Translational selection and molecular evolution. Curr. Opin. Genet. Dev. 1998;8:688–93. doi: 10.1016/s0959-437x(98)80038-5. [DOI] [PubMed] [Google Scholar]
- 12.Gupta S.K., Ghosh T.C. Gene expressivity is the main factor in dictating the codon usage variation among the genes in Pseudomonas aeruginosa. Gene. 2001;273:63–70. doi: 10.1016/s0378-1119(01)00576-5. [DOI] [PubMed] [Google Scholar]
- 13.Hershberg R., Petrov D.A. Selection on codon bias. Annu. Rev. Genet. 2008;42:287–99. doi: 10.1146/annurev.genet.42.110807.091442. [DOI] [PubMed] [Google Scholar]
- 14.Harrison R.J., Charlesworth B. Biased gene conversion affects patterns of codon usage and amino acid usage in the Saccharomyces sensu stricto group of yeasts. Mol. Biol. Evol. 2011;28:117–29. doi: 10.1093/molbev/msq191. [DOI] [PubMed] [Google Scholar]
- 15.Hill W.G., Robertson A. The effect of linkage on limits to artificial selection. Genet. Res. 1966;8:269–94. [PubMed] [Google Scholar]
- 16.McInerney J.O. Replicational and transcriptional selection on codon usage in Borrelia burgdorferi. Proc. Natl. Acad. Sci. USA. 1998;95:10698–703. doi: 10.1073/pnas.95.18.10698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen S.L., Lee W., Hottes A.K., Shapiro L., McAdams H.H. Codon usage between genomes is constrained by genome-wide mutational processes. Proc. Natl. Acad. Sci. USA. 2004;101:3480–85. doi: 10.1073/pnas.0307827100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qin H., Biao W.W., Comeron J.M., Kreitman M., Li W.H. Intragenic spatial patterns of codon usage bias in prokaryotic and eukaryotic genomes. Genetics. 2004;168:2245–60. doi: 10.1534/genetics.104.030866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stoletzki N., Eyre-Walker A. Synonymous codon usage in Escherichia coli: selection for translational accuracy. Mol. Biol. Evol. 2007;24:374–81. doi: 10.1093/molbev/msl166. [DOI] [PubMed] [Google Scholar]
- 20.Lin Y.S., Byrnes J.K., Hwang J.K., Li W.H. Codon-usage bias versus gene conversion in the evolution of yeast duplicate genes. Proc. Natl. Acad. Sci. USA. 2006;103:14412–16. doi: 10.1073/pnas.0606348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou T., Sun X., Lu Z. Synonymous codon usage in environmental Chlamydia UWE25 reflects an evolutional divergence from pathogenic Chlamydiae. Gene. 2006;368:117–25. doi: 10.1016/j.gene.2005.10.035. [DOI] [PubMed] [Google Scholar]
- 22.Sharp P.M., Emery L.R., Zeng K. Forces that influence the evolution of codon bias. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010;365:1203–12. doi: 10.1098/rstb.2009.0305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wright F. The ‘effective number of codons’ used in a gene. Gene. 1990;87:23–29. doi: 10.1016/0378-1119(90)90491-9. [DOI] [PubMed] [Google Scholar]
- 24.Wan X.F., Xu D., Kleinhofs A., Zhou J. Quantitative relationship between synonymous bias and GC composition across unicellular genomes. BMC Evol. Biol. 2004;4:19. doi: 10.1186/1471-2148-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu Q., Chen L.L., Ruan X., et al. The draft genome of sweet orange (Citrus sinensis) Nat. Genet. 2012 doi: 10.1038/ng.2472. doi:10.1038/ng.2472. [DOI] [PubMed] [Google Scholar]
- 26.Huang X., Madan A. CAP3: a DNA sequence assembly program. Genome Res. 1999;9:868–77. doi: 10.1101/gr.9.9.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gouzy J., Carrere S., Schiex T. FrameDP: sensitive peptide detection on noisy matured sequences. Bioinformatics. 2009;25:670–71. doi: 10.1093/bioinformatics/btp024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schiex T., Gouzy J., Moisan A., de Oliveira Y. FrameD: a flexible program for quality check and gene prediction in prokaryotic genomes and noisy matured eukaryotic sequences. Nucleic Acids Res. 2003;31:373–81. doi: 10.1093/nar/gkg610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Altschul S.F., Madden T.L., Schäffer A.A., et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Greenacre M.J. Theory and Application of Correspondence Analysis. London: Academic Press; 1984. p. 223. [Google Scholar]
- 31.Sharp P.M., Li W.H. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Biol. 1986;24:28–38. doi: 10.1007/BF02099948. [DOI] [PubMed] [Google Scholar]
- 32.Kyte J., Doolittle R. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982;157:105–32. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 33.Ikemura T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol. Biol. Evol. 1985;2:13–34. doi: 10.1093/oxfordjournals.molbev.a040335. [DOI] [PubMed] [Google Scholar]
- 34.Sablok G., Nayak K., Vazquez F., Tatarinova T.V. Synonymous codon usage, GC3 and evolutionary patterns across plastomes of three pooid model species – emerging grass genome models for monocots. Mol. Biotechnol. 2011;49:116–28. doi: 10.1007/s12033-011-9383-9. [DOI] [PubMed] [Google Scholar]
- 35.Lechner M., Findeiss S., Steiner L., Marz M., Stadler P.F., Prohaska S.J. Proteinortho: detection of (co-) orthologs in large-scale analysis. BMC Bioinformatics. 2011;12:124. doi: 10.1186/1471-2105-12-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Foster P.G., Jermiin L.S., Hickey D.A. Nucleotide composition bias affects amino acid content in proteins coded by animal mitochondria. J. Mol. Evol. 1997;44:282–88. doi: 10.1007/pl00006145. [DOI] [PubMed] [Google Scholar]
- 37.Singer G.A.C., Hickey D.A. Nucleotide bias causes a genomewide bias in the amino acid composition of proteins. Mol. Biol. Evol. 2000;17:1581–88. doi: 10.1093/oxfordjournals.molbev.a026257. [DOI] [PubMed] [Google Scholar]
- 38.Hu G.B., Zhang S.L., Xu C.J., Lin S.Q. Analysis of codon usage in Citrus. J. Fruit Sci. 2006;23:479–85. [Google Scholar]
- 39.Sueoka N. On the evolution of informational macromolecules. In: Bryson V., Vogel H.J., editors. Evolving Genes and Proteins. New York: Academic Press; 1964. pp. 479–96. [Google Scholar]
- 40.Sueoka N., Kawanishi Y. DNA G+ C content of the third codon position and codon usage biases of human genes. Gene. 2000;261:53–62. doi: 10.1016/s0378-1119(00)00480-7. [DOI] [PubMed] [Google Scholar]
- 41.Sueoka N. Directional mutation pressure and neutral molecular evolution. Proc. Natl. Acad. Sci. USA. 1988;85:2653–57. doi: 10.1073/pnas.85.8.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang H.C., Hickey D.A. Rapid divergence of codon usage patterns within the rice genome. BMC Evol. Biol. 2007;7(Suppl. 1):S6. doi: 10.1186/1471-2148-7-S1-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gupta S.K., Bhattacharyya T.K., Ghosh T.C. Synonymous codon usage in Lactococcus lactis: mutational bias versus translational selection. J. Biomol. Struct. Dyn. 2004;21:527–36. doi: 10.1080/07391102.2004.10506946. [DOI] [PubMed] [Google Scholar]
- 44.Wright F., Bibb M.J. Codon usage in the G+C-rich Streptomyces genome. Gene. 1992;113:55–65. doi: 10.1016/0378-1119(92)90669-g. [DOI] [PubMed] [Google Scholar]
- 45.Tatarinova T., Alexandrov N., Bouck J., Feldmann K. GC3 biology in corn, rice, sorghum and other grasses. BMC Genomics. 2010;11:308. doi: 10.1186/1471-2164-11-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sharp P.M., Cowe E., Higgins D.G., Shields D.C., Wolfe K.H., Wright F. Codon usage in Escherichia coli, Bacillus subtilis, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Drosophila melanogaster and Homo sapiens; a review of the considerable within-species diversity. Nucleic Acids Res. 1988;16:8207–11. doi: 10.1093/nar/16.17.8207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Andersson S.G.E., Kurland C.G. Codon preferences in free-living microorganisms. Microbiol. Rev. 1990;54:198–210. doi: 10.1128/mr.54.2.198-210.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qian W., Yang J-R., Pearson N.M., Maclean C., Zhang J. Balanced codon usage optimizes eukaryotic translational efficiency. PLoS Genet. 2012;8:e1002603. doi: 10.1371/journal.pgen.1002603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sørensen M.A., Pedersen S. Absolute in vivo translation rates of individual codons in Escherichia coli: the two glutamic acid codons GAA and GAG are translated with a threefold difference in rate. J. Mol. Biol. 1991;222:265–80. doi: 10.1016/0022-2836(91)90211-n. [DOI] [PubMed] [Google Scholar]
- 50.Kudla G., Murray A.W., Tollervey D., Plotkin J.B. Coding-sequence determinants of gene expression in Escherichia coli. Science. 2009;324:255–58. doi: 10.1126/science.1170160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Audic S., Claverie J.M. The significance of digital gene expression profiles. Genome Res. 1997;7:986–95. doi: 10.1101/gr.7.10.986. [DOI] [PubMed] [Google Scholar]
- 52.Munoz E.T., Bogarad L.D., Deem M.W. Microarray and EST database estimates of mRNA expression levels differ: the protein length versus expression curve for C. elegans. BMC Genomics. 2004;5:30. doi: 10.1186/1471-2164-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cutter A.D., Wasmuth J.D., Washington N.L. Patterns of molecular evolution in Caenorhabditis preclude ancient origins of selfing. Genetics. 2008;178:2093–104. doi: 10.1534/genetics.107.085787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ingvarsson P.D. Molecular evolution of synonymous codon usage in Populus. BMC Evol. Biol. 2008;8:307. doi: 10.1186/1471-2148-8-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Whittle C.A., Sun Y., Johannesson H. Evolution of synonymous codon usage in Neurospora tetrasperma and Neurospora discrete. Genome Biol. Evol. 2011;3:332–43. doi: 10.1093/gbe/evr018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Knight R.D., Freeland S.J., Landweber L.F. A simple model based on mutation and selection explains trends in codon and amino acid usage and GC composition within and across genomes. Genome Biol. 2001;2:research0010. doi: 10.1186/gb-2001-2-4-research0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mitreva M., Wendl M.C., Martin J., et al. Codon usage patterns in Nematoda: analysis based on over 25 million codons in thirty-two species. Genome Biol. 2006;7:R75. doi: 10.1186/gb-2006-7-8-r75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Palidwor G.A., Perkins T.J., Xia X. A general model of codon bias due to GC mutational bias. PLoS ONE. 2010;5:e13431. doi: 10.1371/journal.pone.0013431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hiwasa-Tanase K., Nyarubona M., Hirai T., Kato K., Ichikawa T., Ezura H. High-level accumulation of recombinant miraculin protein in transgenic tomatoes expressing a synthetic miraculin gene with optimized codon usage terminated by the native miraculin terminator. Plant Cell Rep. 2011;30:113–24. doi: 10.1007/s00299-010-0949-y. [DOI] [PubMed] [Google Scholar]
- 60.Mendez R., Fritsche M., Porto M., Bastolla U. Mutation bias favors protein folding stability in the evolution of small populations. PLoS Comput. Biol. 2010;6:e1000767. doi: 10.1371/journal.pcbi.1000767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jiang N., Fergusona A.A., Slotkinb R.K., Lischc D. Pack-mutator-like transposable elements (Pack-MULEs) induce directional modification of genes through biased insertion and DNA acquisition. Proc. Natl. Acad. Sci. USA. 2011;108:1537–42. doi: 10.1073/pnas.1010814108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stayssman R., Nejman D., Roberts D., et al. Developmental programming of CpG island methylation profiles in the human genome. Nat. Struct. Mol. Biol. 2009;16:564–71. doi: 10.1038/nsmb.1594. [DOI] [PubMed] [Google Scholar]
- 63.Gupta A.B., Sankararamakrishnan R. Genome-wide analysis of major intrinsic proteins in the tree plant Populus trichocarpa: characterization of XIP subfamily of aquaporins from evolutionary perspective. BMC Plant Biol. 2009;9:134. doi: 10.1186/1471-2229-9-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.