

Abstract
Age-related neurodegenerative diseases are associated with alterations in gene expression in affected neurons. One of the mechanisms that could account for this is altered subcellular localization of transcription factors, which has been observed in human post-mortem brains of each of the major neurodegenerative diseases, including Parkinson’s disease (PD). The specific mechanisms are yet to be elucidated; however a potential mechanism involves alterations in nuclear transport. In this study, we examined the nucleocytoplasmic trafficking of select transcription factors in response to a PD-relevant oxidative injury, 6-hydroxydopamine (6OHDA). Utilizing a well-established model of ligand-regulated nucleocytoplasmic shuttling, the glucocorticoid receptor, we found that 6OHDA selectively impaired nuclear import through an oxidative mechanism without affecting nuclear export or nuclear retention. Interestingly, impaired nuclear import was selective as Nrf2 (nuclear factor E2-related factor 2) nuclear localization remained intact in 6OHDA-treated cells. Thus, oxidative stress specifically impacts the subcellular localization of some but not all transcription factors, which is consistent with observations in post-mortem PD brains. Our data further implicate a role for altered microtubule dependent trafficking in the differential effects of 6OHDA on transcription factor import. Oxidative disruption of microtubule-dependent nuclear transport may contribute to selective declines in transcriptional responses of aging or diseased dopaminergic cells.
Keywords: Parkinson’s disease, Nuclear trafficking, 6-Hydroxydopamine, Oxidative stress, Microtubules
1. Introduction
Age-related neurodegenerative diseases, such as Parkinson’s disease (PD), Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS), and Huntington’s disease (HD), are associated with alterations in expression of specific genes in affected neurons [1–3]. Transcription factors play a key role in the regulation of gene expression and the activity of many transcription factors is limited by their localization within the cytoplasm. A number of mechanisms are utilized to direct sequestered transcription factors into the nucleus in response to specific signals [4–11]. Since nucleocytoplasmic protein trafficking can be bidirectional [12], transcription factors exhibiting predominant nuclear localization may transit through the cytoplasm and also function in nongenomic signaling networks [13–16]. Interestingly, alterations in the subcellular localization of a number of transcription factors are documented in human postmortem brains of each of the major neurodegenerative diseases [5]. Examples include cytoplasmic aggregation of phosphorylated (p)CREB (cAMP response element binding protein) and lack of nuclear pCREB in PD [17], reduced nuclear localization of Nrf2 (nuclear factor E2-related factor 2) in AD but not in PD [18], increased cytoplasmic ATF2 (activating transcription factor 2) levels in AD [19], and increased cytoplasmic:nuclear ratios of TDP-43 (TAR DNA-binding protein 43) in frontotemporal lobar dementias and ALS [20].
Extensive evidence of oxidative damage to proteins, lipids, and DNA is found upon analysis of post-mortem brains of various neurodegenerative diseases, including PD [21–31]. Furthermore, genetic and environmental models of various neurodegenerative diseases support the notion that oxidative stress plays a primary role in their pathogenesis [24–26]. In the case of PD, mutations or deficiency of proteins such as Parkin, PINK1 (PTEN-induced putative kinase 1), α-synuclein, DJ-1, and LRRK2 (leucine-rich repeat kinase 2), have been shown to increase susceptibility to oxidative stress-mediated cell death [32–34]. Parkinsonian mimetics such as, 6-hydroxydopamine (6OHDA), 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), rotenone, and paraquat, generate reactive oxygen species (ROS) and cause neuronal death [35]. These studies suggest a key role for oxidative stress in the pathogenesis of these diseases.
6OHDA has been widely used to study neuronal injury responses in PD. It is an analog of catecholamine neurotransmitters that is taken up into cells by dopamine and norepinephrine reuptake transporters. It is commonly used to model PD-relevant oxidative stress as it demonstrates an early autoxidation phase of ROS production and also has a delayed phase of mitochondrial ROS production [36]. Although used as an exogenous neurotoxin, there is evidence of spontaneous ring hydroxylation of dopamine in vivo, with elevated body fluid levels of 6OHDA detected in patients treated with L-Dopa [37,38].
6OHDA treatment leads to increased cytoplasmic accumulation of (p) ERK (extracellular signal-regulated kinase) [39] and decreased nuclear levels of GFP (green fluorescent protein)-ERK2 [40], as well as decreasing the nuclear to cytoplasmic ratios of pCREB in SH-SY5Y cells and in primary midbrain neurons [17]. These findings are consistent with alterations observed in dopaminergic neurons in post-mortem PD brains [5]. CREB-regulated gene transcription, which is important for axon growth, mitochondrial biogenesis, and neuronal survival, was also repressed in 6OHDA treated cells. Given the dynamic shuttling of transcription factors between the nuclear and cytoplasmic compartments, there are a number of mechanisms that could result in altered distribution of transcription factors in degenerating neurons, including compartmentalization, altered expression levels, sequestration in protein aggregates, and nuclear transport deficits [5]. Interestingly, 6OHDA impairment of CREB-mediated transcription (e.g. Bcl2 [B-cell lymphoma 2] and BDNF [brain-derived neurotrophic factor]) was reversed with cAMP treatment, which activates CREB through mechanisms not requiring active nuclear import [17,41]—suggesting a possible impairment in the nucleocytoplasmic trafficking of CREB. The observed steady state alterations in the distribution of transcription factors could result from altered nuclear import/export with significant consequences on neuronal survival. The main goal of this study was to directly examine, in a neuronal cell line, the impact of 6OHDA-induced oxidative stress on distinct steps in the regulated nucleocytoplasmic trafficking of select transcription factors.
2. Materials and methods
2.1. Cell culture
SH-SY5Y neuroblastoma cells (ATCC, Manassas, VA) were grown on 10 cm cell culture dishes containing Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% heat-inactivated fetal calf serum, 2 mM L-glutamine, and 10 mM HEPES. Cells were maintained at 37 °C in a humidified 5% CO2 incubator.
2.2. Plasmids and transfections
SH-SY5Y cells were transfected with Lipofectamine™ 2000 reagent (Invitrogen, Carlsbad, CA) utilizing Opti-MEM® I reduced serum media (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol at a final Lipofectamine concentration of 0.1%. Cells were grown for at least 48 h before transfection and then allowed to express the protein of interest for another 24 h–48 h before treatment. GR-GFP and Nrf2-GFP plasmids were kindly provided by Dr. Ian Macara (University of Virginia) and Dr. Manabu Furukawa (University of Nebraska), respectively.
2.3. Drug treatments
6OHDA (Sigma, St. Louis, MO) was prepared in cold sterile water immediately before use. Dexamethasone (Sigma), a synthetic glucocorticoid hormone, was prepared at a stock concentration of 10−3 M in 95% EtOH and used at a working concentration of 10−6 M unless otherwise stated. Cortisol (Sigma) was prepared at a stock concentration of 10−3 M to 10−4 M in 100% EtOH and used at a working concentration of 10−6 M to 10−7 M unless otherwise stated. For antioxidant treatment, a metalloporphyrin antioxidant, MnTBAP (A.G. Scientific, San Diego, CA), was utilized at a final concentration of 150 μM. Colchicine (Sigma) and Paclitaxel (Sigma) were prepared in dH2O and DMSO, respectively, and used at the concentrations indicated in the text.
2.4. Monomeric tubulin extraction
Cells treated with either vehicle or the drug of interest were washed in 37 °C DPBS (Dulbecco’s Phosphate-Buffered Saline) and then incubated with tubulin extraction buffer (10 mM PIPES, pH 6.8, 50 mM KCl, 2 mM EGTA, 1 mM MgCl2, 2 M glycerol, 0.5% Triton X-100, 1 mM Na3VO4, and protease inhibitor cocktail [Sigma]) for 15 min at room temperature. The resulting lysate was then centrifuged at 37 °C at 16,000×g for 2 min to pellet any polymerized microtubules. The resulting supernatant, containing soluble (monomeric) tubulin, was then subjected to Western blotting. The protocol was modified from [42].
2.5. Immunoblotting
Cell extracts were collected and immunoblots were performed as previously described [43] with the following exception: BioRad (Hercules, CA) gel running system with a 10% gel was used to separate proteins. Primary antibodies used: anti-α-tubulin (1:5000; Sigma) and anti-GAPDH (1:10,000; Abcam). ImageJ (NIH) was utilized for densitometry (from N≥3 experiments).
2.6. Cell toxicity assay
Cells were plated in 96-well plates and treated with 150 μM 6OHDA for varying amounts of time. LDH (lactate dehydrogenase) release assay was carried out according to the protocol provided by the manufacturer (Promega, Madison, WI). Spectramax M2 plate reader was utilized to read the fluorescent signal (Molecular Devices, Sunnyvale, CA). Percent cytotoxicity was calculated by the following formula: 100×[(Experimental−Medium Background)/(Maximum LDH release−Medium Background)]. Maximum LDH release was determined by treatment of cells with lysis buffer as per manufacturer’s protocol.
2.7. Quantitative image analysis and statistics
An inverted epifluorescent microscope (Olympus IX71) was used for imaging and ImageJ was utilized for quantification. Nuclear:cytoplasmic ratios were calculated from at least 50 randomly imaged cells per experiment (and pooled from at least three independent experiments) by measuring the fluorescent intensity within a randomly distributed region of interest of fixed size within the nuclear and the cytoplasmic compartments. Raw fluorescent intensity is shown in the figures. Nrf2 distribution was also scored into two categories: (1) cytoplasmic and (2) cytoplasmic and nuclear. Example images of these categories are shown in the Results section. Student’s t test was used to compare means between two groups. One-way analysis of variance (ANOVA) followed by posthoc Tukey HSD test was used for multiple comparisons. A p-value<0.05 was considered statistically significant.
3. Results
3.1. Subcellular localization of GR in response to hormone
The glucocorticoid receptor (GR) was used to examine the effects of 6OHDA-induced oxidative stress on bidirectional nucleocytoplasmic trafficking. Transport of GR from the cytoplasm to the nucleus is hormone-dependent and bidirectional as hormone withdrawal triggers its export from the nucleus [12,44–46]. Therefore, both steps in the nucleocytoplasmic trafficking of this transcription factor are regulated and can be experimentally isolated. SH-SY5Y human neuroblastoma cells were transfected with an expression vector for GR-GFP and treated for 1 h with either Dexamethasone (Dex), a synthetic glucocorticoid, or Cortisol (Cort), the primary circulating glucocorticoid in humans. In the absence of Dex or Cort, GR-GFP is cytoplasmic in the majority of the cells (~60% on average) with the remainder of cells also showing some nuclear localization. The mean fluorescent intensity within equal size nuclear and cytoplasmic regions (nuclear:cytoplasmic ratio) for untreated cells is on average around 1 (Fig. 1). A dose-dependent increase in nuclear transport of GR-GFP was observed with a significant nuclear localization attained at ≥10−7 M Dex or Cort (Fig. 1).
Fig. 1.

Dexamethasone and cortisol induce GR nuclear import. SH-SY5Y cells expressing GR-GFP were treated for 1 h with Dex or Cort at the indicated concentrations. In the absence of either ligand, GR-GFP is cytoplasmic in the majority of the cells (~60%) with the remainder showing some nuclear localization as well. The raw mean fluorescent intensity within equal size nuclear and cytoplasmic regions (N/C ratio) for untreated cells is approximately 1. After hormone treatment, GR-GFP is primarily localized within the nucleus. Nuclear:cytoplasmic GR-GFP ratios were determined as noted in the Materials and methods section. Representative images from Dex treatment are shown. Mean+/−SEM, *p<0.05 vs. Veh. Cont. Compiled from 3 independent experiments.
3.2. 6OHDA treatment impairs nuclear import of GR
With 150 μM 6OHDA (dose where impaired CREB/pCREB localization is observed [17]), treatment for less than 6 h did not cause cell death (Fig. 2A). To determine if 6OHDA treatment causes an impairment in nuclear import, GR-GFP expressing SH-SY5Y cells were first treated with 150 μM 6OHDA for 1 h and 4 h and then treated with 1 μM Dex for 1 h to induce the nuclear localization of GR-GFP. As seen in Fig. 2B and C, pre-treatment with 6OHDA impaired the hormone-dependent nuclear import of GR-GFP. Significant reduction in the extent of nuclear import was also seen at doses varying from 90 μM to 300 μM and treatment times as early as 30 min (data not shown).
Fig. 2.
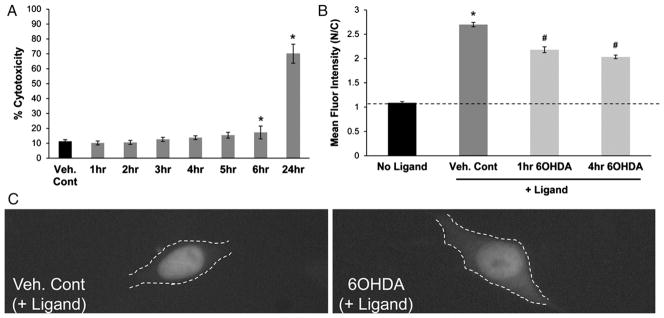
6OHDA reduces GR nuclear import. (A) LDH release assay showed no evidence of toxicity until 6 h of 6OHDA treatment (150 μM). Mean+/−SEM, *p<0.05 vs. Cont. (B and C) GR-GFP expressing SH-SY5Y cells were exposed to 150 μM 6OHDA for 1 h and 4 h and then treated with 1 μM Dex for an additional 1 h. Significant reduction in GR import was observed in 6OHDA-treated cells. Mean+/−SEM, *p<0.05 vs. No Ligand; #p<0.05 vs. Veh. Cont. Compiled from 3 independent experiments.
3.3. Antioxidant treatment reverses 6OHDA-induced impairment in nuclear import
6OHDA has the propensity to produce free radicals [36,47]. To determine whether the nuclear import impairment observed in 6OHDA-treated cells involves oxidative stress, SH-SY5Y cells expressing GR-GFP were treated with MnTBAP, a cell permeable SOD mimetic, 30 min before or 30 min after the start of 6OHDA treatment (150 μM; 4 h) (Fig. 3). When cells were treated with MnTBAP before being exposed to 6OHDA, a near complete rescue in GR-GFP nuclear import was observed (Fig. 3A). When cells were first treated with 6OHDA for 30 min before the addition of MnTBAP, a partial protection was observed (Fig. 3B). This is consistent with the observation that 6OHDA treatment as short as 30 min is enough to cause some impairment in GR nuclear import (data not shown). The rescue observed with MnTBAP treatment suggests that an oxidative mechanism is responsible at least in part for the 6OHDA-mediated reduction in GR nuclear import.
Fig. 3.

MnTBAP rescues 6OHDA-induced impairment in GR nuclear import. SH-SY5Y cells expressing GR-GFP were treated with MnTBAP, a cell permeable SOD mimetic, 30 min before (A) and 30 min after (B) the start of 6OHDA treatment (150 μM for 4 h). A complete and a partial protection was observed with pre- and post-6OHDA treatments with MnTBAP, respectively. Mean+/−SEM, *p<0.05. Compiled from 3 independent experiments.
3.4. Nuclear export of GR is not altered in 6OHDA-treated cells
In addition to impaired nuclear import, enhanced nuclear export could also explain the altered transcription factor distribution observed in neurodegenerative diseases. To study if nuclear export is affected by 6OHDA-induced oxidative stress, a hormone withdrawal study was performed in SH-SY5Y cells expressing GR-GFP. Cort, the naturally occurring ligand for GR, dissociates rapidly from GR and is therefore better suited for studying the effects of hormone withdrawal [46,48]. SH-SY5Y cells expressing GR-GFP were first treated with 10−7 M Cort for 1 h to induce the nuclear localization of GR-GFP. The cells were then withdrawn from the ligand for up to 4 h in the presence of either vehicle or 150 μM 6OHDA. No significant change in nuclear export of GR-GFP was observed in cells exposed to 6OHDA compared to vehicle control (Fig. 4).
Fig. 4.
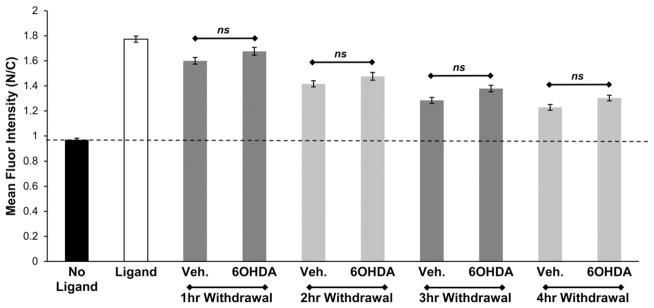
6OHDA does not alter GR nuclear export. SH-SY5Y cells expressing GR-GFP were treated with 10−7 M Cort for 1 h and then withdrawn from the ligand in the presence of vehicle or 6OHDA for up to 4 h. No significant change in nuclear export of GR-GFP was observed in 6OHDA treated cells. Mean+/−SEM, p>0.05. Compiled from 3 independent experiments.
3.5. 6OHDA does not alter GR nuclear retention
6OHDA-induced oxidative stress could damage the nuclear envelope or chromatin binding sites and therefore alter nuclear retention of GR. To investigate this possibility, SH-SY5Y cells expressing GR-GFP were first treated with Dex for 1 h to fully induce the nuclear translocation of GR-GFP and then exposed to 150 μM of 6OHDA from 2 to 6 h in the presence of Dex (Fig. 5). If 6OHDA-induced oxidative stress damaged the nuclear envelope or reduced GR binding to high affinity chromatin binding sites, an increase in ligand-bound cytoplasmic GR-GFP could occur. However, no significant change in subcellular localization was observed—suggesting that 6OHDA does not reduce GR-GFP nuclear retention or trigger its leakage from a “damaged” nucleus. In combination with the results of GR nuclear import and export assays, our data suggest that 6OHDA-induced oxidative stress impairs the hormone-dependent nuclear import of GR.
Fig. 5.
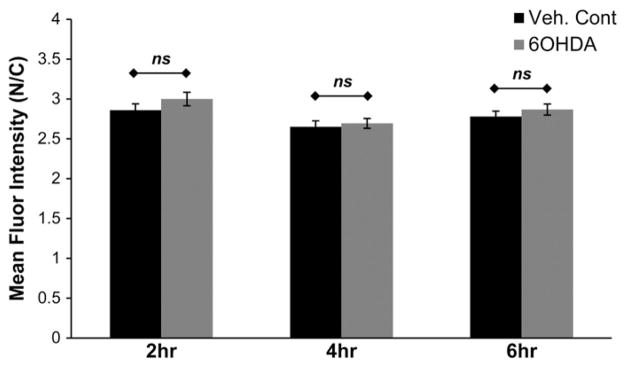
6OHDA does not alter GR nuclear retention. SH-SY5Y cells expressing GR-GFP were first treated with Dex for 1 h and then exposed to 150 μM of 6OHDA from 2 to 6 h in the presence of Dex. No significant change in nuclear retention of GR-GFP was observed. Mean+/−SEM, p>0.05. Compiled from 3 independent experiments.
3.6. Nrf2 translocates into the nucleus in response to 6OHDA
One of the transcription factors that did not show reduced nuclear localization in post-mortem human PD brains is Nrf2, a key protein in the defense against cellular stresses. Under basal metabolic conditions, Nrf2 is sequestered in the cytoplasm and targeted for proteasomal degradation via its association with Keap1 (kelch-like ECH-associated protein 1). Upon exposure to ROS, electrophiles, or serum factors, Nrf2 dissociates from the Keap1 inhibitory complex and translocates into the nucleus, where it upregulates stress response genes and protects against neurotoxic insults [49–55]. For this reason, studies of Nrf2 nuclear translocation typically involve pre-incubation under serum-deprived conditions to shift the basal distribution of Nrf2 toward the cytoplasm [54,56]. SH-SY5Y cells expressing Nrf2-GFP exhibited reduced nuclear:cytoplasmic ratios when grown in low-serum media (1% FBS) for 24 h (Fig. 6A). Serum deprivation also caused an increase in the percentage of cells with cytoplasmic localized Nrf2-GFP and a decrease in the percentage of cells showing both cytoplasmic and nuclear localization (Fig. 6B).
Fig. 6.

6OHDA induces Nrf2 nuclear localization. (A and B) Standard culture conditions elicit Nrf2 nuclear translocation. To desaturate the assay, cells were incubated in low serum media (1% FBS) for 24 h and nuclear localization was quantified by two methods as described in the Materials and methods section. Example images of cells scored as cytoplasmic Nrf2-GFP or cytoplasmic+nuclear Nrf2-GFP are shown. Mean+/−SEM, *p<0.05 vs. 10% FBS. (C and D) Treatment of serum deprived SH-SY5Y cells with 150 μM 6OHDA caused an increased nuclear and a decreased cytoplasmic localization of Nrf2-GFP. Mean+/−SEM, *p<0.05 vs. Veh. Cont. Compiled from 3 independent experiments.
Nrf2 nuclear localization in response to 6OHDA-induced oxidative stress was then studied. Serum deprived SH-SY5Y cells expressing Nrf2-GFP were treated with increasing doses of 6OHDA (from 30 μM to 150 μM), which caused a progressive increase in the nuclear localization of Nrf2. The results from cells treated with 150 μM 6OHDA are shown in Fig. 6C. Quantitation of Nrf2-GFP localization revealed a decrease in the percentage of cells showing predominant cytoplasmic localization and an increase in the percentage of cells showing both cytoplasmic and nuclear Nrf2 localization(Fig. 6D). These results are similar to the studies examining Nrf2 nuclear translocation in response to H2O2 treatment [54]. Thus, inhibitory effects of 6OHDA on nuclear transport are selective and influence the active nuclear import of GR but not of Nrf2.
3.7. 6OHDA increases levels of unpolymerized tubulin
The nucleocytoplasmic trafficking of some transcription factors, including CREB [57] and GR [58,59], is dependent on microtubules (MTs) in some cells. Hence, alterations in MT function due to 6OHDA-induced oxidative stress could help explain the selective trafficking impairments. This is of particular relevance given the alterations in MT function observed in several age-related neurodegenerative diseases [60,61]. In the case of PD, both toxin (e.g. MPP+ and rotenone) and genetic (e.g. α-synuclein and LRRK2) models implicate impaired MT function as a common pathway of neuronal degeneration [62–65].
We investigated the effects of 6OHDA-induced oxidative stress on polymerization of tubulin. We first determined the effects of MT-altering agents, colchicine and paclitaxel, on the levels of unpolymerized tubulin (Fig. 7A). SH-SY5Y cells were treated with either vehicle, 1 μM colchicine (2 h), or 50 nM paclitaxel (1 h) and then incubated with tubulin extraction buffer to extract monomeric tubulin. Colchicine increased and paclitaxel reduced the levels of monomeric tubulin, respectively, while not significantly affecting the levels of total tubulin (Fig. 7A and B). These concentrations of MT-altering agents did not cause cell death (data not shown). We then examined the effects of 6OHDA on the levels of unpolymerized tubulin. SH-SY5Y cells were treated with 150 μM 6OHDA from 1 h to 4 h after which monomeric tubulin was extracted. Cells treated with 6OHDA showed increased levels of monomeric tubulin (Fig. 7C). Levels of total tubulin were not altered in 6OHDA-treated cells (Fig. 7D).
Fig. 7.

6OHDA increases levels of unpolymerized tubulin. (A) SH-SY5Y cells were treated with either vehicle, 1 μM colchicine (2 h), or 50 nM paclitaxel (1 h) and then incubated with tubulin extraction buffer to extract monomeric tubulin and resolved by Western blotting. Colchicine increased and paclitaxel reduced the levels of monomeric tubulin, respectively. Mean+/−SEM, *p<0.05 vs. Veh. Cont. (B) Whole cell extracts of colchicine and paclitaxel treated cells were subjected to Western blotting, which showed no significant changes in total tubulin levels. Mean+/−SEM, p>0.05 vs. Veh. Cont. (C) SH-SY5Y cells were treated with 150 μM 6OHDA for 1 h and 4 h after which monomeric tubulin was extracted. 6OHDA-induced oxidative stress increased the levels of monomeric tubulin. Mean+/−SEM, *p<0.05 vs. Veh. Cont. (D) Whole cell extracts of 6OHDA-treated cells were subjected to Western blotting, which showed no changes in total tubulin levels. Mean+/−SEM, p>0.05 vs. Veh. Cont. Western blot results were quantified and compiled from 3 independent experiments.
3.8. MTs regulate GR but not Nrf2 nuclear trafficking
We then examined the effects of directly modulating MTs on the trafficking of GR and Nrf2. Colchicine and paclitaxel were utilized to disrupt or stabilize, respectively, the MT network in SH-SY5Y cells expressing GR-GFP and Nrf2-GFP (Fig. 8). SH-SY5Y cells expressing GR-GFP were exposed to these agents at doses and times indicated above and then treated with Cort to induce the nuclear translocation of GR-GFP. MT stabilization using paclitaxel did not alter the nuclear import of GR-GFP, however a decrease in nuclear import was observed in cells where the MT network was disrupted by colchicine treatment (Fig. 8A). Neither MT stabilization nor disruption significantly impaired Nrf2-GFP subcellular localization compared to vehicle in response to 6OHDA stress (Fig. 8B). Thus, the effect of 6OHDA on MT stability was similar to that of colchicine, and colchicine recapitulated the selective nuclear import impairment elicited by 6OHDA.
Fig. 8.

MTs regulate GR but not Nrf2 nuclear trafficking. (A) GR-GFP expressing SH-SY5Y cells were treated with either paclitaxel or colchicine and then treated with Cort for 30 min to induce GR-GFP nuclear import. Reduced GR-GFP nuclear import was observed in colchicine treated cells but not in paclitaxel treated cells. Mean+/−SEM, *p<0.05 vs. Veh. Cont. (B) Nrf2-GFP expressing SH-SY5Y cells were treated with either paclitaxel or colchicine and then treated with 150 μM 6OHDA for 30 min to induce Nrf2-GFP nuclear localization. No significant impairment was observed with either MT-altering agent. Mean+/−SEM, p>0.05 vs. Veh. Cont. Compiled from 3 independent experiments.
4. Discussion
In this study, we examined the effects of 6OHDA on the nucleocytoplasmic trafficking of two transcription factors that are sequestered in the cytoplasm, but utilize distinct mechanisms for release from cytoplasmic anchors and import into the nucleus. Previously published work has demonstrated adverse functional effects of altered pCREB or pERK2 subcellular localization on midbrain dopaminergic neuron survival [17,39], but the mechanism underlying such localization changes remained unclear. Utilizing a well-established model of regulated nuclear transport, the glucocorticoid receptor, we found that 6OHDA treatment elicited early impairments in nuclear import without causing reduced nuclear retention or enhanced nuclear export. Treatment with an antioxidant rescued the defect in GR nuclear import, supporting an oxidative mechanism for the 6OHDA-induced effects.
While relatively high doses of H2O2 and other oxidants can affect nuclear transport through global mechanisms in non-neuronal cells [10,66–72], the effects of sublethal/prelethal oxidative stress have not been previously studied in neuronal cells. Interestingly, the impaired nuclear import observed in our system did not result from global nuclear import failure as Nrf2 nuclear localization remained intact in 6OHDA-treated cells. The selective nature of these effects suggests a specific alteration that impacts the subcellular localization of some but not all transcription factors. Our in vitro results are consistent with observations that Nrf2 shows clear nuclear localization in post-mortem PD substantia nigra neurons [18], while kinases and transcription factors activated by trophic signals show abnormal cytoplasmic accumulation [5]. These findings suggest that the impaired neurotrophic response may outweigh the intact antioxidant response after oxidative injury, leading to neuronal degeneration.
Interestingly, GR has been linked to the regulation of the dopaminergic system, including through its interactions with the nuclear receptor related 1 protein (Nurr1)—which is important for dopaminergic differentiation and phenotype maintenance as well as being associated with PD [73–83]. Furthermore, glucocorticoids have been shown to play a protective role in models of PD, although the mechanisms may involve more than one cell type in the brain [74,77]. In any case, selective impairment of a subset of transcription factors in the aging or oxidatively stressed brain may account for downregulation of dopaminergic differentiation markers observed in PD models [84–90] and for alterations in neurotrophic gene expression observed in PD midbrain tissues [1,91–94].
The mechanism(s) underlying the selectivity in nuclear trafficking of proteins observed in this model of parkinsonian injury and in diseased PD brains may be of particular relevance to chronic neurodegeneration. Since not all transcription factors are affected, a global mechanism that would impair all nuclear transport, such as bioenergetic collapse, is unlikely. This is further supported by the early time points at which the trafficking impairment becomes apparent. The current data implicate a role for altered microtubule dependent trafficking. Some transcription factors utilize MTs for their transport. Indeed, a sequence within the parathyroid hormone related protein was recently found to efficiently transport fusion proteins into the nucleus via a MT facilitated “fast track” mechanism [95]. Oxidative stress (e.g. H2O2) has been linked to impaired MT function, such as decreased tubulin polymerization rate and MT growth rate [96–99]. In our model, we found that 6OHDA increased the levels of free monomeric tubulin similar to colchicine, suggesting enhanced depolymerization and/or reduced polymerization. Furthermore, colchicine-mediated disruption but not stabilization of MTs mimicked the effects of 6OHDA in reducing nuclear import of GR without affecting Nrf2 import. These results suggest that 6OHDA modulates transcription factor trafficking through effects on MT-dependent transport of transcription factors.
How could 6OHDA-induced oxidative stress affect the state of tubulin polymerization? A possible mechanism is through the direct oxidative modification of the tubulin heterodimer, which has numerous cysteine residues. Oxidation of cysteine residues has been shown to reduce tubulin polymerization rate and hence could explain the increased levels of unpolymerized tubulin after 6OHDA treatment [100–102]. Furthermore, rates of tubulin polymerization and depolymerization are also regulated by microtubule-associated proteins (MAPs) and the binding of these proteins to MTs could be directly regulated by oxidative modifications [98,103–105]. The binding of MAPs to MTs is also regulated by tubulin post-translational modifications (PTMs), such as acetylation, tyrosination/detyrosination, and polyglutamylation [106,107]. Modulation of tubulin PTMs by oxidative stress therefore represents another means by which 6OHDA could impact the state of tubulin polymerization.
In addition to altered MT dynamics, there are other potential mechanisms that could contribute to selective impairments in nuclear transport. Oxidative injury, e.g. H2O2, has been shown to alter the distribution of the small G protein Ran, which is a key player in the classic import/export cycle [68,70,71,108]. Although such an alteration would be expected to affect nuclear transport of many proteins, it is possible that different transcription factors have differential sensitivity to the breakdown of the Ran gradient across the nucleus or utilize an alternative non-Ran dependent mechanism for their nuclear import [11,109]. Another possibility is the involvement of carrier proteins for nuclear import, importins. Different transcription factors utilize different importins for their nuclear import, as is the case for CREB, GR, and Nrf2—hence oxidative stress-induced alterations in different importins could also help explain the observed selectivity [110–112]. Evidence of altered levels or localization of specific importins has been described in various neurodegenerative diseases [10].
In summary, we have found that 6OHDA selectively impairs nuclear import through an oxidative mechanism that may involve alterations in MT-dependent trafficking of transcription factors. Given the importance of transcription factors for regulating gene expression, alterations in their nucleocytoplasmic transport could have significant consequences for neuronal differentiation, function, and survival. For chronic diseases such as PD, oxidative stress related to aging, the cellular handling of the oxidative catecholamine neurotransmitters, or mitochondrial pathology could contribute to reduced trophic and reparative transcriptional responses through selective impairments in nuclear import.
Acknowledgments
Funding
This work was supported in part by R01 AG026389. VP was supported in part by T32-NS007433 and F31-NS076040.
Abbreviations
- 6OHDA
6-Hydroxydopamine
- AD
Alzheimer’s disease
- ALS
amyotrophic lateral sclerosis
- ATF2
activating transcription factor 2
- Bcl2
B-cell lymphoma 2
- BDNF
brain-derived neurotrophic factor
- Cort
cortisol
- CREB
cAMP response element binding protein
- Dex
dexamethasone
- DMEM
Dulbecco’s Modified Eagle’s Medium
- DPBS
Dulbecco’s Phosphate-Buffered Saline
- ERK
extracellular signal-regulated kinase
- GFP
green fluorescent protein
- GR
glucocorticoid receptor
- HD
Huntington’s disease
- Keap1
kelch-like ECH-associated protein 1
- LDH
lactate dehydrogenase
- LRRK2
leucine-rich repeat kinase 2
- MPTP
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- MT
microtubule
- PD
Parkinson’s disease
- Nrf2
nuclear factor E2-related factor 2
- Nurr1
nuclear receptor related 1 protein
- PINK1
PTEN-induced putative kinase 1
- ROS
reactive oxygen species
- TDP-43
TAR DNA-binding protein 43
Footnotes
The authors declare that they have no actual or potential conflicts of interest related to the content of this paper.
Contributor Information
Vivek P. Patel, Email: vpp3@pitt.edu.
Donald B. DeFranco, Email: dod1@pitt.edu.
Charleen T. Chu, Email: ctc4@pitt.edu.
References
- 1.Courtney E, Kornfeld S, Janitz K, Janitz M. Transcriptome profiling in neurodegenerative disease. J Neurosci Methods. 2010;193:189–202. doi: 10.1016/j.jneumeth.2010.08.018. [DOI] [PubMed] [Google Scholar]
- 2.Malaspina A, Kaushik N, de Belleroche J. Differential expression of 14 genes in amyotrophic lateral sclerosis spinal cord detected using gridded cDNA arrays. J Neurochem. 2001;77:132–145. doi: 10.1046/j.1471-4159.2001.t01-1-00231.x. [DOI] [PubMed] [Google Scholar]
- 3.Tanaka F, Niwa J, Ishigaki S, Katsuno M, Waza M, Yamamoto M, Doyu M, Sobue G. Gene expression profiling toward understanding of ALS pathogenesis. Ann N Y Acad Sci. 2006;1086:1–10. doi: 10.1196/annals.1377.011. [DOI] [PubMed] [Google Scholar]
- 4.Calnan DR, Brunet A. The FoxO code. Oncogene. 2008;27:2276–2288. doi: 10.1038/onc.2008.21. [DOI] [PubMed] [Google Scholar]
- 5.Chu CT, Plowey ED, Wang Y, Patel V, Jordan-Sciutto KL. Location, location, location: altered transcription factor trafficking in neurodegeneration. J Neuropathol Exp Neurol. 2007;66:873–883. doi: 10.1097/nen.0b013e318156a3d7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greene WC, Chen LF. Regulation of NF-kappaB action by reversible acetylation. Novartis Found Symp. 2004;259:208–217. (discussion 218–225) [PubMed] [Google Scholar]
- 7.Harreman MT, Kline TM, Milford HG, Harben MB, Hodel AE, Corbett AH. Regulation of nuclear import by phosphorylation adjacent to nuclear localization signals. J Biol Chem. 2004;279:20613–20621. doi: 10.1074/jbc.M401720200. [DOI] [PubMed] [Google Scholar]
- 8.Jans DA, Xiao CY, Lam MH. Nuclear targeting signal recognition: a key control point in nuclear transport? Bioessays. 2000;22:532–544. doi: 10.1002/(SICI)1521-1878(200006)22:6<532::AID-BIES6>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 9.Kaspar JW, Niture SK, Jaiswal AK. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic Biol Med. 2009;47:1304–1309. doi: 10.1016/j.freeradbiomed.2009.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Patel VP, Chu CT. Nuclear transport, oxidative stress, and neurodegeneration. Int J Clin Exp Pathol. 2011;4:215–229. [PMC free article] [PubMed] [Google Scholar]
- 11.Sorokin AV, Kim ER, Ovchinnikov LP. Nucleocytoplasmic transport of proteins. Biochemistry (Mosc) 2007;72:1439–1457. doi: 10.1134/s0006297907130032. [DOI] [PubMed] [Google Scholar]
- 12.Madan AP, DeFranco DB. Bidirectional transport of glucocorticoid receptors across the nuclear envelope. Proc Natl Acad Sci U S A. 1993;90:3588–3592. doi: 10.1073/pnas.90.8.3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krug AW, Pojoga LH, Williams GH, Adler GK. Cell membrane-associated mineralocorticoid receptors? New evidence. Hypertension. 2011;57:1019–1025. doi: 10.1161/HYPERTENSIONAHA.110.159459. [DOI] [PubMed] [Google Scholar]
- 14.Revollo JR, Cidlowski JA. Mechanisms generating diversity in glucocorticoid receptor signaling. Ann N Y Acad Sci. 2009;1179:167–178. doi: 10.1111/j.1749-6632.2009.04986.x. [DOI] [PubMed] [Google Scholar]
- 15.Watson CS, Jeng YJ, Kochukov MY. Nongenomic signaling pathways of estrogen toxicity. Toxicol Sci. 2010;115:1–11. doi: 10.1093/toxsci/kfp288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Samarasinghe RA, Di Maio R, Volonte D, Galbiati F, Lewis M, Romero G, DeFranco DB. Nongenomic glucocorticoid receptor action regulates gap junction intercellular communication and neural progenitor cell proliferation. Proc Natl Acad Sci U S A. 2011;108:16657–16662. doi: 10.1073/pnas.1102821108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chalovich EM, Zhu JH, Caltagarone J, Bowser R, Chu CT. Functional repression of cAMP response element in 6-hydroxydopamine-treated neuronal cells. J Biol Chem. 2006;281:17870–17881. doi: 10.1074/jbc.M602632200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramsey CP, Glass CA, Montgomery MB, Lindl KA, Ritson GP, Chia LA, Hamilton RL, Chu CT, Jordan-Sciutto KL. Expression of Nrf2 in neurodegenerative diseases. J Neuropathol Exp Neurol. 2007;66:75–85. doi: 10.1097/nen.0b013e31802d6da9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamada T, Yoshiyama Y, Kawaguchi N. Expression of activating transcription factor-2 (ATF-2), one of the cyclic AMP response element (CRE) binding proteins, in Alzheimer disease and non-neurological brain tissues. Brain Res. 1997;749:329–334. doi: 10.1016/S0006-8993(96)01356-X. [DOI] [PubMed] [Google Scholar]
- 20.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 21.Dexter DT, Carter CJ, Wells FR, Javoy-Agid F, Agid Y, Lees A, Jenner P, Marsden CD. Basal lipid peroxidation in substantia nigra is increased in Parkinson’s disease. J Neurochem. 1989;52:381–389. doi: 10.1111/j.1471-4159.1989.tb09133.x. [DOI] [PubMed] [Google Scholar]
- 22.Yoritaka A, Hattori N, Uchida K, Tanaka M, Stadtman ER, Mizuno Y. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc Natl Acad Sci U S A. 1996;93:2696–2701. doi: 10.1073/pnas.93.7.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moreira PI, Nunomura A, Nakamura M, Takeda A, Shenk JC, Aliev G, Smith MA, Perry G. Nucleic acid oxidation in Alzheimer disease. Free Radic Biol Med. 2008;44:1493–1505. doi: 10.1016/j.freeradbiomed.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 24.Nunomura A, Castellani RJ, Zhu X, Moreira PI, Perry G, Smith MA. Involvement of oxidative stress in Alzheimer disease. J Neuropathol Exp Neurol. 2006;65:631–641. doi: 10.1097/01.jnen.0000228136.58062.bf. [DOI] [PubMed] [Google Scholar]
- 25.Browne SE, Beal MF. Oxidative damage in Huntington’s disease pathogenesis. Antioxid Redox Signal. 2006;8:2061–2073. doi: 10.1089/ars.2006.8.2061. [DOI] [PubMed] [Google Scholar]
- 26.Barber SC, Shaw PJ. Oxidative stress in ALS: key role in motor neuron injury and therapeutic target. Free Radic Biol Med. 2010;48:629–641. doi: 10.1016/j.freeradbiomed.2009.11.018. [DOI] [PubMed] [Google Scholar]
- 27.Alam ZI, Daniel SE, Lees AJ, Marsden DC, Jenner P, Halliwell B. A generalised increase in protein carbonyls in the brain in Parkinson’s but not incidental Lewy body disease. J Neurochem. 1997;69:1326–1329. doi: 10.1046/j.1471-4159.1997.69031326.x. [DOI] [PubMed] [Google Scholar]
- 28.Floor E, Wetzel MG. Increased protein oxidation in human substantia nigra pars compacta in comparison with basal ganglia and prefrontal cortex measured with an improved dinitrophenylhydrazine assay. J Neurochem. 1998;70:268–275. doi: 10.1046/j.1471-4159.1998.70010268.x. [DOI] [PubMed] [Google Scholar]
- 29.Alam ZI, Jenner A, Daniel SE, Lees AJ, Cairns N, Marsden CD, Jenner P, Halliwell B. Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J Neurochem. 1997;69:1196–1203. doi: 10.1046/j.1471-4159.1997.69031196.x. [DOI] [PubMed] [Google Scholar]
- 30.Nakabeppu Y, Tsuchimoto D, Yamaguchi H, Sakumi K. Oxidative damage in nucleic acids and Parkinson’s disease. J Neurosci Res. 2007;85:919–934. doi: 10.1002/jnr.21191. [DOI] [PubMed] [Google Scholar]
- 31.Shimura-Miura H, Hattori N, Kang D, Miyako K, Nakabeppu Y, Mizuno Y. Increased 8-oxo-dGTPase in the mitochondria of substantia nigral neurons in Parkinson’s disease. Ann Neurol. 1999;46:920–924. [PubMed] [Google Scholar]
- 32.Tan EK, Skipper LM. Pathogenic mutations in Parkinson disease. Hum Mutat. 2007;28:641–653. doi: 10.1002/humu.20507. [DOI] [PubMed] [Google Scholar]
- 33.Pridgeon JW, Olzmann JA, Chin LS, Li L. PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol. 2007;5:e172. doi: 10.1371/journal.pbio.0050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liou AK, Leak RK, Li L, Zigmond MJ. Wild-type LRRK2 but not its mutant attenuates stress-induced cell death via ERK pathway. Neurobiol Dis. 2008;32:116–124. doi: 10.1016/j.nbd.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Drechsel DA, Patel M. Role of reactive oxygen species in the neurotoxicity of environmental agents implicated in Parkinson’s disease. Free Radic Biol Med. 2008;44:1873–1886. doi: 10.1016/j.freeradbiomed.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kulich SM, Horbinski C, Patel M, Chu CT. 6-Hydroxydopamine induces mitochondrial ERK activation. Free Radic Biol Med. 2007;43:372–383. doi: 10.1016/j.freeradbiomed.2007.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Andrew R, Watson DG, Best SA, Midgley JM, Wenlong H, Petty RK. The determination of hydroxydopamines and other trace amines in the urine of parkinsonian patients and normal controls. Neurochem Res. 1993;18:1175–1177. doi: 10.1007/BF00978370. [DOI] [PubMed] [Google Scholar]
- 38.Linert W, Jameson GN. Redox reactions of neurotransmitters possibly involved in the progression of Parkinson’s Disease. J Inorg Biochem. 2000;79:319–326. doi: 10.1016/s0162-0134(99)00238-x. [DOI] [PubMed] [Google Scholar]
- 39.Zhu JH, Kulich SM, Oury TD, Chu CT. Cytoplasmic aggregates of phosphorylated extracellular signal-regulated protein kinases in Lewy body diseases. Am J Pathol. 2002;161:2087–2098. doi: 10.1016/S0002-9440(10)64487-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dagda RK, Zhu J, Kulich SM, Chu CT. Mitochondrially localized ERK2 regulates mitophagy and autophagic cell stress: implications for Parkinson’s disease. Autophagy. 2008;4:770–782. doi: 10.4161/auto.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stevenson AS, Cartin L, Wellman TL, Dick MH, Nelson MT, Lounsbury KM. Membrane depolarization mediates phosphorylation and nuclear translocation of CREB in vascular smooth muscle cells. Exp Cell Res. 2001;263:118–130. doi: 10.1006/excr.2000.5107. [DOI] [PubMed] [Google Scholar]
- 42.Breitfeld PP, McKinnon WC, Mostov KE. Effect of nocodazole on vesicular traffic to the apical and basolateral surfaces of polarized MDCK cells. J Cell Biol. 1990;111:2365–2373. doi: 10.1083/jcb.111.6.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhu JH, Horbinski C, Guo F, Watkins S, Uchiyama Y, Chu CT. Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am J Pathol. 2007;170:75–86. doi: 10.2353/ajpath.2007.060524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carey KL, Richards SA, Lounsbury KM, Macara IG. Evidence using a green fluorescent protein-glucocorticoid receptor chimera that the Ran/TC4 GTPase mediates an essential function independent of nuclear protein import. J Cell Biol. 1996;133:985–996. doi: 10.1083/jcb.133.5.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heitzer MD, Wolf IM, Sanchez ER, Witchel SF, DeFranco DB. Glucocorticoid receptor physiology. Rev Endocr Metab Disord. 2007;8:321–330. doi: 10.1007/s11154-007-9059-8. [DOI] [PubMed] [Google Scholar]
- 46.Yang J, Liu J, DeFranco DB. Subnuclear trafficking of glucocorticoid receptors in vitro: chromatin recycling and nuclear export. J Cell Biol. 1997;137:523–538. doi: 10.1083/jcb.137.3.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Przedborski S, Ischiropoulos H. Reactive oxygen and nitrogen species: weapons of neuronal destruction in models of Parkinson’s disease. Antioxid Redox Signal. 2005;7:685–693. doi: 10.1089/ars.2005.7.685. [DOI] [PubMed] [Google Scholar]
- 48.Munck A, Foley R. Kinetics of glucocorticoid-receptor complexes in rat thymus cells. J Steroid Biochem Mol Biol. 1976;7:1117–1122. doi: 10.1016/0022-4731(76)90042-x. [DOI] [PubMed] [Google Scholar]
- 49.Zipper LM, Mulcahy RT. The Keap1 BTB/POZ dimerization function is required to sequester Nrf2 in cytoplasm. J Biol Chem. 2002;277:36544–36552. doi: 10.1074/jbc.M206530200. [DOI] [PubMed] [Google Scholar]
- 50.Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23:8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang HC, Nguyen T, Pickett CB. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J Biol Chem. 2002;277:42769–42774. doi: 10.1074/jbc.M206911200. [DOI] [PubMed] [Google Scholar]
- 52.Yu R, Lei W, Mandlekar S, Weber MJ, Der CJ, Wu J, Kong AN. Role of a mitogen-activated protein kinase pathway in the induction of phase II detoxifying enzymes by chemicals. J Biol Chem. 1999;274:27545–27552. doi: 10.1074/jbc.274.39.27545. [DOI] [PubMed] [Google Scholar]
- 53.Zipper LM, Mulcahy RT. Inhibition of ERK and p38 MAP kinases inhibits binding of Nrf2 and induction of GCS genes. Biochem Biophys Res Commun. 2000;278:484–492. doi: 10.1006/bbrc.2000.3830. [DOI] [PubMed] [Google Scholar]
- 54.Rojo AI, Sagarra MR, Cuadrado A. GSK-3beta down-regulates the transcription factor Nrf2 after oxidant damage: relevance to exposure of neuronal cells to oxidative stress. J Neurochem. 2008;105:192–202. doi: 10.1111/j.1471-4159.2007.05124.x. [DOI] [PubMed] [Google Scholar]
- 55.Calkins MJ, Johnson DA, Townsend JA, Vargas MR, Dowell JA, Williamson TP, Kraft AD, Lee JM, Li J, Johnson JA. The Nrf2/ARE pathway as a potential therapeutic target in neurodegenerative disease. Antioxid Redox Signal. 2009;11:497–508. doi: 10.1089/ars.2008.2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Velichkova M, Hasson T. Keap1 regulates the oxidation-sensitive shuttling of Nrf2 into and out of the nucleus via a Crm1-dependent nuclear export mechanism. Mol Cell Biol. 2005;25:4501–4513. doi: 10.1128/MCB.25.11.4501-4513.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cox LJ, Hengst U, Gurskaya NG, Lukyanov KA, Jaffrey SR. Intra-axonal translation and retrograde trafficking of CREB promotes neuronal survival. Nat Cell Biol. 2008;10:149–159. doi: 10.1038/ncb1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dvorak Z, Modriansky M, Pichard-Garcia L, Balaguer P, Vilarem MJ, Ulrichova J, Maurel P, Pascussi JM. Colchicine down-regulates cytochrome P450 2B6, 2C8, 2C9, and 3A4 in human hepatocytes by affecting their glucocorticoid receptor-mediated regulation. Mol Pharmacol. 2003;64:160–169. doi: 10.1124/mol.64.1.160. [DOI] [PubMed] [Google Scholar]
- 59.Harrell JM, Murphy PJ, Morishima Y, Chen H, Mansfield JF, Galigniana MD, Pratt WB. Evidence for glucocorticoid receptor transport on microtubules by dynein. J Biol Chem. 2004;279:54647–54654. doi: 10.1074/jbc.M406863200. [DOI] [PubMed] [Google Scholar]
- 60.Gunawardena S, Goldstein LS. Cargo-carrying motor vehicles on the neuronal highway: transport pathways and neurodegenerative disease. J Neurobiol. 2004;58:258–271. doi: 10.1002/neu.10319. [DOI] [PubMed] [Google Scholar]
- 61.Morfini GA, Burns M, Binder LI, Kanaan NM, LaPointe N, Bosco DA, Brown RH, Jr, Brown H, Tiwari A, Hayward L, Edgar J, Nave KA, Garberrn J, Atagi Y, Song Y, Pigino G, Brady ST. Axonal transport defects in neurodegenerative diseases. J Neurosci. 2009;29:12776–12786. doi: 10.1523/JNEUROSCI.3463-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cartelli D, Ronchi C, Maggioni MG, Rodighiero S, Giavini E, Cappelletti G. Microtubule dysfunction precedes transport impairment and mitochondria damage in MPP+*induced neurodegeneration. J Neurochem. 2010;115:247–258. doi: 10.1111/j.1471-4159.2010.06924.x. [DOI] [PubMed] [Google Scholar]
- 63.Chung CY, Koprich JB, Siddiqi H, Isacson O. Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV alpha-synucleinopathy. J Neurosci. 2009;29:3365–3373. doi: 10.1523/JNEUROSCI.5427-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gillardon F. Leucine-rich repeat kinase 2 phosphorylates brain tubulin-beta isoforms and modulates microtubule stability—a point of convergence in parkinsonian neurodegeneration? J Neurochem. 2009;110:1514–1522. doi: 10.1111/j.1471-4159.2009.06235.x. [DOI] [PubMed] [Google Scholar]
- 65.Ren Y, Liu W, Jiang H, Jiang Q, Feng J. Selective vulnerability of dopaminergic neurons to microtubule depolymerization. J Biol Chem. 2005;280:34105–34112. doi: 10.1074/jbc.M503483200. [DOI] [PubMed] [Google Scholar]
- 66.Crampton N, Kodiha M, Shrivastava S, Umar R, Stochaj U. Oxidative stress inhibits nuclear protein export by multiple mechanisms that target FG nucleoporins and Crm1. Mol Biol Cell. 2009;20:5106–5116. doi: 10.1091/mbc.E09-05-0397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Czubryt MP, Austria JA, Pierce GN. Hydrogen peroxide inhibition of nuclear protein import is mediated by the mitogen-activated protein kinase, ERK2. J Cell Biol. 2000;148:7–16. doi: 10.1083/jcb.148.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kodiha M, Chu A, Matusiewicz N, Stochaj U. Multiple mechanisms promote the inhibition of classical nuclear import upon exposure to severe oxidative stress. Cell Death Differ. 2004;11:862–874. doi: 10.1038/sj.cdd.4401432. [DOI] [PubMed] [Google Scholar]
- 69.Kodiha M, Tran D, Morogan A, Qian C, Stochaj U. Dissecting the signaling events that impact classical nuclear import and target nuclear transport factors. PLoS One. 2009;4:e8420. doi: 10.1371/journal.pone.0008420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kodiha M, Tran D, Qian C, Morogan A, Presley JF, Brown CM, Stochaj U. Oxidative stress mislocalizes and retains transport factor importin-alpha and nucleoporins Nup153 and Nup88 in nuclei where they generate high molecular mass complexes. Biochim Biophys Acta. 2008;1783:405–418. doi: 10.1016/j.bbamcr.2007.10.022. [DOI] [PubMed] [Google Scholar]
- 71.Stochaj U, Rassadi R, Chiu J. Stress-mediated inhibition of the classical nuclear protein import pathway and nuclear accumulation of the small GTPase Gsp1p. FASEB J. 2000;14:2130–2132. doi: 10.1096/fj.99-0751fje. [DOI] [PubMed] [Google Scholar]
- 72.Yasuda Y, Miyamoto Y, Saiwaki T, Yoneda Y. Mechanism of the stress-induced collapse of the Ran distribution. Exp Cell Res. 2006;312:512–520. doi: 10.1016/j.yexcr.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 73.Carpentier R, Sacchetti P, Segard P, Staels B, Lefebvre P. The glucocorticoid receptor is a co-regulator of the orphan nuclear receptor Nurr1. J Neurochem. 2008;104:777–789. doi: 10.1111/j.1471-4159.2007.05055.x. [DOI] [PubMed] [Google Scholar]
- 74.Kurkowska-Jastrzebska I, Litwin T, Joniec I, Ciesielska A, Przybylkowski A, Czlonkowski A, Czlonkowska A. Dexamethasone protects against dopaminergic neurons damage in a mouse model of Parkinson’s disease. Int Immunopharmacol. 2004;4:1307–1318. doi: 10.1016/j.intimp.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 75.Le W, Conneely OM, He Y, Jankovic J, Appel SH. Reduced Nurr1 expression increases the vulnerability of mesencephalic dopamine neurons to MPTP-induced injury. J Neurochem. 1999;73:2218–2221. [PubMed] [Google Scholar]
- 76.Le WD, Xu P, Jankovic J, Jiang H, Appel SH, Smith RG, Vassilatis DK. Mutations in NR4A2 associated with familial Parkinson disease. Nat Genet. 2003;33:85–89. doi: 10.1038/ng1066. [DOI] [PubMed] [Google Scholar]
- 77.Marchetti B, Serra PA, Tirolo C, L’Episcopo F, Caniglia S, Gennuso F, Testa N, Miele E, Desole S, Barden N, Morale MC. Glucocorticoid receptor-nitric oxide crosstalk and vulnerability to experimental parkinsonism: pivotal role for glia–neuron interactions. Brain Res Rev. 2005;48:302–321. doi: 10.1016/j.brainresrev.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 78.Marinelli M, Aouizerate B, Barrot M, Le Moal M, Piazza PV. Dopamine-dependent responses to morphine depend on glucocorticoid receptors. Proc Natl Acad Sci U S A. 1998;95:7742–7747. doi: 10.1073/pnas.95.13.7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ojeda V, Fuentealba JA, Galleguillos D, Andres ME. Rapid increase of Nurr1 expression in the substantia nigra after 6-hydroxydopamine lesion in the striatum of the rat. J Neurosci Res. 2003;73:686–697. doi: 10.1002/jnr.10705. [DOI] [PubMed] [Google Scholar]
- 80.Philips A, Maira M, Mullick A, Chamberland M, Lesage S, Hugo P, Drouin J. Antagonism between Nur77 and glucocorticoid receptor for control of transcription. Mol Cell Biol. 1997;17:5952–5959. doi: 10.1128/mcb.17.10.5952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Piazza PV, Rouge-Pont F, Deroche V, Maccari S, Simon H, Le Moal M. Glucocorticoids have state-dependent stimulant effects on the mesencephalic dopaminergic transmission. Proc Natl Acad Sci U S A. 1996;93:8716–8720. doi: 10.1073/pnas.93.16.8716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sleiman PM, Healy DG, Muqit MM, Yang YX, Van Der Brug M, Holton JL, Revesz T, Quinn NP, Bhatia K, Diss JK, Lees AJ, Cookson MR, Latchman DS, Wood NW. Characterisation of a novel NR4A2 mutation in Parkinson’s disease brain. Neurosci Lett. 2009;457:75–79. doi: 10.1016/j.neulet.2009.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sousa KM, Mira H, Hall AC, Jansson-Sjostrand L, Kusakabe M, Arenas E. Microarray analyses support a role for Nurr1 in resistance to oxidative stress and neuronal differentiation in neural stem cells. Stem Cells. 2007;25:511–519. doi: 10.1634/stemcells.2006-0238. [DOI] [PubMed] [Google Scholar]
- 84.Le W, Pan T, Huang M, Xu P, Xie W, Zhu W, Zhang X, Deng H, Jankovic J. Decreased NURR1 gene expression in patients with Parkinson’s disease. J Neurol Sci. 2008;273:29–33. doi: 10.1016/j.jns.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liu H, Wei L, Tao Q, Deng H, Ming M, Xu P, Le W. Decreased NURR1 and PITX3 gene expression in Chinese patients with Parkinson’s disease. Eur J Neurol. 2012;19:870–875. doi: 10.1111/j.1468-1331.2011.03644.x. [DOI] [PubMed] [Google Scholar]
- 86.Murer MG, Dziewczapolski G, Menalled LB, Garcia MC, Agid Y, Gershanik O, Raisman-Vozari R. Chronic levodopa is not toxic for remaining dopamine neurons, but instead promotes their recovery, in rats with moderate nigrostriatal lesions. Ann Neurol. 1998;43:561–575. doi: 10.1002/ana.410430504. [DOI] [PubMed] [Google Scholar]
- 87.Stephenson DT, Childs MA, Li Q, Carvajal-Gonzalez S, Opsahl A, Tengowski M, Meglasson MD, Merchant K, Emborg ME. Differential loss of presynaptic dopaminergic markers in Parkinsonian monkeys. Cell Transplant. 2007;16:229–244. doi: 10.3727/000000007783464704. [DOI] [PubMed] [Google Scholar]
- 88.Tillerson JL, Caudle WM, Reveron ME, Miller GW. Detection of behavioral impairments correlated to neurochemical deficits in mice treated with moderate doses of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Exp Neurol. 2002;178:80–90. doi: 10.1006/exnr.2002.8021. [DOI] [PubMed] [Google Scholar]
- 89.Vernier P, Moret F, Callier S, Snapyan M, Wersinger C, Sidhu A. The degeneration of dopamine neurons in Parkinson’s disease: insights from embryology and evolution of the mesostriatocortical system. Ann N Y Acad Sci. 2004;1035:231–249. doi: 10.1196/annals.1332.015. [DOI] [PubMed] [Google Scholar]
- 90.Wilson JM, Levey AI, Rajput A, Ang L, Guttman M, Shannak K, Niznik HB, Hornykiewicz O, Pifl C, Kish SJ. Differential changes in neurochemical markers of striatal dopamine nerve terminals in idiopathic Parkinson’s disease. Neurology. 1996;47:718–726. doi: 10.1212/wnl.47.3.718. [DOI] [PubMed] [Google Scholar]
- 91.Howells DW, Porritt MJ, Wong JY, Batchelor PE, Kalnins R, Hughes AJ, Donnan GA. Reduced BDNF mRNA expression in the Parkinson’s disease substantia nigra. Exp Neurol. 2000;166:127–135. doi: 10.1006/exnr.2000.7483. [DOI] [PubMed] [Google Scholar]
- 92.Mogi M, Togari A, Kondo T, Mizuno Y, Komure O, Kuno S, Ichinose H, Nagatsu T. Brain-derived growth factor and nerve growth factor concentrations are decreased in the substantia nigra in Parkinson’s disease. Neurosci Lett. 1999;270:45–48. doi: 10.1016/s0304-3940(99)00463-2. [DOI] [PubMed] [Google Scholar]
- 93.Nagatsu T, Mogi M, Ichinose H, Togari A. Changes in cytokines and neurotrophins in Parkinson’s disease. J Neural Transm. 2000;(Suppl):277–290. doi: 10.1007/978-3-7091-6301-6_19. [DOI] [PubMed] [Google Scholar]
- 94.Parain K, Murer MG, Yan Q, Faucheux B, Agid Y, Hirsch E, Raisman-Vozari R. Reduced expression of brain-derived neurotrophic factor protein in Parkinson’s disease substantia nigra. Neuroreport. 1999;10:557–561. doi: 10.1097/00001756-199902250-00021. [DOI] [PubMed] [Google Scholar]
- 95.Roth DM, Moseley GW, Pouton CW, Jans DA. Mechanism of microtubule-facilitated “fast track” nuclear import. J Biol Chem. 2011;286:14335–14351. doi: 10.1074/jbc.M110.210302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Davison AJ, Legault NA, Steele DW. Effect of 6-hydroxydopamine on polymerization of tubulin. Protection by superoxide dismutase, catalase, or anaerobic conditions. Biochem Pharmacol. 1986;35:1411–1417. doi: 10.1016/0006-2952(86)90104-8. [DOI] [PubMed] [Google Scholar]
- 97.Lee CF, Liu CY, Hsieh RH, Wei YH. Oxidative stress-induced depolymerization of microtubules and alteration of mitochondrial mass in human cells. Ann N Y Acad Sci. 2005;1042:246–254. doi: 10.1196/annals.1338.027. [DOI] [PubMed] [Google Scholar]
- 98.Smyth JW, Hong TT, Gao D, Vogan JM, Jensen BC, Fong TS, Simpson PC, Stainier DY, Chi NC, Shaw RM. Limited forward trafficking of connexin 43 reduces cell–cell coupling in stressed human and mouse myocardium. J Clin Invest. 2010;120:266–279. doi: 10.1172/JCI39740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tomlinson DR, Bennett T. Fluorescence histochemical and ultrastructural observations on preterminal noradrenergic axons following ligation or treatment with 6-hydroxydopamine. Med Biol. 1979;57:39–47. [PubMed] [Google Scholar]
- 100.Landino LM, Hasan R, McGaw A, Cooley S, Smith AW, Masselam K, Kim G. Peroxynitrite oxidation of tubulin sulfhydryls inhibits microtubule polymerization. Arch Biochem Biophys. 2002;398:213–220. doi: 10.1006/abbi.2001.2729. [DOI] [PubMed] [Google Scholar]
- 101.Mellon MG, Rebhun LI. Sulfhydryls and the in vitro polymerization of tubulin. J Cell Biol. 1976;70:226–238. doi: 10.1083/jcb.70.1.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Landino LM, Moynihan KL, Todd JV, Kennett KL. Modulation of the redox state of tubulin by the glutathione/glutaredoxin reductase system. Biochem Biophys Res Commun. 2004;314:555–560. doi: 10.1016/j.bbrc.2003.12.126. [DOI] [PubMed] [Google Scholar]
- 103.Akhmanova A, Steinmetz MO. Tracking the ends: a dynamic protein network controls the fate of microtubule tips. Nat Rev Mol Cell Biol. 2008;9:309–322. doi: 10.1038/nrm2369. [DOI] [PubMed] [Google Scholar]
- 104.Galjart N. Plus-end-tracking proteins and their interactions at microtubule ends. Curr Biol. 2010;20:R528–R537. doi: 10.1016/j.cub.2010.05.022. [DOI] [PubMed] [Google Scholar]
- 105.van der Vaart B, Akhmanova A, Straube A. Regulation of microtubule dynamic instability. Biochem Soc Trans. 2009;37:1007–1013. doi: 10.1042/BST0371007. [DOI] [PubMed] [Google Scholar]
- 106.Janke C, Kneussel M. Tubulin post-translational modifications: encoding functions on the neuronal microtubule cytoskeleton. Trends Neurosci. 2010;33:362–372. doi: 10.1016/j.tins.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 107.Fukushima N, Furuta D, Hidaka Y, Moriyama R, Tsujiuchi T. Post-translational modifications of tubulin in the nervous system. J Neurochem. 2009;109:683–693. doi: 10.1111/j.1471-4159.2009.06013.x. [DOI] [PubMed] [Google Scholar]
- 108.Miyamoto Y, Saiwaki T, Yamashita J, Yasuda Y, Kotera I, Shibata S, Shigeta M, Hiraoka Y, Haraguchi T, Yoneda Y. Cellular stresses induce the nuclear accumulation of importin alpha and cause a conventional nuclear import block. J Cell Biol. 2004;165:617–623. doi: 10.1083/jcb.200312008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wagstaff KM, Jans DA. Importins and beyond: non-conventional nuclear transport mechanisms. Traffic. 2009;10:1188–1198. doi: 10.1111/j.1600-0854.2009.00937.x. [DOI] [PubMed] [Google Scholar]
- 110.Forwood JK, Lam MH, Jans DA. Nuclear import of Creb and AP-1 transcription factors requires importin-beta 1 and Ran but is independent of importin-alpha. Biochemistry. 2001;40:5208–5217. doi: 10.1021/bi002732+. [DOI] [PubMed] [Google Scholar]
- 111.Freedman ND, Yamamoto KR. Importin 7 and importin alpha/importin beta are nuclear import receptors for the glucocorticoid receptor. Mol Biol Cell. 2004;15:2276–2286. doi: 10.1091/mbc.E03-11-0839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Theodore M, Kawai Y, Yang J, Kleshchenko Y, Reddy SP, Villalta F, Arinze IJ. Multiple nuclear localization signals function in the nuclear import of the transcription factor Nrf2. J Biol Chem. 2008;283:8984–8994. doi: 10.1074/jbc.M709040200. [DOI] [PMC free article] [PubMed] [Google Scholar]