

Abstract
Next-generation sequencing (NGS) allows faster acquisition of metagenomic data, but complete exploration of complex ecosystems is hindered by the extraordinary diversity of microorganisms. To reduce the environmental complexity, we created an innovative solution hybrid selection (SHS) method that is combined with NGS to characterize large DNA fragments harbouring biomarkers of interest. The quality of enrichment was evaluated after fragments containing the methyl coenzyme M reductase subunit A gene (mcrA), the biomarker of methanogenesis, were captured from a Methanosarcina strain and a metagenomic sample from a meromictic lake. The methanogen diversity was compared with direct metagenome and mcrA-based amplicon pyrosequencing strategies. The SHS approach resulted in the capture of DNA fragments up to 2.5 kb with an enrichment efficiency between 41 and 100%, depending on the sample complexity. Compared with direct metagenome and amplicons sequencing, SHS detected broader mcrA diversity, and it allowed efficient sampling of the rare biosphere and unknown sequences. In contrast to amplicon-based strategies, SHS is less biased and GC independent, and it recovered complete biomarker sequences in addition to conserved regions. Because this method can also isolate the regions flanking the target sequences, it could facilitate operon reconstructions.
Keywords: α-subunit of the methyl-coenzyme M reductase, metagenomics, sequence capture, 454 pyrosequencing, microbial diversity
1. Introduction
Microorganisms are extremely diverse and crucial for healthy, functioning biospheres.1,2 Although studies of isolated species have produced a great deal of information about microbial genetics, physiology, biotechnology and molecular biology, the diversity and structure of complex microbial communities are still poorly understood. This deficiency results from the inability to culture most microorganisms using standard microbiological techniques.1,3 Consequently, although there are most likely millions of bacterial species on the planet, only a few thousand have been formally described.4
Culture-independent techniques, such as metagenomics,5 circumvent the problem of unculturability and transcend previous studies on individual organisms to focus on microbial communities present in an environment. Metagenomics has enriched our knowledge of environmental microbiology through the structural (gene/species richness and distribution)6 and functional (metabolic)7 profiling of complex environmental microbial communities. Based on unselective (shotgun) or targeted (activity driven and sequence driven) methods, metagenomics links genome information with structure and function relationships within microbial populations.8,9
Recently developed next-generation sequencing (NGS) technologies recover genetic materials from environmental samples without the preparation of metagenomic clone libraries.10 Furthermore, they explore a greater amount of sequence information because they have higher throughput and lower costs than other methods.11 Nevertheless, Quince et al.12 showed that covering 90% of the species richness in some hyper-diverse environments could require 10—1000-fold increases in the current NGS sequencing efforts. In addition, the massive amount of short metagenomic sequence reads (between 20 and 700 bases depending on the platform) can be problematic for assembling and identifying complete coding DNA sequence and/or operon structure.13 One promising alternative is to reduce the environmental sample complexity by enriching the desired genomic target before sequencing.
Currently, several strategies of genomic-scale sequence enrichment have been reported.14 The more efficient methods rely on complementary nucleic acid capture probes that hybridize to the targeted DNA sequences. Two hybridization methods—solid phase15–17 and solution phase, also known as solution hybrid selection (SHS)18,19—can be used to ascertain genetic variation by specifically enriching and resequencing regions from complex eukaryotic genomes.
To the best of our knowledge, only high-throughput enrichment methods based on polymerase chain reaction (PCR) have been applied to target functional genes in complex environments.20 Because no current methods use oligonucleotide capture probes to specifically enrich targeted genes from a complex environmental genomic DNA (gDNA), we applied this methodology in the context of microbial ecology (Fig. 1A) to specifically capture DNA fragments harbouring known or unknown genetic biomarkers of interest (Fig. 1B). We hypothesized that the use of variant specific and explorative probes21,22 would more accurately define the overall biomarker diversity (including the rare biosphere and unknown sequences) and would facilitate the discovery of genes linked to the target sequences via the reconstruction of adjacent DNA regions. This method should lead to better diversity coverage that is not influenced by PCR biases, as generally occurs in amplicon sequencing.23,24 Because it is not limited to a specific DNA region (as in PCR enrichment), this strategy will increase the sequence coverage over target regions and lower the cost per target when compared with shotgun sequencing.
Figure 1.

Schematic comparison of targeted capture methods applied to classical direct selection method of individual genomic loci (human for instance) (A) and our new approach for metagenomics targeting (B). The enrichment through microarray and the SHS of large genomic regions within complex eukaryotic genomes, as described in A, uses specific tiling probes to target resequencing genomic loci for copy number variation (CNV) and single nucleotide polymorphism detection. Our SHS method (B) uses the design of specific variants and explorative probes across a targeted biomarker to specifically enrich large DNA fragments from complex metagenomic DNA. Captured DNA fragments are sequenced to explore biomarker diversity and adjacent flanking regions. The red rectangles indicate the targeted regions.
In the present study, we describe the first adaptation of the SHS capture method for the selective enrichment of a target-specific biomarker from a complex environmental metagenome. Methane (CH4) is an important radiative trace gas responsible for the greenhouse effect, and a significant proportion (6–16%) of the global natural methane emissions are released from freshwater lakes.25 We surveyed the methanogen diversity in a permanently stratified crater lake located in the French Massif Central (Lake Pavin). This original freshwater ecosystem is composed of an anoxic deep water layer (monimolimnion, ∼60–90 m depth) separated from the oxygenated upper layer (mixolimnion) by an intermediate layer (mesolimnion),26 where both the sediments and the anoxic water column contribute to methane production.27 We targeted the gene coding for the α-subunit of the methyl coenzyme M reductase (mcrA) that is involved in the final step of methanogenesis. This gene is arranged in a single transcriptional unit—the mcr operon—that is highly conserved among all methanogens.28,29 To highlight the broad benefits of the gene capture approach when compared with the more classical sequencing methods, three methods were used for pyrosequencing of an environmental sample: the SHS method, a classical random-shotgun metagenomic approach and an mcrA-targeted amplicon sequencing survey.
2. Materials and methods
2.1. Capture probe design and synthesis
Two sets of capture probes were designed. The first set targeted the mcrA gene from the Methanosarcina acetivorans C2A genome (GenBank accession no. AE010299), and the second set targeted the mcrA sequences pooled from environmental samples. The first set of capture probes consisted of six high specific 50-mer probes (P1–P6) targeting six distinct regions of the M. acetivorans C2A mcrA gene (Fig. 2, Supplementary Table S1). These probes were designed with HiSpOD software.30 Adaptor sequences were added at each end, resulting in 80-mer hybrid probes consisting of 5′-ATCGCACCAGCGTGT(X)50CACTGCGGCTCCTCA-3′, with X50 indicating the specific capture probe.
Figure 2.

Schematic representation of mcr operon fragments on (A) M. acetivorans C2A gDNA and (B) Lake Pavin metagenomic DNA. Primer pairs used for fmdA (1) and mcrA (2) quantification as well as mcrA-fmdC region (3) amplification are symbolized. Dashed arrows indicate the sequence coverage of each of the five clones retrieved from the environmental sample (B). P1–P6: Positions of the six capture probes in the mcrA gene of M. acetivorans (see Supplementary Table S1 for probe sequences).
The second set of capture probes was 26 oligos (1 49-mer and 25 50-mers) designed to target mcrA and mrtA (encoding the α-subunit of the methyl coenzyme M reductase isoform II), but not the mcrA of anaerobic methanotrophs (Supplementary Table S2, Supplementary Fig. S1).
Oligonucleotides were purchased from Eurogentec S.A. (Belgium). The RNA probe was prepared as described by Gnirke et al.19
2.2. Preparation of biological samples and libraries
The two biological models used in this study were the M. acetivorans C2A strain (DSM 2834) and Lake Pavin, located in the French Massif Central (45°29′74″N, 2°53′28″E). The M. acetivorans C2A strain was cultivated using the medium 304 (http://www.dsmz.de/microorganisms/medium/pdf/DSMZ_Medium304.pdf) according to the manufacturer's instructions. gDNA from the strain was extracted using the Easy DNA kit (Invitrogen), whereas environmental DNA was extracted from 350 ml of freshwater collected from Lake Pavin at a 90-m depth, as described by Dugat-Bony et al.30
Libraries were prepared using Roche's GS FLX Titanium General Library Preparation Kit (Roche Applied Science) according to the manufacturer's instructions. First, 5 µg of DNA was sheared by nebulization. DNA fragments were size selected with AMPure beads (Beckman Coulter Genomics). After purification, fragment end polishing, adaptor ligation (A and B adapter keys; Supplementary Table S1) and fill-in reactions, the libraries were PCR amplified with the 454 Ti-A and 454 Ti-B primers (Supplementary Table S1). The cycle conditions were 3 min at 93°C followed by 20 cycles of 15 s at 93°C, 1 min at 58°C and 8 min at 68°C and a final elongation step at 68°C for 6 min. The amplified libraries were purified with AMPure beads and stored at −20°C until use.
For the amplicon library, mcrA fragments were PCR amplified from total community DNA with the mcrA-specific primer pair MM_01/MM_0231 (Supplementary Table S1). The amplicon was run on a 2% (wt/vol) agarose gel, and the ∼500 bp product was purified with a QIAquick gel extraction kit (Qiagen) and AMPure beads. Each DNA library was quantified by fluorometry with a Quant-iT PicoGreen dsDNA assay kit (Invitrogen). The DNA quality and size distribution were assessed on an Agilent Bioanalyzer High Sensitivity DNA chip (Agilent Technologies).
2.3. Hybridization capture and elution
For each SHS-capture method library, 2.5 µg of salmon sperm DNA (Ambion) and 500 ng of DNA library were mixed (7 µl final volume), denatured for 5 min at 95°C, incubated for 5 min at 65°C before adding 13 µl of prewarmed (65°C) hybridization buffer (10X SSPE, 10X Denhardt's Solution, 10 mM EDTA and 0.2% SDS) and 6 µl freshly prepared, prewarmed (2 min at 65°C) biotinylated RNA probes (500 ng). After 24 h at 65°C, 500 ng of washed M-280 Dynabeads coated with streptavidin (Invitrogen) were added to the hybridization mix that was incubated for 30 min at room temperature (RT). The beads were precipitated with a magnetic stand (Ambion) and washed once for 15 min at RT with 500 µl 1X SSC/0.1% SDS and three times for 10 min at 65°C with 500 µl prewarmed 0.1X SSC/0.1% SDS. The captured DNA was eluted with 50 µl 0.1 M NaOH for 10 min at RT. After magnetic bead precipitation, the DNA supernatant was transferred to a sterile tube containing 70 µl of 1 M Tris–HCl pH 7.5, purified on a QIAquick column (Qiagen) and eluted in a final volume of 20 µl. A 2.5 µl aliquot was subjected to 15 cycles of PCR amplification using the 454 Ti-A and Ti-B primers as described above. After purification, a second round of capture was performed from each first-round PCR product. To increase the DNA yield, a final PCR amplification consisting of 20 cycles was performed. The final product was purified on a QIAquick column (Qiagen) and quantified with a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies).
2.4. Sanger sequencing and data analysis
PCR products were cloned using the TOPO TA cloning kit (Invitrogen). Plasmids were screened for high-size inserts by digestion with EcoRI, and positive clones were Sanger sequenced at MWG DNA sequencing services (Ebersberg, Germany). Sequences were processed and joined using the Staden package program,32 and primer sequences were removed from paired-end consensus sequences. The mcr sequence data retrieved from Lake Pavin by the SHS method were deposited in the GenBank database under accession numbers JQ404494, JQ404495, JQ404496, JQ404497 and JQ404498, and the sequence of the mcrA-fmd region-spanning fragment was deposited under accession number JQ425691.
2.5. 454 GS FLX Titanium sequencing and data analysis
DNA samples were sequenced using the GS FLX Titanium system on the ‘GINA’ platform (part of the GENTYANE platform, labelled IBISA since 2009; BP 392, 63 011 Clermont-Ferrand, France) at the Centre Jean Perrin, according to the manufacturer's specifications. For quality filtering and de-replication of reads, sequences were trimmed with the PRINSEQ-lite PERL script33 using the parameters described in the preprocessing chart (http://prinseq.sourceforge.net/Preprocessing_454_SFF_chart.pdf).
Functional assignment and enrichment were assessed with a BLASTX query34 against a database containing 12 603 McrA protein sequences downloaded from the Genbank database (http://www.ncbi.nlm.nih.gov/), using WWW-Query (http://pbil.univ-lyon1.fr/search/query_fam.php) to perform an advanced keyword search. Reads showing >40% identity over 100 or more amino acids were classified as McrA sequences. Chimaera detection was performed with the UCHIME program35 with a stringent threshold score of five. Sequences containing possible frameshifts were identified with the ‘–w 20’ BLAST option and disabled low complexity filters. Amino acid sequences without frameshifts were extracted from the BLAST results, and only the sequences that passed this filter were chosen for further phylogenetic analysis.
The sequence data were deposited in the NCBI as a Short Read Archive (SRA) project under accession no. SRA049219.
2.6. Phylogenetic analysis and tree construction
All McrA sequences derived from the SHS method and metagenomic libraries were aligned to a sequence obtained from the amplicon library. The amino acid alignment used the ClustalW2 alignment method36 driven by the Seaview version 4 program37 to select the reads having at least 100 amino acids in common with this reference sequence. The overlapping regions of the remaining amino acid sequences, all amplicon pyrosequences and 29 McrA sequences previously identified from the same sampling depth and downloaded from GenBank (http://www.ncbi.nlm.nih.gov/genbank/) were fed to CD-HIT38 that assigned them to operational taxonomic units (OTUs) using a complete linkage clustering method at a 91% cut-off value.27,39
One representative sequence of each OTU was chosen to build a phylogenetic tree (Seaview 4)37 using the neighbour-joining method40,41 and 1000 bootstrapped trials. Closely related sequences available from GenBank (http://www.ncbi.nlm.nih.gov/) were included in the phylogenetic trees to decipher the microbial community diversity. A final tree was drawn in MEGA version 5.42
2.7. qPCR assays for enrichment and methanogen abundance
The assays were conducted in 20 µl with 5 µl of DNA sample or mcrA PCR product standards (covering a dynamic range of 5 × 107 to 50 copies), 10 µl of 2X MESA Green quantitative Polymerase Chain Reaction (qPCR) for SYBR assay mixture (Eurogentec S.A) and 0.2 μM forward and reverse primers. The thermo cycling protocol included an initial step of 95°C for 5 min, followed by 40 cycles of denaturation at 95°C for 15 s, annealing at the melting temperature of each primer set for 15 s and elongation at 68°C for 30 s. The samples and each point of the standard curve were quantified in triplicate. The primer sets are described in Supplementary Table S3. The data were analysed with Realplex software version 1.5 (Eppendorf Inc.) and MxPro qPCR software 4.10d (Agilent technologies). Based on the ΔΔCt method,43 relative enrichments (R) were calculated according to R = 2−ΔΔCt. The relative quantification method established a mean Ct value comparison (ΔCt) between mcrA (target gene) and fmdA (non-target gene 500 kb upstream from mcrA). The relative capture enrichment was determined by the comparison of ΔCt before and after capture, and this result described the fold change or ΔΔCt.
2.8. SHS de novo read assembly
The filtered SHS reads were assembled with Newbler version 2.6 (Roche Applied Science) using stringent assembly parameters (60 bases overlap and 95% overlap identity) and the ‘- rip’ option that forces Newbler to place each read into one unique contig. The functional assignment of contigs and singletons was performed by a BLASTX query34 against our database containing 12 603 McrA protein sequences. Chimaeras were detected in the mcrA contigs and singletons with the UCHIME program35 and a stringent threshold score of five. Prediction of the mcrA gene location within contigs and singletons was performed by BLASTN44 against the reference genomes of Candidatus Methanoregula boonei 6A8 (Methanomicrobiales order, accession no. NC_009712), Methanosaeta concilii GP-6 (Methanosarcinales order, accession no. CP002565) and Methanosphaera stadtmanae DSM 3091 (Methanobacteriales order, accession no. CP000102). Contigs extending at least 100 nucleotides beyond mcrA were segregated for BLASTX34 analysis against the non-redundant (nr) protein sequences database to identify putative open-reading frames within the flanking regions.
The sequence data from homologous mcrA contigs (without chimaeras or frameshifts) were deposited in the GenBank database under accession no. KC184908 to KC185399.
3. Results
3.1. Development of an SHS method for genomic-scale sequence enrichment
3.1.1. Method validation: mcrA gene enrichment from M. acetivorans C2A gDNA
We performed the initial validation of our enrichment strategy by capturing the mcrA gene from a 1 to 3 kb fragment library of the completely sequenced methanogenic M. acetivorans C2A strain. The minimal probe set spanned different non-overlapping regions of the gene (Fig. 2A). The qPCR reactions revealed a 461-fold relative enrichment of mcrA sequences after the first cycle of capture and at least 175 365-fold enrichment after the second cycle. Furthermore, as the M. acetivorans C2A genome consists of 5751 kb with a single mcrA gene copy, the probability of randomly sequencing this gene from a 1 to 3 kb fragment size clone library is 0.02–0.05%. Using our solution-based DNA capture-enrichment method and working on an isolated species, the likelihood increased from 7.8 to 23% after the first cycle and could reach 100% after the second.
The DNA sequence of fragments retrieved after the second cycle of capture was controlled by the cloning-sequencing method. Six clones were sequenced, and all had a perfect correspondence to the mcrA gene from M. acetivorans C2A, reinforcing the efficiency of the two iterative cycles of capture. The captured fragments were assembled into a 1834-bp contig containing the nearly complete mcrA gene (1645 bp) and its 3′ non-coding region (189 bp). After validating this approach, we further tested the performance of the method by enriching mcrA sequences from a complex methanogenic freshwater environment.
3.1.2. Environmental application: mcrA sequence enrichment from a methanogenic lacustrine environment (Lake Pavin)
The freshwater sample was collected in the anoxic zone at 90 m depth, where the highest methanogen diversity was available in the lacustrine environment.27 An improved mcrA probe set included all known mcrA sequences and targeted new variants with explorative probes (Supplementary Table S2). The efficiency of the mcrA enrichment was determined by cloning and sequencing the second capture product. Five out of the ten clones with large inserts (2041–2493 bp) included mcrA sequences. All positive clones had a ∼1500 bp common zone corresponding to the mcrA gene, but they also harboured upstream or downstream regions containing other genes (Fig. 2B). BLAST analysis of the cloned sequences revealed that they are very similar (99% similarity) to mcrA sequences previously retrieved from this ecosystem (accession nos. GQ389949, GQ389912 and GQ389806).27 The closest relative to the mcrA, mcrG and partial mcrC sequences were from a cultured methanogen, Candidatus Methanoregula boonei 6A8 (>85, 84 and 81% similarity, respectively). This hydrogenotrophic species belongs to the Methanomicrobiales order, and it was isolated from an acidic peat bog.45 Furthermore, the fmdC gene fragment identified 821 bp downstream the target gene (Fig. 2B) that shared 77% identity with subunit C of the formyl methanofuran dehydrogenase gene of this species. This gene has been located in the reference genome (GenBank: CP000780.1) at almost 300 kb from the mcr operon. It should be noted that this genome organization—with the fmd operon located just downstream from the mcr operon—has not been described previously in methanogens. To exclude the possibility of chimaera formation during metagenomic library amplification, a PCR fragment spanning the mcrA–fmdC region was obtained directly from the initial metagenomic DNA sample, using two specific primers (Fig. 2B, Supplementary Table S1). The sequenced 821 bp PCR product (JQ425691) confirmed the organization revealed by the SHS method (100% identity with the captured DNA fragment).
Our results showed that the capture method not only efficiently enriched targets out of a complex environmental genomic mixture, but also recovered sequences adjacent to the targeted biomarker gene. Additionally, the SHS method was coupled with NGS technologies to assess the coverage of archaeal mcrA diversity in a complex ecosystem.
3.2. Metagenome exploration with genome-scale sequence enrichment and NGS
The benefit of the SHS method in terms of diversity coverage, when compared with more classical approaches, was further examined by sequencing the SHS capture products. A new random-shotgun DNA metagenomic library adapted for pyrosequencing (fragment sizes ∼500 bp) was prepared for the SHS products and for direct sequencing (shotgun metagenomics approach). From the same metagenomic DNA sample, mcrA PCR products were also amplified with the primer set MM_01-MM_02,31 generating amplicons of ∼500 bp. Sequencing (captured DNA fragments, metagenome and amplicons) was performed with the 454 GS FLX Titanium technology, generating a slightly different amount of raw data with an average read length of 414–471 bases. After pre-processing, sequencing datasets from all three approaches had nearly equivalent numbers of reads (Table 1).
Table 1.
Summary statistics from 454 pyrosequencing
| Metagenome | Amplicons | SHS | |
|---|---|---|---|
| Total number of raw reads | 136 256 | 121 665 | 177 977 |
| Number of reads after pre-processing | 116 365 | 119 437 | 122 772 |
| Average length of cleaned reads (bases) | 471 | 414 | 454 |
| mcrA homologous sequencesa | 3 | 119 409 | 50 727 |
| Enrichment performance (%) | 0.003 | 99.98 | 41.32 |
| Number of chimaeras | 0 | 150 | 30 |
| Number of reads containing frameshifts | 1 | 80 390 | 21 855 |
| Number of high-quality mcrA homologous sequences (without chimaera and frameshifts) | 2 | 38 869 | 28 842 |
| McrA sequences used for methanogenic diversity and abundance (comparison of a common region) | 1 | 38 807 | 11 442 |
| Number of OTUs | 1 | 40 | 44 |
| McrA sequences related to OTUs | 1 | 38 784b | 11 324b |
| Relative abundance of mcrA sequences affiliated with Methanomicrobiales (%) | 0 | 98.57 | 98.82 |
| Relative abundance of mcrA sequences affiliated with Methanosarcinales (%) | 0 | 0.005 | 0.86 |
| Relative abundance of mcrA sequences affiliated with the Novel Order (%) | 100 | 1.43 | 0.13 |
| Relative abundance of mcrA sequences affiliated with Methanobacteriales (%) | 0 | 0 | 0.19 |
aBLASTX parameters: percentage of identity: 40%; E-value cut-off: 10.
bMcrA sequences related to OTUs containing more than one sequence.
3.2.1. Functional assignment and enrichment performance
Only three reads (0.003% of total reads) from the random-shotgun sequencing approach corresponded to the mcrA gene. For the SHS method, 50 727 reads were identified as mcrA sequences (41.32%), and almost all the amplicon approach sequences were from mcrA (119 409 reads, 99.98%).
For mcrA diversity evaluation, however, we only analysed high-quality sequences (no chimaeras or frameshifts), and all the problematic reads were subsequently excluded.
3.2.2. Methanogen diversity and abundance
The phylogeny of the methanogenic McrA protein sequences was investigated and compared for each of the three approaches. We used ClustalW236 to determine a common reference region of 143 amino acids shared by the largest number of McrA sequences retrieved from the 3 approaches. All McrA sequences that included this region were truncated so that at least 100 amino acids aligned with this reference. The resulting sequences, which included 1 read from the shotgun library, 11 442 reads from the SHS method library and 38 807 reads from the amplicon library, were used for further analysis. Furthermore, 29 additional sequences (referred to as Pavin90m) from a previous study27 were included in the analysis.
Following the clustering method, 127 distinct OTUs (longer than 300 bp) were observed, and the 58 OTUs that contained more than 1 sequence were included in a more detailed phylogenetic analysis. The shotgun library sequence, which contained a single final read, was also included. Among these 58 OTUs, 44 were detected from the SHS method, 40 from the amplicon approach, 1 from the metagenomic shotgun library and 3 from Pavin90m sequences. The SHS method and amplicons shared 27 OTUs, including 3 from the Pavin90m sequences (Fig. 3). The remaining 31 OTUs were specific to a single method, with 1 for the metagenome, 17 for the SHS and 13 for the amplicons (Fig. 3).
Figure 3.
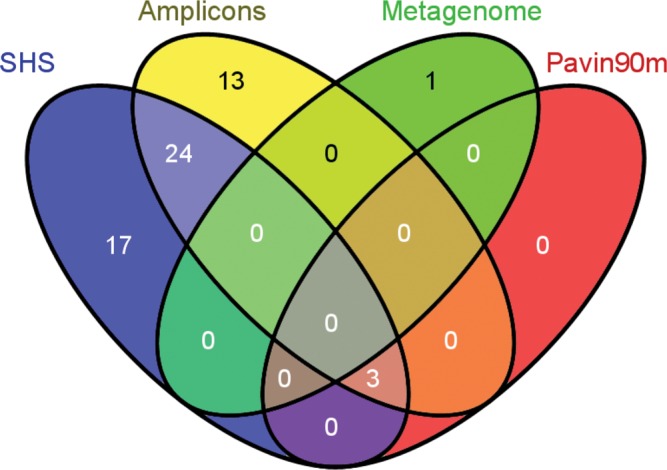
Venn diagram showing the number of unique and shared OTUs for the in-solution capture method (SHS), PCR-based strategy (Amplicons) and sequences isolated at 90 m depth from a previous PCR-based study of Lake Pavin (Pavin90m).27 The Venn diagram was generated with Venny (http://bioinfogp.cnb.csic.es/tools/venny/index.html).
The 58 OTUs covered four lineages including Methanobacteriales, Methanomicrobiales, Methanosarcinales and a putative fourth lineage called ‘Novel Order’. Most OTUs were closely related to the Methanomicrobiales order (48 OTUs, 98.6% of the total input sequences). OTU3, OTU10 and OTU17 formed a distinct branch within this cluster (Fig. 4A), and they were closely related to cultured methanogenic species that also have an insertion in their McrA protein sequence (Supplementary Fig. S2). Both the SHS and amplicon strategies clustered sequences in the most abundant OTUs (Fig. 5). These abundant OTUs represented 94 and 98%, respectively, of the total sequences for each approach. The Methanosarcinales (two OTUs; Fig. 4B) grouped into two distinct branches were related to the reference acetoclastic species M. concilii GP6 (85 and 87% similarity with OTU9 and OTU18, respectively). The most abundant cluster was OTU9 that represented 0.83% of the total SHS reads and 0.005% for the total amplicon reads (Fig. 5). In contrast, the putative Novel Order (five OTUs; Fig. 4C) was dominated by OTU13 clustering with 1.39% of the total amplicons sequences, but only 0.13% of the total SHS reads (Fig. 5). Even if we did not include the more recently described sequences of Methanomassiliicoccus luminyensis46 and Candidatus Methanomethylophilus alvus47 belonging to the novel order for the probe design, distant sequences could be captured with probes by a mismatched nucleotide pairing. We cannot exclude that the sequences captured by specific probes allow indirect hybridization of other mcrA sequences as described for DNA microarrays experiments and referred to as ‘hitchhiking’.48 Despite the substantial sequencing effort for amplicons, no sequences belonging to the Methanobacteriales order were recovered from this approach. These sequences were obtained only from the SHS sample (Fig. 4D), and they were clustered in three OTUs such that one was 90% similar to MrtA (MCR isoenzyme encoded by the mrt operon) from M. stadtmanae DSM 309149 and the remaining two were 77 and 79% identical to MrtA sequences from Methanobacterium lacus that is in the Methanobacteriales order and has been isolated from Lake Pavin sediments.50 These sequences represented 0.19% of total SHS mcrA-related sequences, with the most abundant OTU78 clustering 0.11% of the total SHS reads (Fig. 5).
Figure 4.

Phylogenetic analysis of deduced McrA amino acid sequences obtained from the PCR, SHS and Pavin90m datasets showing evolutionary distances within the orders Methanomicrobiales (A), Methanosarcinales (B), Novel Order (C) and Methanobacteriales (D). Evolutionary history was inferred using the neighbour-joining method40,41 (Poisson distance model) using Seaview software.37 The final tree was drawn in MEGA 5.42 The bars represent a 5% sequence divergence. Numbers at the nodes represent bootstrap values >60% (1000 resamplings). The number of amino acid sequences assigned to each OTU is given in brackets, together with the name of the strategies for obtaining them. McrA amino acid sequence from Methanosarcina barkeri (AAZ69867), uncultured methanogenic archaeon clone Lak19-ML (CAH68744) and Methanobrevibacter smithii (ABQ87220) were used as outgroups, and Methanopyrus kandleri (AAM01870) was an outgroup for rooting the tree. Bold arrows indicate dominant OTUs.
Figure 5.
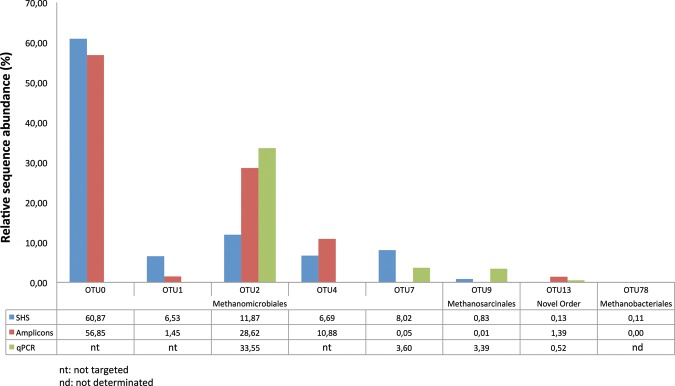
The relative abundances of dominant OTUs from four methanogenic bacterial orders identified by the targeted capture method (SHS), PCR-based strategy (amplicons) and qPCR experiments (qPCR). The relative abundances calculated by qPCR were computed using mcrA copy number as reference obtained using a primers pair targeting all OTUs (Supplementary Table S3).
The GC content of the mcrA genes ranged from 50.4 to 61.1% for amplicons and from 37 to 63.2% for SHS. In the mcrA database, the GC content ranges from 36.2 to 67.2%, indicating that the SHS method is most likely less affected by GC composition than PCR approaches. Furthermore, we evaluated the presence of mismatch residues between PCR primers and probes on mcrA genes in both SHS and amplicon approaches. We identified 99.10, 0.77 and 0.13% of mcrA sequences for amplicons versus 37.68, 50.22 and 12.10% for SHS with 0, 1 and 2 mismatch residues, respectively, between probes (or primers) and sequences. This trend highlights the potential advantage of the SHS approach with long capture probes that tolerate more mismatches, allowing access to new mcrA gene variants.
In parallel, qPCR was used to precisely describe the methanogen abundance in Lake Pavin with regard to the most abundant OTUs and bacterial orders (primers are listed in Supplementary Table S3). The results were compared with the relative sequence abundance calculated previously for the selected OTUs with amplicons and SHS (Fig. 5). The abundance of OTU2, which included the Methanomicrobiales order, was similar in qPCR and amplicons (33.5 and 28.62%), but not SHS (11.87%). In contrast, the second Methanomicrobiales OTU (OTU7) was more abundant in SHS (8.02%) and qPCR (3.6%), but not amplicons (0.05%). The same trend was observed for OTU9 (Methanosarcinales). No significant difference was observed for OTU13 (Novel Order). Finally, no qPCR amplification of OTU78 (Methanobacteriales) occurred. However, we validated the presence of this OTU in Lake Pavin by successive PCR cycles, cloning and sequencing (100% identity). This result indicates that Methanobacteriales are rare in this ecosystem.
3.2.3. De novo assembly of SHS reads
To reconstruct contigs with sequences flanking the targeted mcrA gene, de novo assembly was performed using the pyrosequencing reads obtained by the SHS method (Table 2). We identified 691 contigs (301–1639 bases) with mcrA sequences. By mapping these sequences to complete reference genomes for the Methanomicrobiales, Methanosarcinales and Methanobacteriales orders (no genome was available for the Novel Order), we identified contigs extending into the mcrA flanking regions. The upstream sequences were all part of the mcrG gene. We also characterized two adjacent ORFs located at 200 bases downstream from the mcrA gene and in the same orientation; these ORFs encoded a DtxR family iron (metal)-dependent repressor and a DOMON domain-containing protein. The DtxR sequences were closely related (76–83% identity) to Methanosphaerula palustris E1-9C (accession no. ACL16981) of the Methanomicrobiales order. In the reference genome of this species, the gene is located ∼700 kb downstream of the mcr operon. The sequences of DOMON domain-containing protein are closely related (74–80% identity) to M. concilii GP-6 (accession no. AEB67518) that belongs to the Methanosarcinales order. In the reference genome of this species, the gene is located ∼50 kb downstream of the mcr operon.
Table 2.
Summary statistics from de novo assembly
| Newbler version 2.6 | SHS |
|---|---|
| No. of reads used for assembly | 122 772 |
| No. of reads assembled into contigs | 53 307 |
| No. of singletons | 56 834 |
| Outliersa | 12 631 |
| No. of contigs assembled | 1916 |
| N50 contig size (bases) | 820 |
| No. of mcrA homologous contigs | 693 |
| No. of mcrA homologous singletons | 1142 |
| Number of chimaeras | 5 |
| Number of high-quality mcrA homologous contigs (without chimaeras) | 691 |
| Number of high-quality mcrA homologous singletons (without chimaeras) | 1139 |
| Average mcrA homologous contig length (bases) | 589 |
| Largest mcrA homologous contig length (bases) | 1639 |
aReads were discarded due to quality control by Newbler.
4. Discussion
We captured specific target DNA from a complex environmental metagenome using a novel SHS capture method and NGS. We showed that the relative enrichment of the target sequence was increased to 175 365-fold with 2 cycles of capture, and this result was superior to previous studies using a single cycle18,19 and microarray-based capture.51 We applied this strategy to the anoxic layer of Lake Pavin, where Archaea account for 17% of 4,6-diamidino-2-phenylindole-stained cells52 and only a fraction of these microbes are methanogens. Our SHS strategy specifically enriched mcrA sequences from the environmental sample. In comparison with the random-shotgun metagenomic approach (0.003% recovery of mcrA sequences), the SHS method was superior (41.32% mcrA sequence enrichment). However, the capture efficiency is also likely influenced by the number of probes used per region and the mismatched residues between the probes and their targets. Consequently, two rounds of capture and multiple long RNA probes are advantageous for efficient enrichment.
With a random-shotgun metagenomics approach, many hundreds of thousands of additional single reads would have been necessary to estimate the biodiversity of the methanogen community in this environment. The SHS experiment contained much more mcrA data and provided a solid taxonomic basis for studying methanogens diversity. Finally, PCR was the most effective enrichment approach; with ∼100% of the amplicons corresponding to the biomarker, the primers used were very specific and efficient.31
The SHS and amplicon strategies both revealed similar patterns in methanogen communities such as the high abundance and diversity of Methanomicrobiales sequences (more than 98% of the total sequences representing 48 OTUs). These data confirm a previous study by Biderre-Petit et al.27 High-throughput sequencing, however, reveals that methanogen diversity is much higher than previously estimated by amplicon libraries and Sanger sequencing.27 Importantly, the amplicon sequencing approach missed all the Methanobacteriales taxonomic groups and some Methanosarcinales, possibly due to mcrA primer bias. PCR undersampling often leads to significant underestimation of true community diversity.24,53 SHS efficiently targets rare sequences, as demonstrated for Methanobacteriales, and does not appear to be influenced by GC content. As previously demonstrated for microarray approaches,21,22,54 more extensive explorative capture probe sets could recover rare sequences, leading to the detection of many uncharacterized microbial populations. Moreover, the SHS and amplicon library results were correlated by qPCR.
We also used de novo assembly of SHS sequence reads to explore the regions flanking the target gene, and we identified two ORFs (dtxR and DOMON domain) at previously unknown positions downstream of mcrA. Because this genomic organization may link methanogenesis to electron transfer and Fe homeostasis in organisms living in the anoxic layer of the Lake Pavin, it could reflect adaptation to this particular environment. More experiments are needed, however, to validate this hypothesis.
In this study, we present a novel enrichment method that, when coupled to NGS, expands our knowledge of the diversity of a target gene within a complex microbial community. The method was successfully applied to a lacustrine environment using the mcrA gene, and it revealed higher methanogen community diversity than observed with other methods. To some extent, this method could be applied to phylogenetic studies to explore the diversity of commonly conserved genes such as the 16S rRNA biomarker. The main limitation is the design of high quality probes sets to expect a full coverage of 16S rDNA sequences as complete as possible. New algorithms, such as KASpOD,55 can be used to design highly specific and explorative probes (i.e. targeting sequences not already included in databases) based on oligonucleotide k-mer signatures. These probe designs would be extremely suitable and beneficial to the SHS approach.
With the emergence of third generation sequencing platforms and the capability to sequence longer DNA sequences without library construction,56,57 the SHS strategy could link genomic structure and function in microbial communities.
Supplementary data
Supplementary data are available at www.dnaresearch.oxfordjournals.org.
Funding
This work was supported by the ANR-09-EBIO-009 project (Agence Nationale de la Recherche). J.D. was supported by a studentship from the Centre National de la Recherche Scientifique (CNRS, grant number 163588) and the Région Auvergne. N.P. was funded by Direction Générale de l'Armement (DGA).
Supplementary Material
Acknowledgements
We would like to thank Yannick Bidet and Maud Privat from the Centre Jean Perrin for their help with sample processing on the 454 GS FLX pyrosequencing platform. We also thank Sarah Orlhac and Nicolas Gallois for their efficient technical assistance and David Tottey for reviewing the English version of the manuscript.
Footnotes
Edited by Prof. Masahira Hattori
References
- 1.Whitman W.B., Coleman D.C., Wiebe W.J. Prokaryotes: the unseen majority. Proc. Natl. Acad. Sci. USA. 1998;95:6578–83. doi: 10.1073/pnas.95.12.6578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Curtis T.P., Head I.M., Lunn M., Woodcock S., Schloss P.D., Sloan W.T. What is the extent of prokaryotic diversity? Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2006;361:2023–37. doi: 10.1098/rstb.2006.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amann R., Ludwig W., Schleifer K.-H. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 1995;59:143–69. doi: 10.1128/mr.59.1.143-169.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eisen J.A. Environmental shotgun sequencing: its potential and challenges for studying the hidden world of microbes. PLoS Biol. 2007;5:e82. doi: 10.1371/journal.pbio.0050082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Handelsman J., Rondon M.R., Brady S.F., Clardy J., Goodman R.M. Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products. Chem. Biol. 1998;5:R245–249. doi: 10.1016/s1074-5521(98)90108-9. [DOI] [PubMed] [Google Scholar]
- 6.Biddle J.F., Fitz-Gibbon S., Schuster S.C., Brenchley J.E., House C.H. Metagenomic signatures of the Peru Margin subseafloor biosphere show a genetically distinct environment. Proc. Natl. Acad. Sci. USA. 2008;105:10583–88. doi: 10.1073/pnas.0709942105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tringe S.G., von Mering C., Kobayashi A., et al. Comparative metagenomics of microbial communities. Science. 2005;308:554–7. doi: 10.1126/science.1107851. [DOI] [PubMed] [Google Scholar]
- 8.Riesenfeld C.S., Schloss P.D., Handelsman J. Metagenomics: genomic analysis of microbial communities. Annu. Rev. Genet. 2004;38:525–52. doi: 10.1146/annurev.genet.38.072902.091216. [DOI] [PubMed] [Google Scholar]
- 9.Suenaga H. Targeted metagenomics: a high-resolution metagenomics approach for specific gene clusters in complex microbial communities. Environ. Microbiol. 2011;14:13–22. doi: 10.1111/j.1462-2920.2011.02438.x. [DOI] [PubMed] [Google Scholar]
- 10.Edwards R.A., Rodriguez-Brito B., Wegley L., et al. Using pyrosequencing to shed light on deep mine microbial ecology. BMC Genomics. 2006;7:57. doi: 10.1186/1471-2164-7-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mardis E.R. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008;24:133–41. doi: 10.1016/j.tig.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 12.Quince C., Curtis T.P., Sloan W.T. The rational exploration of microbial diversity. ISME J. 2008;2:997–1006. doi: 10.1038/ismej.2008.69. [DOI] [PubMed] [Google Scholar]
- 13.Hoff K.J. The effect of sequencing errors on metagenomic gene prediction. BMC Genomics. 2009;10:520. doi: 10.1186/1471-2164-10-520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Summerer D. Enabling technologies of genomic-scale sequence enrichment for targeted high-throughput sequencing. Genomics. 2009;94:363–8. doi: 10.1016/j.ygeno.2009.08.012. [DOI] [PubMed] [Google Scholar]
- 15.Albert T.J., Molla M.N., Muzny D.M., et al. Direct selection of human genomic loci by microarray hybridization. Nat. Methods. 2007;4:903–5. doi: 10.1038/nmeth1111. [DOI] [PubMed] [Google Scholar]
- 16.Okou D.T., Steinberg K.M., Middle C., Cutler D.J., Albert T.J., Zwick M.E. Microarray-based genomic selection for high-throughput resequencing. Nat. Methods. 2007;4:907–9. doi: 10.1038/nmeth1109. [DOI] [PubMed] [Google Scholar]
- 17.Mokry M., Feitsma H., Nijman I.J., et al. Accurate SNP and mutation detection by targeted custom microarray-based genomic enrichment of short-fragment sequencing libraries. Nucleic Acids Res. 2010;38:e116. doi: 10.1093/nar/gkq072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tewhey R., Nakano M., Wang X., et al. Enrichment of sequencing targets from the human genome by solution hybridization. Genome Biol. 2009;10:R116. doi: 10.1186/gb-2009-10-10-r116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gnirke A., Melnikov A., Maguire J., et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol. 2009;27:182–9. doi: 10.1038/nbt.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iwai S., Chai B., Sul W.J., Cole J.R., Hashsham S.A., Tiedje J.M. Gene-targeted-metagenomics reveals extensive diversity of aromatic dioxygenase genes in the environment. ISME J. 2010;4:279–85. doi: 10.1038/ismej.2009.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Terrat S., Peyretaillade E., Goncalves O., et al. Detecting variants with Metabolic Design, a new software tool to design probes for explorative functional DNA microarray development. BMC Bioinformatics. 2010;11:478. doi: 10.1186/1471-2105-11-478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dugat-Bony E., Peyretaillade E., Parisot N., et al. Detecting unknown sequences with DNA microarrays: explorative probe design strategies. Environ. Microbiol. 2011;14:356–371. doi: 10.1111/j.1462-2920.2011.02559.x. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki M., Giovannoni S. Bias caused by template annealing in the amplification of mixtures of 16S rRNA genes by PCR. Appl. Environ. Microbiol. 1996;62:625–30. doi: 10.1128/aem.62.2.625-630.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hong S., Bunge J., Leslin C., Jeon S., Epstein S.S. Polymerase chain reaction primers miss half of rRNA microbial diversity. ISME J. 2009;3:1365–73. doi: 10.1038/ismej.2009.89. [DOI] [PubMed] [Google Scholar]
- 25.Bastviken D., Cole J., Pace M., Tranvik L. Methane emissions from lakes: dependence of lake characteristics, two regional assessments, and a global estimate. Global Biogeochem. Cycles. 2004;18 GB4009, doi:10.1029/2004GB002238. [Google Scholar]
- 26.Aeschbach-Hertig W., Hofer M., Kipfer R., Imboden D.M., Wieler R. Accumulation of mantle gases in a permanently stratified volcanic lake (Lake Pavin, France) Geochim. Cosmochim. Acta. 1999;63:3357–72. [Google Scholar]
- 27.Biderre-Petit C., Jezequel D., Dugat-Bony E., et al. Identification of microbial communities involved in the methane cycle of a freshwater meromictic lake. FEMS Microbiol. Ecol. 2011;77:533–45. doi: 10.1111/j.1574-6941.2011.01134.x. [DOI] [PubMed] [Google Scholar]
- 28.Reeve J.N. Molecular biology of methanogens. Annu. Rev. Microbiol. 1992;46:165–91. doi: 10.1146/annurev.mi.46.100192.001121. [DOI] [PubMed] [Google Scholar]
- 29.Klein A., Allmansberger R., Bokranz M., Knaub S., Müller B., Muth E. Comparative analysis of genes encoding methyl coenzyme M reductase in methanogenic bacteria. Mol. Gen. Genet. 1988;213:409–20. doi: 10.1007/BF00339610. [DOI] [PubMed] [Google Scholar]
- 30.Dugat-Bony E., Missaoui M., Peyretaillade E., et al. HiSpOD: probe design for functional DNA microarrays. Bioinformatics. 2011;27:641–8. doi: 10.1093/bioinformatics/btq712. [DOI] [PubMed] [Google Scholar]
- 31.Mihajlovski A., Alric M., Brugere J.F. A putative new order of methanogenic Archaea inhabiting the human gut, as revealed by molecular analyses of the mcrA gene. Res. Microbiol. 2008;159:516–21. doi: 10.1016/j.resmic.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 32.Staden R. The Staden sequence analysis package. Mol. Biotechnol. 1996;5:233–41. doi: 10.1007/BF02900361. [DOI] [PubMed] [Google Scholar]
- 33.Schmieder R., Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27:863–4. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Altschul S.F., Madden T.L., Schäffer A.A., et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Edgar R.C., Haas B.J., Clemente J.C., Quince C., Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Larkin M.A., Blackshields G., Brown N.P., et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–8. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 37.Gouy M., Guindon S., Gascuel O. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2009;27:221–4. doi: 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]
- 38.Li W., Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–9. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 39.Luton P.E., Wayne J.M., Sharp R.J., Riley P.W. The mcrA gene as an alternative to 16S rRNA in the phylogenetic analysis of methanogen populations in landfill. Microbiology. 2002;148:3521–30. doi: 10.1099/00221287-148-11-3521. [DOI] [PubMed] [Google Scholar]
- 40.Studier J., Keppler K. A note on the neighbor-joining algorithm of Saitou and Nei. Mol. Biol. Evol. 1988;5:729–31. doi: 10.1093/oxfordjournals.molbev.a040527. [DOI] [PubMed] [Google Scholar]
- 41.Saitou N., Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987;4:406–25. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 42.Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011;28:2731–9. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 44.Altschul S., Gish W., Miller W., Myers E., Lipman D. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 45.Brauer S.L., Cadillo-Quiroz H., Yashiro E., Yavitt J.B., Zinder S.H. Isolation of a novel acidiphilic methanogen from an acidic peat bog. Nature. 2006;442:192–4. doi: 10.1038/nature04810. [DOI] [PubMed] [Google Scholar]
- 46.Dridi B., Fardeau M.L., Ollivier B., Raoult D., Drancourt M. Methanomassiliicoccus luminyensis gen. nov. sp. nov. a methanogenic archaeon isolated from human faeces. Int. J. Syst. Evol. Microbiol. 2012;62:1902–7. doi: 10.1099/ijs.0.033712-0. doi:1910.1099/ijs.1900.033712-033710. [DOI] [PubMed] [Google Scholar]
- 47.Borrel G., Harris H.M.B., Tottey W., et al. Genome sequence of ‘Candidatus Methanomethylophilus alvus’ Mx1201, a methanogenic archaeon from the human gut belonging to a seventh order of methanogens. J. Bacteriol. 2012;194:6944–5. doi: 10.1128/JB.01867-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Palmer C. Rapid quantitative profiling of complex microbial populations. Nucleic Acids Res. 2006;34:e5. doi: 10.1093/nar/gnj007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fricke W.F., Seedorf H., Henne A., et al. The genome sequence of Methanosphaera stadtmanae reveals why this human intestinal archaeon is restricted to methanol and H2 for methane formation and ATP synthesis. J. Bacteriol. 2005;188:642–58. doi: 10.1128/JB.188.2.642-658.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Borrel G., Joblin K., Guedon A., et al. Methanobacterium lacus sp. nov., a novel hydrogenotrophic methanogen from the deep cold sediment of a meromictic lake. Int. J. Syst. Evol. Microbiol. 2011;62:1625–1629. doi: 10.1099/ijs.0.034538-0. [DOI] [PubMed] [Google Scholar]
- 51.Summerer D., Wu H., Haase B., et al. Microarray-based multicycle-enrichment of genomic subsets for targeted next-generation sequencing. Genome Res. 2009;19:1616–21. doi: 10.1101/gr.091942.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lehours A.C., Bardot C., Thenot A., Debroas D., Fonty G. Anaerobic microbial communities in Lake Pavin, a unique meromictic lake in France. Appl. Environ. Microbiol. 2005;71:7389–400. doi: 10.1128/AEM.71.11.7389-7400.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jeon S., Bunge J., Leslin C., Stoeck T., Hong S., Epstein S.S. Environmental rRNA inventories miss over half of protistan diversity. BMC Microbiol. 2008;8:222. doi: 10.1186/1471-2180-8-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Militon C., Rimour S., Missaoui M., et al. PhylArray: phylogenetic probe design algorithm for microarray. Bioinformatics. 2007;23:2550–7. doi: 10.1093/bioinformatics/btm392. [DOI] [PubMed] [Google Scholar]
- 55.Parisot N., Denonfoux J., Dugat-Bony E., Peyret P., Peyretaillade E. KASpOD – a web service for highly specific and explorative oligonucleotide design. Bioinformatics. 2012;28:3161–3162. doi: 10.1093/bioinformatics/bts597. [DOI] [PubMed] [Google Scholar]
- 56.McCarthy A. Third generation DNA sequencing: pacific biosciences’ single molecule real time technology. Chem. Biol. 2010;17:675–6. doi: 10.1016/j.chembiol.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 57.Schadt E.E., Turner S., Kasarskis A. A window into third-generation sequencing. Hum. Mol. Genet. 2010;19:R227–240. doi: 10.1093/hmg/ddq416. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.