

Abstract
Diffuse pulmonary lymphangiomatosis (DPL) is a rare disease characterized by infiltration of the lung, pleura and mediastinum with thin-walled lymphangiomas. DPL can result in mass effect from infiltrative disease, restrictive and obstructive pulmonary physiology, chylous effusions and respiratory failure. The present article discusses clinical, radiographic and pathological features, and treatment options for DPL.
Keywords: Chylous effusions, Diffuse pulmonary lymphangiomatosis, Interstitial lung disease, Lymphangiomatosis
Abstract
La lymphangiomatose pulmonaire diffuse (LPD) est une maladie rare caractérisée par l’infiltration de lymphangiomes à paroi mince dans les poumons, la plèvre et le médiastin. La LPD peut entraîner un effet de masse causé par la maladie infiltrante, la physiologie pulmonaire restrictive et obstructive, les effusions chyleuses et l’insuffisance respiratoire. Le présent article aborde les caractéristiques cliniques, radiographiques et pathologiques, de même que les possibilités thérapeutiques, de la LPD.
Learning objective:
Recognize the clinical, radiographic and pathological features of diffuse pulmonary lymphangiomatosis (DPL).
CanMEDS competency: Medical Expert
Pre-test:
What are the common clinical features of DPL?
What treatments are available for the management of this disease?
CASE PRESENTATION
A 48-year-old previously healthy woman was referred for management of dyspnea on exertion of more than nine months’ duration. Before referral, she was evaluated by her local physician for the complaint of odynophagia and pharyngeal irritation. These issues prompted a computed tomography (CT) scan of her neck, which was normal. However, patchy infiltrates were incidentally noted in the upper lung fields bilaterally, for which she was treated with an empirical course of oral antibiotics for presumptive community-acquired pneumonia. Her pharyngeal symptoms abated shortly thereafter. However, her dyspnea persisted and prompted referral. On discussion with the patient, she denied cough, sputum production, hemoptysis, chest pain, orthopnea, paroxysmal nocturnal dyspnea, recent travel or constitutional symptoms. Her medical history was notable for hypothyroidism and a remote history of pneumonia 30 years previously that required a decortication. She was a former smoker of approximately 20 years, and denied significant alcohol or illicit drug use. A comprehensive review of systems was otherwise negative.
On examination, the patient exhibited normal vital signs. Inspiratory crackles without wheezes in the posterior lung bases bilaterally were the only notable findings. Laboratory evaluation demonstrated a normal erythrocyte sedimentation rate, renal function, liver function tests, electrolytes and complete blood count. A chest x-ray demonstrated patchy infiltrates in the mid to upper lung fields, bilateral pleural effusions and pleural thickening (Figure 1). High-resolution CT of the chest showed patchy bilateral lung parenchymal abnormalities with interlobular septal thickening, ground-glass opacities, thickening of the pleura bilaterally and diffuse infiltration of the mediastinum by cystic fluid densities (Figures 2 and 3). Pulmonary function tests revealed moderate-severe restriction (total lung capacity 2.77 L, 54% predicted), with a normal forced expiratory volume in 1 s/forced vital capacity ratio of 77.2, and a decreased diffusing capacity of carbon monoxide (51% predicted). A transthoracic echocardiogram was normal without suggestion of pulmonary hypertension or cardiac dysfunction. Thoracentesis was performed and was consistent with a chylous effusion (ie, triglyceride level of 7.05 mmol/L).
Figure 1).
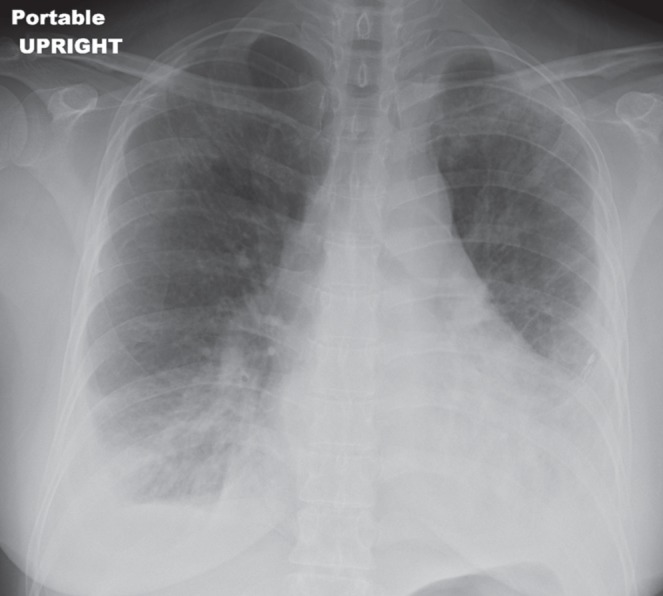
Anteroposterior chest x-ray showing upper-lobe predominant interstitial infiltrates and bilateral pleural effusions
Figure 2).
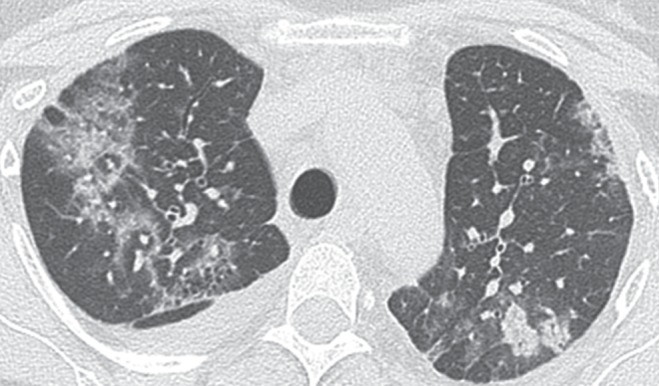
Computed tomography scan of the chest revealing peribronchovascular and interlobular thickening, interstitial infiltrates and pleural thickening with effusions
Figure 3).
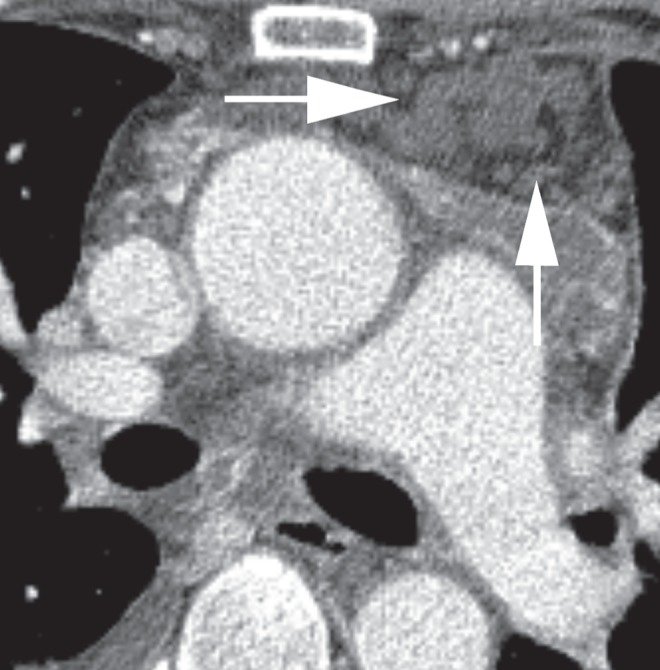
Computed tomography of the chest (mediastinal window) showing diffuse infiltration of the mediastinum by cystic fluid densities (arrows)
Given the progressive dyspnea, chylous effusions and indeterminate infiltrates, the patient underwent bronchoscopy with transbronchial biopsy, which demonstrated diffuse nodular thickening and mucosal edema throughout the airways, with a nondiagnostic biopsy. Subsequently, the patient underwent CT-guided mediastinal lymph node biopsy, which again yielded nondiagnostic results. Due to concerns of possible malignancy, a mediastinoscopy with biopsy was performed and revealed fibroadenomatous hyperplasia without other abnormalities. Given the low diagnostic yield of the biopsies, the patient was advised to undergo wedge resections in addition to a pleural biopsy for a definitive diagnosis. The biopsies demonstrated findings consistent with DPL. Dilated lymphatic channels and anastomosing structures lined with epithelial cells were observed. The cells stained positively for D2-40 and CD31, as is typical of DPL (Figure 4).
Figure 4).
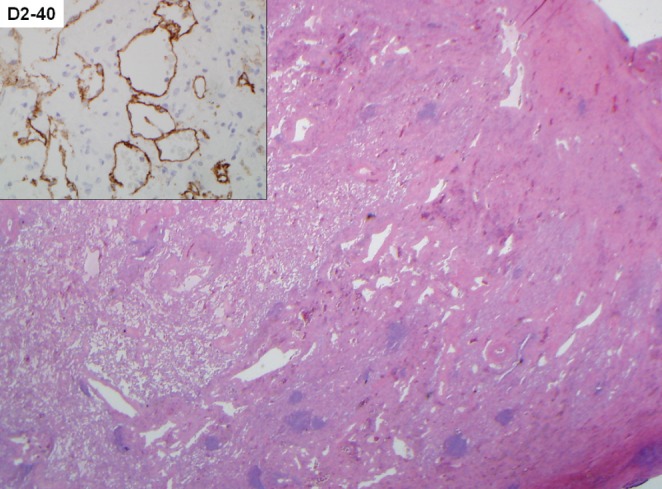
Low-power view demonstrates a markedly thickened visceral pleura and interlobular septa due to abnormal lymphatic vascular proliferation (hematoxylin and eosin stain, original magnification ×20), as supported by D2-40 immunohistochemical staining (inset, original magnification ×200)
DISCUSSION
Lymphangiomas are congenital, benign, cystic, focal areas of lymphatic proliferation that can occur in any part of the body containing lymphatics. Widespread proliferation of these anastomosing lymphatic spaces (ie, lymphangiomas) limited to the thorax leads to the rare syndrome of DPL (1). This pathological process can result in mass effect from infiltrative disease, restrictive and/or obstructive physiology, chylous effusions and respiratory failure. The disease is predominantly detected in children and young adults, and affects both sexes equally (2).
DPL can often have a progressive course leading to respiratory failure and death in pediatric populations (3,4). Depending on the location and degree of thoracic involvement, the clinical presentation in adults varies from mild wheezing and a nonproductive cough to marked respiratory insufficiency due to progressive infiltrative disease and recurrent pleural effusions. Pleural effusions are often chylous and patients may have associated chyloptysis (5), hemoptysis (6) or chylopericardium (7). Disseminated intravascular coagulation has been rarely associated with DPL (4,8).
Chest radiographs are nonspecific and can reveal diffuse interstitial infiltrates and pleural effusions. Findings on chest CT are more distinct and include peribronchovascular and interlobular septal thickening, diffuse liquid-like infiltration of the mediastinal soft tissue and pleural thickening with effusions (3,9,10). These findings are suggestive – but are not pathognomonic – for DPL. Pulmonary function testing often reveals a mixed pattern; however, restrictive features predominate. Bronchoscopy is nonspecific and can reveal airway mucosal erythema and edema, bronchial narrowing and, in advanced cases, thin-walled vesicles containing chylous fluid (10). Although bronchoscopy with transbronchial biopsy has been reported to yield a diagnosis, most cases in the literature were confirmed through open lung biopsy (11). Given the clinical and radiographic features, the differential diagnosis of DPL includes lymphangioleiomyomatosis (LAM), lymphangiomyomatosis, hemangiomatosis, lymphangiectasis and pulmonary involvement by Kaposi’s sarcoma.
Pathologically, DPL is distinct from LAM because there is no infiltration into the lung parenchyma or the typical HMB45-positive LAM cells. Instead, complex anastomosing lymphatic channels are contained within the normal lymphatic pathways of the lung (2). The small portion of mature smooth muscle cells further distinguishes DPL from LAM. Finally, the presence of endothelial markers such as CD31, factor VIII-related antigen and D2-40 on immunohistochemical staining is characteristic of DPL.
Prognosis is generally poor and treatment is largely supportive because there are no established curative treatments available for this disorder. For localized lung or mediastinal lesions, thoracotomy or thorascopic resection has been reported to be effective (12). Other surgical interventions that have been attempted include pleurodesis, parietal pleurectomy and ligation of the thoracic duct (13,14). Albumin transfusions and a low-fat diet with medium-chain triglycerides have been attempted but are minimally successful. Therapeutic thoracentesis and pleurodesis for recurrent pleural effusions are the mainstay of therapy for advanced disease. Systemic glucocorticoids, recombinant interferon therapy, cyclophosphamide, tamoxifen and radiation therapy may decrease parenchymal involvement and effusion burden. These therapies have been attempted with varied success but are largely limited due to toxicity (15). More recently, treatment with propranolol, sirolimus and bevacizumab have been shown to decrease recurrent pleural effusions and lymphatic proliferation with less severe toxicity (16). Finally, one case of successful lung transplantation for DPL has been reported (17).
The patient experienced a relatively uncomplicated postoperative course. Additional information about potential treatment options was provided; however, she elected to defer treatment given her symptomatic improvement following repeat thoracentesis. She continues to undergo clinical monitoring. This case exemplified the typical radiographic and pathological findings of DPL. However, the patient’s clinical presentation of mild respiratory limitations despite significant radiographic findings were quite unusual. This further supports the diagnostic and therapeutic challenges that a patient with DPL typifies.
Post-test:
-
What are the common clinical features of DPL?
Clinically, DPL can present as dyspnea and a nonproductive cough. However, in younger patients, a more aggressive course is often observed. Radiographic features include bilateral interstitial infiltrates and pleural effusions on chest roentogram, and peribronchovascular and interlobular septal thickening, liquid-like infiltration of the mediastinal fat and pleural thickening on chest CT.
-
What treatment modalities are available for the management of this disease?
Almost all treatments are palliative and, at this time, based on anecdotal data. Less toxic and more recent reports suggest that propranolol, sirolimus and bevacizumab can decrease pleural effusions and lymphatic proliferation. Therapeutic thoracentesis and pleurodesis remain mainstays of symptom control in advanced disease.
Footnotes
DISCLOSURES: This work was supported by funds from the Mayo Foundation. KCK and AHL identified, managed the case, and assisted with the generation and the review of the manuscript. SMP assisted with the literature review and with the generation and the review of the manuscript. ESY assisted with the interpretation of the histology and the review of the manuscript. The authors have no financial disclosures.
REFERENCES
- 1.Tazelaar HD, Kerr D, Yousem SA, Saldana MJ, Langston C, Colby TV. Diffuse pulmonary lymphangiomatosis. Hum Pathol. 1993;24:1313–22. doi: 10.1016/0046-8177(93)90265-i. [DOI] [PubMed] [Google Scholar]
- 2.Faul JL, Berry GJ, Colby TV, et al. Thoracic lymphangiomas, lymphangiectasis, lymphangiomatosis, and lymphatic dysplasia syndrome. Am J Respir Crit Care Med. 2000;161:1037–46. doi: 10.1164/ajrccm.161.3.9904056. [DOI] [PubMed] [Google Scholar]
- 3.Swensen SJ, Hartman TE, Mayo JR, et al. Diffuse pulmonary lymphangiomatosis: CT findings. J Comput Assist Tomog. 1995;19:348–352. doi: 10.1097/00004728-199505000-00002. [DOI] [PubMed] [Google Scholar]
- 4.Alvarez OA, Kjellin I, Zuppan CW. Thoracic lymphangiomatosis in a child. J Pediatr Hematol Oncol. 2004;26:136–41. doi: 10.1097/00043426-200402000-00018. [DOI] [PubMed] [Google Scholar]
- 5.Sanders JS, Rosenow EC, III, Piehler JM, Gloviczki P, Brown LR. Chyloptysis (chylous sputum) due to thoracic lymphangiectasis with successful surgical correction. Arch Intern Med. 1988;148:1465–6. [PubMed] [Google Scholar]
- 6.Nair LG, Kurtz CP. Lymphangiomatosis presenting with bronchial cast formation. Thorax. 1996;51:765–6. doi: 10.1136/thx.51.7.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen YL, Lee CC, Yeh ML, Lee JS, Sung TC. Generalized lymphangiomatosis presenting as cardiomegaly. J Formos Med Assoc. 2007;106(3 Suppl):S10–14. doi: 10.1016/s0929-6646(09)60359-4. [DOI] [PubMed] [Google Scholar]
- 8.Fukahori S, Tsuru T, Asagiri K, et al. Thoracic lymphangiomatosis with massive chylothorax after a tumor biopsy and with disseminated intravenous coagulation – lymphoscintigraphy, an alternative minimally invasive imaging technique: Report of a case. Surg Today. 2011;41:978–82. doi: 10.1007/s00595-010-4383-0. [DOI] [PubMed] [Google Scholar]
- 9.Raman SP, Pipavath SN, Raghu G, Schmidt RA, Godwin JD. Imaging of thoracic lymphatic diseases. Am J Roentgenol. 2009;193:1504–13. doi: 10.2214/AJR.09.2532. [DOI] [PubMed] [Google Scholar]
- 10.Du MH, Ye RJ, Sun KK, et al. Diffuse pulmonary lymphangiomatosis: A case report with literature review. Chin Med J (Engl) 2011;124:797–800. [PubMed] [Google Scholar]
- 11.El Hajj L, Mazieres J, Rouquette I, et al. Diagnostic value of bronchoscopy, CT and transbronchial biopsies in diffuse pulmonary lymphangiomatosis: Case report and review of the literature. Clin Radiol. 2005;60:921–5. doi: 10.1016/j.crad.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 12.Takemura T, Watanabe M, Takagi K, Tanaka S, Aida S. Thoracoscopic resection of a solitary pulmonary lymphangioma: Report of a case. Surg Today. 1995;25:651–3. doi: 10.1007/BF00311443. [DOI] [PubMed] [Google Scholar]
- 13.Browse NL, Allen DR, Wilson NM. Management of chylothorax. Br J Surg. 1997;84:1711–6. [PubMed] [Google Scholar]
- 14.Rostom AY. Treatment of thoracic lymphangiomatosis. Arch Dis Child. 2000;83:138–9. doi: 10.1136/adc.83.2.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blei F. Lymphangiomatosis: clinical overview. Lymphat Res Biol. 2011;9:185–90. doi: 10.1089/lrb.2011.0020. [DOI] [PubMed] [Google Scholar]
- 16.Ozeki M, Fukao T, Kondo N. Propranolol for intractable diffuse lymphangiomatosis. N Engl J Med. 2011;364:1380–2. doi: 10.1056/NEJMc1013217. [DOI] [PubMed] [Google Scholar]
- 17.Kinnier CV, Eu JP, Davis RD, Howell DN, Sheets J, Palmer SM. Successful bilateral lung transplantation for lymphangiomatosis. Am J Transplant. 2008;8:1946–50. doi: 10.1111/j.1600-6143.2008.02340.x. [DOI] [PMC free article] [PubMed] [Google Scholar]